

Abstract
Activins regulate bone formation by controlling osteoclasts and osteoblasts. We investigated Activin‐A mechanism of action on human osteoblast mineralization, RNA and microRNA (miRNA) expression profile. A single 2‐day treatment of Activin‐A at Day 5 of osteoblast differentiation significantly reduced matrix mineralization. Activin A‐treated osteoblasts responded with transient change in gene expression, in a 2‐wave‐fashion. The 38 genes differentially regulated during the first wave (within 8 hr after Activin A start) were involved in transcription regulation. In the second wave (1–2 days after Activin A start), 65 genes were differentially regulated and related to extracellular matrix. Differentially expressed genes in both waves were associated to transforming growth factor beta signaling. We identified which microRNAs modulating osteoblast differentiation were regulated by Activin‐A. In summary, 2‐day treatment with Activin‐A in premineralization period of osteoblast cultures influenced miRNAs, gene transcription, and reduced matrix mineralization. Modulation of Activin A signaling might be useful to control bone quality for therapeutic purposes.
Keywords: Activin‐A, bone, extracellular matrix, mineralization, osteoblasts
A single 2‐day treatment of Activin‐A in the premineralization period of osteoblast cultures significantly reduced extracellular matrix mineralization 5–7 days later.
Activin‐A treatment induced a transient change in osteoblast gene expression in a 2‐wave‐fashion over time. Differentially regulated genes were involved in transcription regulation, extracellular matrix structure, and associated to transforming growth factor beta signaling. Activin‐A modulated osteoblast microRNA profiles.

1. INTRODUCTION
Activins belong to the transforming growth factor‐β (TGF‐β) superfamily and are formed by dimerization of two inhibin β subunits (Massague, 1998; Vale et al., 2004). Activin‐A binds to transmembrane serine/threonine kinase receptor type 2 (ACVR2A and ACVR2B), and leads to the recruitment and activation of receptor type 1B (ACVR1B=ALK4). The activated receptor induces a cytoplasmic signal transduction involving SMAD2/3, eventually recruiting the common mediator SMAD4. Only this phosphorylated complex (SMAD2/3/4) can then translocate into the nucleus and regulate target gene transcription (Chen et al., 2006; Derynck, 1998). The Activin‐A signaling cascade modulates several biological functions, such as cell proliferation, differentiation, and apoptosis (Chen et al., 2006; Chen, Lui, Lin, Lee, & Ying, 2002). Activin‐A signaling is modulated by several regulatory proteins, such as Activin receptors interacting proteins (ARIPs), and eventually shut off by SMAD ubiquitin regulatory factors (Smurf1 and Smurf2), that interact with inhibitory SMADs (SMAD6 and SMAD7), and mediate the ubiquitin‐dependent degradation of the receptors (Inoue & Imamura, 2008). SMAD2/3 belongs also to TGF‐β‐signaling, whereas bone morphogenic protein (BMP) signaling involves SMAD1,5,8 but it is transduced by receptor type 2, highlighting a crosstalk between these pathways (Harrison et al., 2004; Tsuchida et al., 2009). In addition, Activin‐A also signals through SMAD‐independent pathways, such as p38 mitogen‐activated protein kinase (MAPK), extracellular signal‐regulated kinase (ERK1/2), and Jun N‐terminal kinase (JNK) pathways, increasing the complexity of this intracellular signal (Chen et al., 2006; de Guise et al., 2006). Endogenous inhibitors such as Follistatin (FST), inhibins and β‐glycan antagonize Activin‐A signaling (Harrison, Gray, Vale, & Robertson, 2005).
The role of activins and inhibins in bone formation and metabolism has been extensively studied in the last years. They regulate skeletal metabolism, by acting on activins, TGF‐βs and BMPs in bone (Nicks, Perrien, Akel, Suva, & Gaddy, 2009; Perrien et al., 2006). However, the role of activins in bone metabolism is still not fully clear. Activin‐A has been detected in human and bovine bone matrix (Eijken et al., 2007; Nicks et al., 2009; Ogawa et al., 1992). It has been shown to be secreted by both osteoblasts and bone marrow cultures during osteoblastogenesis, and during bone matrix resorption by osteoclasts (Eijken et al., 2007; Funaba et al., 1996; Gaddy‐Kurten, Coker, Abe, Jilka, & Manolagas, 2002). Activin‐A has been shown to modulate bone‐cell behavior and having a pro‐osteoclastogenic effect in vitro (Fuller, Bayley, & Chambers, 2000; Gaddy‐Kurten et al., 2002; Nicks et al., 2009; Sakai, Eto, Ohtsuka, Hirafuji, & Shinoda, 1993). Despite this, its effect on osteoblasts is still controversial. Several studies report Activin‐A to promote osteogenic differentiation in vitro, and bone formation and fracture healing in in vivo systems (Gaddy‐Kurten et al., 2002; Ogawa et al., 1992; Sakai et al., 2000; Sakai, Miwa, & Eto, 1999). However, we and others have shown the inhibitory effect of Activin‐A on osteoblast as well as vascular smooth muscle cells‐mediated mineralization in vitro (Eijken et al., 2007; Hashimoto et al., 1992; Ikenoue, Jingushi, Urabe, Okazaki, & Iwamoto, 1999). These findings were also supported by in vivo studies, in which increased bone mass has been observed after blocking Activin‐A in murine and primate models (Lotinun et al., 2010; Pearsall et al., 2008). These controversial findings might be related to differences in cell‐culture system or in the species that were used (Ikenoue et al., 1999; Nicks et al., 2009).
We have shown that Activin‐A affected the expression of extracellular matrix (ECM)‐related genes, influencing the ECM maturation phase. This resulted in an altered ECM protein composition that was unable to mineralize. This effect was stronger when Activin‐A was present during the last days before the onset of extracellular matrix mineralization (Alves, Eijken, Bezstarosti, Demmers, & van Leeuwen, 2013; Eijken et al., 2007). In addition, Activin‐A impaired matrix vesicle secretion at the onset of extracellular matrix mineralization (Alves et al., 2013). This highlights the importance of Activin‐A signaling in bone metabolism, but despite this, the molecular processes underlying Activin A‐driven inhibition of osteogenic differentiation and mineralization are still unclear.
The aim of the present study was to investigate the impact of a temporary 2‐day treatment of Activin‐A on osteoblast extracellular matrix mineralization and to unravel the molecular mechanisms of Activin‐A signaling during differentiation of human osteoblasts.
2. MATERIALS AND METHODS
2.1. Osteoblast cultures
Human SV‐HFO cells (Simian virus 40‐immortalized osteoblast precursors) were cultured (1 × 104 vital cells/cm2) in α‐MEM (pH 7.5, phenol‐red free; GIBCO, Paisley, UK) supplemented with streptomycin/penicillin, 1.8 mM CaCl2 (Sigma, St. Louis, MO), HEPES, and 2% charcoal‐treated and heat inactivated fetal bovine serum (GIBCO). After 2 days, medium was supplemented with dexamethasone (100 nM) and β‐glycerophosphate (10 mM; Sigma, St. Louis, MO) to induce osteogenic differentiation. Activin‐A (50 ng/ml; R&D System, Minneapolis, MN) was added at Day 5 of osteogenic differentiation and removed after 2 days, keeping cultures in osteogenic medium. Medium was replaced every 2–3 days.
To assess the specificity of the Activin‐A signaling, osteoblasts were treated with a SMAD inhibitor (SB‐505124; Sigma‐Aldrich, St. Louis, MO). SB‐505124 selectively inhibits ALK4 kinase activity, repressing Activin A‐ and TGF‐β‐induced SMAD2 and SMAD3 signaling (Harrison et al., 2005). SB‐505124 (0.125 µM in dimethyl sulfoxide) was added 30 min before Activin‐A, and removed at the same time. Osteoblasts treated only with SB‐505124 were used as control.
2.2. Analysis of osteogenic differentiation and mineralization
Alkaline phosphatase (ALP) activity and extracellular matrix mineralization were measured in cell extracts of Activin‐A treated and untreated osteoblasts at late stages of culture (Day 10, 12, and 14, see Figure 1 a), as previously described (Eijken et al., 2007).
Figure 1 .
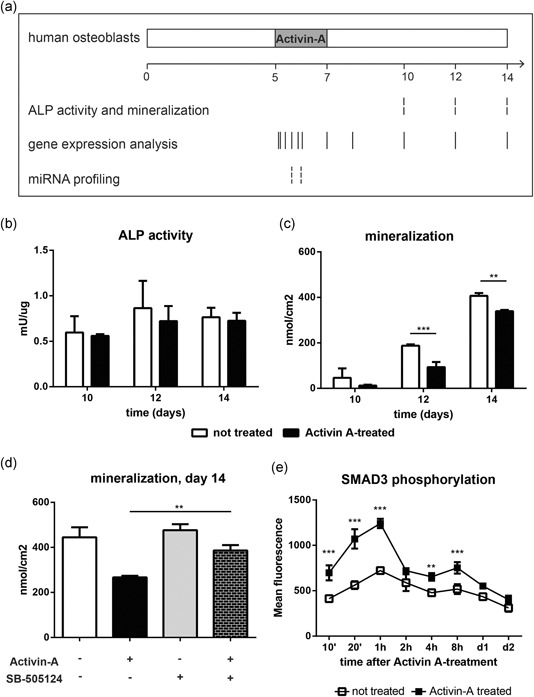
A single 2‐day pulse of Activin‐A reduced osteoblast mineralization. (a) Schematic overview of culture conditions. Human SV‐HFO cells were treated with Activin‐A from Day 5 to Day 7 of osteogenic differentiation. ALP activity, mineralization, gene expression, and miRNA profile were checked at the indicated time points. (b) ALP activity of Activin A‐treated and untreated osteoblasts at Day 10, 12, and 14 of culture. ALP activity was corrected for protein content at each time point. (c) Calcium deposition by Activin A‐treated and untreated osteoblasts at Day 10, 12, and 14 of culture. (d) Calcium deposition at Day 14 of culture, in cell extracts of untreated osteoblasts, osteoblasts treated with Activin‐A, SMAD inhibitor (SB‐505124), and Activin‐A and SMAD inhibitor. Bars indicate mean±SD. (**p<.01; ***p<.001). (e) Phosphorylation of SMAD3 followed by flow cytometry at the indicated time points, in Activin‐A treated and untreated osteoblasts. Values: mean±SD. (**p<.01; ***p<.001 relative to untreated cells). ALP, alkaline phosphatase; miRNA, microRNA; SD, standard deviation
2.3. Phospho‐flow cytometry of SMAD3 phosphorylation
Human SV‐HFO cells were seeded in a density of 4,210 vital cells/cm2 and cultured as described above, and SMAD3 phosphorylation was measured by flow cytometry. Samples were collected from Activin A‐treated and untreated osteoblasts, at 10 min, 20 min, 1 hr, 2 hr, 4 hr, 8 hr, 24 hr, and 48 hr after the start of Activin‐A treatment. Cell extracts were fixed with 4% paraformaldehyde, and permeabilized with 90% ice‐cold methanol. Samples were incubated with anti‐SMAD3 antibody (anti‐SMAD3, pS423 + S425; clone ab52903, Abcam, Cambridge, UK; secondary antibody: anti rabbit IgG, Alexa Fluor® 488 conjugated, #4412, Cell Signaling Technology) and SMAD3 phosphorylation was followed over time by flow cytometry (Becton Dickinson FACS‐Canto and DIVA Flow Cytometry System [BD Bioscience]).
2.4. Illumina gene chip‐based expression
Illumina HumanHT‐12 v3 BeadChip (Illumina, Inc.) human whole‐genome expression arrays were used to analyze gene expression of Activin A‐treated and untreated osteoblasts. RNA (100 ng) was collected from three biological replicates for each condition (at 20 min, 1 hr, 2 hr, 4 hr, 8 hr, 1 day, 2 days, 3 days, 5 days, 7 days, and 9 days after starting of Activin A treatment). RNA integrity was checked by RNA 6000 Nano assay on a 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA). RNA was amplified by Illumina TotalPrep RNA amplification kit (Ambion, Austin, TX), according to manufacturer's instruction. Briefly, single stranded complementary DNA (cDNA) was generated by using T7oligo (dT) primer, then followed by second strand synthesis to generate double stranded cDNA. Biotin‐labeled cRNA was synthesized by in vitro transcription using T7 RNA polymerase and column purified. cRNA quality was assessed on a Bioanalyzer and its concentration by NanoDrop (Thermo Fisher Scientific). A total of 750 ng of cRNA was hybridized for each array and detected using standard Illumina protocol with Streptavidin‐Cy3 (GE). Slides were scanned on iScan and analyzed by GenomeStudio v2010.1 (both from Illumina, Inc.)
2.5. Microarray analysis
Raw gene expression data was background subtracted using Genome Studio v2010 (gene expression module 1.6), and further processed using the Bioconductor R2.10 lumi package (Du, Kibbe, & Lin, 2008). Data was transformed by variance stabilization and quantile normalized.
Probes that were present in at least three samples (Illumina detection p< .01) were considered to be expressed and further analyzed. Differentially expressed probes were identified using the Bioconductor package “limma” (Smyth, 2004) with adjusted p value (q‐value) to reduce false discovery rate. Differentially expressed probes in Activin A‐treated samples relative to untreated samples at the same time point (q< .01) were considered. Selected genes were further analyzed for enrichment of gene ontology (GO) terms using Database for Annotation, Visualization and Integrated Discovery (DAVID) Bioinformatics Resource v6.7 (Huang da, Sherman, & Lempicki, 2009), against the whole human genome as background, and by QIAGEN's Ingenuity® Pathway Analysis (IPA®, QIAGEN Redwood City http://www.qiagen.com/ingenuity) using the Expression Analysis tool (Canonical Pathways, Upstream Analysis and Disease and Function).
2.6. Quantification of mRNA expression
Total RNA isolation, cDNA synthesis and quantitative polymerase chain reaction (qPCR) were performed as previously described (Eijken et al., 2007). Primer sequences (Sigma‐Aldrich) are listed in Table S1.
2.7. microRNA array and analysis
Total RNA and small RNA populations were isolated from Activin A‐treated and untreated osteoblasts, at 4 hr and 1 day after the start of Activin‐A pulse. RNA (250 ng) was collected from two biological replicates of each condition. Illumina microRNA Expression Profiling Assay for BeadChip array (Illumina, Inc.) was used to analyze microRNA (miRNA) profiling of Activin A‐treated and untreated osteoblasts, following manufacturer's instructions.
Probes that were detected above background (detection p < .01), in at least one time point, in both biological replicates and annotated to a known miRNA (miRbase http://www.mirbase.org; Griffiths‐Jones, Grocock, van Dongen, Bateman, & Enright, 2006), were considered for further analysis. Probes that were detected only in Activin A‐treated or untreated osteoblasts and probes more than two‐fold upregulated or downregulated were further analyzed (Table 1). miRNAs were analyzed by using DIANA miRpath v3 (Vlachos et al., 2015), IPA and Targetscan Release 7.1 (Agarwal, Bell, Nam, & Bartel, 2015) as prediction tools. Human species and predicted interactions derived from TarBase v7.0 were used in DIANA miRpath v3. In Targetscan, all miRNAs were queried against human genome as background, if possible, otherwise mouse, and only predicted targets with conserved sites were considered.
Table 1.
miRNAs modulated by Activin‐A in osteoblasts, 4 hr and 1 day after the start of Activin‐A treatment. Intensities are indicated as AVG signal
| Mature miRNAs | A‐OBs (AVG signal) | NT‐OBs (AVG signal) | Mature miRNAs | A‐OBs (AVG signal) | NT‐OBs (AVG signal) | |
|---|---|---|---|---|---|---|
| 4 hr | >2‐Fold upregulated in A‐OBs | >2‐Fold downregulated in A‐OBs | ||||
| miR‐590‐3p | 790.81 | 351.31 | miR‐486‐3p | 1,788.62 | 4,489.87 | |
| miR‐142‐3p | 386.24 | 178.37 | miR‐19a | 1,209.74 | 2,925.18 | |
| miR‐18b | 609.81 | 300.37 | miR‐33a | 122.18 | 1,942.43 | |
| miR‐32 | 171.06 | 439.93 | ||||
| miR‐150 | 142.06 | 358.49 | ||||
| Detected only in A‐OBs | Not detected in A‐OBs | |||||
| miR‐432* | 106.87 | miR‐573 | 84.24 | |||
| miR‐337:9.1 | 88.12 | miR‐483‐5p | 76.06 | |||
| miR‐33b* | 84.37 | miR‐1263 | 75.37 | |||
| miR‐96 | 77.87 | miR‐371‐3p | 70.37 | |||
| miR‐592 | 76.87 | miR‐186* | 69.06 | |||
| miR‐744* | 68.56 | miR‐765 | 66.74 | |||
| miR‐338‐3p* | 67.99 | miR‐198 | 65.06 | |||
| miR‐517a, b | 61.68 | miR‐563 | 63.81 | |||
| miR‐20b | 60.93 | miR‐10b | 62.18 | |||
| miR‐645 | 59.31 | miR‐891a | 59.18 | |||
| miR‐580 | 58.93 | |||||
| miR‐744* | 58.68 | |||||
| 1 day | >2‐Fold upregulated in A‐OBs | >2‐Fold downregulated in A‐OBs | ||||
| miR‐486‐3p | 4,929.56 | 2,055.87 | miR‐193a‐3p | 963.43 | 2,079.74 | |
| miR‐100* | 993.68 | 453.87 | miR‐590‐3p | 126.06 | 432.18 | |
| miR‐150 | 390.12 | 184.31 | ||||
| miR‐24‐1* | 258.74 | 122.43 | ||||
| Detected only in A‐OBs | Not detected in A‐OBs | |||||
| miR‐18b | 203.18 | miR‐220b | 94.87 | |||
| miR‐217 | 107.74 | let‐7f‐2* | 74.37 | |||
| miR‐548o | 83.43 | miR‐431* | 69.99 | |||
| miR‐589* | 79.99 | miR‐610 | 67.62 | |||
| miR‐548f | 75.74 | miR‐518e | 66.06 | |||
| miR‐181d | 71.93 | miR‐632 | 65.74 | |||
| miR‐218‐2 | 66.56 | miR‐520d‐5p | 63.06 | |||
| miR‐1208 | 64.37 | |||||
| miR‐921 | 64.31 | |||||
| miR‐432* | 63.56 | |||||
| miR‐563 | 61.74 | |||||
| miR‐559 | 57.43 | |||||
| miR‐645 | 55.68 | |||||
| miR‐517a,b | 54.31 | |||||
| miR‐1236 | 54.06 | |||||
Abbreviations: A‐OBs, Activin‐A treated osteoblasts; miRNAs, microRNAs; NT‐OBs, non‐treated osteoblasts.
2.8. Statistical analysis
Data for biochemical analysis were representative of independent experiments. All values are presented as mean ± (SD) of technical replicates. Significance was calculated by two‐way analysis of variance (ANOVA), followed by Bonferroni Post Hoc test, otherwise indicated elsewhere.
3. RESULTS
3.1. A single pulse of Activin‐A is sufficient to reduce SV‐HFO osteoblast mineralization
Previously, we have shown that Activin‐A reduced matrix mineralization of osteoblasts that were continuously treated with Activin‐A and that the inhibition of mineralization was most effective when Activin‐A was present in the final 7 days in the premineralization period (considered up to Day 10 of culture; Eijken et al., 2007). Here we investigated if a short‐term incubation with Activin‐A from Day 5 to 7 of culture in the premineralization period resulted in the same effect. Human SV‐HFO osteoblasts were treated for 2 days with Activin‐A starting at Day 5, and ALP activity and extracellular matrix mineralization were analyzed at later stages, as represented in Figure 1 a. ALP activity was not affected by Activin‐A (Figure 1 b). Nevertheless, a single 2‐day‐treatment with Activin‐A was able to reduce the extracellular matrix mineralization at later stages of culture. Calcium deposition at Day 12 of culture was two‐fold lower in Activin A‐treated osteoblasts than untreated cells, as shown in Figure 1 c (p = .0003).
To assess the specificity of Activin A signaling, osteoblasts were treated with the SMAD‐signaling inhibitor SB‐505124. SB‐505124 counteracted the effect of Activin‐A on mineralization. Matrix mineralization by SV‐HFO osteoblasts treated with both Activin‐A and SMAD inhibitor was significantly higher than in the cells treated with Activin‐A only (p = .004) and at comparable levels as that of untreated osteoblasts (Figure 1 d). SMAD inhibitor did not affect differentiation of the cells, as SV‐HFO osteoblasts cultured in the presence of SMAD inhibitor mineralize at similar extend than the untreated cells.
3.2. SMAD3 phosphorylation reached the maximum 1 hr after Activin‐A treatment
As SMAD2/3 are downstream signal transducers of Activin‐A, we analyzed SMAD3 phosphorylation by flow cytometry. Phosphorylation of SMAD3 was significantly higher in Activin A‐treated SV‐HFO osteoblasts than in untreated cells, as shown in Figure 1 e. The Activin‐A pulse increased SMAD3 phosphorylation, with a peak after 1 hr of treatment, which was almost 2‐fold higher in Activin A‐treated osteoblasts than in untreated cells (p < .001).
3.3. Activin‐A treatment induced changes in gene expression in a two‐wave fashion
As a 2‐day treatment of Activin‐A at early stages of osteoblast differentiation was able to reduce extracellular matrix mineralization, we investigated the molecular mechanism by which Activin‐A would affect osteoblast gene expression. We performed comparative gene expression profiling of Activin‐A treated and untreated osteoblasts at various time points using Illumina Human HT‐12 v3 BeadChip array. Activin‐A treatment induced a transient change in SV‐HFO osteoblast gene expression in a two‐wave fashion over time, as shown in Figure 2a. The first wave consisted of 38 differentially regulated genes (q< .01) and occurred from 1 hr till 8 hr after the start of Activin‐A treatment (Table S2). The second wave of differentially regulated genes occurred between 1 and 2 days after the Activin‐A pulse (Table S3). Moreover, differentially regulated genes were detected at each time point until Day 2, but once Activin‐A was replaced with control growth media, no gene expression differences were observed. The TGF‐β signaling pathway was within the top 10 most enriched canonical pathways of the differentially regulated genes in both waves (Tables S4 and S5). Moreover, TGF‐β was within the most enriched upstream regulators in both first and second wave, as analyzed by IPA and shown in Figure 2b,c by the predicted target genes (Table S6).
Figure 2.
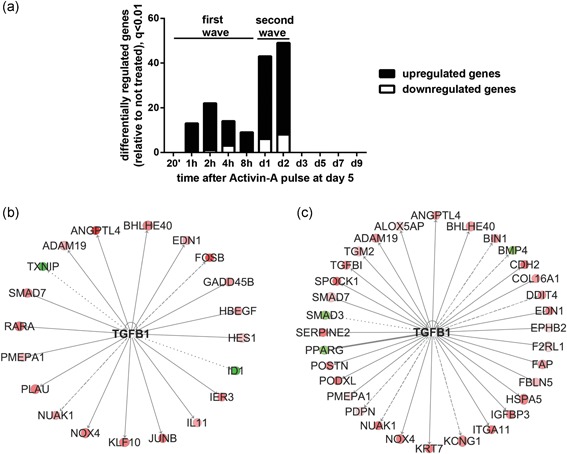
Activin‐A treatment induced a transient change in osteoblast gene expression, in a two‐wave‐fashion over time. (a) Number of significantly differentially regulated genes in Activin A‐treated osteoblasts compared to untreated cells at each time point (q< .01). (b) TGF‐β is predicted to target 20 genes within those differentially regulated in the first wave (IPA analysis). (c) TGFβ is the most enriched upstream regulator, targeting 32 genes within those differentially regulated in the second wave of gene expression changes. In (b) and (c) shades of red: upregulated genes by Activin‐A (log ratio of Activin A‐treated/untreated cells). Shades of green: downregulated genes. Solid line: predicted activation; thick solid line: predicted inhibition; dashed line: not predicted effect; dotted line: findings inconsistent with the state of downstream molecule by IPA. IPA, Ingenuity® Pathway Analysis; TGF‐β, transforming growth factor‐β
GO analysis of the 38 genes that are differentially regulated in the first wave showed that these changes were related to transcription regulation (GO:0045892) and vasculature development (GO:0001944; p < .05; Figure 3a). In addition, these 38 genes were analyzed using IPA (Figure 3b and Table S7). “Binding of DNA,” including genes such as SMAD7, JUNB, and FOSB, was among the most significantly enriched terms (p = 6.49 × 10−9; Figure 3b). Most of these genes were upregulated by Activin‐A. Furthermore “Vasculogenesis” (SMAD7, JUNB, and ANGPTL4) was identified to be significantly enriched (p = 5.37 × 10−9) among the regulated genes. Interestingly, “differentiation of connective tissue cells,” including genes such as SMAD7, KLF10, and JUNB, was significantly enriched (p = 9.06 × 10−8). As SMAD7, JUNB, and KLF10 were involved in most of the identified pathways and have SMAD‐responsive elements, their regulation was further confirmed by qPCR (Figure 3c).
Figure 3.
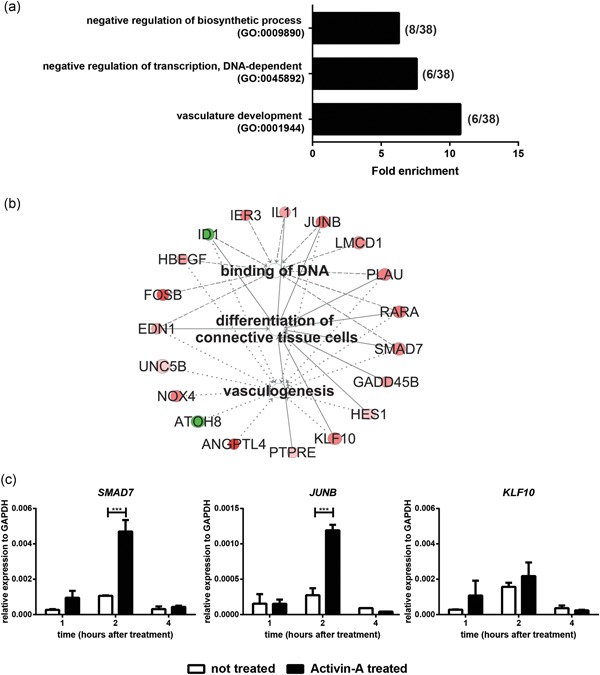
Activin‐A pulse induced changes in gene transcription. (a) GO analysis of the 38 genes of the first wave of gene expression changes (1–8 hr after Activin A treatment start), that were differentially regulated in Activin A‐treated cells compared to untreated ones. Only significantly enriched GO terms are shown (Benjamini p < .05). In brackets: number of genes for each GO term. (b) Pathway analysis of these 38 genes: vasculogenesis, binding of DNA, and differentiation of connective tissue cells were significantly enriched. Shades of red: upregulated genes by Activin‐A (log ratio of Activin A‐treated/untreated cells). Shades of green: downregulated genes. (c) qPCR analysis of selected transcription factors in Activin‐A treated and untreated osteoblasts at the indicated time points. Bars indicate mean ± SD. GO, Gene Ontology; SD, standard deviation. (***p < .001)
In the second wave (1–2 days after Activin‐A start), 65 genes were differentially regulated (q< .01; Table S3). These genes are involved in vasculature development (GO:0001944), ECM structure (GO:0031012), cell migration (GO:0030334), and adhesion (GO:0007155; Figure 4a). Ingenuity Pathway analysis of these 65 genes confirmed our findings by DAVID, as shown in Figure 4b and Table S8. The functional categories Vasculogenesis (p = 8.85 × 10−8) including genes such as IGFBP3, FBLN5, and SMAD3, but also Adhesion of connective tissue cells (p = 2.47 × 10−5) (POSTN, TGFBI, and BMP4) and Differentiation of osteoblasts (p = 7.56 × 10−6) (SMAD3, PPARG, POSTN, TGFBI, and CTHRC1), were significantly enriched. ECM‐related genes involved in these pathways such as POSTN, TGFB1, upregulated by Activin‐A, and the downregulated BMP4 were further confirmed by qPCR (Figure 4c).
Figure 4.
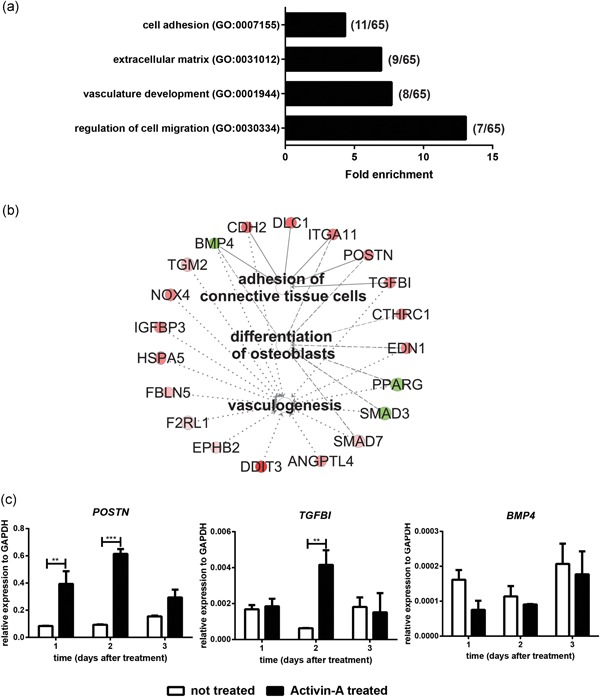
Activin A pulse induced changes in ECM composition. (a) GO analysis of the 65 genes that were differentially regulated in the second wave of gene expression (1 and 2 days after Activin A pulse start) in Activin A‐treated cells. Only significantly enriched GO terms are shown (Benjamini p < .05). The number of genes for each enriched GO term are reported in brackets. (b) Significantly enriched pathways of the IPA analysis of the 65 genes of second wave. Shades of red: upregulated genes by Activin‐A (log ratio of Activin A‐treated/untreated cells). Shades of green: downregulated genes. (c) qPCR analysis of selected ECM proteins in Activin A‐treated and untreated osteoblasts at 1, 2, and 3 days after the start of Activin‐A treatment. Bars indicate mean ± SD. ECM, extracellular matrix; IPA, Ingenuity® Pathway Analysis; GO, Gene Ontology; qPCR, quantitative polymerase chain reaction; SD, standard deviation. (**p < .01; ***p < .001)
3.4. Activin‐A treatment modified the microRNA profile of SV‐HFO osteoblasts
In addition to the mRNA changes that occurred upon Activin‐A treatment we analyzed the microRNA gene expression profile in SV‐HFO osteoblasts, 4 hr and 1 day after addition of Activin‐A at Day 5 of culture. Assuming miRNAs as “upstream” regulators of mRNA expression, and focusing on the second wave of gene expression changes (Day 1 and Day 2 after Activin‐A addition), we chose 4 hr and Day 1 respectively to assess the miRNA profiles. A detailed flowchart of the miRNA profile analysis is presented in Figure S1.
Four hours after starting the Activin‐A pulse, 12 of the 561 miRNAs were uniquely detected in the Activin A‐treated osteoblasts (Figure 5a). Three miRNAs (miR‐18b, miR‐142‐3p, and miR‐590‐3p) were > 2‐fold upregulated in Activin A‐treated osteoblasts compared with the untreated cells (Figure 5b; Table 1). Furthermore, Activin‐A decreased the expression of some miRNAs. Of these, 10 miRNAs were absent in the Activin A‐treated osteoblasts, and five miRNAs (miR‐19a, miR‐32, miR‐33a, miR‐150, and miR‐486‐3p) were > 2‐fold downregulated in these cells compared with untreated cells (Figure 5b; Table 1). The miRNAs that were uniquely detected in each condition displayed very low expression. We analyzed in which pathways the targets of these miRNAs were involved using Diana pathway analysis (Vlachos et al., 2015). Within the enriched pathways, we focused on TGF‐β signaling, as it is known to guide osteoblast differentiation and as shown in Figure 2 gene expression analyses identified this signaling pathway. TGF‐β signaling was significantly enriched (p = 1.54 × 10−6), as 9 of the 15 miRNAs upregulated by Activin‐A targeted 35 genes involved in TGF‐β signaling. This is supported by the observation that also 29 mRNAs targeted by eight miRNAs downregulated by Activin‐A, were involved in the TGF‐β pathway (Table 2). Thus, target genes that are involved in TGF‐β signaling were targets of miRNAs that were both upregulated and downregulated by Activin‐A within 4 hr of treatment.
Figure 5.
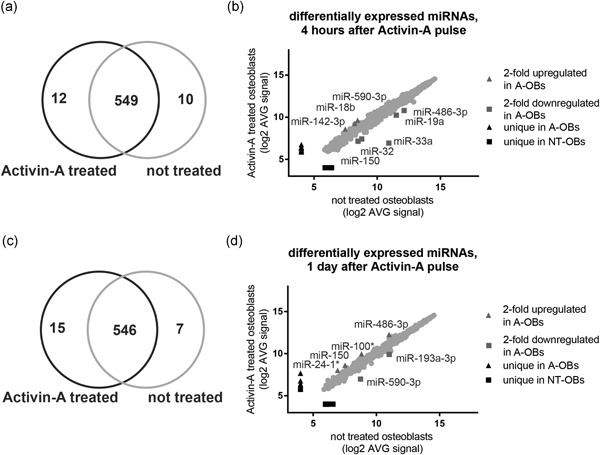
Osteoblast miRNA expression profiles were modulated by Activin‐A treatment. (a) 12 miRNAs were uniquely detected in Activin A‐treated osteoblasts, 10 miRNAs uniquely in the untreated cells, and 549 were detected in both conditions, 4 hr after the start of Activin‐A treatment. (b) miRNAs uniquely detected in Activin A‐treated osteoblasts, untreated osteoblasts, and with more than 2‐fold difference in detection, 4 hr after the start of Activin‐A treatment. (c) One day after the start of Activin‐A treatment, 15 miRNAs were uniquely detected in Activin A‐treated osteoblasts, 7 miRNAs uniquely in the untreated ones, and 546 in both conditions. (d) Number of miRNAs uniquely detected in Activin A‐treated osteoblasts, untreated, and with more than 2‐fold difference in detection, 1 day after the start of Activin‐A. Only probes detected above background and annotated to known miRNAs are shown (p < .01). A‐OBs, Activin A‐treated osteoblasts; miRNAs, microRNAs; NT‐OBs, not treated osteoblasts
Table 2.
Activin‐A modulated miRNAs that are predicted to target genes involved in TGF‐β signaling pathway (DIANA miRpath v3; p < .01)
| Time after Activin‐A pulse | KEGG pathway | p value | # Involved miRNAs | # Involved genes | |
|---|---|---|---|---|---|
| miRNAs unique and upregulated in A‐OBs | 4 hr | TGFβ signaling | 1.54 × 10−5 | 9(/15) | 35 |
| miRNAs absent and downregulated in A‐OBs | 5.30 × 10−5 | 8(/15) | 29 | ||
| miRNAs unique and upregulated in A‐OBs | 1 day | 1.15 × 10−4 | 8(/19) | 17 | |
| miRNAs absent and downregulated in A‐OBs | 2.70 × 10−2 | 5(/9) | 19 |
Abbreviations: A‐OBs, Activin‐A treated osteoblasts; KEGG, Kyoto Encyclopedia of Genes and Genomes; miRNAs, microRNAs; TGF‐β, transforming growth factor‐β.
One day after the start of Activin‐A treatment, 15 miRNAs were detected being unique in Activin A‐treated osteoblasts, and 546 were shared with the untreated cells (Figure 5c). Of these 546, two were more than two‐fold downregulated in Activin A‐treated osteoblasts (Figure 5d; Table 1). In line with the 4‐hr‐treatment data, TGF‐β signaling was significantly enriched (p = .0001), as Activin‐A upregulated 8 miRNAs that targeted 17 genes involved in this pathway, as well as downregulating 5 miRNAs that target 19 genes related to this signaling pathway (Table 2). Overall, within 1 day of treatment, Activin‐A modulated miRNAs that are predicted to target genes involved in TGFβ signaling.
Next, we combined the data of miRNA and mRNA profiling, hypothesizing that miRNAs that were upregulated by Activin A treatment (4 hr and 1 day), would target mRNAs that were downregulated after 1 and 2 days in the Activin A‐treated osteoblasts (for analysis scheme see Figure S1). As Activin‐A also downregulated some miRNAs, we checked if they could target genes found upregulated in Activin A‐treated osteoblasts.
We considered the genes differentially regulated in the second wave of gene expression, and that were involved in the top 10 most enriched canonical pathways (Table S5). Within these pathways, we analyzed whether the genes that were downregulated by Activin‐A were also predicted targets of miRNAs upregulated by Activin‐A (Table S9). Indeed, we identified two miRNAs (miR‐142‐3p and miR‐432*) that were upregulated by Activin‐A and were predicted to target the downregulated gene BMP4 (Table 3). In addition, seven miRNAs (miR‐24‐1*, miR‐181d, miR‐548f, miR‐559, miR‐589*, miR‐645, and miR‐1236) were identified that downregulated the expression of SMAD3 (Table 3).
Table 3.
Genes downregulated by Activin‐A in the second wave of gene expression changes (Day 1 and Day 2) and targets of the indicated miRNAs upregulated by Activin‐A (IPA, Targetscan analysis)
| Downregulated genes | Target of miRNAs 4 hr | Target of miRNAs 1 day |
|---|---|---|
| BMP4 | miR‐142‐3p | miR‐432* |
| miR‐432* | ||
| PPARG | miR‐590‐3p | miR‐559 |
| SMAD3 | miR‐24‐1* | |
| miR‐181d | ||
| miR‐548f | ||
| miR‐559 | ||
| miR‐589* | ||
| miR‐645 | ||
| miR‐1236 |
Abbreviation: miRNAs, microRNAs.
Similarly, we investigated if the genes that were upregulated by Activin‐A in the second wave of gene expression, were also predicted targets of miRNAs downregulated by Activin‐A (Table S10). Interestingly, Activin‐A downregulated miRNAs that targeted genes being upregulated in Activin A‐treated osteoblasts (Table 4). For example, miR‐32, miR‐486‐3p, miR‐573, and miR‐1263 were downregulated by Activin‐A, and are predicted to target SMAD7, which was upregulated by Activin A treatment.
Table 4.
Genes upregulated by Activin‐A (Day 1 and Day 2) and predicted as target of the indicated miRNAs that are downregulated by Activin‐A (IPA, Targetscan analysis)
| Upregulated genes | Target of miRNAs 4 hr | Target of miRNAs 1 day |
|---|---|---|
| DDIT4 | miR‐590‐3p | |
| EDN1 | miR‐19a | miR‐590‐3p |
| miR‐33a | ||
| miR‐486‐3p | ||
| miR‐765 | ||
| HSPA5 | miR‐590‐3p | |
| IGFBP3 | miR‐371b‐3p | |
| miR‐563 | ||
| NOX4 | miR‐10b | miR‐590‐3p |
| miR‐32 | ||
| miR‐33a | ||
| PMEPA1 | miR‐10b | |
| miR‐23 | ||
| miR‐186* | ||
| miR‐765 | ||
| SMAD7 | miR‐32 | |
| miR‐486‐3p | ||
| miR‐573 | ||
| miR‐1263 |
Abbreviation: miRNAs, microRNAs.
4. DISCUSSION
In this study, we demonstrated that a short‐term Activin‐A treatment for only 2 days in the SV‐HFO osteoblast differentiation period preceding mineralization inhibits matrix mineralization 5–7 days later. Activin‐A exerts this by affecting mRNA expression in a biphasic manner as well as regulating miRNA expression within these 2 days.
We took advantage of our previous work in which we demonstrated that Activin A‐inhibition of matrix mineralization was most effective when Activin‐A was present in the final 7 days of premineralization period (Eijken et al., 2007). In line with this, in this study we provided evidence that Activin A treatment only between Day 5 and 7 of osteoblast cultures reduced mineralization, highlighting the importance of timing of Activin‐A presence during osteoblast differentiation and bone formation. ALP activity was not affected by Activin‐A at the time points that were analyzed, in contrast with our previous findings (Alves et al., 2013). Possibly, the timing of Activin A treatment should be earlier during differentiation to influence ALP activity. Nevertheless, mineralization was significantly reduced by this Activin‐A treatment regimen, implicating that the effect is independent of changes in ALP activity. This is supported by earlier observations of Eijken et al. (2007), that the Activin‐A treatment leads to a change in ECM composition.
The Activin‐A pulse altered osteoblast gene expression in a biphasic fashion, by modulating genes involved in transcription regulation and ECM structure. Between 1 and 8 hr after the treatment, SMAD‐responsive transcription factors, such as ID1, KLF10, JUNB, and SMAD7 were regulated, highlighting the specificity of Activin A signaling. The inhibitory SMAD7 was upregulated by Activin‐A, in line with our previous findings (Eijken et al., 2007). As SMAD7 is a known inhibitor of BMP‐ and TGFβ‐signaling, and was shown to reduce mouse osteoblast mineralization (Massague, 1998; Yano et al., 2012), this confirms our findings in human osteoblast cultures. SMAD7 was also predicted as target of four miRNAs upregulated by Activin‐A, thus representing an interesting candidate for functional analyses of auto‐regulation by Activin‐A signaling. Unexpectedly, other SMAD‐responsive transcription factors were upregulated by Activin‐A. KLF10 and JUNB, which are known inducers of osteogenic differentiation (Kenner et al., 2004; Long, 2012; Subramaniam et al., 2005), were upregulated in human osteoblasts that were continuously treated with Activin‐A (Eijken et al., 2007). Yet an explanation is unclear, however, it is tempting to speculate that these are part of an intricate regulatory network of genes of which the concerted action eventually leads to the inhibition of mineralization. In line, Activin‐A was shown to act on many different proteins, revealing the complexity of its signaling (Alves et al., 2013), thus maybe explaining these findings.
In the second wave of gene expression changes we detected genes involved in osteoblast differentiation and ECM composition. Interestingly, Activin‐A upregulated matricellular proteins such as POSTN, SPARC/osteonectin (SPOCK1), and growth factors such as TGF‐β, that promote osteoblast adhesion and bone structure (Delany, Kalajzic, Bradshaw, Sage, & Canalis, 2003; Janssens, ten Dijke, Janssens, & Van Hul, 2005; Merle & Garnero, 2012), in line with previous findings showing that Activin‐A induced ECM‐related genes (Alves et al., 2013; Eijken et al., 2007). Activin‐A might stimulate initial stages of osteogenesis, but creating an ECM compartment that fails to induce mineralization at later stages. In line with this, the osteogenic stimulator BMP4 was downregulated by Activin‐A and also predicted as target of miR‐142 and miR‐432*, that were upregulated by Activin‐A. Moreover, Heat shock protein family A member 5 (HSPA5) was upregulated in Activin A‐treated osteoblasts and predicted as target of Activin A modulated miRNAs. HSPA5 is involved in unfolded protein response (UPR) to counteract endoplasmic reticulum (ER) stress. In physiological conditions ER stress is elevated in osteoblasts, and UPR has been related to osteoblast differentiation, playing a role during bone homeostasis and skeletal disorders (Horiuchi, Tohmonda, & Morioka, 2016). However, further studies are needed to investigate the impact of Activin‐A in matrix secretion and its relation to osteoblast management of stress response.
Genes involved in vasculogenesis, such as ANGPTL4, NADPH oxidase 4 (NOX4), and endothelin 1 (EDN1), were upregulated by Activin‐A in both waves of gene expression changes, indicating consistent regulation over time. ANGPTL4 is a target of TGFβ and Hypoxia inducible factor (HIF), and it mediates HIF‐driven bone resorption in the angiogenic‐osteogenic coupling (Knowles, Cleton‐Jansen, Korsching, & Athanasou, 2010). Conversely, it also promotes osteoblast differentiation in fracture repair and stimulates vascular endothelial growth factor expression (Wilson, Wong, Toupadakis, & Yellowley, 2015). Activin‐A is considered as commitment factor for the differentiation of erythroid progenitors (Yu, Shao, Vaughan, Vale, & Yu, 1989). Therefore, the role of Activin‐A in vasculogenesis needs further investigation for future clinical applications in fracture repair or control of metastatic bone diseases, also based on the importance of erythropoietin (EPO) on bone formation (Shiozawa et al., 2010), but also on the HIF‐mediated EPO production by osteoblasts that contributes to erythropoiesis and hematopoietic stem cell expansion (Rankin et al., 2012). Our findings showed that Activin‐A upregulated miRNAs involved in erythropoiesis. For instance, miR‐486‐3p was upregulated in untreated osteoblasts 4 hr after the treatment, but became very abundant in Activin A‐treated osteoblasts 1 day after the treatment. miR‐486‐3p was shown to regulate γ‐globin expression in erythroid cells, thus maybe representing and interesting candidate for Activin‐A involvement in vasculogenesis (Lulli et al., 2013). miR‐24 was shown to directly target ALK4 modulating Activin‐mediated erythropoiesis (Wang et al., 2008), and indeed miR‐24‐1* was upregulated in Activin A‐treated osteoblasts.
Activin‐A was shown to modulate miRNA profile in human prostate cancer cell lines and human embryonic stem cells (hESCs) (Ottley, Nicholson, & Gold, 2016; Tsai et al., 2010). Our study showed that Activin‐A modified also the miRNA profile of SV‐HFO osteoblasts. For instance, 1 day after the start of the treatment, miR‐217 was upregulated by Activin‐A. miR‐217 was also upregulated by Activin‐A in hESCs (Tsai et al., 2010) and it reduced murine osteoblast mineralization by targeting RUNX2 (Zhang et al., 2012), thus representing an important target for Activin A‐mediated inhibition of mineralization. Also other miRNAs, such as miR‐20b, were upregulated by Activin‐A, but were shown to enhance mineralization (He et al., 2010). The number of miRNAs that were altered by Activin‐A in osteoblasts reflects the complexity of Activin‐A signaling. A miRNA profile at even more time points than the ones we selected may help us unraveling Activin‐A mechanism in more detail and describe the Activin A induced intracellular regulatory network.
In summary, we showed that a single two‐day‐pulse of Activin‐A between Day 5 and 7 of SV‐HFO osteoblast differentiation was able to reduce extracellular matrix mineralization 5–7 days later. Activin‐A altered osteoblast gene expression profile in a biphasic fashion, first acting at transcription level, and subsequently altering ECM‐related genes. Moreover, Activin A pulse was able to modify the miRNA profile of SV‐HFO osteoblasts that could be linked to changes in mRNA expression. The results gave further insights into the mechanism by which Activin‐A modulates osteoblast behavior and matrix mineralization. Activin‐A and/or its mRNA and miRNA targets represent potential candidates for stimulation of bone formation and future clinical treatment of bone‐related diseases, to control bone formation and bone quality, but also conditions of ectopic calcification such as atherosclerosis (Eijken et al., 2007).
CONFLICT OF INTERESTS
The authors declare that there are no conflict of interests.
AUTHOR CONTRIBUTIONS
Study design and conduct: M. B., K. D., M. E., J. P., and J. L. Data collection: M. B. and K. D. Data analysis and interpretation: M. B., K. D., B. E., M. E., J. P., and J. L. Drafting manuscript, revising manuscript content and approving final version of manuscript: M. B., J. P., B. E., and J. L.
Supporting information
Supporting information
Supporting information
Supporting information
ACKNOWLEDGMENTS
The authors thank Marijke Koedam and Iris Robbesom for technical assistance, Prof H. Chiba at Fukushima Medical University for kindly providing SV‐HFO cells.
Baroncelli M, Drabek K, Eijken M, van der Eerden BCJ, van de Peppel J, van Leeuwen JP. Two‐day‐treatment of Activin‐A leads to transient change in SV‐HFO osteoblast gene expression and reduction in matrix mineralization. J Cell Physiol. 2020;235:4865–4877. 10.1002/jcp.29365
DATA AVAILABILITY STATEMENT
Data available on request to the authors.
REFERENCES
- Agarwal, V. , Bell, G. W. , Nam, J. W. , & Bartel, D. P. (2015). Predicting effective microRNA target sites in mammalian mRNAs. Elife, 4, e05005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alves, R. D. , Eijken, M. , Bezstarosti, K. , Demmers, J. A. , & van Leeuwen, J. P. (2013). Activin A suppresses osteoblast mineralization capacity by altering extracellular matrix (ECM) composition and impairing matrix vesicle (MV) production. Molecular & Cellular Proteomics, 12, 2890–2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, Y. G. , Lui, H. M. , Lin, S. L. , Lee, J. M. , & Ying, S. Y. (2002). Regulation of cell proliferation, apoptosis, and carcinogenesis by activin. Experimental Biology and Medicine (Maywood), 227, 75–87. [DOI] [PubMed] [Google Scholar]
- Chen, Y. G. , Wang, Q. , Lin, S. L. , Chang, C. D. , Chuang, J. , & Ying, S. Y. (2006). Activin signaling and its role in regulation of cell proliferation, apoptosis, and carcinogenesis. Experimental Biology and Medicine (Maywood), 231, 534–544. [DOI] [PubMed] [Google Scholar]
- Delany, A. M. , Kalajzic, I. , Bradshaw, A. D. , Sage, E. H. , & Canalis, E. (2003). Osteonectin‐null mutation compromises osteoblast formation, maturation, and survival. Endocrinology, 144, 2588–2596. [DOI] [PubMed] [Google Scholar]
- Derynck, R. (1998). SMAD proteins and mammalian anatomy. Nature, 393, 737–739. [DOI] [PubMed] [Google Scholar]
- Du, P. , Kibbe, W. A. , & Lin, S. M. (2008). lumi: A pipeline for processing Illumina microarray. Bioinformatics, 24, 1547–1548. [DOI] [PubMed] [Google Scholar]
- Eijken, M. , Swagemakers, S. , Koedam, M. , Steenbergen, C. , Derkx, P. , Uitterlinden, A. G. , … van Leeuwen, J. P. (2007). The activin A‐follistatin system: Potent regulator of human extracellular matrix mineralization. FASEB Journal, 21, 2949–2960. [DOI] [PubMed] [Google Scholar]
- Fuller, K. , Bayley, K. E. , & Chambers, T. J. (2000). Activin A is an essential cofactor for osteoclast induction. Biochemical and Biophysical Research Communications, 268, 2–7. [DOI] [PubMed] [Google Scholar]
- Funaba, M. , Ogawa, K. , Murata, T. , Fujimura, H. , Murata, E. , Abe, M. , … Torii, K. (1996). Follistatin and activin in bone: Expression and localization during endochondral bone development. Endocrinology, 137, 4250–4259. [DOI] [PubMed] [Google Scholar]
- Gaddy‐Kurten, D. , Coker, J. K. , Abe, E. , Jilka, R. L. , & Manolagas, S. C. (2002). Inhibin suppresses and activin stimulates osteoblastogenesis and osteoclastogenesis in murine bone marrow cultures. Endocrinology, 143, 74–83. [DOI] [PubMed] [Google Scholar]
- Griffiths‐Jones, S. , Grocock, R. J. , van Dongen, S. , Bateman, A. , & Enright, A. J. (2006). miRBase: MicroRNA sequences, targets and gene nomenclature. Nucleic Acids Research, 34, D140–D144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Guise, C. , Lacerte, A. , Rafiei, S. , Reynaud, R. , Roy, M. , Brue, T. , & Lebrun, J. J. (2006). Activin inhibits the human Pit‐1 gene promoter through the p38 kinase pathway in a Smad‐independent manner. Endocrinology, 147, 4351–4362. [DOI] [PubMed] [Google Scholar]
- Harrison, C. A. , Wiater, E. , Gray, P. C. , Greenwald, J. , Choe, S. , & Vale, W. (2004). Modulation of activin and BMP signaling. Molecular and Cellular Endocrinology, 225, 19–24. [DOI] [PubMed] [Google Scholar]
- Harrison, C. A. , Gray, P. C. , Vale, W. W. , & Robertson, D. M. (2005). Antagonists of activin signaling: Mechanisms and potential biological applications. Trends in Endocrinology and Metabolism, 16, 73–78. [DOI] [PubMed] [Google Scholar]
- Hashimoto, M. , Shoda, A. , Inoue, S. , Yamada, R. , Kondo, T. , Sakurai, T. , … Muramatsu, M. (1992). Functional regulation of osteoblastic cells by the interaction of activin‐A with follistatin. Journal of Biological Chemistry, 267, 4999–5004. [PubMed] [Google Scholar]
- He, J. , Zhang, J. F. , Yi, C. , Lv, Q. , Xie, W. D. , Li, J. N. , … Zhang, Y. (2010). miRNA‐mediated functional changes through co‐regulating function related genes. PLOS One, 5, e13558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horiuchi, K. , Tohmonda, T. , & Morioka, H. (2016). The unfolded protein response in skeletal development and homeostasis. Cellular and Molecular Life Science, 73, 2851–2869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Huang , W. , Sherman, B. T. , & Lempicki., R. A. (2009). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature Protocols, 4, 44–57. [DOI] [PubMed] [Google Scholar]
- Ikenoue, T. , Jingushi, S. , Urabe, K. , Okazaki, K. , & Iwamoto, Y. (1999). Inhibitory effects of activin‐A on osteoblast differentiation during cultures of fetal rat calvarial cells. Journal of Cellular Biochemistry, 75, 206–214. [DOI] [PubMed] [Google Scholar]
- Inoue, Y. , & Imamura, T. (2008). Regulation of TGF‐beta family signaling by E3 ubiquitin ligases. Cancer Prevention Research, 99, 2107–2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssens, K. , ten Dijke, P. , Janssens, S. , & Van Hul, W. (2005). Transforming growth factor‐beta1 to the bone. Endocrine Reviews, 26, 743–774. [DOI] [PubMed] [Google Scholar]
- Kenner, L. , Hoebertz, A. , Beil, F. T. , Keon, N. , Karreth, F. , Eferl, R. , … Wagner, E. F. (2004). Mice lacking JunB are osteopenic due to cell‐autonomous osteoblast and osteoclast defects. Journal of Cell Biology, 164, 613–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowles, H. J. , Cleton‐Jansen, A. M. , Korsching, E. , & Athanasou, N. A. (2010). Hypoxia‐inducible factor regulates osteoclast‐mediated bone resorption: Role of angiopoietin‐like 4. FASEB Journal, 24, 4648–4659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long, F. (2012). Building strong bones: Molecular regulation of the osteoblast lineage. Nature Reviews Molecular Cell Biology, 13, 27–38. [DOI] [PubMed] [Google Scholar]
- Lotinun, S. , Pearsall, R. S. , Davies, M. V. , Marvell, T. H. , Monnell, T. E. , Ucran, J. , … Baron, R. (2010). A soluble activin receptor Type IIA fusion protein (ACE‐011) increases bone mass via a dual anabolic‐antiresorptive effect in Cynomolgus monkeys. Bone, 46, 1082–1088. [DOI] [PubMed] [Google Scholar]
- Lulli, V. , Romania, P. , Morsilli, O. , Cianciulli, P. , Gabbianelli, M. , Testa, U. , … Marziali., G. (2013). MicroRNA‐486‐3p regulates gamma‐globin expression in human erythroid cells by directly modulating BCL11A. PLOS One, 8, e60436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massague, J. (1998). TGF‐beta signal transduction. Annual Review of Biochemistry, 67, 753–791. [DOI] [PubMed] [Google Scholar]
- Merle, B. , & Garnero, P. (2012). The multiple facets of periostin in bone metabolism. Osteoporosis International, 23, 1199–1212. [DOI] [PubMed] [Google Scholar]
- Nicks, K. M. , Perrien, D. S. , Akel, N. S. , Suva, L. J. , & Gaddy, D. (2009). Regulation of osteoblastogenesis and osteoclastogenesis by the other reproductive hormones, activin and inhibin. Molecular and Cellular Endocrinology, 310, 11–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa, Y. , Schmidt, D. K. , Nathan, R. M. , Armstrong, R. M. , Miller, K. L. , Sawamura, S. J. , … Rosen, D. M. (1992). Bovine bone activin enhances bone morphogenetic protein‐induced ectopic bone formation. Journal of Biological Chemistry, 267, 14233–14237. [PubMed] [Google Scholar]
- Ottley, E. C. , Nicholson, H. D. , & Gold, E. J. (2016). Activin A regulates microRNAs and gene expression in LNCaP cells. Prostate, 76, 951–963. [DOI] [PubMed] [Google Scholar]
- Pearsall, R. S. , Canalis, E. , Cornwall‐Brady, M. , Underwood, K. W. , Haigis, B. , Ucran, J. , … Bouxsein, M. L. (2008). A soluble activin type IIA receptor induces bone formation and improves skeletal integrity. Proceedings of the National Academy of Sciences of the United States of America, 105, 7082–7087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrien, D. S. , Achenbach, S. J. , Bledsoe, S. E. , Walser, B. , Suva, L. J. , Khosla, S. , & Gaddy, D. (2006). Bone turnover across the menopause transition: Correlations with inhibins and follicle‐stimulating hormone. Journal of Clinical Endocrinology and Metabolism, 91, 1848–1854. [DOI] [PubMed] [Google Scholar]
- Rankin, E. B. , Wu, C. , Khatri, R. , Wilson, T. L. , Andersen, R. , Araldi, E. , … Giaccia, A. J. (2012). The HIF signaling pathway in osteoblasts directly modulates erythropoiesis through the production of EPO. Cell, 149, 63–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai, R. , Miwa, K. , & Eto, Y. (1999). Local administration of activin promotes fracture healing in the rat fibula fracture model. Bone, 25, 191–196. [DOI] [PubMed] [Google Scholar]
- Sakai, R. , Fujita, S. , Horie, T. , Ohyama, T. , Miwa, K. , Maki, T. , … Eto, Y. (2000). Activin increases bone mass and mechanical strength of lumbar vertebrae in aged ovariectomized rats. Bone, 27, 91–96. [DOI] [PubMed] [Google Scholar]
- Sakai, R. , Eto, Y. , Ohtsuka, M. , Hirafuji, M. , & Shinoda, H. (1993). Activin enhances osteoclast‐like cell formation in vitro. Biochemical and Biophysical Research Communications, 195, 39–46. [DOI] [PubMed] [Google Scholar]
- Shiozawa, Y. , Jung, Y. , Ziegler, A. M. , Pedersen, E. A. , Wang, J. , Wang, Z. , … Taichman, R. S. (2010). Erythropoietin couples hematopoiesis with bone formation. PLOS One, 5, e10853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth, G. K. (2004). Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Statistical Applications in Genetics and Molecular Biology, 3(1), 1–25. [DOI] [PubMed] [Google Scholar]
- Subramaniam, M. , Gorny, G. , Johnsen, S. A. , Monroe, D. G. , Evans, G. L. , Fraser, D. G. , … Spelsberg, T. C. (2005). TIEG1 null mouse‐derived osteoblasts are defective in mineralization and in support of osteoclast differentiation in vitro. Molecular and Cellular Biology, 25, 1191–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai, Z. Y. , Singh, S. , Yu, S. L. , Kao, L. P. , Chen, B. Z. , Ho, B. C. , … Li, S. S. (2010). Identification of microRNAs regulated by activin A in human embryonic stem cells. Journal of Cellular Biochemistry, 109, 93–102. [DOI] [PubMed] [Google Scholar]
- Tsuchida, K. , Nakatani, M. , Hitachi, K. , Uezumi, A. , Sunada, Y. , Ageta, H. , & Inokuchi, K. (2009). Activin signaling as an emerging target for therapeutic interventions. Cell Communication and Signaling: CCS, 7(15), 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vale, W. , Wiater, E. , Gray, P. , Harrison, C. , Bilezikjian, L. , & Choe, S. (2004). Activins and inhibins and their signaling. Annals of the New York Academy of Sciences, 1038, 142–147. [DOI] [PubMed] [Google Scholar]
- Vlachos, I. S. , Zagganas, K. , Paraskevopoulou, M. D. , Georgakilas, G. , Karagkouni, D. , Vergoulis, T. , … Hatzigeorgiou, A. G. (2015). DIANA‐miRPath v3.0: Deciphering microRNA function with experimental support. Nucleic Acids Research, 43, W460–W466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Q. , Huang, Z. , Xue, H. , Jin, C. , Ju, X. L. , Han, J. D. , & Chen, Y. G. (2008). MicroRNA miR‐24 inhibits erythropoiesis by targeting activin type I receptor ALK4. Blood, 111, 588–595. [DOI] [PubMed] [Google Scholar]
- Wilson, S. S. , Wong, A. , Toupadakis, C. A. , & Yellowley, C. E. (2015). Expression of angiopoietin‐like protein 4 at the fracture site: Regulation by hypoxia and osteoblastic differentiation. Journal of Orthopaedic Research, 33, 1364–1373. [DOI] [PubMed] [Google Scholar]
- Yano, M. , Inoue, Y. , Tobimatsu, T. , Hendy, G. , Canaff, L. , Sugimoto, T. , … Kaji, H. (2012). Smad7 inhibits differentiation and mineralization of mouse osteoblastic cells. Endocrine Journal (Kyoto Japan), 59, 653–662. [DOI] [PubMed] [Google Scholar]
- Yu, J. , Shao, L. , Vaughan, J. , Vale, W. , & Yu, A. L. (1989). Characterization of the potentiation effect of activin on human erythroid colony formation in vitro. Blood, 73, 952–960. [PubMed] [Google Scholar]
- Zhang, Y. , Xie, R. L. , Gordon, J. , LeBlanc, K. , Stein, J. L. , Lian, J. B. , … Stein, G. S. (2012). Control of mesenchymal lineage progression by microRNAs targeting skeletal gene regulators Trps1 and Runx2. Journal of Biological Chemistry, 287, 21926–21935. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information
Supporting information
Supporting information
Data Availability Statement
Data available on request to the authors.