

Abstract
Caveolae are flask‐shaped invaginations of the cell membrane rich in cholesterol and sphingomyelin, with caveolin proteins acting as their primary structural components that allow compartmentalization and orchestration of various signalling molecules. In this review, we discuss how pleiotropic functions of caveolin‐1 (Cav1) and its intricate roles in numerous cellular functions including lipid trafficking, signalling, cell migration and proliferation, as well as cellular senescence, infection and inflammation, are integral for normal development and functioning of skin and its appendages. We then examine how disruption of the homeostatic levels of Cav1 can lead to development of various cutaneous pathophysiologies including skin cancers, cutaneous fibroses, psoriasis, alopecia, age‐related changes in skin and aberrant wound healing and propose how levels of Cav1 may have theragnostic value in skin physiology/pathophysiology.
Keywords: caveolae, caveolin‐1, fibrosis, melanoma, psoriasis, squamous cell carcinoma, wound healing
1. INTRODUCTION TO CAVEOLINS
Although caveolae were identified as subcellular structures in the 1950s using electron microscopy, it has taken decades of study to unravel the complex biology regulated by caveolae.1, 2 They have initially been characterized as flask‐, or omega‐shaped, membrane invaginations that can be seen in most cell types.3, 4 Caveolins are cytoplasmic proteins that were discovered in the 1990s which serve as the major structural constituents of the caveolar membrane. Much of what we fundamentally know regarding caveolins can be attributed to the work of Richard Anderson's (who discovered caveolin‐1 (Cav1)), Michael Lisanti's, Robert Parton's and Tim Thompson's groups, who developed Cav1 knockout mice and made seminal discoveries regarding its structure and oligomerization into caveolae.3, 5, 6, 7, 8, 9 In addition to localizing to the cell membrane, Cav1 has been shown to be localized to ER, Golgi, mitochondria, endosomes, as well as lipid droplets, and nucleus and extracellular vesicles.10, 11, 12, 13, 14, 15, 16 Cellular organelles, such as the ER and Golgi apparatus, usually express both Cav1 and Cav2 which then organize into heterooligomers, that form caveolae.17, 18 It should be noted that although it is widely accepted that caveolin expression is ubiquitous in mammalian tissues, levels of expression vary considerably between the different caveolin isoforms as well as tissue types. For example, cells with highest level of Cav1 expression tend to be those which are terminally differentiated (ie adipocytes, endothelia, smooth muscle cells, among others), while Cav2 usually colocalizes and mirrors expression of Cav1, whereas Cav3 expression is mainly localized to muscle cells.19, 20 This review will specifically focus on Cav1 protein and its respective functions including lipid trafficking, membrane trafficking, signal transduction, cell migration and cell proliferation, all of which have crucial roles in normal skin functioning. We will then focus on the role of Cav1 in pathologic skin disorders related to Cav1 malfunction, such as skin cancer, scleroderma, psoriasis, alopecia, ageing‐related changes in skin, as well as in non‐healing chronic wounds.
2. ROLE OF CAV1 IN CELLULAR PROCESSES CRUCIAL FOR NORMAL SKIN FUNCTION
Caveolae and Cav1 have long been known to be involved in endocytosis through various mechanisms, including regulation of early endosome migration, as well as EGF receptor (EGFR) and extracellular membrane‐binding protein turnover.21, 22 Moreover, Cav1 is extremely stable at the cellular membrane and only some Cav1 rich vesicles actually become internalized,23, 24, 25 which suggests that Cav1 may also act to stabilize cellular membrane and slow down membrane invagination, budding, and vesicle internalization. In other words, instead of being only structurally involved, Cav1 may take on a regulatory function of endocytosis through several different mechanisms including regulation of cholesterol content of raft domains,26 slowing down caveolar budding,27, 28 or by isolation of signalling molecules necessary for caveolar endocytosis (including G proteins).28 Cav1 may also function in sorting proteins and lipids into distinct vesicles where Cav1 may be a platform that regulates the content of caveolar invaginations.29 Below, we will outline how Cav1 regulates some basic cellular functions crucial in skin physiology.
2.1. Lipid transport
Caveolae are commonly found in areas of the cell membrane enriched in cholesterol and sphingomyelin,6, 30 with some early in vitro studies pointing to interaction of Cav1 and cholesterol,31 which potentially allows its arrangement in and curvature of caveolar and cell membranes32, 33, 34 and to act as cholesterol transporter.35, 36, 37, 38 For example, immediately after being synthesized in the ER, cholesterol migrates directly to the caveolar surface, from which it then localizes to different areas of the plasma membrane and extracellular space in a Cav1‐dependent manner.39 Consequently, it is unsurprising that Cav1 is required for extracellular and intracellular lipid trafficking, and this could explain the relatively high expression of Cav1 by adipocytes,40 as well as the lipid metabolism defects observed in mice completely lacking Cav1.41, 42 Thus, it appears that Cav1 does not act as a single molecule, but rather exists in a complex involving intracellular lipids that act to transport cholesterol and potentially other lipids between different cellular compartments.43 If one considers the elaborate lipidomic profile of the stratified epidermis, elucidating the role Cav1 plays in its development and maintenance will undoubtably provide tremendous insights into epidermal permeability barrier and treatment of various barrier function defects including atopic dermatitis, ichthyosis and psoriasis, among others.
2.2. Infection and inflammation
Persistence of infection due to intracellular pathogen colonization is starting to garner a lot of interest in wound healing especially since it is now widely accepted that all chronic wounds should be considered infected, even if they do not exhibit clinical signs of infection. Interestingly, caveolae‐dependent endocytosis is upregulated in chronologically aged and senescent cells, which may be one of the reasons for why the elderly are more susceptible to infections.44 Once vesicles are endocytosed, instead of going to the lysosomes, they are trafficked directly to the Golgi apparatus or endoplasmic reticulum44, 45, 46 and thus protected from degradation. As this process appears to be non‐selective and is utilized by many bacteria and viruses, including Salmonela typhimurium, Vibrio cholera, Pseudomonal aeruginosa, Leishmania, HIV and coronavirus, this endocytic route may provide protection for these pathogens.44, 47, 48, 49, 50, 51, 52, 53 For example, S typhimurium delivers SopE protein into the host cell via the type III secretion system, which binds to Cav1 and leads to Cav1‐dependent Rac1 protein recruitment, which in turn promotes actin fibre rearrangement, phagocytosis and bacterial invasion of the host cell.44, 54 Interestingly, other cells which express low levels of Cav1 exhibit increased internalization of Staphylococcus aureus due to enhanced membrane mobility of Cav1‐deficient cells, thus arguing for more of a context‐dependent role.55 It remains to be seen whether commensal bacteria interact with host Cav1 and how Cav1 expression correlates with development of biofilms (which comprise of diverse array of bacteria); however, considering that non‐healing chronic wounds exhibit elevated levels of Cav1,56 understanding the role Cav1 plays in bacterial colonization will provide invaluable insights into treatment of chronic wounds.
With regard to inflammation, Cav1‐null mice exhibit a systemic proinflammatory state, with a noticeable increase in cytokines including IL‐6, TNF‐alpha and IL‐12p7057 (however, these results are yet to be validated in human samples). Additionally, Cav1‐null mice exhibited greater chemokine‐dependent immune cell recruitment when compared to controls.58 Interestingly, transcriptional profiling of human keratinocytes after cholesterol depletion by methyl‐β‐cyclodextrin (MβCD) has been shown to promote secretion of an inflammatory marker IL‐8 together with plasminogen activator urokinase (PLAU), similarly to human atopic dermatitis (AD) samples.59 It should be noted that although the authors of this study did not observe diminished levels of Cav1 as a result of cholesterol depletion, this could be ascribed to the relatively stable nature of Cav1 and their short administration of MβCD (up to 8hrs); thus, prolonged caveolae disruption would undoubtedly lead to Cav1 downregulation as well. Consequently, it is yet to be determined whether Cav1 levels correlate with AD; however, if downregulation of Cav1 is present in AD samples, it would be interesting to see whether topical application of caveolin scaffolding domain (CSD) peptide could alleviate IL‐8 secretion and thus be used as a potential therapeutic target for treatment of AD. Cav1 has also been shown to interact with TLR4 receptor in endothelial cells and mediate activation of MyD88 signalling cascade in sepsis‐induced lung inflammation,60 whereby Cav1 inhibition could prevent sepsis‐induced lung inflammation. Thus, it becomes clear that Cav1's role in both infection and inflammation, just like the other process, is complex and further studies are necessary to elucidate these intricacies.
2.3. Signal transduction
Shortly after the discovery of Cav1, numerous Cav1‐binding partners have been discovered and attempts at understanding how they impact various cell signalling cascades have been undertaken.21 The tendency of Cav1 to oligomerize via N‐terminal side‐chain amino acids (aa 82‐101) suggests that Cav1 could be a recruiter of the signalling cascade molecules via its CSD,61, 62 leading to the Cav1 signalling hypothesis which posits that Cav1 could perhaps function as a scaffolding protein that recruits numerous signalling molecules, although that depends on the accessibility of the caveolin‐binding motifs of the signalling proteins proposed to interact with Cav1.63
Altogether, more than 50 studies found that Cav1 interaction with other molecules via its scaffolding domain results in deactivation of these molecules in the signalling cascade.64, 65 There is also evidence that Cav1 may stimulate some signalling events.66 However, it has yet to be confirmed whether the scaffolding domain of Cav1 is directly related to its primarily suppressing and occasionally activating function, since when this region is mutated, Cav1 is unable to leave the Golgi apparatus and cell signalling transduction process in this sense becomes impossible to study.67 Regardless, via its scaffolding domain, Cav1 has been shown to interact with numerous receptor tyrosine kinases (EGF, TGF‐β1, Her2, PDGF, VEGF and insulin receptors),68, 69, 70, 71, 72, 73, 74, 75, 76, 77, 78, 79, 80, 81 steroid hormone receptors (oestrogen receptor alpha, glucocorticoid and androgen receptor), G protein‐coupled receptors (angiotensin II receptor and glutamate receptor),56, 82, 83, 84, 85, 86, 87, 88, 89 as well as with numerous intracellular signalling molecules (including but not limited to PKA, PKC, PLC, PLD, ERK1/2, Akt, Ras, Csk, Src, Irs1 and Grb2).26, 63, 66, 71, 90, 91, 92, 93, 94, 95, 96, 97, 98, 99, 100, 101, 102, 103, 104, 105, 106, 107 Thus, it is evident that Cav1 has a tremendous network of signalling factors that may regulate numerous cellular processes. In line with its role in cholesterol recruitment, Cav1 may regulate cell signalling through lipid organization more so than through proteins. Considering that Cav1 has its role in maintaining and regulating cholesterol levels, it becomes evident how it would indirectly regulate signal cell transduction via lipids.
2.4. Cell migration
Due to its ability to bind to and interact with such a diverse array of signalling molecules, it is unsurprising that Cav1 has been implicated as a major regulator of cell migration.108, 109, 110, 111 Immunostaining experiments show polarized Cav1 localization may be necessary for migration of endothelial cells,112, 113, 114, 115, 116 where it binds to actin cross‐linking protein filamin, suggesting that Cav1 may be modulating the cytoskeleton. In line with these findings, Cav1 has been shown to affect cytoskeletal remodelling and focal adhesion assembly by differentially affecting activity Rho family of GTPases, namely RhoA, Rac1 and Cdc42 through interaction with various guanine nucleotide exchange factors (GEFs) and GTPase activating proteins (GAPs).117 However, whether Cav1 promotes or inhibits cell migration is still controversial and seems to be cell‐ and context‐dependent. For instance, Cav1 downregulation increases, while upregulation decreases the ability of EGF‐stimulated cells to migrate56, 70, 118, 119 and Cav1 knockdown accelerates directional cell migration.56 Furthermore, Cav1 has been shown to downregulate c‐Src and c‐met signalling, and metastatic potential of osteosarcoma cells.120 On the other hand, Cav1 was shown to interact with eNOS as well as integrin β1 and urokinase receptor uPAR, resulting in cytoskeletal reorganization and cell migration.121, 122, 123, 124 Moreover, Cav1Y14 phosphorylation leads organization of caveolae in the focal adhesions, by rearranging and co‐localizing important signalling molecules necessary for cell migration.125, 126, 127 Its role within the extracellular matrix is evident from studies that found that Cav1 enhances cell surface plasminogen activation,128 and regulation of MMP‐1.129 Therefore, the migratory potential of Cav1 is undoubtably very complex and most likely depends not only on Cav1, but on the array of migratory proteins in its environment that directly or indirectly interact with Cav1.
2.5. Cell proliferation
Similar to cell migration, due to its ability to orchestrate compartmentalization of numerous signalling molecules, numerous studies have implicated Cav1 in regulation of cell proliferation. In general, Cav1 expression reduces cell proliferation in normal and cancer cells in early stages of cancer disease and has generally been considered a tumor suppressor.11 Specifically, Cav1 was found to have antiproliferative properties, causing G0/G1 cell cycle arrest, through upregulation of p53 and p21, and downregulation of cyclin D1130 (which is commonly upregulated downstream of many pathways including ERK1/2, PI3K and β‐catenin).131, 132, 133 Thus, Cav1 deregulation leads to increased ERK1/2 activation, upregulation of cyclin D1, downregulation of p21 and increased proliferation.134 Similarly, Cav1 acts as a negative regulator of PI3K/Akt pathway135, 136, 137 where depletion of Cav1 leads to higher levels of Rac and Akt and increase in Cyclin D1 function.138 Cav1 also acts as a negative regulator of Wnt/β‐catenin canonical pathway, where Cav1 sequesters β‐catenin and prevents its nuclear translocation and interaction with TCF/LEF to decrease cyclin D1 levels.139 Since Cav1 generally exhibits antiproliferative properties, its aberrant expression has tremendous ramifications on cutaneous hyperproliferative conditions, which will be discussed later.
2.6. Expression of Cav1 in Skin
Due to their orchestration of a vast array of signalling molecules, it is therefore unsurprising that caveolins have been shown to have extensive roles not only in development and homeostasis of skin, but also in pathophysiology associated with skin disorders. First, Cav1 and Cav2 have been shown to exhibit differential expression in the epidermis, where Cav1 localizes primarily to undifferentiated basal keratinocytes,56, 140, 141 while Cav2 localizes to suprabasal, differentiated keratinocytes,140, 141 suggesting possibly distinct roles in keratinocyte proliferation and differentiation for different caveolin variants. Furthermore, in vitro induction of keratinocyte differentiation has been shown to stimulate a transient drop in Cav1 expression (consistent with its absence from suprabasal levels) in the epidermis, and interestingly treatment of keratinocytes with Cav1 scaffolding domain peptide can inhibit calcium‐induced keratinocyte differentiation by blocking calcium‐induced increases in phosphatidylglycerol.142 Together these data support the role of Cav1 in inhibiting keratinocyte differentiation and thus raise intriguing possibilities for targeting Cav1 in cutaneous disorders manifested by abnormal differentiation and proliferation, including psoriasis and hypertrophic scarring.143, 144, 145, 146 Interestingly, Cav1 also localizes to melanocytes and its expression can be induced by UV exposure, where it leads to changes in cell morphology and leading to increased melanin transfer and skin pigmentation as a result of changes in cAMP production.147
2.7. Expression of Cav1 in hair
Although there have been some recent studies of Cav1 expression and potential function in the skin, not much is known about Cav1 in hair development. Current knowledge comes from mouse models and studying pathologic disorders, such as different types of alopecia. For example, Selleri et al have confirmed Cav1's expression in the bulge area of the hair follicle, and its expression was not affected by doxorubicin (DXR) chemotherapy treatment. While expression of β‐catenin, a marker of cell proliferation, was absent from the bulge cells post‐treatment with DXR, Cav1 remained visible in all stages of hair cycle, thus suggesting a potential role in protection from permanent chemotherapy‐induced alopecia, and allowing for future hair regrowth.148 This study also confirmed that the cells of the bulge area express Cav1 consistently regardless of the phase of the hair cycle, which posits that when cells leave the bulge area, they potentially stop expressing Cav1. Since β‐catenin was shown to have differential expression in different phases of the hair cycle, and as discussed previously, Cav1 sequesters β‐catenin, proper balance between Cav1 and β‐catenin may be the key to future therapies for hair regrowth. Paus' group has previously shown that selected immunophilin ligands (IPLs) and cyclosporine (CsA) also act to protect from the chemotherapy‐induced alopecia and follicle dystrophy, possibly by shifting the follicle into the anagen phase. Since CsA has been previously shown to destabilize caveolae by decreasing their cholesterol content and by inhibiting the interaction of Cav1 with cyclophilin A,59 it would be interesting to see whether CsA treatment affects expression of Cav1, interaction of Cav1/cyclophilin A, and whether less toxic alternatives to CsA (like WAY‐316606)149 could be used to alter Cav1 expression levels and thus promote hair growth, without eliciting the known side effects of CsA. This mechanism could also be modulating the Cav1/β‐catenin balance, and further studies are necessary to better elucidate their interplay.150 Further, differential expression of genes between bolding (BAB) and non‐bolding scalp (BAN) in relation to androgen‐induced alopecia found that Cav1 is downregulated in BAB in comparison to BAN.151 Recently, it has also been proposed that reduction of Cav1 in the hair follicle may be due to enrichment of preadipocytes during catagen phase of the hair follicle cycle, since preadipocytes have been previously shown to exhibit diminished levels of Cav1.152 However, it has also been demonstrated that upregulation of miR‐199a‐5p (miRNA that has been shown to target Cav1) can prevent lipid accumulation during preadipocyte differentiation.153 Thus, whether downregulation of Cav1 is the cause or consequence of balding is yet to be fully delineated. Together, these studies reveal potential therapeutic targets in clinically challenging conditions such as alopecia and further research in this area could only better our current knowledge and treatment options.
3. ROLE OF CAVEOLINS IN SKIN CANCERS
The function of caveolins in cancer pathogenesis has been increasingly examined over the past decade. However, the role of Cav1 in carcinogenesis appears to vary with the tumor type and tumor progression. For example, its expression is associated not only with cancer suppression in oesophageal adenocarcinoma, lung adenocarcinoma, and ovarian cancer, but also with progression of prostate cancer, renal cell carcinoma, and lung squamous cell carcinoma.154, 155, 156 This argues for a cell‐specific role where Cav1 can interact with cell type–specific proteins and thus either hinder or promote tumor progression. Below, we will outline the current understanding for the role of Cav1 in skin cancers (Tables 1 & 2).
Table 1.
Role of caveolins in NMSCs
| Tumor Suppressing or Promoting | Model | Translational relevance | Reference |
|---|---|---|---|
| Suppressing | In vitro | Predictive biomarker of disease severity and progression | 157 |
|
In vitro In vivo |
Targeted therapy inducing Cav1 gene expression in cancer cells to attenuate tumor growth and metastasis | 158 |
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
Table 2.
Role of caveolins in melanoma
| Tumor promoting or suppressing | Model | Translational relevance | Reference |
|---|---|---|---|
| N/A |
In vitro Ex vivo |
Diagnostic and/or predictive and prognostic biomarker | 159, 160, 162 |
| Suppressing | In vitro | Targeted therapy inducing Cav1 expression in cancer cells to attenuate tumor growth and metastasis | 161, 162 |
| Both (1° tumor promoting, metastasis suppressing) | In vitro | Targeted therapy towards balanced Cav1 expression in cancer and surrounding cells to attenuate tumor growth and metastasis | 163, 164, 165 |
|
In vitro In vivo |
Biomarker of poor prognosis in melanoma patients undergoing surgery. Predictor of likelihood of malignancy | 165 | |
| Promoting |
In vitro In vivo |
Targeted therapy suppressing Cav1 expression in cancer cells to attenuate tumor growth and metastasis | 202, 203 |
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
3.1. Role of caveolins in non‐melanoma skin cancers (NMSCs)
3.1.1. Cav1 in cutaneous squamous cell carcinomas (SCCs)
Although the role of caveolins in oesophageal, lung and other non‐cutaneous forms of SCCs has been considerably explored, there are only a few studies assessing caveolin expression in cutaneous SCCs. One study compared immunohistochemical expression of Cav1 in patients with cutaneous SCC with normal control skin specimens and observed a statistically significant downregulation of Cav1 expression in SCCs compared to the control group, in both intensity and pattern of expression.157 Furthermore, poorly differentiated SCCs showed significantly reduced Cav1 expression compared to moderately and well‐differentiated SCCs, suggesting that Cav1 downregulation not only plays a role in the promotion of SCC tumorigenesis, but also in its progression. The study went on to explore the potential role of Cav1 in the pathogenesis on non‐melanoma skin cancers (NMSCs) and ultimately found that levels of Cav1 expression could be used as predictive biomarkers for patients at risk of cancer progression.157 Murine models of cutaneous SCC demonstrate similar relationship between Cav1 and tumor development, where mice overexpressing Cav1 exhibit decreased in vitro cell proliferation, as well as decreased in vivo tumor incidence, volume and weight.158 Conversely, Cav1‐null mice exhibit increased cell and tumor growth, in addition to increased invasive ability and incidence of spontaneous lymph node metastasis, which is likely due to hyperactivation of the MAPK pathway in the Cav1‐null mice,158 thus introducing Cav1 as a potential regulator of invasion and metastasis in cutaneous SCCs.
3.1.2. Cav1 in basal cell carcinoma (BCCs)
Although the association between Cav1‐BCC development has been largely unexplored, a recent study correlated Cav1 expression with the clinicopathological parameters of BCCs.157 BCC samples demonstrated a statistically significant downregulation of Cav1 expression compared to specimens from healthy individuals. Additionally, localization differed between the two groups, with BCC group exhibiting only membranous localization, while the control group demonstrated both membranous and cytoplasmic. Cav1 expression also varied in BCC depending on the histopathological type, with significant downregulation being seen in aggressive types (including micronodular, infiltrative and metatypical BCC) compared to the non‐aggressive types (including nodular and superficial BCC). These findings provide evidence that Cav1 expression is significantly decreased in aggressive types of BCC compared to the non‐aggressive types and ultimately suggests that Cav1 may be a predictor of the biological behaviour of BCCs that could be useful in the detection of high‐risk patients.
3.2. Role of caveolins in melanoma
Whereas the literature on the role of Cav1 in NMSCs largely supports a tumor‐suppressing role, research on its role in melanoma is more controversial. Some studies have demonstrated increased levels of Cav1 in melanoma patients, where baseline serum Cav1 levels were found to be significantly higher in patients with melanoma than those in the control group (0.47 vs. 0.37 ng/ml, respectively, P = .05).159 However, the same study did not find a correlation between Cav1 levels and other known clinical variables, such as sex, location of the lesion, histology, stage of disease, or response to chemotherapy, and levels of Cav1 had no prognostic role in survival (P = .44). Similarly, another study examined Cav1 expression in exosomes from SCID mice engrafted with human melanoma cells and observed significantly increased levels of plasma exosomes expressing Cav1 in the melanoma group compared to healthy donors160; thus, measuring Cav1 levels in exosomes in human plasma could be another non‐invasive tool for melanoma screening and follow‐up.
Although various studies have observed an association between caveolin and melanoma, whether this protein acts as a tumor suppressor or tumor promotor is still controversial, where in one study overexpression of Cav1 in human melanoma cell lines caused decreased cell proliferation and migration while other studies support the tumor‐suppressing role of Cav1 in melanoma specifically assess its regulation of metastasis.161 For example, loss of Cav1 expression in melanoma cells predicts decreased survival in primary malignant melanoma as evidenced when the association of Cav1 levels and survival in both primary melanomas and melanoma lymph node metastases was examined. Furthermore, it was found that levels of Cav1 in the stroma, but not in melanoma cells, strongly predicted the clinical outcome once the tumor had metastasized and the absence of stromal Cav1 was associated with aggressive melanoma behaviour, including invasion and metastasis.162 This study was one of the first to propose that Cav1 may function as a metastasis suppressor in the stromal compartment of malignant melanoma, which not only suggests its potential as a new biomarker of melanoma development, but also for stromal‐targeted therapies in preventing tumor progression. In another study, in vitro overexpression of Cav1 enhanced cell growth and increased DNA synthesis163; however, when melanoma cells were implanted into mice, Cav1 overexpression appeared to suppress the ability of melanoma cells to form lung metastases by antagonizing FAK, Src and integrin β3. Furthermore, levels of Cav1 were found to be significantly lower in human metastatic melanoma cell lines. Together, these findings suggest that Cav1 expression controls melanoma formation and progression in a stage‐specific manner.
Interestingly, the opposite scenario has also been demonstrated, where injecting Cav1‐deficient dermal fibroblasts with melanoma cells into mice led to increased melanoma cell growth via enhanced paracrine cytokine signalling and ShhN expression.164 Of note, lack of Cav2 did not have this effect, suggesting that Cav1 specifically plays a tumor‐suppressing role in primary melanoma. However, the same study showed that in contrast to primary tumor growth, the Cav1‐deficient mice also had reduced lung metastases. Similarly, another study specifically addressed the postsurgical setting and found that Cav1 expression suppressed tumor formation, but enhanced lung metastasis.165 These two studies thereby support a primary tumor‐suppressing role, with a metastases‐promoting role of Cav1 in melanoma.
Altogether, our current understanding of Cav1 with regard to melanoma is still not very clear. Therefore, it may not be possible to classify the protein as simply tumor suppressing or oncogenic. As many of the aforementioned studies suggested, its function may depend on timing, tumor stage, location or its interaction with other cellular proteins. Nonetheless, the established association between Cav1 and cancer stresses the need for further elucidation, as its diagnostic and therapeutic potential may be transformative in cutaneous oncology.
4. ROLE OF CAVEOLIN‐1 IN LOCALIZED AND DIFFUSE CUTANEOUS FIBROSIS
Systemic sclerosis, or scleroderma (SSc), is a connective tissue disorder of unknown aetiology characterized by localized or diffuse cutaneous fibrosis and obliterated blood vessels that may spread to internal organs, leading to death.166 Reduced Cav1 protein expression has been demonstrated in multiple immunohistochemical analyses of full‐thickness biopsies of SSc patients,167, 168 as well as in vitro studies of cultured fibroblasts from SSc patients.166, 167, 169 Similarly, the observation that Cav1 dysregulation promotes fibrosis suggests that Cav1 may also have implications in other fibrotic skin disorders, including keloids, which are characterized by excessive collagen deposition in the dermis and their formation is highly dependent on skin tension.170 In fact, one study found that cultured fibroblasts obtained from keloids exhibited reduced Cav1 levels compared to healthy skin.171 Furthermore, Cav1 downregulation has been observed in bleomycin‐induced lung, and postwounding skin fibrosis, while allogeneic adipose‐derived mesenchymal stromal cells (ASCs) injected mice exhibited attenuated bleomycin‐induced lung and skin fibrosis and promoted faster wound healing.172 Reduction in Cav1 expression has been implicated in reversing several profibrotic signalling cascades contributing to fibrosis in SSc and other fibrotic skin diseases, since Cav1 downregulates TGF‐β signalling by inhibiting Smad2 and Smad3 phosphorylation, ultimately inhibiting downstream ECM production and profibrotic effects.167, 171, 173 Additionally, TGF‐β receptors may be inhibited directly by caveolae‐mediated internalization of the receptor, subsequent degradation and turnover.166, 171 Cav1 downregulation in keloid‐derived fibroblasts has also been demonstrated to enhance TGF‐β‐mediated production of α‐SMA, collagen type I and fibronectin, contributing to profibrotic phenotype.171 In vitro studies of human dermal fibroblasts have also demonstrated Cav1 modulation of both the activity and function of various matrix metalloproteinases (MMPs) in the skin, notably MMP1 and MMP14.174, 175 MMP1 deregulation has been associated with collagen deposition, tissue fibrosis, impaired wound healing and cancer metastases.174 To this end, it has been demonstrated that an inverse relationship between Cav1 expression and increased MMP1 that was associated with activated MAP kinase pathway specifically phosphorylated Erk1/2 and Ets1.174 One of the recent studies argues that the plasticity of dermal adipocytes to undergo de‐ and re‐differentiation under physiological conditions can be disrupted by bleomycin treatment, thus leading to differentiation of preadipocytes into myofibroblasts (which have previously been shown to exhibit diminished levels of Cav1). Interestingly, myofibroblasts are commonly observed in hypertrophic and keloid scars and this new finding thus offers a novel explanation regrading pathogenesis of these cutaneous fibroses.152
The pathogenesis of SSc may also involve chemotaxis of bone marrow‐derived fibroblasts migrating to the dermis and expressing a profibrotic phenotype. One such study identified an increased number of monocyte‐derived, Cav1 deficient, fibrocytes in the fibrotic dermis of SSc patient biopsies, compared control tissue.166 Additionally, SSc fibrocytes had increased expression of CCR5 and CXCR4 chemokine receptors and hypermigratory phenotype towards respective ligands MIPIa and MIPIb.166, 176 Additionally, this trend was also seen at a lesser extent in healthy African American dermal fibroblasts, suggesting a genetic susceptibility towards developing cutaneous fibrotic disease in this ethnic population. Early studies with CSD peptide have shown promising results in treatment of lung fibrosis,177, 178 and as such, in vitro and in vivo assays investigating reversal effects of CSD peptide in SSc found that CSD blocked hypermigration of SSc monocytes via a reduction in CXCR, CCR5, CCR1, CCR2 and CCR3 expression, indicating a potential role of Cav1 in inhibiting monocyte recruitment and migration, possibly via chemokine receptor degradation.166, 176 Downregulation of Cav1 may also be associated with myofibroblast‐mediated fibrosis contributing to vasculopathy in SSc as one study demonstrated decreased Cav1 expression was correlated with increased pigment epithelium‐derived factor (PEDF) expression mediated by TGF‐β induction in SSc fibroblasts, which has been associated with anti‐angiogenic properties including anti‐vasopermeability and neurotrophic activities.169
5. ROLE OF CAVEOLIN‐1 IN PSORIASIS
Campbell et al first hypothesized the role of Cav1 in pathogenesis of psoriasis back in 2000, when his group recognized the plausible connection between Cav1 regulation of proper keratinocyte proliferation, differentiation, calcium homeostasis and angiogenesis.143 Soon after, evidence began to emerge demonstrating the connection between downregulated Cav1 and excessive epidermal hyperplasia that is classically seen in psoriasis.179, 180 Several immunohistochemical and histological analyses of psoriatic skin lesions demonstrated decreased expression of Cav1 compared to unaffected skin.143, 144, 145, 181 Additionally, one study found markedly decreased expression of Cav1 in different types of psoriasis, including psoriasis vulgaris, localized pustular psoriasis and erythrodermic psoriasis, with psoriasis vulgaris having the most significant downregulation of Cav1 expression compared to the other two types.145 Other studies found Cav1 expression to be present throughout the full thickness of the epidermis of healthy skin, with more intense expression in the basal, upper granular and spinous layers, whereas psoriasis skin had little to no Cav1 staining present.143, 144 Additionally, there was intense cytoplasmic Cav1 staining in psoriatic keratinocytes.143 The same study looked at Cav1 expression in lesional, advancing edge and non‐lesional psoriatic skin. Though there was a significant reduction in Cav1 expression in lesional skin compared to non‐lesional skin, the difference in Cav1 expression in the advancing edge was equivocal, with 3 of the 9 patient samples demonstrating differential expression, and 6 of the 9 samples demonstrating no discernible difference.143 Another study also demonstrated an inverse relationship between Cav1 expression and psoriasis severity as measured by the PASI score, proliferation index, microvascular density at dermal papilla.144 Thus, the role of Cav1 in development and progression of the different forms of psoriasis needs to be explored further and higher sample numbers need to be assayed before any meaningful results can be interpreted.
6. ROLE OF CAV1 IN CELLULAR SENESCENCE AND SKIN AGEING
In addition to previously mentioned functions, several studies have found that Cav1 may also be related to stress‐induced premature senescence in a biphasic manner. Initially, it appeared that Cav1 can be induced by sub‐cytotoxic levels of H2O2, to accelerate premature senescence and mitochondrial dysfunction182, 183, 184, 185; however, when Cav1 expression is inhibited either in Cav1‐null mice, or by using antisense Cav1, premature senescence by H2O2 does not occur.182, 186 Recently, it has also been shown that strong suppression of Cav1 induces premature senescence in a p53‐p21‐dependent manner.187 Multiple studies have found Cav1 to be downregulated in fibrotic skin and lung disease where bleomycin (BLM) treated mice exhibited downregulation of Cav1, developed lung and skin fibrosis and displayed delayed wound healing.172, 188 However, ASC injection reduced BLM‐induced lung and skin fibrosis and sped up the wound healing process. Additionally, Cav1 downregulation characteristic of fibrotic tissue was also stagnated post‐ASC injection.172 Another example is sublethal UV‐C light acting to increase Cav1 expression and this way lead to premature senescence of fibroblasts.182 Further, Cav1 is commonly used as a marker of ageing.37, 189, 190 Specifically, senescent fibroblasts exhibit a greater level of Cav1, where it localizes in proximity to EGFR.69 Preliminary data from our laboratory indicate that chronologically aged human skin exhibits elevated levels of Cav1 (Figure 1); however, it should be noted that exposure to sun was not controlled for in these samples. Interestingly, other studies have recently shown that in the skin of the elderly, Cav1 promotes skin ageing via TGF‐β pathway that ultimately leads to decreased collagen production by dermal fibroblasts.191 For further comprehensive description on the role of Cav1 in macro‐ and mesoscopic alterations during in skin ageing and other ageing‐related diseases, please refer to previously published review articles.192, 193 With further investigation, targeting Cav1 could potentially be beneficial in decelerating skin ageing and thus used for cosmetic purposes in the future as well.
Figure 1.
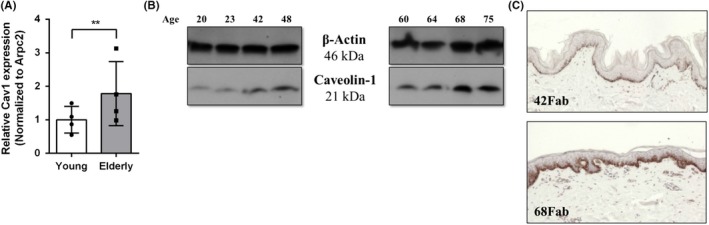
Elevated expression of Cav1 in chronologically aged skin. Levels of Cav1 from 4 young (<50 years of age) and 4 elderly (>60 years of age) female abdominal skin (Fab) skin were assessed by qRT‐PCR (A), Western blotting (B) with Arpc2 and β‐actin serving as normalizing and loading controls, respectively. Briefly, sex‐matched skin (N = 8) from patients undergoing routine reduction surgeries (abdominoplasties), composed of both dermis and epidermis, was used to assess Cav1 levels which were determined by qRT‐PCR and Western, blotting, respectively (Arpc2 forward primer (5'‐TCCGGGACTACCTGCACTAC‐3') and reverse primer (5'‐GGTTCAGCACCTTGAGGAAG‐3'); Cav1 forward primer (5'‐GCGACCCTAAACACCTCAAC −3') and reverse primer (5'‐ATGCCGTCAAAACTGTGTGTC‐3'). Protein levels were validated by immunoperoxidase staining using Cav1 antibody (Sigma HPA049326) in chronologically aged skin. Expression of Cav1 was found to positively correlate with increasing age (Pearson's correlation coefficient r(6)=0.7509, P=.031). Error bars correspond to standard deviation from 4 biological samples from each category. **P < .01 (Student's t test). (C). Immunoperoxidase staining of representative young (42‐year‐old) and elderly (68‐year‐old) skin was used to validate upregulation of Cav1 in chronologically aged skin. Control healthy human skin specimens were obtained as discarded tissue from reduction surgery procedures in accordance to institutional approvals. Specifically, protocol to obtain unidentified skin specimens was submitted to University of Miami Human Subject Research Office (HSRO). Upon review conducted by University of Miami Institutional Review Board (IRB), it was determined that such protocol does not constitute Human Subject Research per 45 CFR46.101.2
7. ROLE OF CAV1 IN WOUND HEALING
In one of the first studies on the role of Cav1 in wound healing, Rhim et al looked at Cav1 expression in corneal epithelium and its effect on wound healing and observed Cav1 overexpression in aged compared to young corneal epithelium, which was associated with delayed wound healing post‐LASEK surgery.194, 195 Along these lines, Cav1 knockout mice have also shown increased concentration of NO, leading to greater MMP‐13 nitration, and faster wound healing.196 Interestingly, our team has also shown significant Cav1 overexpression in wound edge biopsies of patients with non‐healing chronic wounds. Likewise, we have shown that Cav1 negatively correlates with healing in acute wounds, where it needs to be spatiotemporally downregulated in the migrating epithelial tongue in order to allow for proliferation and migration of keratinocytes from the wound edge into the wound bed using in vitro, in vivo (mouse and porcine) and human ex vivo models of wound healing.56 Interestingly, we observed that Cav1 can be used as a bona fide marker of the wound healing edge, where it localizes to the basal keratinocytes proximal to the wound. When co‐stained with keratin 6 (which demarcates activated migrating keratinocytes of the healing migrating epithelial tongue), we observed a distinct localization and absence of Cav1 from the migrating epithelial tongue using skin equivalent organotypics, human ex vivo wound, as well as murine and porcine in vivo wound models. Upregulation of Cav1, as seen in non‐healing chronic wounds (diabetic foot ulcers and venous leg ulcers), results in increased Cav1 interaction with membranous glucocorticoid receptor (which potentiates wound healing‐inhibitory signalling events), as well as sequestration of EGFR signalling, which altogether results in inhibition of keratinocyte migration and subsequent wound closure. Interestingly, topical administration of cholesterol depleting agents, including (MβCD and Mevasatin), reversed the Cav1‐mediated inhibition of migration and resulted in accelerated wound closure.56, 197 However, it is yet to be established whether the same is true for pressure ulcers.
Other studies however have shown Cav1 to be crucial as a promoter of the wound healing process. Specifically, overexpression of Cav1 in epidermal stem cells (EpiSCs) enhanced wound re‐epithelization and cellularity, and improved wound vasculature and overall healing scores. As such, Cav1 appears to be crucial in future wound healing therapies involving EpiSCs.198 Furthermore, another study demonstrated that disruption of either syndecan‐4 or Cav1 could lead to impaired wound closure in mice.199 Thus, it should be noted that therapeutic targeting of Cav1 in wound healing should be interpreted in the context of each type of wound. For example, burns and other types of acute wounds will require temporal downregulation of Cav1 expression, whereas inability to normalize homeostatic levels could result in aberrant inflammatory response, infection, hyperproliferation as well as excessive collagen deposition and thus yield undesirable healing outcomes (chronic wounds or hypertrophic scars). Therefore, as with other roles discussed above, Cav1's role in wound healing is complex and environment‐dependent, but it is clear how understanding its role is important for developing potential therapies with many patients with non‐healing chronic wounds, considering that the last biologic for treatment of chronic wounds was approved by the FDA over 20 years ago.
8. CONCLUSIONS AND TRANSLATIONAL PERSPECTIVES
The signalling pathways that regulate various cellular functions ranging from migration, proliferation and differentiation, to endocytosis and cellular senescence, are of integral importance to development and normal functioning of skin. There is abundant evidence reviewed here that supports the role of Cav1 in all the aforementioned cellular processes and thus brings about a new spotlight on Cav1 in development and progression of numerous cutaneous disorders (Figure 2). Specifically, it is the fine balance of Cav1 that is key to physiologic skin structure and function, as the slight imbalance in one direction or another affects localization and activation of various key signalling molecules and can tip the scale towards one of the skin pathologies (Figure 3). Thus, by studying the cellular and molecular mechanisms of Cav1 balance and imbalance in physiologic and pathologic skin conditions, respectively, one can arrive at a better understanding of commonly encountered and challenging to manage skin diseases, including skin cancer, psoriasis, scleroderma, skin ageing and senescence, wound healing and others. For example, spatiotemporal downregulation of Cav1 may be beneficial for proper wound closure, but sustained downregulation of Cav1 may bring about prominent changes in cell proliferation and collagen production and thus yield undesirable outcomes in line with non‐melanoma skin cancers, psoriasis and cutaneous fibroses. On the other hand, overexpression of Cav1 can also alter cellular migration and lead to decreased collagen deposition and thus have unfavourable outcomes in wound closure, cellular senescence and the resulting age‐related changes in skin as well as in progression of melanoma. Not only would it be of interested to target Cav1 expression in various cutaneous pathologies outlined above, but if Cav1 expression is confirmed to be either up‐ or downregulated in specific conditions by studies with larger samples sizes, Cav1 may become a very useful theragnostic (therapeutic and diagnostic) tool in dermatology.
Figure 2.
Pleiotropic Roles of Cav1 in Skin Physiology
Figure 3.
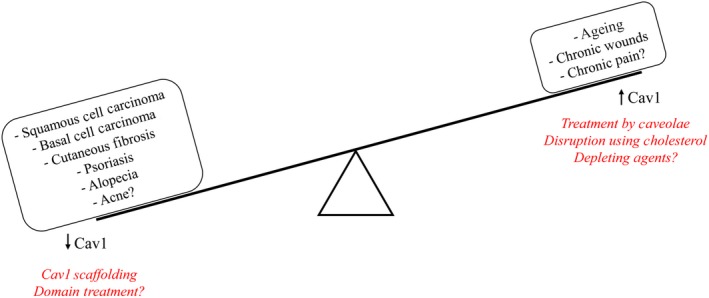
Skin Pathophysiology associated with deregulation of homeostatic Cav1 levels. It is the fine balance of Cav1 that is key to physiologic skin structure and function, as the slight imbalance in one direction or another affects localization and activation of various key signalling molecules and can tip the scale towards one of numerous skin pathologies. On one side, upregulation of Cav1 has been implicated in skin ageing, development of non‐healing chronic wounds and, whereas downregulation has been associated with squamous cell carcinoma, basal cell carcinoma, cutaneous fibroses, psoriasis and alopecia
It is also important to note that some of the observed effects of Cav1 on certain cutaneous disorders come from animal models (ie Cav1‐null mice) which do not always recapitulate human conditions and thus should be taken cautiously. Therefore, our understanding of the role Cav1 plays in development and progression of numerous cutaneous disorders may be limited by those models and highlights the need for development of better models which more closely mirror the human condition. Further, current therapeutic strategies in conditions such as psoriasis, scleroderma, ageing and especially wound healing have their limitations and beget newly discovered targets. At the same time, current strategies do not target Cav1, which evidently plays a role in these pathologies, and has now become the spotlight as a potential new and exciting therapeutic target candidate. While many of the aforementioned functions may not be confirmed in humans, are not fully elucidated and in some cases even controversial, the vast preclinical data discussed are certainly promising and exciting for both scientists as well as clinicians and should serve as a large stepping stone towards more confirmatory and useful clinical and therapeutic innovations. Likewise, there are still many unanswered questions regarding the role of Cav1 in development of other cutaneous pathophysiologies not highlighted in this review. For example, although there is ample evidence supporting the role of Cav1 in internalization of various aforementioned bacteria, does Cav1 also regulate internalization of Cutibacterium acnes, and thus can Cav1 be targeted in treatment of acne? Also, why is Cav1 expression consistently low in most neuronal cells and what implications does this have on development of pruritus and/or various cutaneous neuropathies? There is some evidence from mouse studies that point to persistent upregulation and activation of Cav1 in the anterior cingulate cortex neurons after chronic constriction injury,200 suggesting a possible role for Cav1 in neuronal transmission pathways associated with pain modulation; however, these are still very preliminary. Lastly, Tim Thompson's group has identified differentially expressed levels of Cav1 in White American vs African American prostate cancer patients201; thus, it would be of great interest to elucidate whether such ethnic/racial disparities of Cav1 expression are present in cutaneous disorders that are known to disproportionally affect ethnic/racial minorities including melanoma and non‐melanoma skin cancers as well as atopic dermatitis (among others). In all, the vast aforementioned preclinical data on Cav1 are certainly inspiring, and as it becomes more clear‐cut, and is recapitulated in humans in the future, it should serve as a large stepping stone towards more confirmatory and useful clinical and therapeutic innovations, ultimately leading to a more effective and successful management of cancerous, psoriatic, senescent, fibrotic and non‐healing skin lesions.
CONFLICT OF INTEREST STATEMENT
The authors declare no competing conflicts of interest.
AUTHOR CONTRIBUTIONS
IJ designed the manuscript and ANE, AR, NMW, SRR, JDF, LLW and IJ wrote the manuscript cooperatively.
ACKNOWLEDGEMENTS
This work was funded in part by a PhRMA Foundation Research Starter Grant and a Medline Wound Healing Foundation Innovation Grant (to IJ).
Egger AN, Rajabi‐Estarabadi A, Williams NM, et al. The importance of caveolins and caveolae to dermatology: Lessons from the caves and beyond. Exp Dermatol. 2020;29:136–148. 10.1111/exd.14068
Andjela N. Egger and Ali Rajabi‐Estarabadi contributed equally to this work.
[Correction added on 9 March 2020, after first online publication: The author name was incorrect and has been changed from Ali Rajabiestarabadi to Ali Rajabi‐Estarabadi in this current version.]
REFERENCES
- 1. Yamada E, J. Biophys. Biochem. Cytol. 1955, 1(5), 445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Palade G, Fine structure of blood capillaries. J. Appl. Phys. 1953, 24, 1424. [Google Scholar]
- 3. Couet J, Belanger MM, Roussel E, Drolet MC, Adv. Drug. Deliv. Rev. 2001, 49(3), 223. [DOI] [PubMed] [Google Scholar]
- 4. Park S, Glover KJ, Im W, J. Comput. Chem. 2019, 40(16), 1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lisanti MP, Scherer PE, Vidugiriene J, Tang Z, Hermanowski‐Vosatka A, Tu YH, Cook RF, Sargiacomo M, J. Cell Biol. 1994, 126(1), 111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Anderson RG, Kamen BA, Rothberg KG, Lacey SW, Science 1992, 255(5043), 410. [DOI] [PubMed] [Google Scholar]
- 7. Fernandez I, Ying Y, Albanesi J, Anderson RG, Proc. Natl. Acad. Sci. USA. 2002, 99(17), 11193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Root KT, Julien JA, Glover KJ, Biochem. Soc. Trans. 2019, 47, 1489. [DOI] [PubMed] [Google Scholar]
- 9. Cao G, Yang G, Timme TL, Saika T, Truong LD, Satoh T, Goltsov A, Park SH, Men T, Kusaka N, Tian W, Am. J. Pathol. 2003, 162(4), 1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Quest AF, Gutierrez‐Pajares JL, Torres VA, J. Cell Mol. Med. 2008, 12(4), 1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Razani B, Schlegel A, Liu J, Lisanti MP, Biochem. Soc. Trans. 2001, 29(Pt 4), 494. [DOI] [PubMed] [Google Scholar]
- 12. Fielding PE, Chau P, Liu D, Spencer TA, Fielding CJ, Biochemistry 2004, 43(9), 2578. [DOI] [PubMed] [Google Scholar]
- 13. Parton RG, Hanzal‐Bayer M, Hancock JF, J. Cell Sci. 2006, 119(Pt 5), 787. [DOI] [PubMed] [Google Scholar]
- 14. Parton RG, Howes MT, J. Cell Biol. 2010, 191, 439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hayer A, Stoeber M, Ritz D, Engel S, Meyer HH, Helenius A, J. Cell Biol. 2010, 191(3), 615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ostermeyer AG, Paci JM, Zeng Y, Lublin DM, Munro S, Brown DA, J. Cell Biol. 2001, 152(5), 1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Scheiffele P, Verkade P, Fra AM, Virta H, Simons K, Ikonen E, J. Cell Biol. 1998, 140(4), 795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Khater IM, Liu Q, Chou KC, Hamarneh G, Nabi IR, Sci. Rep. 2019, 9(1), 9888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Graf GA, Matveev SV, Smart EJ, Trends Cardiovasc. Med. 1999, 9(8), 221. [DOI] [PubMed] [Google Scholar]
- 20. Williams TM, Lisanti MP, Genome Biol. 2004, 5(3), 214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Anderson RG, Annu. Rev. Biochem. 1998, 67, 199. [DOI] [PubMed] [Google Scholar]
- 22. Pelkmans L, Burli T, Zerial M, Helenius A, Cell 2004, 118(6), 767. [DOI] [PubMed] [Google Scholar]
- 23. Pelkmans L, Helenius A, Traffic 2002, 3(5), 311. [DOI] [PubMed] [Google Scholar]
- 24. Mundy DI, Machleidt T, Ying YS, Anderson RG, Bloom GS, J. Cell Sci. 2002, 115(Pt 22), 4327. [DOI] [PubMed] [Google Scholar]
- 25. Thomsen P, Roepstorff K, Stahlhut M, van Deurs B, Mol. Biol. Cell. 2002, 13(1), 238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Roy S, Luetterforst R, Harding A, Apolloni A, Etheridge M, Stang E, Rolls B, Hancock JF, Parton RG, Nat. Cell Biol. 1999, 1(2), 98. [DOI] [PubMed] [Google Scholar]
- 27. Henley JR, Krueger EW, Oswald BJ, McNiven MA, J. Cell Biol. 1998, 141(1), 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Le PU, Guay G, Altschuler Y, Nabi IR, J. Biol. Chem. 2002, 277(5), 3371. [DOI] [PubMed] [Google Scholar]
- 29. Liu P, Rudick M, Anderson RG, J. Biol. Chem. 2002, 277, 41295. [DOI] [PubMed] [Google Scholar]
- 30. Chang WJ, Rothberg KG, Kamen BA, J. Cell Biol. 1992, 118(1), 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Murata M, Peranen J, Schreiner R, Wieland F, Kurzchalia TV, Simons K, Proc. Natl. Acad. Sci. USA. 1995, 92(22), 10339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Golani G, Ariotti N, Parton RG, Kozlov MM, Dev. Cell. 2019, 48(4), 523. [DOI] [PubMed] [Google Scholar]
- 33. Krishna A, Sengupta D, Biophys. J. 2019, 116(1), 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Raggi C, Diociaiuti M, Caracciolo G, Fratini F, Fantozzi L, Piccaro G, Fecchi K, Pizzi E, Marano G, Ciaffoni F, Bravo E, Biomolecules. 2019, 9(7), 287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Otis JP, Shen MC, Quinlivan V, Anderson JL, Farber SA, Dis. Model Mech. 2017, 10(3), 283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sohn J, Lin H, Fritch MR, Tuan RS, Stem Cell Res. Ther. 2018, 9(1), 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bist A, Fielding PE, Fielding CJ, Proc. Natl. Acad. Sci. USA 1997, 94(20), 10693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Smart EJ, Ying YS, Conrad PA, Anderson RG, J. Cell. Biol. 1994, 127(5), 1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mineo C, James GL, Smart EJ, Anderson RG, J. Biol. Chem. 1996, 271(20), 11930. [DOI] [PubMed] [Google Scholar]
- 40. Li WP, Liu P, Pilcher BK, J. Cell Sci. 2001, 114(Pt 7), 1397. [DOI] [PubMed] [Google Scholar]
- 41. Frank PG, Pavlides S, Cheung MW, Daumer K, Lisanti MP, Am. J. Physiol. Cell Physiol. 2008, 295(1), C242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Heimerl S, Liebisch G, Le Lay S, Böttcher A, Wiesner P, Lindtner S, Kurzchalia TV, Simons K, Schmitz G, Biochem. Biophys. Res. Commun. 2008, 367(4), 826. [DOI] [PubMed] [Google Scholar]
- 43. Bai X, Yang X, Jia X, Rong Y, Chen L, Zeng T, Deng X, Li W, Wu G, Wang L, Li Y, Autophagy 2019, 1 10.1080/15548627.2019.1659613 [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lim JS, Choy HE, Park SC, Han JM, Jang IS, Cho KA, Aging Cell 2010, 9(2), 243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kiss AL, Botos E, J. Cell Mol. Med. 2009, 13(7), 1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gruenberg J, van der Goot FG, Nat. Rev. Mol. Cell Biol. 2006, 7(7), 495. [DOI] [PubMed] [Google Scholar]
- 47. Pang H, Le PU, Nabi IR, J. Cell Sci. 2004, 117(Pt 8), 1421. [DOI] [PubMed] [Google Scholar]
- 48. Ferrari A, Pellegrini V, Arcangeli C, Fittipaldi A, Giacca M, Beltram F, Mol. Ther. 2003, 8(2), 284. [DOI] [PubMed] [Google Scholar]
- 49. Zaas DW, Swan Z, Brown BJ, Wright JR, Abraham SN, Commun. Integr. Biol. 2009, 2(6), 535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kumar GA, Karmakar J, Mandal C, Chattopadhyay A, Sci. Rep. 2019, 9(1), 12636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Owczarek K, Szczepanski A, Milewska A, Baster Z, Rajfur Z, Sarna M, Pyrc K, Sci. Rep. 2018, 8(1), 7124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sanderlin AG, Vondrak C, Scricco AJ, Fedrigo I, Ahyong V, Lamason RL, Mol. Biol. Cell. 2019, 30(17), 2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Szczepanski A, Owczarek K, Milewska A, Baster Z, Rajfur Z, Mitchell JA, Pyrc K, Vet. Res. 2018, 49(1), 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lim JS, Shin M, Kim HJ, Kim KS, Choy HE, Cho KA, J. Infect. Dis. 2014, 210(5), 793. [DOI] [PubMed] [Google Scholar]
- 55. Hoffmann C, Berking A, Agerer F, Buntru A, Neske F, Chhatwal GS, Ohlsen K, Hauck CR, J. Cell Sci. 2010, 123(Pt 24), 4280. [DOI] [PubMed] [Google Scholar]
- 56. Jozic I, Sawaya AP, Pastar I, Head CR, Wong LL, Glinos GD, Wikramanayake TC, Brem H, Kirsner RS, Tomic‐Canic M, Mol. Ther. 2019, 27(1), 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Codrici E, Albulescu L, Popescu ID, Mihai S, Enciu AM, Albulescu R, Tanase C, Hinescu ME, J. Immunol. Res. 2018, 2018, 2498576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Li X, Gu X, Boyce TM, Zheng M, Reagan AM, Qi H, Mandal N, Cohen AW, Callegan MC, Carr DJ, Elliott MH, Invest. Ophthalmol. Vis. Sci. 2014, 55(10), 6224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Mathay C, Pierre M, Pittelkow MR, Depiereux E, Nikkels AF, Colige A, Poumay Y, J. Invest. Dermatol. 2011, 131(1), 46‐58. [DOI] [PubMed] [Google Scholar]
- 60. Jiao H, Zhang Y, Yan Z, Wang ZG, Liu G, Minshall RD, Malik AB, Hu G, J. Immunol. 2013, 191(12), 6191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Li S, Couet J, Lisanti MP, J. Biol. Chem. 1996, 271(46), 29182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Khater IM, Aroca‐Ouellette ST, Meng F, Nabi IR, Hamarneh G, PLoS ONE 2019, 14(8), e0211659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Couet J, Li S, Okamoto T, Ikezu T, Lisanti MP, J. Biol. Chem. 1997, 272(10), 6525. [DOI] [PubMed] [Google Scholar]
- 64. Okamoto T, Schlegel A, Scherer PE, Lisanti MP, J. Biol. Chem. 1998, 273(10), 5419. [DOI] [PubMed] [Google Scholar]
- 65. Cornejo G, Fuentes L, Jacob E, Subphrenic abscess (author's transl). Rev. Med. Chil. 1975, 103(1), 17. [PubMed] [Google Scholar]
- 66. Czarny M, Lavie Y, Fiucci G, Liscovitch M, J. Biol. Chem. 1999, 274(5), 2717. [DOI] [PubMed] [Google Scholar]
- 67. Machleidt T, Li WP, Liu P, Anderson RG, J. Cell Biol. 2000, 148(1), 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Yamamoto M, Toya Y, Schwencke C, Lisanti MP, Myers MG Jr, Ishikawa Y, J. Biol. Chem. 1998, 273(41), 26962. [DOI] [PubMed] [Google Scholar]
- 69. Park WY, Park JS, Cho KA, Kim DI, Ko YG, Seo JS, Park SC, J. Biol. Chem. 2000, 275(27), 20847. [DOI] [PubMed] [Google Scholar]
- 70. Zhang W, Razani B, Altschuler Y, Bouzahzah B, Mostov KE, Pestell RG, Lisanti MP, J. Biol. Chem. 2000, 275(27), 20717. [DOI] [PubMed] [Google Scholar]
- 71. Jang IH, Kim JH, Lee BD, Bae SS, Park MH, Suh PG, Ryu SH, FEBS Lett. 2001, 491(1–2), 4. [DOI] [PubMed] [Google Scholar]
- 72. Razani B, Zhang XL, Bitzer M, von Gersdorff G, Bottinger EP, Lisanti MP, J. Biol. Chem. 2001, 276(9), 6727. [DOI] [PubMed] [Google Scholar]
- 73. Ushio‐Fukai M, Griendling KK, Becker PL, Hilenski L, Halleran S, Alexander RW, Arterioscler. Thromb. Vasc. Biol. 2001, 21(4), 489. [DOI] [PubMed] [Google Scholar]
- 74. Cohen AW, Razani B, Wang XB, Combs TP, Williams TM, Scherer PE, Lisanti MP, Am. J. Physiol. Cell Physiol. 2003, 285(1), C222. [DOI] [PubMed] [Google Scholar]
- 75. Lajoie P, Partridge EA, Guay G, Goetz JG, Pawling J, Lagana A, Joshi B, Dennis JW, Nabi IR, J. Cell Biol. 2007, 179(2), 341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Zhang B, Peng F, Wu D, Ingram AJ, Gao B, Krepinsky JC, Cell Signal. 2007, 19(8), 1690. [DOI] [PubMed] [Google Scholar]
- 77. Agelaki S, Spiliotaki M, Markomanolaki H, Kallergi G, Mavroudis D, Georgoulias V, Stournaras C, Cancer Biol. Ther. 2009, 8(15), 1470. [DOI] [PubMed] [Google Scholar]
- 78. Park JH, Han HJ, Am. J. Physiol. Cell Physiol. 2009, 297(4), C935. [DOI] [PubMed] [Google Scholar]
- 79. Yamamoto M, Toya Y, Jensen RA, Ishikawa Y, Exp. Cell Res. 1999, 247(2), 380. [DOI] [PubMed] [Google Scholar]
- 80. Labrecque L, Royal I, Surprenant DS, Patterson C, Gingras D, Beliveau R, Mol. Biol. Cell. 2003, 14(1), 334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Pereira PMR, Sharma SK, Carter LM, Edwards KJ, Pourat J, Ragupathi A, Janjigian YY, Durack JC, Lewis JS, Nat. Commun. 2018, 9(1), 5137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Ishizaka N, Griendling KK, Lassegue B, Alexander RW, Hypertension 1998, 32(3), 459. [DOI] [PubMed] [Google Scholar]
- 83. Schlegel A, Wang C, Pestell RG, Lisanti MP, Biochem. J. 2001, 359(Pt 1), 203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Razandi M, Alton G, Pedram A, Ghonshani S, Webb P, Levin ER, Mol. Cell Biol. 2003, 23(5), 1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Wyse BD, Prior IA, Qian H, Morrow IC, Nixon S, Muncke C, Kurzchalia TV, Thomas WG, Parton RG, Hancock JF, J. Biol. Chem. 2003, 278(26), 23738. [DOI] [PubMed] [Google Scholar]
- 86. Hong YH, Kim JY, Lee JH, Chae HG, Jang SS, Jeon JH, Kim CH, Kim J, Kim SJ, J. Neurochem. 2009, 111(1), 61. [DOI] [PubMed] [Google Scholar]
- 87. Matthews L, Berry A, Ohanian V, Ohanian J, Garside H, Ray D, Mol. Endocrinol. 2008, 22(6), 1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Lu ML, Schneider MC, Zheng Y, Zhang X, Richie JP, J. Biol. Chem. 2001, 276(16), 13442. [DOI] [PubMed] [Google Scholar]
- 89. Chinnakkannu P, Reese C, Gaspar JA, Panneerselvam S, Pleasant‐Jenkins D, Mukherjee R, Baicu C, Tourkina E, Hoffman S, Kuppuswamy D, PLoS ONE 2018, 13(12), e0207844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Li S, Seitz R, Lisanti MP, J. Biol. Chem. 1996, 271(7), 3863. [PubMed] [Google Scholar]
- 91. Song KS, Li S, Okamoto T, Quilliam LA, Sargiacomo M, Lisanti MP, J. Biol. Chem. 1996, 271(16), 9690. [DOI] [PubMed] [Google Scholar]
- 92. Engelman JA, Chu C, Lin A, Jo H, Ikezu T, Okamoto T, Kohtz DS, Lisanti MP, FEBS Lett. 1998, 428(3), 205. [DOI] [PubMed] [Google Scholar]
- 93. Galbiati F, Volonte D, Engelman JA, Watanabe G, Burk R, Pestell RG, Lisanti MP, EMBO J. 1998, 17(22), 6633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Kim JH, Han JM, Lee S, Kim Y, Lee TG, Park JB, Lee SD, Suh PG, Ryu SH, Biochemistry 1999, 38(12), 3763. [DOI] [PubMed] [Google Scholar]
- 95. Razani B, Rubin CS, Lisanti MP, J. Biol. Chem. 1999, 274(37), 26353. [DOI] [PubMed] [Google Scholar]
- 96. Czarny M, Fiucci G, Lavie Y, Banno Y, Nozawa Y, Liscovitch M, FEBS Lett. 2000, 467(2–3), 326. [DOI] [PubMed] [Google Scholar]
- 97. Prevostel C, Alice V, Joubert D, Parker PJ, J. Cell Sci. 2000, 11(Pt 14), 2575. [DOI] [PubMed] [Google Scholar]
- 98. Kranenburg O, Verlaan I, Moolenaar WH, Curr. Biol. 2001, 11(23), 1880. [DOI] [PubMed] [Google Scholar]
- 99. Razani B, Lisanti MP, Am. J. Physiol. Cell Physiol. 2001, 281(4), C1241. [DOI] [PubMed] [Google Scholar]
- 100. Cao H, Courchesne WE, Mastick CC, J. Biol. Chem. 2002, 277(11), 8771. [DOI] [PubMed] [Google Scholar]
- 101. Zhuang L, Lin J, Lu ML, Solomon KR, Freeman MR, Cancer Res. 2002, 62(8), 2227. [PubMed] [Google Scholar]
- 102. Biedi C, Panetta D, Segat D, Cordera R, Maggi D, Endocrinology 2003, 144(12), 5497. [DOI] [PubMed] [Google Scholar]
- 103. Cao H, Sanguinetti AR, Mastick CC, Exp. Cell Res. 2004, 294(1), 159. [DOI] [PubMed] [Google Scholar]
- 104. Panetta D, Biedi C, Repetto S, Cordera R, Maggi D, Biochem. Biophys. Res. Commun. 2004, 316(1), 240. [DOI] [PubMed] [Google Scholar]
- 105. Waschke J, Golenhofen N, Kurzchalia TV, Drenckhahn D, Histochem. Cell Biol. 2006, 126(1), 17. [DOI] [PubMed] [Google Scholar]
- 106. Gottlieb‐Abraham E, Shvartsman DE, Donaldson JC, Ehrlich M, Gutman O, Martin GS, Henis YI, Mol. Biol. Cell. 2013, 24, 3881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Jozic I, Vukelic S, Stojadinovic O, Liang L, Ramirez HA, Pastar I, Canic MT, J. Invest. Dermatol. 2017, 137(5), 1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Lin F, Pei L, Zhang Q, Han W, Jiang S, Lin Y, Dong B, Cui L, Li M, J. Cell Physiol. 2018, 233(10), 6683. [DOI] [PubMed] [Google Scholar]
- 109. Llanses Martinez M, Rainero E, Essays Biochem. 2019, 63(5), 469. [DOI] [PubMed] [Google Scholar]
- 110. Park SY, Park JW, Lee GW, Li L, Chun YS, BMC Cancer 2018, 18(1), 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Reppetti J, Reca A, Seyahian EA, Medina Y, Martínez N, Szpilbarg N, Damiano AE, J. Cell Physiol. 2019, 1 10.1002/jcp.29226 [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 112. Isshiki M, Ando J, Yamamoto K, Fujita T, Ying Y, Anderson RG, J. Cell Sci. 2002, 115(Pt 3), 475. [DOI] [PubMed] [Google Scholar]
- 113. Isshiki M, Ying YS, Fujita T, Anderson RG, J. Biol. Chem. 2002, 277(45), 43389. [DOI] [PubMed] [Google Scholar]
- 114. Parat MO, Anand‐Apte B, Fox PL, Mol. Biol. Cell. 2003, 14(8), 3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Beardsley A, Fang K, Mertz H, Castranova V, Friend S, Liu J, J. Biol. Chem. 2005, 280(5), 3541. [DOI] [PubMed] [Google Scholar]
- 116. Sun XH, Flynn DC, Castranova V, Millecchia LL, Beardsley AR, Liu J, J. Biol. Chem. 2007, 282(10), 7232. [DOI] [PubMed] [Google Scholar]
- 117. Grande‐Garcia A, Echarri A, de Rooij J, Alderson NB, Waterman‐Storer CM, Valdivielso JM, del Pozo MA, J. Cell Biol. 2007, 177(4), 683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Gonzalez E, Nagiel A, Lin AJ, Golan DE, Michel T, J. Biol. Chem. 2004, 279(39), 40659. [DOI] [PubMed] [Google Scholar]
- 119. Brouet A, DeWever J, Martinive P, Havaux X, Bouzin C, Sonveaux P, Feron O, FASEB J. 2005, 19(6), 602. [DOI] [PubMed] [Google Scholar]
- 120. Cantiani L, Manara MC, Zucchini C, De Sanctis P, Zuntini M, Valvassori L, Serra M, Olivero M, Di Renzo MF, Colombo MP, Picci P, Cancer Res. 2007, 67(16), 7675. [DOI] [PubMed] [Google Scholar]
- 121. Feron O, Saldana F, Michel JB, Michel T, J. Biol. Chem. 1998, 273(6), 3125. [DOI] [PubMed] [Google Scholar]
- 122. Garcia‐Cardena G, Martasek P, Masters BS, Skidd PM, Couet J, Li S, Lisanti MP, Sessa WC, J. Biol. Chem. 1997, 272(41), 25437. [DOI] [PubMed] [Google Scholar]
- 123. Wary KK, Mainiero F, Isakoff SJ, Marcantonio EE, Giancotti FG, Cell 1996, 87(4), 733. [DOI] [PubMed] [Google Scholar]
- 124. Wei Y, Yang X, Liu Q, Wilkins JA, Chapman HA, J. Cell Biol. 1999, 144(6), 1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Goetz JG, Joshi B, Lajoie P, Strugnell SS, Scudamore T, Kojic LD, Nabi IR, J. Cell Biol. 2008, 180(6), 1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Lee H, Park DS, Razani B, Russell RG, Pestell RG, Lisanti MP, Am. J. Pathol. 2002, 161(4), 1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Lingwood D, Simons K, Science 2010, 327(5961), 46. [DOI] [PubMed] [Google Scholar]
- 128. Stahl A, Mueller BM, J. Cell Biol. 1995, 129(2), 335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Tang W, Hemler ME, J. Biol. Chem. 2004, 279(12), 11112. [DOI] [PubMed] [Google Scholar]
- 130. Galbiati F, Volonte D, Liu J, Capozza F, Frank PG, Zhu L, Pestell RG, Lisanti MP, Mol. Biol. Cell. 2001, 12(8), 2229‐2244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Gille H, Downward J, J. Biol. Chem. 1999, 274(31), 22033. [DOI] [PubMed] [Google Scholar]
- 132. Lavoie JN, L'Allemain G, Brunet A, Muller R, Pouyssegur J, J. Biol. Chem. 1996, 271(34), 20608. [DOI] [PubMed] [Google Scholar]
- 133. Shtutman M, Zhurinsky J, Simcha I, Albanese C, D'Amico M, Pestell R, Ben‐Ze'ev A. Proc. Natl. Acad. Sci. USA 1999, 96(10), 5522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Geraci MW, Moore M, Gesell T, Yeager ME, Alger L, Golpon H, Gao B, Loyd JE, Tuder RM, Voelkel NF, Circ. Res. 2001, 88(6), 555. [DOI] [PubMed] [Google Scholar]
- 135. Camoretti‐Mercado B, Transl. Res. 2009, 154(4), 165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Di Guglielmo GM, Le Roy C, Goodfellow AF, Wrana JL, Nat. Cell Biol. 2003, 5(5), 410. [DOI] [PubMed] [Google Scholar]
- 137. Murphy DM, O'Byrne PM, Chest 2010, 137(6), 1417. [DOI] [PubMed] [Google Scholar]
- 138. Cerezo A, Guadamillas MC, Goetz JG, Sánchez‐Perales S, Klein E, Assoian RK, Del Pozo MA, Mol. Cell Biol. 2009, 29(18), 5046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Hulit J, Bash T, Fu M, Galbiati F, Albanese C, Sage DR, Schlegel A, Zhurinsky J, Shtutman M, Ben‐Ze'ev A, Lisanti MP, J. Biol. Chem. 2000, 275(28), 21203. [DOI] [PubMed] [Google Scholar]
- 140. Gassmann MG, Werner S, Exp. Cell Res. 2000, 258(1), 23. [DOI] [PubMed] [Google Scholar]
- 141. Sando GN, Zhu H, Weis JM, Richman JT, Wertz PW, Madison KC, J. Invest. Dermatol. 2003, 120(4), 531. [DOI] [PubMed] [Google Scholar]
- 142. Qin H, Bollag WB, PLoS ONE 2013, 8(11), e80946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Campbell L, Gumbleton M, IUBMB Life 2000, 50(6), 361. [DOI] [PubMed] [Google Scholar]
- 144. Ma WY, Zhuang L, Cai DX, Zhong H, Zhao C, Sun Q, J. Int. Med. Res. 2012, 40(5), 1745. [DOI] [PubMed] [Google Scholar]
- 145. Zhang F, Li H, Zhou Y, Gu Y, Wang L, Indian J. Dermatol. 2014, 59(3), 225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Kruglikov IL, Scherer PE, NPJ. Regen. Med. 2019, 4, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Domingues L, Hurbain I, Gilles‐Marsens F, Andre N, Dewulf M, Romao M, de Lesegno CV, Blouin C, Guere C, Vie K, Raposo G. bioRxiv. 2019, 666388 10.1101/666388 [Epub ahead of print]. [DOI] [Google Scholar]
- 148. Selleri S, Arnaboldi F, Palazzo M, Hussein U, Balsari A, Rumio C, Br. J. Dermatol. 2005, 153(3), 506. [DOI] [PubMed] [Google Scholar]
- 149. Hawkshaw NJ, Hardman JA, Haslam IS, Shahmalak A, Gilhar A, Lim X, Paus R, PLoS Biol. 2018, 16(5), e2003705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Maurer M, Handjiski B, Paus R, Am. J. Pathol. 1997, 150(4), 1433. [PMC free article] [PubMed] [Google Scholar]
- 151. Chew EGY, Tan JHJ, Bahta AW, Ho BS, Liu X, Lim TC, Sia YY, Bigliardi PL, Heilmann S, Wan AC, Nöthen MM, J. Invest. Dermatol. 2016, 136(8), 1559. [DOI] [PubMed] [Google Scholar]
- 152. Zhang Z, Shao M, Hepler C, Zi Z, Zhao S, An YA, Zhu Y, Ghaben AL, Wang MY, Li N, Onodera T, J. Clin. Invest. 2019, 129, 5327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Shi XE, Li YF, Jia L, Ji HL, Song ZY, Cheng J, Wu GF, Song CC, Zhang QL, Zhu JY, Yang GS, Int. J. Mol. Sci. 2014, 15(5), 8526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Fu P, Chen F, Pan Q, Zhao X, Zhao C, Cho WC, Chen H, Onco Targets Ther. 2017, 10, 819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Wiechen K, Sers C, Agoulnik A, Arlt K, Dietel M, Schlag PM, Schneider U, Am. J. Pathol. 2001, 158(3), 833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Yang G, Truong LD, Timme TL, Ren C, Wheeler TM, Park SH, Nasu Y, Bangma CH, Kattan MW, Scardino PT, Thompson TC, Clin. Cancer Res. 1998, 4(8), 1873. [PubMed] [Google Scholar]
- 157. Gheida SF, Neinaa YM, Mohammed DA, Dermatologic Sinica. 2018, 36, 179. [Google Scholar]
- 158. Trimmer C, Bonuccelli G, Katiyar S, Sotgia F, Pestell RG, Lisanti MP, Capozza F, Am. J. Pathol. 2013, 182(3), 992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Tas F, Karabulut S, Tilgen Yasasever C, Duranyildiz D, Int. J. Dermatol. 2016, 55(5), 558. [DOI] [PubMed] [Google Scholar]
- 160. Logozzi M, De Milito A, Lugini L, Borghi M, Calabro L, Spada M, Perdicchio M, Marino ML, Federici C, Iessi E, Brambilla D, PLoS ONE 2009, 4(4), e5219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Nakashima H, Hamamura K, Houjou T, Taguchi R, Yamamoto N, Mitsudo K, Tohnai I, Ueda M, Urano T, Furukawa K, Furukawa K, Cancer Sci. 2007, 98(4), 512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Wu KN, Queenan M, Brody JR, Potoczek M, Sotgia F, Lisanti MP, Witkiewicz AK, Cell Cycle 2011, 10(24), 4250. [DOI] [PubMed] [Google Scholar]
- 163. Trimmer C, Whitaker‐Menezes D, Bonuccelli G, Milliman JN, Daumer KM, Aplin AE, Pestell RG, Sotgia F, Lisanti MP, Capozza F, Cancer Res. 2010, 70(19), 7489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Capozza F, Trimmer C, Castello‐Cros R, Katiyar S, Whitaker‐Menezes D, Follenzi A, Crosariol M, Llaverias G, Sotgia F, Pestell RG, Lisanti MP, Cancer Res. 2012, 72(9), 2262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Lobos‐Gonzalez L, Aguilar‐Guzman L, Fernandez JG, Muñoz N, Hossain M, Bieneck S, Silva V, Burzio V, Sviderskaya EV, Bennett DC, Leyton L, Melanoma Res. 2014, 24(2), 108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Lee R, Perry B, Heywood J, Reese C, Bonner M, Hatfield CM, Silver RM, Visconti RP, Hoffman S, Tourkina E, Front. Pharmacol. 2014, 5, 140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Del Galdo F, Sotgia F, de Almeida CJ, Jasmin JF, Musick M, Lisanti MP, Jiménez SA, Arthritis Rheum. 2008, 58(9), 2854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Manetti M, Allanore Y, Saad M, Fatini C, Cohignac V, Guiducci S, Romano E, Airó P, Caramaschi P, Tinazzi I, Riccieri V, Ann. Rheum. Dis. 2012, 71(6), 1034. [DOI] [PubMed] [Google Scholar]
- 169. Liakouli V, Elies J, El‐Sherbiny YM, Scarcia M, Grant G, Abignano G, Derrett‐Smith EC, Esteves F, Cipriani P, Emery P, Denton CP, Ann. Rheum. Dis. 2018, 77(3), 431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Harn HI, Ogawa R, Hsu CK, Hughes MW, Tang MJ, Chuong CM, Exp. Dermatol. 2019, 28(4), 464. [DOI] [PubMed] [Google Scholar]
- 171. Zhang GY, Yu Q, Cheng T, Liao T, Nie CL, Wang AY, Zheng X, Xie XG, Albers AE, Gao WY, Br. J. Dermatol. 2011, 164(3), 623. [DOI] [PubMed] [Google Scholar]
- 172. Rubio GA, Elliot SJ, Wikramanayake TC, Xia X, Pereira‐Simon S, Thaller SR, Glinos GD, Jozic I, Hirt P, Pastar I, Tomic‐Canic M, J. Cell Physiol. 2018, 233(8), 5503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Qian N, Ueno T, Arch Med Res. 2010, 41(4), 297. [DOI] [PubMed] [Google Scholar]
- 174. Haines P, Samuel GH, Cohen H, Trojanowska M, Bujor AM, J. Dermatol. Sci. 2011, 64(3), 210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Kim S, Han J, Lee DH, Cho KH, Kim KH, Chung JH, Ann. Dermatol. 2010, 22(4), 379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Lee R, Reese C, Perry B, Heywood J, Bonner M, Zemskova M, Silver RM, Hoffman S, Tourkina E, Fibrogenesis Tissue Repair. 2015, 8, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Tourkina E, Gooz P, Pannu J, Bonner M, Scholz D, Hacker S, Silver RM, Trojanowska M, Hoffman S, J. Biol. Chem. 2005, 280(14), 13879. [DOI] [PubMed] [Google Scholar]
- 178. Tourkina E, Richard M, Gooz P, Bonner M, Pannu J, Harley R, Bernatchez PN, Sessa WC, Silver RM, Hoffman S, Am. J. Physiol. Lung Cell Mol. Physiol. 2008, 294(5), L843. [DOI] [PubMed] [Google Scholar]
- 179. Kruglikov IL, Scherer PE, NPJ. Aging Mech. Dis. 2019, 5, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Takamura N, Yamaguchi Y, Watanabe Y, Asami M, Komitsu N, Aihara M, Sci. Rep. 2019, 9(1), 125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Roelandt T, Giddelo C, Heughebaert C, Denecker G, Hupe M, Crumrine D, Kusuma A, Haftek M, Roseeuw D, Declercq W, Feingold KR, J. Invest. Dermatol. 2009, 129(4), 927. [DOI] [PubMed] [Google Scholar]
- 182. Volonte D, Zhang K, Lisanti MP, Galbiati F, Mol. Biol. Cell. 2002, 13(7), 2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Chen YH, Lin WW, Liu CS, Su SL, Mol Med Rep. 2017, 16(5), 7841. [DOI] [PubMed] [Google Scholar]
- 184. Sun S, Cai B, Li Y, Su W, Zhao X, Gong B, Li Z, Zhang X, Wu Y, Chen C, Tsang SH, 2019, 11(13), 4323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Yamao T, Yamashita YI, Yamamura K, Nakao Y, Tsukamoto M, Nakagawa S, Okabe H, Hayashi H, Imai K, Baba H, Ann. Surg. Oncol. 2019, 26(5), 1552. [DOI] [PubMed] [Google Scholar]
- 186. Bartholomew JN, Volonte D, Galbiati F, Cancer Res. 2009, 69(7), 2878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Yu DM, Jung SH, An HT, Lee S, Hong J, Park JS, Lee H, Lee H, Bahn MS, Lee HC, Han NK, Aging Cell 2017, 16(4), 773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Castello‐Cros R, Whitaker‐Menezes D, Molchansky A, Purkins G, Soslowsky LJ, Beason DP, Sotgia F, Iozzo RV, Lisanti MP, Cell Cycle 2011, 10(13), 2140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Boopathi E, Gomes CM, Goldfarb R, John M, Srinivasan VG, Alanzi J, Malkowicz SB, Kathuria H, Zderic SA, Wein AJ, Chacko S, Am. J. Pathol. 2011, 178(5), 2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. van den Heuvel AP, Schulze A, Burgering BM, Biochem. J. 2005, 385(Pt 3), 795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Lee JA, Choi DI, Choi JY, Kim SO, Cho KA, Lee JB, Yun SJ, Lee SC, Oncotarget. 2015, 6(4), 1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Kruglikov IL, Zhang Z, Scherer PE, Ageing Res. Rev. 2019, 55, 100959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Nguyen KC, Cho KA, Chonnam. Med. J. 2017, 53(1), 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Rhim JH, Kim JH, Yeo EJ, Kim JC, Park SC, Mol Med. 2010, 16(11–12), 527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Gu X, Reagan AM, McClellan ME, Elliott MH, Prog. Retin. Eye Res. 2017, 56, 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Lizarbe TR, Garcia‐Rama C, Tarin C, Saura M, Calvo E, López JA, López‐Otín C, Folgueras AR, Lamas S, Zaragoza C, FASEB J. 2008, 22(9), 3207. [DOI] [PubMed] [Google Scholar]
- 197. Sawaya AP, Jozic I, Stone RC, Pastar I, Egger AN, Stojadinovic O, Glinos GD, Kirsner RS, Tomic‐Canic MJCI, Insight. 2019, 4, e129320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Yang R, Wang J, Zhou Z, Qi S, Ruan S, Lin Z, Xin Q, Lin Y, Chen X, Xie J, Dev. Biol. 2019, 445(2), 271. [DOI] [PubMed] [Google Scholar]
- 199. Bass MD, Williamson RC, Nunan RD, Humphries JD, Byron A, Morgan MR, Martin P, Humphries MJ, Dev. Cell. 2011, 21(4), 681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Yang JX, Hua L, Li YQ, Jiang YY, Han D, Liu H, Tang QQ, Yang XN, Yin C, Hao LY, Yu L, J. Neurosci. 2015, 35(1), 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Yang G, Addai J, Ittmann M, Wheeler TM, Thompson TC, Clin. Cancer Res. 2000, 6(9), 3430. [PubMed] [Google Scholar]
- 202. Ortiz R, Diaz J, Diaz N, Lobos‐Gonzalez L, Cárdenas A, Contreras P, Díaz MI, Otte E, Cooper‐White J, Torres V, Leyton L, Oncotarget. 2016, 7(26), 40571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Guerrero S, Diaz‐Garcia VM, Contreras‐Orellana P, Lara P, Palma S, Guzman F, Lobos‐Gonzalez L, Cárdenas A, Rojas‐Silva X, Muñoz L, Leyton L, Nanomedicine (Lond). 2018, 13(12), 1447. [DOI] [PubMed] [Google Scholar]