

Abstract
A systematic study on the reactivity of the triple‐decker complex [(Cp’’’Co)2(μ,η4:η4‐C7H8)] (A) (Cp’’’=1,2,4‐tritertbutyl‐cyclopentadienyl) towards sandwich complexes containing cyclo‐P3, cyclo‐P4, and cyclo‐P5 ligands under mild conditions is presented. The heterobimetallic triple‐decker sandwich complexes [(Cp*Fe)(Cp’’’Co)(μ,η5:η4‐P5)] (1) and [(Cp’’’Co)(Cp’’’Ni)(μ,η3:η3‐P3)] (3) (Cp*=1,2,3,4,5‐pentamethylcyclopentadienyl) were synthesized and fully characterized. In solution, these complexes exhibit a unique fluxional behavior, which was investigated by variable temperature NMR spectroscopy. The dynamic processes can be blocked by coordination to {W(CO)5} fragments, leading to the complexes [(Cp*Fe)(Cp’’’Co)(μ3,η5:η4:η1‐P5){W(CO)5}] (2 a), [(Cp*Fe)(Cp’’’Co)(μ4,η5:η4:η1:η1‐P5){(W(CO)5)2}] (2 b), and [(Cp’’’Co)(Cp’’’Ni)(μ3,η3:η2:η1‐P3){W(CO)5}] (4), respectively. The thermolysis of 3 leads to the tetrahedrane complex [(Cp’’’Ni)2(μ,η2:η2‐P2)] (5). All compounds were fully characterized using single‐crystal X‐ray structure analysis, NMR spectroscopy, mass spectrometry, and elemental analysis.
Keywords: cobalt, cyclo-Pn ligands, iron, nickel, triple-decker complexes
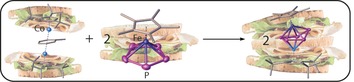
From double to triple: A synthetic route to otherwise inaccessible heterometallic triple‐decker complexes is reported (see scheme). The cobalt complex [(Cp’’’Co)2(toluene)] (Cp’’’=1,2,4‐tritertbutyl‐cyclopentadienyl) as a source for unsaturated {Cp’’’Co} fragments was reacted with the sandwich complexes [Cp*Fe(η5‐P5)], [Cp’’’Co((η4‐P4)] and [Cp’’’Ni(η3‐P3)] to form novel heterobimetallic triple‐decker complexes.
The transformation of the P4 tetrahedron of white phosphorus in the coordination sphere of transition‐metal complexes yielding functionalized phosphorus compounds is a widely investigated field of research. Most reactions proceed by thermolysis of carbonyl complexes such as [(CpRM(CO)n)2] (e.g. M=Cr,1 Mo,2, 3 W,4 Fe,5, 6 Co,7 Ni,8 Cp=cyclopentadienyl) or photolysis of [CpRM(CO)4] (e.g. M=V,9 Nb,10, 11 Ta11) with P4 under harsh reaction conditions, yielding the thermodynamically most stable products. In contrast, organometallic compounds bearing labile ligands can be used to perform reactions with white phosphorus under much milder conditions, even at very low temperatures. Here, in general, kinetic products or metastable compounds can be obtained. As an example, the [(nacnac)M(L)] (nacnac=β‐diketiminate) complexes (M=Cu, L=MeCN,12 M=Fe, L=toluene,13 M=Co, L=toluene,14 cf. Figure 1) have been used in the reaction with P4 yielding a plethora of kinetic products. Another type of complex used in the activation of P4 is the reactive cobalt toluene triple‐decker complex [(Cp′′′Co)2(μ,η4:η4‐C7H8)] (A) (Cp′′′=η5‐C5H2 tBu3), which dissociates in solution into a {Cp’’’Co(C7H8)} and {Cp’’’Co} fragment.15 Its reactivity towards white phosphorus strongly depends on the reaction conditions. Thus, at room temperature, the cleavage of the P4 tetrahedron into two P2 units to form [(Cp′′′Co)2(μ,η2:η2‐P2)2] (D) or, at low temperatures, the metal‐controlled aggregation of P4 molecules occur to form large neutral polyphosphorus cages with, for example, a P12, P16, and P24 ligand, respectively.16 All reactions of the mentioned carbonyl or nacnac complexes with P4 lead to either sandwich complexes with a Pn ligand as an end deck (e.g. [Cp*Fe(η5‐P5)]5) (Cp*=1,2,3,4,5‐pentamethylcyclopentadienyl) or a middle deck in homometallic triple‐decker complexes (e.g. [(Cp*Mo)2(μ,η6:η6‐P6)]3). But no heterobimetallic compounds can be obtained that way. Moreover, there are only very few examples known that are accessible from reactions of [Cp*M(η5‐P5)] (M=Fe, Ru) with cationic fragments17 or carbonyl compounds.18 Most of them are based on rare or expensive metals such as tantalum, rhodium, or iridium and are synthesized in only moderate yields, partially under harsh conditions (Figure 1). On the other hand, substituted derivatives of heterobimetallic triple‐decker complexes can be obtained from the electrophilic quenching of pentaphosphaferrocene ([Cp*Fe(η4‐P5R)]−) functionalized by main‐group nucleophiles, resulting in e.g. [(Cp*Fe)(Cp’’’Co)(μ,η4:η3‐P5NMe2)]19 (Figure 1).
Figure 1.
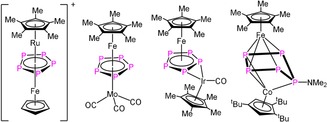
Selected examples of heterobimetallic triple‐decker complexes.
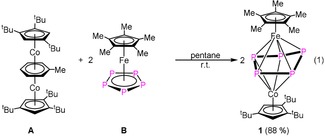
The question arises as to how heterobimetallic triple‐decker complexes can be synthesized easier and in high yields under mild conditions including cheap and abundant metals. Therefore we investigated the reaction of [(Cp′′′Co)2(μ,η4:η4‐C7H8)] (A) with compounds containing cyclo‐Pn ligands as end‐decks, such as [Cp*Fe(η5‐P5)] (B), [Cp’’’Co(η4‐P4)] (C) and [Cp’’’Ni(η3‐P3)] (E) at room temperature. Herein, we report the synthesis and properties of two new mixed metal triple‐decker sandwich complexes comprising cyclo‐Pn (n=3 and 5) middle decks, respectively, as well as their reaction behavior towards white phosphorus and their thermal stability.
The reaction of [(Cp′′′Co)2(μ,η4:η4‐C7H8)] (A) with pentaphosphaferrocene ([Cp*Fe(η5‐P5)] (B)) in pentane at room temperature leads to the quantitative formation of [(Cp*Fe)(Cp’’’Co)(μ,η5:η4‐P5)] (1), which is obtained as a red air‐sensitive solid in crystalline yields of 88 % [Eq. 1]. In contrast to the thermolysis of B with cobalt carbonyl compounds,20 the formation of 1 proceeds without the fragmentation of the cyclo‐P5 ligand (the thermolysis leads to the formation of P4, P2, and P1 ligand‐containing complexes). Alternatively, 1 can be also obtained by the reaction of [K2(dme)3][(Cp*Fe)2(μ,η4:η4‐P10)] with [(Cp’’’CoCl)2] in lower yield (cf. Supporting Information).
The crystal structure of 1 reveals a dinuclear complex bearing a cyclo‐P5 ligand in an envelope conformation, coordinating in η4 fashion to the Cp’’’Co fragment and η5 fashion to the Cp*Fe fragment (Figure 2). Both CpRM fragments are twisted by approximately 16° to each other and P1 is bent out of the plane by approx. 34.7°. Three of the P−P bond lengths in 1 are almost identical to those in B (P1‐P2 2.1459(10) Å, P1−P5 2.1498(10) Å, P3‐P4 2.1283(10) Å), in B average 2.120(2) Å), whereas two are elongated (P2−P3 2.2441(11) Å, P4−P5 2.2407(10) Å), which is confirmed by the WBIs of P1−P2, P3−P4 and P1−P5 (0.99, 0.95, 0.99) and P2−P3 and P4−P5 (0.79, 0.80). Compound 1 shows a dynamic behavior in solution, which was studied by variable temperature NMR spectroscopy. At room temperature, the 1H NMR spectrum reveals four sharp singlets corresponding to one Cp* and one freely rotating Cp’’’ ligand, whereas the 31P{1H} NMR spectrum shows one very broad signal at δ=−160 ppm (ω 1/2=16 000 Hz). Upon cooling to −80 °C, the signals in the 1H NMR spectra remain unchanged, while the broad signal observed in the 31P{1H} NMR spectrum at room temperature splits into three sharp multiplets centered at δ=288.7, −207.3 and −250.3 ppm, respectively, with an integral ratio of 1:2:2 displaying an AMM'XX’ spin system. The corresponding coupling constants were obtained from the simulation of the 31P{1H} NMR spectrum at 213 K (cf. Supporting Information). The magnitude of the 1 J PP coupling correlates nicely with the P−P bond lengths observed in the solid‐state structure, indicating that this structure is presumably present in solution at 213 K. On the other hand, when warming the sample to 90 °C, the signals in the 1H NMR spectra do not change, while the broad signal in the 31P{1H} NMR spectrum sharpens to a singlet at δ=−115.3 ppm (ω 1/2=500 Hz). At this temperature, the dynamic process (Figure 3) speeds up and all five P atoms become equivalent in the NMR. The free activation enthalpy of this process at the coalescence temperature (T c=293 K) amounts to 48.2 kJ mol−1 (cf. Supporting Information).21 All these findings indicate that the dynamic process proceeds at the cyclo‐P5 ligand. Compound 1 is thermally stable. Refluxing 1 in toluene for one day leads to only minor decomposition. As a decomposition product, [Cp*Fe(η5‐P5)] has been identified by 31P{1H} NMR spectroscopy.
Figure 2.
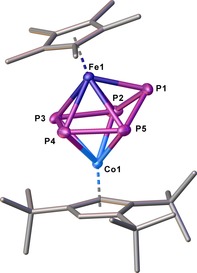
Structure of one independent molecule of 1 in the solid state. Thermal ellipsoids are shown at 50 % probability level. Hydrogen atoms are omitted for clarity, with one independent molecule of 1 being depicted.
Figure 3.
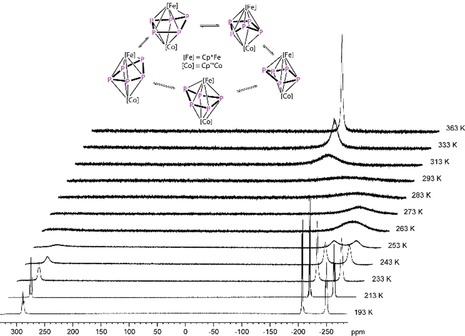
VT 31P{1H} NMR spectra of 1 in [D8]toluene at different temperatures between 193 and 363 K and the proposed dynamic process.
To block the dynamic process in solution at room temperature, 1 was reacted with an excess of [W(CO)5(thf)] (2.2 equiv) and the mono‐ and dicoordinated compounds [(Cp*Fe)(Cp’’’Co)(μ3,η5:η4:η1‐P5){W(CO)5}] (2 a) and [(Cp*Fe)(Cp’’’Co)(μ4,η5:η4:η1:η1‐P5){W(CO)5}2] (2 b) are formed [Eq. 2]. The stoichiometric reaction (1:1) leads to a selective formation of 2 a (according to 31P{1H} NMR).
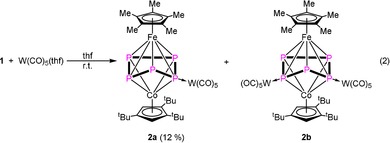
According to the 31P{1H} NMR spectrum of the reaction mixture, 2 a and 2 b are formed in an approximately 1:1 ratio. After workup of the reaction mixture by thin layer chromatography, only 2 a can be isolated as a pure product, in addition to some minor amounts of 2 b. It is assumed that one {W(CO)5} fragment is removed during the chromatography. Single crystals of 2 a suitable for X‐ray diffraction could be obtained from a concentrated solution in pentane stored at −30 °C. For 2 b, few crystals could be obtained by evaporating the solvent from a toluene solution of the crude reaction mixture. The structure of 2 a in the solid state reveals a cyclo‐P5 ligand in an envelope conformation, whereas a {W(CO)5} fragment is coordinated to the P2 atom (Figure 4). For 2 b, an additional coordination of a {W(CO)5} fragment at P5 is observed (Figure 4). The 31P{1H} NMR spectrum of the reaction mixture at room temperature reveals three sharp multiplets for 2 b and four broad signals for 2 a. These four broad signals indicate a dynamic process for 2 a in solution. VT 31P{1H} NMR spectroscopic investigations of 2 a reveal that the broad signals sharpen into five multiplets displaying an AMNXY spin system upon cooling down to 193 K (cf. Supporting Information).
Figure 4.
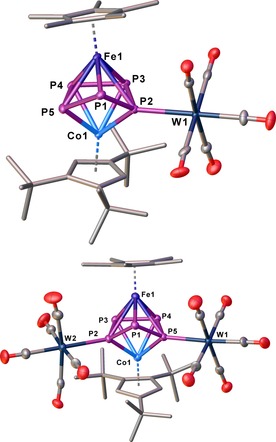
Molecular structure of 2 a (top) and 2 b (bottom) in the solid state. Thermal ellipsoids are shown at 50 % probability level. Hydrogen atoms are omitted for clarity.
The reaction of [(Cp′′′Co)2(μ,η4:η4‐C7H8)] (A) with [Cp’’’Co(η4‐P4)] (C) in toluene at room temperature leads to the quantitative formation of [(Cp’’’Co)2(μ,η2:η2‐P2)2] (D) [Eq. 3].7
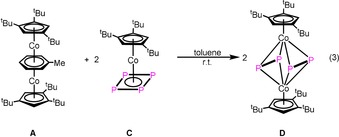
The cyclo‐P4 ligand in C is selectively fragmented into two separated P2 units upon coordination to another Cp’’’Co fragment in D. Compound D was already reported.7
Stirring a pentane solution of A with [Cp’’’Ni(η3‐P3)] (E) at room temperature for few minutes results in the quantitative formation of the dinuclear complex [(Cp’’’Co)(Cp’’’Ni)(μ,η3:η3‐P3)] (3) [Eq. 4].
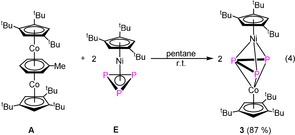
Compound 3 is an orange‐brown air‐sensitive solid. The crystal structure analysis of 3 (Figure 5) reveals a dinuclear complex bearing an allylic P3 ligand. Since both metal fragments contain the Cp’’’ ligand, the attribution of the atom types of the metal was made based on their anisotropic displacement parameters. Due to the similarity of the bond lengths, a mixed‐site occupation cannot be excluded.
Figure 5.
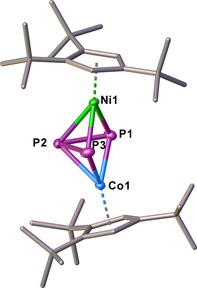
Structure of 3 in the solid state. Thermal ellipsoids are shown at 50 % probability level. Hydrogen atoms are omitted for clarity.
The P3 ligand coordinates to the metals with two short M−P bonds to P1/P3 (between 2.1826(5) and 2.1931(5) Å) and one elongated M−P bond to P2 (2.3457(4) and 2.3538(5) Å). The Cp’’’ ligands are tilted by 18° to each other. Compared to the P−P bond lengths in E 8 (average: 2.098(2) Å), the P1−P2 (2.1895(6) Å) and P2−P3 (2.1945(6) Å) bond lengths in 3 are elongated upon coordination to another metal fragment and lie in the range of P−P single bonds.22 The Wiberg Bond Indices for P1–P2 and P2–P3 with values of 0.96 and 0.96 underline this description. The P1–P3 distance of 2.7619(6) Å is considerably longer than a P−P single bond, but lies below the sum of the van der Waals radii of phosphorus (Σ vdW=3.80 Å).23 The Wiberg Bond Index of 0.22 indicates a weak interaction between the two phosphorus atoms P1 and P3. In the 1H and 31P{1H} NMR spectra of 3, a dynamic process can be observed that is dependent on the temperature and the solvent used (CD2Cl2 or [D8]toluene) (Figure 6). The 31P{1H} NMR in CD2Cl2 spectrum shows three broad signals centered at δ=377.3, 249.7 and 133.1 ppm with an integral ratio of 0.19:1:0.12. Upon cooling, the relative intensity of the broad signals centered at δ=366.8 and 126.2 ppm increases in comparison to the singlet at δ=237.8 ppm, while they sharpen and split into a doublet and a triplet with a 1 J PP of 357 Hz. This clearly confirms the presence of a fixed allylic P3 ligand (3 b) at low temperature (Figure 6). The singlet at δ=237.8 ppm can be assigned to the compound 3 a bearing a cyclo‐P3 ligand. The ratio of 3 a and 3 b is dependent on the temperature, but also on the polarity of the solvent used (cf. Figure 6).
Figure 6.
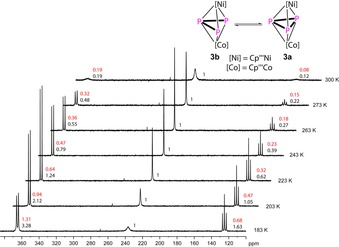
VT 31P{1H} NMR spectra of 3 in CD2Cl2 at different temperatures between 183 and 300 K and the proposed temperature‐dependent equilibrium. Integral ratio given for measurement in CD2Cl2 (black) and [D8]toluene (red).
At low temperatures, compound 3 b is predominant (63 % in toluene and 83 % in dichloromethane at 183 K according to the 31P{1H} NMR spectra, Figure 6). The situation can also be monitored in the VT 1H NMR spectra (cf. Supporting Information). Warming up a solution of 3 in [D8]toluene to 363 K, in the 31P{1H} NMR spectrum, the signals for 3 b disappear completely while the singlet assigned to 3 a broadens. Above 323 K, decomposition is observed and two new sharp singlets at δ=285.8 and −43.7 ppm arise, which can be assigned to a new Ni complex (5, vide infra) and D.7 All these spectroscopic data clearly show that, in solution, there is an equilibrium between 3 a and 3 b. The phosphorus atoms in 3 contain a lone pair of electrons, possibly available for coordination (cf. Figure S23, Supporting Information), which raises the question as to whether the equilibrium between 3 a and 3 b can be shifted by coordination to a {W(CO)5} fragment [Eq. 5].
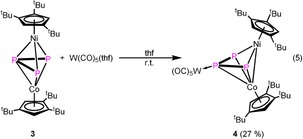
Reacting 3 with an excess of [W(CO)5(thf)] (2.2 equiv) yields [(Cp’’’Co)(Cp’’’Ni)(μ3,η3:η2:η1‐P3){W(CO)5}] (4) as a major product (coordination of {W(CO)5} at the central P atom (P2)) beside small amounts of I4‐B/I4‐C (coordination of {W(CO)5} at the outer P atom (P1 or P3). According to the 31P{1H} NMR spectrum of the reaction mixture 4 and I4‐B/I4‐C are present in a ratio of 1:0.04:0.1. Despite many attempts, only 4 could be isolated and fully characterized. Single crystals of 4 suitable for X‐ray structure analysis can be obtained from a concentrated solution in CH2Cl2 layered with MeCN at room temperature. In 4, the {W(CO)5} fragment is coordinated to the central P atom of the three‐membered chain (Figure 7). Upon the coordination of 3 to {W(CO)5}, the Cp’’’ metal fragments are further tilted towards each other (from 18 to 52°) and the Co–Ni distance (2.6182(7) Å) is significantly shortened compared to 3 (3.3941(5) Å), indicating the presence of a metal–metal bond (WBI of 0.43, cf. Figure S24, Supporting Information). The P3 ligand is coordinated in η3 fashion to Co and in η2 fashion to Ni. The assignment of the Co and Ni positions in the structure of 4 is based on the anisotropic displacement parameters and is also supported by DFT calculations at the B3LYP/def2‐SVP level, showing that this isomer is energetically more favored (by 45.31 kJ mol−1) compared to the isomer with exchanged Co and Ni positions.
Figure 7.
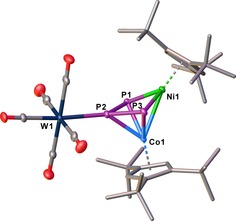
Structure of 4 in the solid state. Thermal ellipsoids are shown at 50 % probability level. Hydrogen atoms are omitted for clarity. Detailed bond lengths and angles are given in the Supporting Information.
The P1−P2 and P2−P3 bonds (2.1214(11) and 2.1234(12) Å) are shortened, whereas the P1–P3 distance is lengthened (3.0011(12) Å) compared to 3. This is also in line with the WBIs (P1−P2 (1.09), P2−P3 (1.08) and P1−P3 (0.12)) indicating the presence of an allylic P3 ligand. The 31P{1H} NMR spectrum of 4 in C6D6 at room temperature reveals a doublet at δ=354.3 ppm (1 J PP=408 Hz) and a triplet at δ=114.6 ppm (1 J PP=408 Hz), the latter with tungsten satellites (1 J PW=203 Hz). According to DFT calculations (Figure 8), the coordination on the central phosphorus atom in 3 to a {W(CO)5} fragment (I4‐A) is energetically disfavored (by 15.01 kJ mol−1), but the final rearrangement of I4‐A to 4 is with −34.36 kJ mol−1 exothermic. In contrast, the coordination of the outer phosphorus atoms in 3 to the {W(CO)5} fragment (I4‐B/I4‐C) is exothermic with −15.39 and −17.96 kJ mol−1, respectively, showing that I4‐B/I4‐C represents the kinetic products, while 4 is the thermodynamic product of the reaction.
Figure 8.
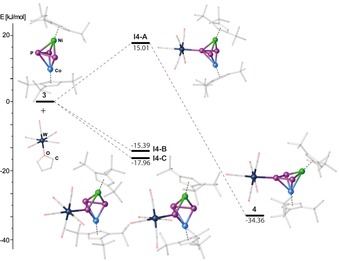
Energetic profile of the reaction of 3 with [W(CO)5(thf)]. B3LYP/def2‐SVP level of theory.
The VT NMR spectra of 3 reveal a decomposition starting at 50 °C (Figure S11, Supporting Information). The thermolysis in boiling toluene for one day leads to the formation of [(Cp’’’Co)2(μ,η2:η2‐P2)2] (D) and [(Cp’’’Ni)2(μ,η2:η2‐P2)] (5) [Eq. 6]. This result shows a ready fragmentation into [Cp’’’Co(P2)] and [Cp’’’Ni≡P], which dimerize to D and 5, respectively. The latter fragment was already proposed to dimerize as a {Cr(CO)5} coordinated species in the formation of [(Cp*Ni)2(μ,η2:η2:η1:η1‐P2){Cr(CO)5}2].24
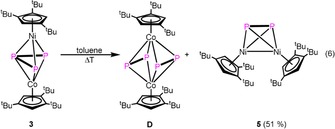
Crystals suitable for X‐ray single‐crystal structure analysis can be obtained by storing a concentrated solution of 5 in hexane at −30 °C.
The structure of 5 in the solid state (Figure 9) reveals a tetrahedral moiety consisting of two Ni and two P atoms. The P−P bond length (2.0805(8) Å) lies in the range of an elongated P−P double bond with a WBI of 1.35.22, 25 The Ni–Ni distance in 5 is 2.5848(5) Å, representing a single bond (WBI of 0.52). Similar P–P and Ni–Ni distances of 2.053(5) and 2.571(1) Å, respectively, have been reported for the related complex [(CpiPrNi)2(μ,η2:η2‐P2)] (CpiPr=C5HiPr4).26 The 31P{1H} NMR spectrum of 5 contains a sharp singlet at δ=282.7 ppm.
Figure 9.
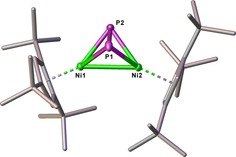
Structure of 5 in the solid state. Thermal ellipsoids are shown at 50 % probability level. Hydrogen atoms are omitted for clarity. Detailed bond lengths and angles are given in the Supporting Information.
Due to the dynamic behavior of 1 and 3 in solution, we assumed that the Cp’’’Co fragment is labile and could be removed in the presence of a suitable reaction partner. Hence, a solution of 1 in hexane was stirred with an excess of P4 at room temperature. After column chromatographic workup, [Cp*Fe(η5‐P5)] (B) could be isolated as a byproduct and separated from the additionally formed [(Cp’’’Co)nPm] complexes [(Cp’’’Co)2(μ,η4:η2:η1‐P8)] (isomer one, 29 %, isomer two, 6 %), [Cp’’’Co(η4‐P4)] (C, 14 %) and [(Cp’’’Co)3(μ3,η4:η2:η2:η2:η1‐P12)] (11 %) (based on cobalt). All these Co complexes can also be obtained from the reaction of white phosphorus with A at room temperature as previously reported.16 In an analogous manner, 3 was reacted with P4. According to the 31P{1H} NMR spectrum, [Cp’’’Ni(η3‐P3)] (E) is quantitatively formed, next to the cobalt complexes [Cp’’’Co(η4‐P4)] (C, 40 %) and [(Cp’’’Co)2(μ,η2:η2‐P2)2] (D, 42 %), beside traces (<1 %) of other P‐rich compounds.
The results show, that a novel synthetic route to two new heterobimetallic triple‐decker complexes was developed. Both can be obtained in gram scale under mild reaction conditions. [(Cp′′′Co)2(μ,η4:η4‐C7H8)] (A) serves as a source of unsaturated {Cp’’’Co} fragments, reacting with [Cp*Fe(η5‐P5)] (B) and [Cp’’’Ni(η3‐P3)] (E) to yield [(Cp*Fe)(Cp’’’Co)(μ,η5:η4‐P5)] (1) and [(Cp’’’Co)(Cp’’’Ni)(μ,η3:η3‐P3)] (3), respectively. Both compounds exhibit unique fluxionalities in solution, which can be inhibited by coordination to {W(CO)5} fragments resulting in [(Cp*Fe)(Cp’’’Co)(μ3,η5:η4:η1‐P5){W(CO)5}] (2 a) or [(Cp*Fe)(Cp’’’Co)(μ4,η5:η4:η1:η1‐P5){W(CO)5}2] (2 b) and [(Cp’’’Co)(Cp’’’Ni)(μ3,η3:η2:η1‐P3){W(CO)5}] (4), respectively. Additionally, 3 is thermally unstable and fragmentizes upon warming into [(Cp′′′Co)2(μ,η2:η2‐P2)2] (D) and [(Cp’’’Ni)2(μ,η2:η2‐P2)] (5). The {Cp’’’Co} fragment in 1 and 3 is still labile bound and can be released to undergo further reactions for instance with P4.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was supported by the Deutsche Forschungsgemeinschaft within the project Sche 384/38‐1. M.P. is grateful to the Fonds der Chemischen Industrie and F.D. to the Studienstiftung des Deutschen Volkes for PhD fellowships.
M. Piesch, F. Dielmann, S. Reichl, M. Scheer, Chem. Eur. J. 2020, 26, 1518.
Dedicated to Professor H. Brunner on the occasion of his 85th birthday
Contributor Information
Martin Piesch, https://www.uni-regensburg.de/chemie-pharmazie/anorganische-chemie-scheer/.
Prof. Dr. Manfred Scheer, Email: manfred.scheer@ur.de.
References
- 1. Sekar P., Umbarkar S., Scheer M., Voigt A., Kirmse R., Eur. J. Inorg. Chem. 2000, 2585–2589. [Google Scholar]
- 2. Scherer O. J., Sitzmann H., Wolmershäuser G., J. Organomet. Chem. 1984, 268, C9–C12. [Google Scholar]
- 3.
- 3a. Scherer O. J., Sitzmann H., Wolmershäuser G., Angew. Chem. Int. Ed. Engl. 1985, 24, 351–353; [Google Scholar]; Angew. Chem. 1985, 97, 358–359; [Google Scholar]
- 3b. Fleischmann M., Dielmann F., Gregoriades L. J., Peresypkina E. V., Virovets A. V., Huber S., Timoshkin A. Y., Balázs G., Scheer M., Angew. Chem. Int. Ed. 2015, 54, 13110–13115; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 13303–13308. [Google Scholar]
- 4. Scherer O. J., Schwalb J., Swarowsky H., Wolmershäuser G., Kaim W., Gross R., Chem. Ber. 1988, 121, 443–449. [Google Scholar]
- 5. Scherer O. J., Brück T., Angew. Chem. Int. Ed. Engl. 1987, 26, 59; [Google Scholar]; Angew. Chem. 1987, 99, 59. [Google Scholar]
- 6. Scherer O. J., Brueck T., Wolmershäuser G., Chem. Ber. 1988, 121, 935–938. [Google Scholar]
- 7. Scherer O. J., Berg G., Wolmershäuser G., Chem. Ber. 1995, 128, 635–639. [Google Scholar]
- 8. Scherer O. J., Dave T., Braun J., Wolmershäuser G., J. Organomet. Chem. 1988, 350, C20–C24. [Google Scholar]
- 9. Herberhold M., Frohmader G., Milius W., J. Organomet. Chem. 1996, 522, 185–196. [Google Scholar]
- 10. Scherer O. J., Vondung J., Wolmershäuser G., Angew. Chem. Int. Ed. Engl. 1989, 28, 1355–1357; [Google Scholar]; Angew. Chem. 1989, 101, 1395–1397. [Google Scholar]
- 11. Scherer O. J., Winter R., Wolmershäuser G., Z. anorg. allg. Chem. 1993, 619, 827–835. [Google Scholar]
- 12. Spitzer F., Sierka M., Latronico M., Mastrorilli P., Virovets A. V., Scheer M., Angew. Chem. Int. Ed. 2015, 54, 4392–4396; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 4467–4472. [Google Scholar]
- 13. Spitzer F., Graßl C., Balázs G., Zolnhofer E. M., Meyer K., Scheer M., Angew. Chem. Int. Ed. 2016, 55, 4340–4344; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 4412–4416. [Google Scholar]
- 14. Spitzer F., Graßl C., Balázs G., Mädl E., Keilwerth M., Zolnhofer E. M., Meyer K., Scheer M., Chem. Eur. J. 2017, 23, 2716–2721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Schneider J. J., Wolf D., Janiak C., Heinemann O., Rust J., Krüger C., Chem. Eur. J. 1998, 4, 1982–1991. [Google Scholar]
- 16. Dielmann F., Timoshkin A., Piesch M., Balázs G., Scheer M., Angew. Chem. Int. Ed. 2017, 56, 1671–1675; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 1693–1698. [Google Scholar]
- 17. Kudinov A. R., Loginov D. A., Starikova Z. A., Petrovskii P. V., Corsini M., Zanello P., Eur. J. Inorg. Chem. 2002, 3018–3027. [Google Scholar]
- 18.
- 18a. Rink B., Scherer O. J., Heckmann G., Wolmershäuser G., Chem. Ber. 1992, 125, 1011–1016; [Google Scholar]
- 18b. Detzel M., Mohr T., Scherer O. J., Wolmershäuser G., Angew. Chem. 1994, 106, 1142–1144; [Google Scholar]
- 18c. Detzel M., Friedrich G., Scherer O. J., Wolmershäuser G., Angew. Chem. Int. Ed. Engl. 1995, 34, 1321–1323; [Google Scholar]; Angew. Chem. 1995, 107, 1454–1456. [Google Scholar]
- 19. Mädl E., Peresypkina E., Timoshkin A. Y., Scheer M., Chem. Commun. 2016, 52, 12298–12301. [DOI] [PubMed] [Google Scholar]
- 20.
- 20a. Scherer O. J., Weigel S., Wolmershäuser G., Chem. Eur. J. 1998, 4, 1910–1916; [Google Scholar]
- 20b. Friedrich G., Scherer O. J., Wolmershäuser G., Z. Anorg. Allg. Chem. 1996, 622, 1478–1486. [Google Scholar]
- 21. Friebolin H., Ein- und zweidimensionale NMR Spektroskopie, VCH, Weinheim, 1992. [Google Scholar]
- 22. Pyykkö P., Atsumi M., Chem. Eur. J. 2009, 15, 186–197. [DOI] [PubMed] [Google Scholar]
- 23. Alvarez S., Dalton Trans. 2013, 42, 8617–8636. [DOI] [PubMed] [Google Scholar]
- 24. Scheer M., Becker U., Chem. Ber. 1996, 129, 1307–1310. [Google Scholar]
- 25. Pyykkö P., Atsumi M., Chem. Eur. J. 2009, 15, 12770–12779. [DOI] [PubMed] [Google Scholar]
- 26. Scherer O. J., Braun J., Walther P., Heckmann C., Wolmershäuser G., Angew. Chem. Int. Ed. Engl. 1991, 30, 852–854; [Google Scholar]; Angew. Chem. 1991, 103, 861–863. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary