

Abstract
Background
Psoriasis, a chronic disease usually requires long‐term disease management.
Objective
This study evaluates the efficacy and safety of recommended ixekizumab (IXE) dose over 4 years (204 weeks) from UNCOVER‐3 study.
Methods
UNCOVER‐3 was a randomised, double‐blind, multicenter, phase 3 study wherein patients with moderate‐to‐severe plaque psoriasis received placebo, IXE 80 mg every 2 weeks (Q2W), IXE 80 mg every 4 weeks (Q4W) (both IXE groups had 160 mg starting dose) or etanercept 50 mg twice weekly. At week 12, all patients switched to IXE Q4W dose for the long‐term extension (264 weeks). After week 60 and at investigator's discretion, patients could receive dose adjustment to IXE Q2W. The efficacy endpoints at week 204 were percentage of patients achieving PASI 75/90/100, sPGA score of 1 or 0, and those achieving PSSI = 0, NAPSI = 0 and PPASI 100. Efficacy data were summarised through 204 weeks using as‐observed, multiple imputation (MI) and modified non‐responder imputation (mNRI) methods.
Results
The proportion of patients achieving PASI 75/90/100 at week 204 using mNRI method were 82.8%, 66.4% and 48.3%, respectively. Using as‐observed and MI methods, 98.2% and 94.8% patients achieved PASI 75, 87.8% and 73.3% achieved PASI 90, and 67.1% and 52.7% achieved PASI 100 response, respectively, at week 204. The response rates for sPGA (0, 1) were 88.7%, 76.2% and 68.5% and for sPGA (0) were 68.9%, 54.6% and 49.7% using as‐observed, MI and mNRI methods, respectively. Similar trends were observed with NAPSI = 0, PSSI = 0, PPASI 100 and itch NRS = 0. There were no new safety concerns through year 4.
Conclusions
This study demonstrated sustained high‐efficacy response through 4 years of continuous treatment with ixekizumab in patients with moderate‐to‐severe plaque psoriasis. The safety profile remained consistent with prior findings, with no new or unexpected safety concerns.
Introduction
Psoriasis is a chronic disease and usually requires long‐term disease management. The efficacy and safety of ixekizumab (IXE), a high‐affinity monoclonal antibody that selectively targets interleukin (IL)‐17A,1 is well established in patients with moderate‐to‐severe plaque psoriasis,2, 3, 4, 5 including from 1,2, 5 25 and 36 years data from UNCOVER‐3 study. Given the chronicity of psoriasis, it is important to understand the long‐term efficacy and safety of IXE in patients with moderate‐to‐severe plaque psoriasis. Therefore, here we report the efficacy and safety over 4 years (204 weeks) of treatment with a recommended IXE dose from the UNCOVER‐3 study.
Methods
Study design and participants
The details of the study design and patient population of UNCOVER‐3 (NCT01646177), a randomised, double‐blind, multicenter, phase 3 study in patients with moderate‐to‐severe plaque psoriasis, were reported previously.2, 3 In the current study, patients randomly received either placebo, 80‐mg IXE every 2 weeks (Q2W), 80‐mg IXE every 4 weeks (Q4W; both IXE groups had a starting dose of 160 mg) or etanercept 50 mg twice weekly. At week 12, all patients were switched to IXE Q4W dose for the long‐term extension (LTE) period. After week 60 and at the investigator's discretion, patients could receive a dose adjustment to IXE Q2W. This report primarily focuses on the data from patients who received the clinically recommended dosing regimen (initial 160 mg, IXE Q2W up to week 12 and IXE Q4W thereafter). Additionally, the data from patients on Q2W/Q4W dosing regimen through 204 weeks, including data from visits where dose was adjusted to IXE Q2W, were also analysed.
The study protocol was approved by the institutional review board or ethics committee at each participating site and was conducted according to the principles of the Declaration of Helsinki. All eligible patients provided written informed consent before undergoing study‐related procedures.
Endpoints and outcome assessment
The efficacy endpoints at week 204 were percentage of patients who achieved at least 75%, 90% or 100% improvement from baseline in the Psoriasis Area and Severity Index (PASI 75/90/100, respectively), percentage of patients with static Physician's Global Assessment (sPGA) score of 1 (clear or minimal) or 0 (clear) and absolute PASI score. Efficacy was also evaluated in patients who had baseline scalp, nail or palmoplantar involvement, by the percentage of patients achieving complete resolution based on the Psoriasis Scalp Severity Index (PSSI = 0), the Nail Psoriasis Severity Index (NAPSI = 0) or the Palmoplantar Psoriasis Area and Severity Index (PPASI 100).
Safety was evaluated in all patients who received at least one dose of IXE and was reported as treatment‐emergent adverse events (TEAEs) by severity and frequency, serious adverse events (SAEs) and deaths. Selected adverse events (AEs) of special interest included infections, malignancies, neutropenia, Crohn's disease, ulcerative colitis and cerebrocardiovascular events (adjudicated in a blinded manner by an independent committee of experts).
Statistical analysis
The intent‐to‐treat population, all randomised patients regardless of treatment compliance, were analysed for efficacy. For the primary efficacy analysis, only data from visits in which patients were treated with the recommended IXE dosing regimen were included. Data collected at visits with IXE Q2W dose adjustments were excluded prior to any summary or applying imputations. Three methods were used to summarise efficacy data through 204 weeks: as‐observed, multiple imputation (MI) and a modified non‐responder imputation (mNRI; combined MI and non‐responder imputation (NRI)5). In addition, the data from patients on Q2W/Q4W dosing regimen through 204 weeks, including data from visits where dose was adjusted to IXE Q2W, were also analysed.
The as‐observed method did not require any imputation for missing values; only data from completers at a visit were relevant. This method estimates the effect of treatment conditional on completion of treatment through the time point of interest.
The MI, a method of imputing missing data estimated what observations would have been if the patient had continued with the hypothetical strategy7 handling missing data. For this study, the monotone missing data were imputed using a sequential predictive mean matching method with the baseline score as covariate.
The mNRI method analysed patient response status with a composite strategy7 based on both the clinical requirements for response and the treatment‐related contraindication reasons of discontinuation. In this study, patients who discontinued the study drug due to AEs, lack of efficacy or relapse were considered non‐responders and were imputed as NRI; all other cases of missing data, considered unrelated to the treatment, were imputed using the MI method as described above.
The TEAEs, SAEs and AEs of special interest were summarised in terms of cumulative frequencies through the LTE period for the LTE population who had at least one dose during the LTE period.
Results
A total of 1346 patients were randomised in this study, of whom 1275 (94.7%), 1185 (88.0%), 1091 (81.1%), 1036 (77.0%) and 984 (73.1%) patients completed 12, 52, 108, 156 and 204 weeks, respectively. In recommended dosing regimen (IXE Q2W/Q4W) group, 385 patients were initially randomised, of whom, 363 (94.3%), 340 (88.3%), 315 (81.8%), 300 (77.9%) and 285 (74.0%) patients completed week 12, 52, 108, 156 and 204 weeks, respectively. Reasons for discontinuation were AEs [n (%), 34 (8.8%)], decision of patients [17 (4.4%)], lack of efficacy [11 (2.9%)], lost to follow‐up [13 (3.4%)], protocol violation [12 (3.1%)] and others [12 (3.1%); Table 1]. Baseline demographics and patient characteristics of the study population have been reported previously2, 3, 5 (Table 2).
Table 1.
Patient randomisation and study completion (ITT and long‐term extension period populations)
| PBO/IXE Q4W N = 193, n (%) | ETN/IXE Q4W N = 382, n (%) | IXE Q4W/IXE Q4W N = 386, n (%) | IXE Q2W/IXE Q4W N = 385, n (%) | |
|---|---|---|---|---|
| Patients completed at week 12 and entered into LTE | 183 (95) | 369 (97) | 360 (93) | 362 (94) |
| Patients completed week 204 | 153 (79.3) | 275 (72.0) | 271 (70.2) | 285 (74.0) |
| Reasons for discontinuation (LTE) at week 204 | ||||
| Adverse event | 16 (8.3) | 35 (9.2) | 40 (10.4) | 34 (8.8) |
| Subject decision | 6 (3.1) | 25 (6.5) | 27 (7.0) | 17 (4.4) |
| Lost to follow‐up | 6 (3.1) | 13 (3.4) | 18 (4.7) | 13 (3.4) |
| Lack of efficacy | 5 (2.6) | 14 (3.7) | 12 (3.1) | 11 (2.9) |
| Investigator decision | 3 (1.6) | 7 (1.8) | 5 (1.3) | 6 (1.6) |
| Protocol violation | 1 (0.5) | 7 (1.8) | 8 (2.1) | 12 (3.1) |
| Clinical relapse | 1 (0.5) | 1 (0.3) | 1 (0.3) | 4 (1.0) |
| Death | 1 (0.5) | 2 (0.5) | 1 (0.3) | 2 (0.5) |
| Sponsor decision | 1 (0.5) | 1 (0.3) | 1 (0.3) | 0 |
ETN, etanercept; ITT, intent‐to‐treat; IXE, ixekizumab; LTE, long‐term extension period; PBO, placebo; Q2W, every 2 weeks; Q4W, every 4 weeks.
Table 2.
Patient demographics and baseline characteristics
| PBO/IXE Q4W N = 183 | ETN/IXE Q4W N = 369 | IXE Q4W/IXE Q4W N = 360 | IXE Q2W/IXE Q4W N = 362 | |
|---|---|---|---|---|
| Age (years), mean (SD) | 46 (12.1) | 46 (13.9) | 45 (12.6) | 45 (13.2) |
| Age < 65 years, n (%) | 172 (94.0) | 337 (91.3) | 335 (93.1) | 329 (90.9) |
| Male, n (%) | 129 (70.5) | 260 (70.5) | 241 (66.9) | 241 (66.6) |
| Weight (kg), mean (SD) | 91.6 (21.3) | 92.3 (24.1) | 91.9 (24.6) | 90.5 (23.6) |
| <80 kg, n (%) | 59 (32.2) | 113 (30.6) | 120 (33.3) | 128 (35.4) |
| ≥80 to < 100 kg, n (%) | 68 (37.2) | 132 (35.8) | 139 (38.6) | 133 (36.7) |
| ≥100 kg, n (%) | 56 (30.6) | 124 (33.6) | 101 (28.1) | 101 (27.9) |
| Psoriasis duration (years), mean (SD) | 18 (12.5) | 18 (11.9) | 18 (12.2) | 18 (12.2) |
| sPGA, n (%) | ||||
| =3 | 89 (48.6) | 184 (49.9) | 193 (53.6) | 192 (53.0) |
| =4 | 84 (45.9) | 168 (45.5) | 151 (41.9) | 150 (41.4) |
| =5 | 10 (5.5) | 17 (4.6) | 16 (4.4) | 20 (5.5) |
| BSA, mean (SD) | 29 (17.8) | 28 (17.6) | 29 (16.6) | 28 (17.0) |
| PASI, mean (SD) | 21 (8.4) | 21 (8.2) | 21 (8.2) | 21 (8.0) |
| PSSI, mean (SD) | 18 (12.9) | 20 (13.4) | 20 (14.6) | 20 (13.7) |
| PPASI, mean (SD) | 11 (13.5) | 8 (9.9) | 10 (13.2) | 10 (11.6) |
| NAPSI, mean (SD) | 25 (19.2) | 25 (20.0) | 27 (20.2) | 27 (20.3) |
| Itch NRS, mean (SD) | 6.4 (2.6) | 6.2 (2.6) | 6.3 (2.6) | 6.4 (2.6) |
| Psoriatic arthritis, n (%) | 38 (20.8) | 66 (17.9) | 78 (21.7) | 74 (20.4) |
| Systemic therapy | 101 (55.2) | 215 (58.3) | 212 (58.9) | 202 (55.8) |
| Non‐biologic systemic therapy | 85 (46.4) | 189 (51.2) | 189 (52.5) | 178 (49.2) |
| Biologic agent | 32 (17.5) | 58 (15.7) | 55 (15.3) | 55 (15.2) |
| Phototherapy | 58 (31.7) | 149 (40.4) | 144 (40.0) | 143 (39.5) |
| History of inadequate response, intolerance or contraindication to ≥3 non‐biologic systemic therapies | 6 (3.3) | 28 (7.6) | 25 (6.9) | 24 (6.6) |
BSA, body surface area; ETN, etanercept; IXE, ixekizumab; NAPSI, Nail Psoriasis Severity Index; NRS, numeric rating scale; PASI, Psoriasis Area and Severity Index; PBO, placebo; PPASI, Palmoplantar Psoriasis Area and Severity Index; PSSI, Psoriasis Scalp Severity Index; Q2W, every 2 weeks; Q4W, every 4 weeks; SD, standard deviation; sPGA, static Physician's Global Assessment.
The clinical responses sustained throughout the 4‐year treatment period. In the IXE Q2W/Q4W group (N = 385), the proportion of patients achieving PASI 75, 90 and 100 at week 204 using mNRI method were 82.8%, 66.4% and 48.3%, respectively (Fig. 1). Using as‐observed and MI methods, PASI 75 was achieved in 98.2% and 94.8%, respectively (Fig. S1a). Similarly, 87.8% and 73.3% of patients achieved PASI 90 (Fig. S1b) and 67.1% and 52.7% achieved PASI 100 (Fig. S1c) response at week 204, respectively.
Figure 1.
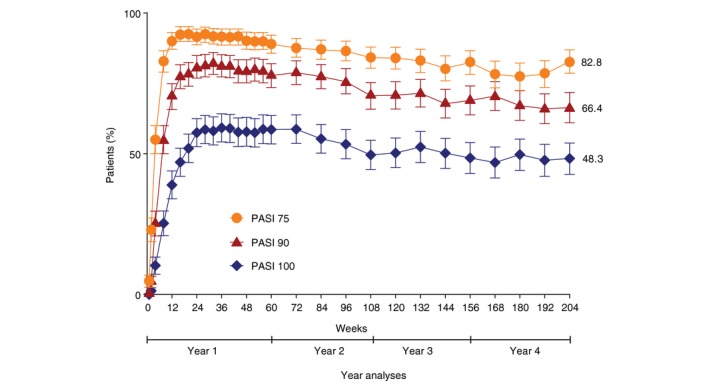
Percentage of patients achieving a psoriasis area and severity index (PASI) 75, 90, and 100 responses over time through 204 weeks with the recommended ixekizumab dosing regiment (modified non‐responder imputation; ITT).
The response rates were also consistently high for sPGA (0, 1) (Fig. 2a) and sPGA (0) (Fig. 2b) throughout the 4‐year treatment period using as‐observed, MI and mNRI methods, respectively [sPGA (0, 1): 88.7%, 76.2% and 68.5%; sPGA (0): 68.9%, 54.6% and 49.7%]. The percentage of patients with absolute PASI scores at week 204 (as‐observed, MI and mNRI, respectively) were 98.2%, 94.3% and 82.3% for PASI ≤ 5; 93.7%, 82.0% and 73.2% for PASI ≤ 3; 89.2%, 74.7% and 67.3% for PASI ≤ 2; and 80.6%, 65.6% and 59.6% for PASI ≤ 1. Sustained efficacy responses were also observed in patients with IXE Q2W/Q4W dosing regimen (204 weeks), including data from visits where the dose was adjusted to IXE Q2W (Fig. 3).
Figure 2.
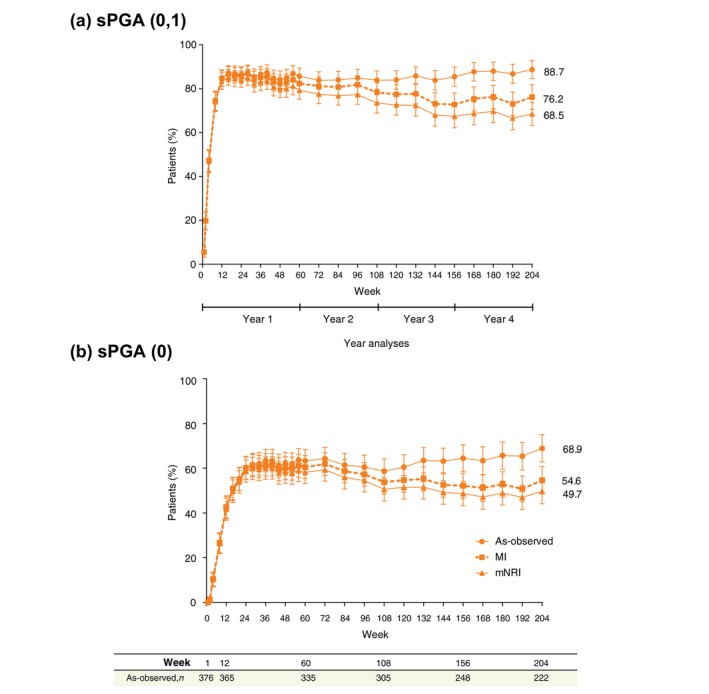
Static Physician's Global Assessment (sPGA) response rates through 204 weeks of treatment with the recommended ixekizumab dosing regimen. Rates of sPGA score of 0 or 1 (a) and sPGA score of 0 (b) at each postbaseline visit for the dosing regimen of ixekizumab every 2 weeks/every 4 weeks. The as‐observed sample size is provided as a table at the bottom of the figure for the Week 1, 12, 60, 108, 156, and 204. Error bars represent 95% confidence intervals. n, number of patients with non‐missing data.
Figure 3.
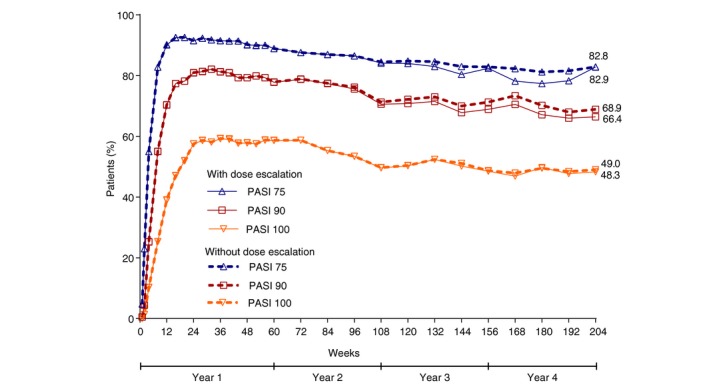
Percentage of patients achieving a psoriasis area and severity index (PASI) response over time through 204 weeks with ixekizumab Q2W/Q4W dosing regimen (204 weeks), including data from visits where the dose was adjusted to ixekizumab Q2W (modified non‐responder imputation; ITT).
Complete resolution throughout the 4‐year treatment period was also observed in patients with baseline nail, scalp and palmoplantar involvement (NAPSI = 0: 75.9%, 74.6% and 67.0%, PSSI = 0: 87.1%, 82.2%, 73.0% and PPASI 100: 95.8%, 92.1%, 77.1% using as‐observed, MI, mNRI methods, respectively) (Fig. 4a–c). The percentage of patients reporting no itch [itch numeric rating scale (NRS) = 0] was 63.1%, 53.6%, 48.6% using as‐observed, MI and mNRI methods, respectively.
Figure 4.
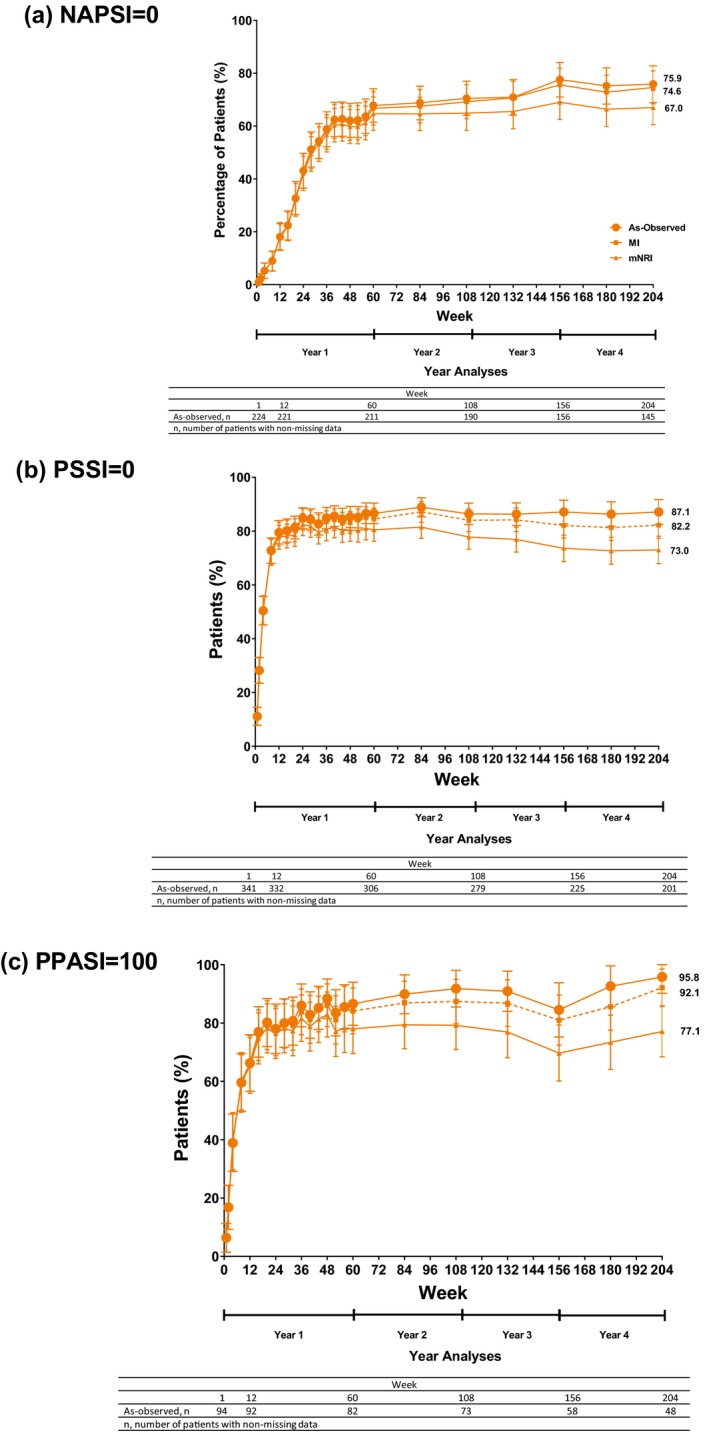
Percentages of patients achieving a Nail Psoriasis Severity Index (NAPSI) score of 0 (a) Psoriasis Scalp Severity Index (PSSI) score of 0 (b), and 100% improvement from baseline in the Palmoplantar Psoriasis Area and Severity Index (PPASI 100) (c) through 204 weeks with the recommended ixekizumab dosing regimen (as‐observed, multiple imputation, and modified non‐responder imputation; intent‐to‐treat population). Filled circles indicate use of the as‐observed method, filled squares indicate use of the multiple imputation method, and filled triangles indicate use of the modified non‐responder imputation method. Response rates are determined for patients with baseline fingernail, scalp, or non‐pustular palmoplantar psoriasis. The as‐observed sample size for each measure is provided (as a table at the bottom of the figure) for the Week 1, 12, 60, 108, 156, and 204 visits. Error bars represent 95% confidence intervals.
Between week 60 and week 204, 254 of 1274 (19.9%) LTE patients were adjusted to IXE Q2W dosing regimen, including 74 of 362 (20.4%) LTE patients on recommended dose (IXE Q2W/Q4W). At the time point when the investigator decided to adjust the dose (n = 74), 54.1% (n = 40) of patients on IXE Q2W/Q4W were PASI 75 responders; 40.5% (n = 30) patients had mild disease activity (sPGA ≤ 2) and PASI 90 and 100 responses were recorded in 18.9% (n = 14) and 0% (n = 0) of patients, respectively. Twenty seven (79.4%) of 34 PASI 75 non‐responder on IXE Q2W/Q4W achieved PASI 75 response at least once after adjusting to Q2W. Nine (12.2%) of the 74 patients discontinued the study treatment before or at week 204; only 2 (2.7%) were due to lack of efficacy.
Safety
During the LTE period, the overall safety profile of IXE treatment over 4 years is shown in Table 3. Majority (n = 1129, 88.6%) of TEAEs were mild or moderate (n = 901, 70.7%) in severity. Among the AEs of special interest, infections were most frequently reported [n = 856, 67.2%; mainly nasopharyngitis (n = 363, 28.5%) and upper respiratory tract infections (n = 138, 10.8%)]. Adverse events associated with duration, such as malignancies (n = 28, 2.2%), were rare. Non‐melanoma skin cancer was reported in 0.7% (n = 9) patients. Clinically significant (grade 3 or 4) neutropenia was also infrequent, occurring in nine patients (0.7%). The overall rate of Crohn's disease (n = 4) and ulcerative colitis (n = 4) was 0.3% for both. During the LTE period, 18.1% (n = 230) of total patients reported SAEs; 13.0% (n = 47) of the incidences were reported in patients on IXE Q2W/Q4W. One death due to acute cardiac failure occurred in the fourth year of the assessment period.
Table 3.
Summary of safety events during the LTE period
| Adverse events† | PBO/IXE Q4W (N = 183) | ETN/IXE Q4W (N = 369) | IXE Q4W/IXE Q4W (N = 360) | IXE Q2W/IXE Q4W (N = 362) | Total LTE Population (N = 1274) |
|---|---|---|---|---|---|
| Treatment‐emergent adverse events, total, n (%) ‡ | 163 (89.1) | 328 (88.9) | 316 (87.8) | 322 (89.0) | 1129 (88.6) |
| Mild | 35 (19.1) | 93 (25.2) | 78 (21.7) | 86 (23.8) | 292 (22.9) |
| Moderate | 86 (47.0) | 168 (45.5) | 168 (46.7) | 187 (51.7) | 609 (47.8) |
| Severe | 42 (23.0) | 67 (18.2) | 70 (19.4) | 49 (13.5) | 228 (17.9) |
| Serious adverse events, n (%) | 44 (24.0) | 69 (18.7) | 70 (19.4) | 47 (13.0) | 230 (18.1) |
| Discontinuations from the study drug due to AEs (including death), n (%) | 17 (9.3) | 34 (9.2) | 37 (10.3) | 32 (8.8) | 120 (9.4) |
| Death | 2 (1.1) | 3 (0.8) | 1 (0.3) | 2 (0.6) | 8 (0.6) |
| Frequently (≥8%) reported treatment‐emergent adverse events, n (%) § | |||||
| Nasopharyngitis | 53 (29.0) | 101 (27.4) | 104 (28.9) | 105 (29.0) | 363 (28.5) |
| Upper respiratory tract infection | 20 (10.9) | 32 (8.7) | 46 (12.8) | 40 (11.0) | 138 (10.8) |
| Injection‐site reaction | 18 (9.8) | 24 (6.5) | 34 (9.4) | 24 (6.6) | 100 (7.8) |
| Headache | 9 (4.9) | 30 (8.1) | 22 (6.1) | 26 (7.2) | 87 (6.8) |
| Back pain | 18 (9.8) | 27 (7.3) | 23 (6.4) | 28 (7.7) | 96 (7.5) |
| Bronchitis | 16 (8.7) | 19 (5.1) | 24 (6.7) | 36 (9.9) | 95 (7.5) |
| Sinusitis | 16 (8.7) | 10 (2.7) | 28 (7.8) | 26 (7.2) | 80 (6.3) |
| Hypertension | 15 (8.2) | 28 (7.6) | 27 (7.5) | 26 (7.2) | 96 (7.5) |
| Arthralgia | 15 (8.2) | 18 (4.9) | 27 (7.5) | 39 (10.8) | 99 (7.8) |
| Selected treatment‐emergent adverse events of special interest, n (%) | |||||
| Malignancies | 2 (1.1) | 8 (2.2) | 9 (2.5) | 9 (2.5) | 28 (2.2) |
| Cerebrocardiovascular events¶ | 7 (3.8) | 16 (4.3) | 6 (1.7) | 11 (3.0) | 40 (3.1) |
| Candida infections†† | 10 (5.5) | 16 (4.3) | 23 (6.4) | 17 (4.7) | 66 (5.2) |
| Crohn's disease | 1 (0.5) | 1 (0.3) | 2 (0.6) | 0 | 4 (0.3) |
| Ulcerative colitis | 1 (0.5) | 1 (0.3) | 1 (0.3) | 1 (0.3) | 4 (0.3) |
| Neutropenia ‡‡ | |||||
| Grade 3 | 1 (0.5) | 2 (0.5) | 4 (1.1) | 0 | 7 (0.5) |
| Grade 4 | 0 | 2 (0.5) | 0 | 0 | 2 (0.2) |
†Adverse events include events that first occurred or worsened during the LTE treatment period. Adverse events reported here are cumulative and include events from the LTE population that occurred during the treatment period before the 204‐week database lock. ‡For patients with >1 occurrence of the same event, severity was categorised based on the event of highest severity. §Frequently reported adverse events included any adverse event occurring at a rate of ≥8% in the total LTE population. ¶Cerebrocardiovascular events were only included if they were confirmed by adjudication. ††Candida infections include MedDRA high level terms and MedDRA preferred terms for events likely representing Candida infections. Specific terms include vulvovaginal candidiasis, vulvovaginal mycotic infection, oral candidiasis, oropharyngeal candidiasis, Candida infection, skin Candida, oral fungal infection, balanitis Candida, nail Candida, and genital candidiasis. ‡‡Treatment‐emergent neutropenia is reported for patients who experienced a worsening from baseline neutrophil counts at any time during LTE. Percentages are calculated based on the number of patients with a baseline and at least 1 postbaseline neutrophil measurement (PBO/IXE Q4W = 183, ETN/IXE Q4W = 369, IXE Q4W/IXE Q4W = 360, IXE Q2W/Q4W = 362, and total = 1274). Neutropenia grades are defined as follows: Grade 1, <2000/mm3 and ≥1500/mm3; Grade 2, <1500/mm3 and ≥1000/mm3; Grade 3, <1000/mm3 and ≥500/mm3; Grade 4, <500/mm3.
AE, adverse event; ETN, etanercept 50 mg twice weekly; IXE, ixekizumab; LTE, long‐term extension; MedDRA, Medical Dictionary for Regulatory Activities; PBO, placebo administered with 2 injections on Week 0, then every 2 weeks; Q2W, every 2 weeks; Q4W, every 4 weeks.
Discussion
Many factors must be considered in selecting the most appropriate therapy for patients with psoriasis. Durability of response and absence of serious side‐effects with long‐term use are important factors.8, 9 The present analyses evaluated the efficacy of the treatment with a recommended IXE dose over 4 years from the UNCOVER‐3 study. Overall, results showed a sustained high‐efficacy response through 4 years of continuous treatment with IXE in patients with moderate‐to‐severe plaque psoriasis. These results corroborate the results that were reported previously in patients with moderate‐to‐severe psoriasis.2, 3, 5, 6 Sustained high response was observed with all the efficacy parameters such as PASI 75, 90, 100 and sPGA (0) or (0, 1), regardless of the statistical analyses (as‐observed, MI and mNRI) performed. Similar results were also observed in patients with baseline nail, scalp and palmoplantar involvement. Furthermore, majority of patients also reported no itch (itch NRS = 0), which has significant impact on quality of life.10
There was no difference in the efficacy response between patients with or without dose escalation; however, 27 of 34 PASI 75 non‐responders achieved PASI 75 at least once after dose adjustment. The significant disease improvements by new classes of biological therapies, such as with IL‐17A inhibition, could have potentially increased the expectations of patients and clinicians on the treatment goals, raising the bar for treatment success to levels beyond the traditional goal of PASI 75. This could be the reason that few PASI 75 responders increased their dose to IXE Q2W despite being responders on the recommended dose. At week 204, about 88% (n = 65) of patients were retained on the IXE treatment, which was similar to that observed previously in patients with moderate‐to‐severe psoriasis, suggesting a clinical benefit with IXE long‐term treatment.
During the LTE period, the overall safety profile of IXE treatment was similar to that reported previously during the induction period,3 week 1085 and week 156.6 Overall, 88.6% (n = 1129) patients reported TEAEs. Of which, 70.7% (n = 901) were mild‐to‐moderate in severity. Adverse events of special interest were low and manageable. As expected, infections were most frequently reported (67.2%, n = 856); the rate of Candida infection during the LTE was 5.2% (n = 66). The overall rate of Crohn's disease (n = 4) and ulcerative colitis (n = 4) were 0.3% for both. These safety findings support the consistency in the safety profile of IXE treatment with no new signals occurring even after the longer exposure.
Conclusions
This study demonstrated a sustained high‐efficacy response through 4 years of continuous treatment with IXE in patients with moderate‐to‐severe plaque psoriasis. The safety profile remained consistent with prior findings, with no new or unexpected safety concerns.
Availability of data and materials
Lilly provides access to all individual participant data collected during the trial, after anonymisation, with the exception of pharmacokinetic or genetic data. Data are available to request in a timely fashion after the indication studied has been approved in the US and EU and after primary publication acceptance. No expiration date of data requests is currently set once they are made available. Access is provided after a proposal has been approved by an independent review committee identified for this purpose and after receipt of a signed data‐sharing agreement. Data and documents, including the study protocol, statistical analysis plan, clinical study report, blank or annotated case report forms, will be provided in a secure data‐sharing environment for up to 2 years per proposal. For details on submitting a request, see the instructions provided at http://www.clinicalstudydatarequest.com. Data are also available on clinicaltrials.gov: NCT01646177.
Supporting information
Figure S1. Percentage of patients achieving a Psoriasis Area and Severity Index (PASI) response over time through 204 weeks with the recommended ixekizumab dosing regimen (as‐observed and multiple imputation; intent‐to‐treat population). Rate of achievement of a 75% improvement from baseline in PASI score (PASI 75) (a), 90% improvement from baseline in PASI score (PASI 90) (b), and 100% improvement from baseline in PASI score (PASI 100) (c) at each postbaseline visit for the dosing regimen of ixekizumab every 2 weeks/every 4 weeks. The as‐observed sample size is provided (as a table at the bottom of the figure) for the week 1, 12, 60, 108, 156, and 204 weeks. Filled circles indicate use of the as‐observed method, filled squares indicate use of the MI method, and filled triangles indicate use of the mMI method. Error bars represent 95% confidence intervals.
{kind=link}
Acknowledgement
The authors would like to thank Kalyan Pulipaka, an employee of Eli Lilly and Company for writing support.
Conflict of interest
Mark Lebwohl is an employee of Mount Sinai and receives research funds from AbbVie, Amgen, AstraZeneca, Boehringer Ingelheim, Celgene, Eli Lilly and Company, Incyte, Janssen/Johnson & Johnson, Kadmon, Leo Pharmaceuticals, MedImmune, Novartis, Pfizer, Sciderm, UCB, Ortho‐dermatologics and ViDac. He is also a consultant for Allergan, Almirall, Arcutis, Boehringer Ingelheim, Bristol Myers Squibb, Castle Biosciences, LEO Pharma, Menlo, Mitsubishi Pharma, Neuroderm LTD, Promius/Dr. Reddy, Theravance Biopharma and Verrica. Kenneth Gordon has consulting relationships with AbbVie, Amgen Inc., Celgene, Eli Lilly and Company, Janssen Pharmaceuticals, Inc., Novartis, Pfizer Inc., Dermira and Boehringer Ingelheim and has received grants from AbbVie, Amgen Inc., Celgene and Janssen. Gaia Gallo and Lu Zhang are employees of Eli Lilly and Company and own stocks. Dr. Carle Paul is a consultant and investigator for Amgen, AbbVie, Almirall, Boehringer Ingelheim, Celgene, Dermira, Janssen‐Cilag, Leo Pharma, Eli Lilly and Company, GlaxoSmithKline, Novartis, Pfizer, Pierre Fabre, Regeneron, Sanofi Genzyme and UCB Pharma.
Funding source
This work was funded by Eli Lilly and Company. Portions of this work have previously been presented in the form of an abstract and manuscript.
Clinical trial registration: A study in participants with moderate‐to‐severe psoriasis (UNCOVER‐3), (NCT01646177).
All patients have provided written informed consent. The protocol was approved by the institutional review board or ethics committee at each participating site.
References
- 1. Gaffen SL, Jain R, Garg AV, Cua DJ. The IL‐23‐IL‐17 immune axis: from mechanisms to therapeutic testing. Nat Rev Immunol 2014; 14: 585–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Griffiths CE, Reich K, Lebwohl M et al Comparison of ixekizumab with etanercept or placebo in moderate‐to‐severe psoriasis (UNCOVER‐2 and UNCOVER‐3): results from two phase 3 randomised trials. Lancet 2015; 386: 541–551. [DOI] [PubMed] [Google Scholar]
- 3. Gordon KB, Blauvelt A, Papp KA et al Phase 3 trials of ixekizumab in moderate‐to‐severe plaque psoriasis. N Engl J Med 2016; 375: 345–356. [DOI] [PubMed] [Google Scholar]
- 4. Strober B, Leonardi C, Papp KA et al Short‐ and long‐term safety outcomes with ixekizumab from 7 clinical trials in psoriasis: etanercept comparisons and integrated data. J Am Acad Dermatol 2017; 76: 432–440.e417. [DOI] [PubMed] [Google Scholar]
- 5. Blauvelt A, Gooderham M, Iversen L et al Efficacy and safety of ixekizumab for the treatment of moderate‐to‐severe plaque psoriasis: results through 108 weeks of a randomized, controlled phase 3 clinical trial (UNCOVER‐3). J Am Acad Dermatol 2017; 77: 855–862. [DOI] [PubMed] [Google Scholar]
- 6. Leonardi C, Maari C, Philipp S et al Maintenance of skin clearance with ixekizumab treatment of psoriasis: three‐year results from the UNCOVER‐3 study. J Am Acad Dermatol 2018; 79: 824–830.e822. [DOI] [PubMed] [Google Scholar]
- 7. International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use . ICH Harmonised Guideline. Estimands and Sensitivity Analysis in Clinical Trials E9 (R1), 2017. URL http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E9/E9-R1EWG_Step2_Guideline_2017_0616.pdf (last accessed: 09 July 2019).
- 8. Kaushik SB, Lebwohl MG. Psoriasis: which therapy for which patient: psoriasis comorbidities and preferred systemic agents. J Am Acad Dermatol 2019; 80: 27–40. [DOI] [PubMed] [Google Scholar]
- 9. Kaushik SB, Lebwohl MG. Psoriasis: which therapy for which patient: focus on special populations and chronic infections. J Am Acad Dermatol 2019; 80: 43–53. [DOI] [PubMed] [Google Scholar]
- 10. Leonardi CL, Blauvelt A, Sofen HL et al Rapid improvements in health‐related quality of life and itch with ixekizumab treatment in randomized phase 3 trials: results from UNCOVER‐2 and UNCOVER‐3. J Eur Acad Dermatol Venereol 2017; 31: 1483–1490. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Percentage of patients achieving a Psoriasis Area and Severity Index (PASI) response over time through 204 weeks with the recommended ixekizumab dosing regimen (as‐observed and multiple imputation; intent‐to‐treat population). Rate of achievement of a 75% improvement from baseline in PASI score (PASI 75) (a), 90% improvement from baseline in PASI score (PASI 90) (b), and 100% improvement from baseline in PASI score (PASI 100) (c) at each postbaseline visit for the dosing regimen of ixekizumab every 2 weeks/every 4 weeks. The as‐observed sample size is provided (as a table at the bottom of the figure) for the week 1, 12, 60, 108, 156, and 204 weeks. Filled circles indicate use of the as‐observed method, filled squares indicate use of the MI method, and filled triangles indicate use of the mMI method. Error bars represent 95% confidence intervals.
Data Availability Statement
Lilly provides access to all individual participant data collected during the trial, after anonymisation, with the exception of pharmacokinetic or genetic data. Data are available to request in a timely fashion after the indication studied has been approved in the US and EU and after primary publication acceptance. No expiration date of data requests is currently set once they are made available. Access is provided after a proposal has been approved by an independent review committee identified for this purpose and after receipt of a signed data‐sharing agreement. Data and documents, including the study protocol, statistical analysis plan, clinical study report, blank or annotated case report forms, will be provided in a secure data‐sharing environment for up to 2 years per proposal. For details on submitting a request, see the instructions provided at http://www.clinicalstudydatarequest.com. Data are also available on clinicaltrials.gov: NCT01646177.