

Abstract
Alzheimer’s disease (AD) is the most common neurodegenerative disorder. Neuroinflammation is a prevalent pathogenic stress leading to neuronal death in AD. Targeting neuroinflammation to keep neurons alive is an attractive strategy for AD therapy. 1-Trifluoromethoxyphenyl-3-(1-propionylpiperidin-4-yl) urea (TPPU) is a potent inhibitor of soluble epoxide hydrolase (sEH) and can enter into the brain. It has a good efficacy on a wide range of chronic inflammatory diseases in preclinical animal models. However, the anti-neuroinflammatory effects and molecular mechanisms of TPPU for potential AD interventions remain elusive. With an aim to develop multi-target therapeutics for neurodegenerative diseases, we screened TPPU against sEH from different vertebrate species and a broad panel of human kinases in vitro for potential new targets relevant to neuroinflammation in AD. TPPU inhibits both human sEH and p38β kinase, two key regulators of inflammation, with nanomolar potencies and distinct selectivity. To further elucidate the molecular mechanisms, differentiated SH-SY5Y human neuroblastoma cells were used as an AD cell model and investigated the neuroprotection of TPPU against amyloid oligomers. We found that TPPU effectively prevents neuronal death by mitigating amyloid neurotoxicity, tau hyperphosphorylation and mitochondrial dysfunction, promoting neurite outgrowth, and suppressing activation and nuclear translocation of NF-κB for inflammatory responses in human nerve cells. The results indicate that TPPU is a potent and selective dual inhibitor of sEH and p38β kinase, showing a synergistic action in multiple AD signaling pathways. Our study sheds light upon TPPU and other sEH/p38β dual inhibitors for potential pharmacological interventions in AD.
Keywords: Alzheimer’s disease, soluble epoxide hydrolase, p38 mitogen-activated protein kinase, dual inhibitor, neuroinflammation, neuroprotection
Graphical Abstract
INTRODUCTION
Alzheimer’s disease (AD) is the most common cause of dementia and is the fifth leading cause of global deaths. To date, no effective drugs can prevent, cure, or even slow this devastating disease.1 The accrued drug failures in AD clinical trials have amplified the calls for a more diverse drug pipeline. AD arises from a complexity of multi-faceted mechanisms, including amyloid neurotoxicity, tau hyperphosphorylation, neuroinflammation, mitochondrial dysfunction, oxidative stress, synaptic loss, and ultimate death of neurons in the brain of AD patients.2–4 Hence, developing a multi-target drug to modulate diverse rather than a single pathogenic process of AD would increase a chance to find effective disease-modifying therapies.5
Inflammation associated with neurodegenerative conditions is a prevalent cellular stress in AD.3, 6–7 Targeting neuroinflammation to keep neurons alive is an attractive therapeutic strategy. The soluble epoxide hydrolase (sEH) has been implicated in many chronic inflammatory conditions.8–9 The sEH is responsible for the conversion of epoxy fatty acids to their corresponding diols in lipid signaling pathways.8 In the arachidonic acid cascade, sEH hydrolyzes epoxyeicosatrienoic acids (EETs) to dihydroxyeicosatrienoic acids (DHETs). EETs are known to mediate vascular relaxation responses and possess anti-inflammatory activities via attenuating endoplasmic reticulum (ER) stress and oxidative stress in vivo,10 while DHETs are relatively inactive in comparison to the EETs in most biological systems.8, 11–12 Therefore, sEH inhibition can increase the endogenous concentration of EETs, leading to mitigation of inflammation. In recent years, there is ample evidence proving the therapeutic effects of sEH inhibitors in many rodent models for neuropsychiatric syndromes associated with inflammation such as stroke, seizure, depression, autism, and Parkinson’s disease.9–10, 13–16 However, questions concerning whether sEH inhibitors can be used for AD intervention and the underlying molecular mechanisms remain elusive.
p38 mitogen-activated protein kinases (MAPK) are another key regulator responsive to a variety of cellular stresses, and the p38 MAPK signaling plays an important role in neuroinflammation.6 Previous studies revealed that p38 kinases are abnormally active in glial cells of AD brains, which enhances inflammatory gene expression and upregulates proinflammatory cytokines.17–18 In addition, an elevated p38 kinase activity in neurons contributes to tau hyperphosphorylation and tau-mediated neuroinflammation in AD.19–20 Hence, inhibiting aberrant p38 MAPK is also a promising approach for treating neurological diseases relevant to inflammation such as AD.18, 21–22
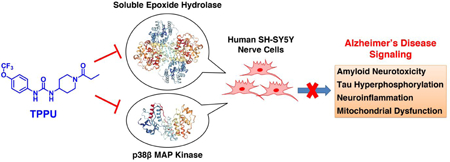
With a goal of developing novel therapeutics to target neuroinflammation for AD, we here tested a hypothesis that simultaneously modulating sEH and p38 MAPK pathways in neurons by a urea-based inhibitor, 1-trifluoromethoxyphenyl-3-(1-propionylpiperidin-4-yl) urea (TPPU), would provide synergistic outcomes of neuroprotection. TPPU (Figure 1) was originally developed as an inhibitor to sEH with a high potency and selectivity.23 Prior studies have shown that TPPU is effective in several animal models of neurological diseases.10, 13–16 Given that the urea-based scaffold is a key pharmacophore in many inhibitors of sEH and protein kinases,24–26 we postulated that TPPU would also inhibit kinases relevant to neuroinflammation. Such dual inhibitory properties of TPPU and the resulting pharmacological effects have not been previously investigated.
Figure 1.

Chemical structure and abbreviation of the sEH inhibitor, 1-trifluoromethoxyphenyl-3-(1-propionylpiperidin-4-yl) urea (TPPU).
We recently established a cell model of extracellular β-amyloid (Aβ) toxicity using differentiated SH-SY5Y human neuroblastoma cells, which is specifically associated with AD.27–28 Herein, we tested pharmacological effects of TPPU in this cell model and demonstrated that TPPU is a highly potent and selective dual inhibitor of human sEH and p38β kinase with multi-functional actions in cell signaling of AD. The results support the studies of TPPU and development of new dual inhibitors of sEH/p38β kinase for potential AD therapy.
RESULTS
TPPU is a Potent Inhibitor Selectively Targeting Primate and Rodent sEHs.
We previously demonstrated that TPPU is a slow and tight-binding inhibitor showing a high target occupancy with the sEH. It is highly potent to human and mouse sEHs.23 To further elaborate its species selectivity, we screened TPPU against sEHs in liver S-9 fractions from different mammals, including human, monkey, mouse, rat, dog, and mini-pig (Table 1). The results substantiated that TPPU is a potent and selective inhibitor for primate and rodent sEHs. Particularly, it has IC50 values less than 50 nM for the human, monkey and rat sEH, and an IC50 of 90 nM for the mouse sEH. In contrast, TPPU is relatively less potent for the dog and mini pig sEHs with IC50 values of 1800 nM and 220 nM, respectively.
Table 1.
Inhibition of sEH activity by TPPU in liver S-9 fractions from various species.
| Species | IC50 (nM)a |
|---|---|
| Human (male) | 45 ± 3 |
| Monkey (male Cynomolgus) | 16 ± 2 |
| Mouse (male C57BL/6) | 90 ± 5 |
| Rat (male Sprague Dawley) | 41 ± 3 |
| Dog (male Beagle) | 1800 ± 300 |
| Mini pig (male Yutacan) | 220 ± 40 |
IC50 was determined with t-DPPO as a substrate. Results were the average ± SD (n =3).
TPPU Selectively Inhibits Kinase Activities of p38β and p38γ Isoforms.
Many urea-based compounds have been developed to target kinases because they can mimic ATP to block phosphate transfer in kinase-catalyzed reactions.24–26 Aberrant kinase activities have been implicated in many tauopathies including AD. With an aim at investigating whether TPPU could be a multi-target therapeutic for AD intervention, we screened TPPU at 1 μM against 40 human kinases (Figure 2A) that are pathologically relevant to AD.19, 28 TPPU selectively inhibited kinase activities of p38β and p38γ isoforms (remaining kinase activities of 36% and 49%, respectively) among other kinases tested. In contrast, its inhibitory effects on p38α and p38δ isoforms were not significant. The results indicate a good isoform-selectivity of TPPU to p38β (IC50, 270 nM) and p38γ (IC50, 890 nM) where p38β is the most selective (approximately 3.3fold over p38γ) (Figure 2B–C). TPPU weakly inhibited a handful of kinases such as GSK-3β, AMPKA2, and CK1α1 in comparison with p38β.
Figure 2.

(A) Inhibitory effects of TPPU on the activities of 40 kinases relevant to AD. Kinases were assayed in the presence of 1 μM TPPU or control (0.2% PEG400 vehicle). Data were the mean of duplicate of each of six independent experiments with ± SEM (n = 6). The data were analyzed by one-way ANOVA with Tukey’s multiple comparison test. **p < 0.01, ***p < 0.001, ****p < 0.0001 relative to the control. (B) TPPU inhibited p38β kinase activity with an IC50 value of 0.27 μM. (C) TPPU inhibited p38γ kinase activity with an IC50 value of 0.89 μM.
SH-SY5Y Human Nerve Cells are a Valid Neuronal Model for the Study of sEH and p38 MAPK.
SH-SY5Y Human neuroblastoma cells are commonly used for the study of neurodegenerative diseases in vitro because they can be differentiated with morphological, biochemical, and functional features resembling human mature neurons.27, 29–30 Western blotting on a whole-cell lysate showed that differentiated SH-SY5Y cells express a reasonable level of sEH and p38β kinase (Figure 3A) in comparison to the housekeeping protein β-actin. Treatment of TPPU (10, 100 and 1000 nM) to the cells for 24 h significantly decreased cellular sEH activities in a dose-dependent manner (Figure 3B). The results indicated that SH-SY5Y cells were a valid in vitro cell model suitable for the present study.
Figure 3.

Differentiated SH-SY5Y cells were a valid neuronal model. (A) Western blotting on a whole-cell lysate. Analysis was performed with antibodies against sEH (EPHX2), p38β kinase, and β-actin (loading control). Optical densities were normalized to β-actin. (B) Treatment with various concentrations of TPPU (10 to 1000 nM) for 24 h significantly decreased cellular sEH activities in SH-SY5Y cells. Analysis was performed with a sEH enzyme assay using a radiolabeled substrate t-DPPO. Data were the mean of three independent experiments with ± SEM (n = 3), *p < 0.05, ***p < 0.001.
TPPU Protects Neurite Outgrowth against Aβ42 Neurotoxicity in SH-SY5Y Cells.
Chronic Aβ exposure in neuronal cells triggers AD-mimic pathologies such as tau hyperphosphorylation, Ca2+ homeostatic dysregulation, activation of MAPK-linked toxicity, mitochondrial dysfunction, production of inflammatory proteins, and the ultimate loss of neuronal integrity.27–28, 31–32 Because SH-SY5Y human neuronal cells express functional sEH and p38β kinase as well as mature tau isoforms with proper neuronal distribution in microtubules,29 we used differentiated SH-SY5Y cells under Aβ42 insults as a defined cell model of AD and evaluated the in vitro pharmacological effects of TPPU.
The results showed that treatment with 10 μM Aβ42 induced detrimental changes in neuronal morphology as many dying and nondifferentiated cells with retracted neurites in comparison to the untreated control (Figure 4A–B). However, pretreatment of 100 nM TPPU effectively relieved Aβ42 toxicity in SH-SY5Y cells (Figure 4C–D). TPPU-treated cells maintained a healthy neuronal morphology for which they were well differentiated with extended neurites. Besides, the TPPU-treated cells tend to have a more pyramidal shaped soma and become distinctly polarized. The cells also had longer and branched neurites and a detectable neuronal network in comparison to the control cells (Figure 4A versus 4C). Being consistent with our prior study in the rat primary sensory and cortical neurons,33 observations of the neuron-like phenotype of SH-SY5Y cells upon TPPU treatment implicated that sEH inhibition promoted axonogenesis. Because sEH is predominantly localized to axons in mature neurons, its inhibition could regulate bioactive EETs to induce axonal regeneration and outgrowth.33 Moreover, maintaining healthy tau−microtubule interactions via intervening the p38 MAPK pathway by TPPU could synergistically contribute to neurite outgrowth.
Figure 4.

Morphological changes of SH-SY5Y cells upon treatments for 72 h. (A) 0.2% PEG 400 vehicle control. Differentiated cells with extended neurites. (B) 10 μM Aβ42 treatment. Dying and nondifferentiated cells with retracted neurites. (C) Pretreatment of 100 nM TPPU followed by 10 μM Aβ42 treatment. (D) Zoomed image showing protected well-differentiated neurons with extended neurites (arrow pointing). Micrographs represent the average morphologic characteristics of cell cultures under a given condition of 5–8 independent experimental replicates (n = 5–8). Scale bar = 100 μm.
TPPU and EETs Prevent Aβ-induced Cytotoxicity in SH-SY5Y Cells.
To demonstrate that TPPU exerts neuroprotection against Aβ neurotoxicity, the cell viability assay was conducted. TPPU alone was tolerable and nontoxic to SH-SY5Y cells up to a dose at 1000 nM (Figure 5A). Treatment with 10 μM Aβ42 decreased cell viability to 30% in comparison with the control cells (Figure 5B). In contrast, pretreated cells with varying concentrations of TPPU ranging from 0.1 to 1000 nM for 1 h followed by 10 μM Aβ42 coincubation for 72 h regained cell viability from 30% to 100% in a dose-dependent manner. TPPU showed an effective neuroprotection with an EC50 value of 48.6 nM (Figure 5B–C).
Figure 5.

TPPU inhibited Aβ42 neurotoxicity in SH-SY5Y cells. Cell viability was determined with the MTS assay. Data were the mean of duplicate of six independent experiments with ± SEM (n = 6). Data were analyzed by one-way ANOVA with Tukey’s multiple comparison test. #p < 0.05, ####p < 0.0001 relative to the vehicle control; ****p < 0.0001 relative to the 10 μM Aβ42 treatment. (A) Cytotoxicity assessment of TPPU in SH-SY5Y cells. Cells were treated with varying concentrations of TPPU or 0.2% PEG 400 vehicle and incubated for 72 h. (B) Cells were pretreated with varying concentrations of TPPU or 0.2% PEG 400 vehicle for 1 h followed by 10 μM Aβ42 treatment for 72 h. (C) TPPU inhibited neurotoxicity induced by 10 μM Aβ42 with an EC50 value of 48.6 nM. The results were normalized as the percentage of the neuroprotective activity relative to the control (100%) and the 10 μM Aβ42 treatment (0%). Neuroprotection curve was analyzed by four-parameter regression. (D) Cotreatment of EETs with TPPU enhanced neuroprotection.
EETs are lipid metabolites and substrates of sEH. Our previous investigations demonstrated that EETs are neuroprotective and are capable of stimulating axonogenesis in rat PC-12 pheochromocytoma cells13 and rat primary sensory and cortical neurons.33 In the present study, treatment of TPPU stimulated the SH-SY5Y cell proliferation (Figure 5A). Cotreatment of EETs and TPPU at 0.1 μM significantly increased cell proliferation by approximately 30% (p < 0.0001) (Figure 5D). Our results also demonstrated that pretreatment of either 0.1 μM EETs or 0.1 μM TPPU alone for 1 h followed by 10 μM Aβ42 exposure for 72 h showed effective neuroprotection in SH-SY5Y cells (p < 0.0001). As expected, cotreatment of 0.1 μM EETs with 0.1 μM TPPU showed an enhanced effect that almost fully protected cell viability.
Pairwise Treatment of Selective Inhibitors of sEH and p38 Kinase Synergizes Neuroprotection in SH-SY5Y Cells.
To further prove that TPPU is a dual inhibitor and simultaneous inhibition of both sEH and p38β kinase would result in a synergistic neuroprotection, we conducted a drug combination screening. A selective sEH inhibitor (t-AUCB) and a selective p38α/β kinase inhibitor (SB202190) were cotreated in a pairwise manner in SH-SY5Y cells under Aβ42 exposure. It is noteworthy that t-AUCB is a very potent urea-based sEH inhibitor (IC50, 2 nM) structurally distinct from TPPU. t-AUCB neither inhibits p38 MAPKs nor other kinases.24, 34 SB202190 is a potent p38α/β inhibitor (IC50, 50 nM) capable of blocking MAPK pathways in cells.35–36 The combination effects of the two compounds were then assessed by the cell viability assay according to a screening method described by He et al.37
As shown in Figure 6, when SB202190 at a very low concentration (1 nM), treatment of t-AUCB from 0.1 to 100 nM protected the cells from Aβ42 toxicity in a dose-dependent manner. In parallel, when t-AUCB at 0.1 nM, treatment of SB202190 from 1 to 1000 nM increased neuroprotection in a dose-dependent manner as well. However, at the highest dosage tested, t-AUCB (100 nM) showed only a 27.3% neuroprotection, while SB202190 (1000 nM) was about 37.5%. Interestingly, an analysis on the combination dose-response matrices revealed that cotreatment of both compounds at certain ratios dramatically improved neuroprotection at different levels (e.g., cotreatment of t-AUCB at 1 nM with SB202190 at 1000 nM resulted in a 54.3% neuroprotection), which is a typical synergistic effect. The data provide a compelling evidence that simultaneous inhibition against both sEH and p38β MAPK would offer synergistic benefits for potential AD therapy.
Figure 6.

A drug combination data analysis. Pairwise treatments of a selective sEH inhibitor (t-AUCB) and a selective p38α/β kinase inhibitor (SB202190) in differentiated SH-SY5Y cells. Each compound was neuroprotective against 10 μM Aβ42 in a dose-dependent manner. Combinations of both compounds in the dose-response matrices showed a synergistic effect. Colors in 3D mesh showed different levels of neuroprotection that were presented in percentage relative to the Aβ42 free control (100%) and the Aβ42 treatment (0%). Data were the mean of three independent experiments (n = 3).
TPPU Attenuates Tau Hyperphosphorylation Induced by Aβ42 in SH-SY5Y Cells.
Aβ oligomers directly bind to the receptor for advanced glycation end-products (RAGE)38–39 or nicotinic acetylcholine receptors (e.g., α7 and α4β2 nAChRs)31 resulting in activation of p38 MAPK signaling and tau hyperphosphorylation in neurons.27–28, 31 Given that TPPU selectively inhibits p38β kinase in vitro (Figure 2), we further evaluated its effects on alleviating Aβ42-induced tau hyperphosphorylation in human SH-SY5Y nerve cells. The tau phosphorylation level at the site S396 was monitored because it is a pathologic p-tau site in AD and is known to be susceptible to p38 MAPK.19 Figure 7 shows that treatment of 10 μM Aβ42 for 72 h increased the p-tau S396 levels in SH-SY5Y cells by approximately 2.8-fold relative to the untreated cells.
Figure 7.

TPPU attenuated Aβ42 induced tau phosphorylation at the site S396 in a dose dependent manner. Differentiated SH-SY5Y cells were pretreated with various concentrations of TPPU or 0.2% PEG 400 vehicle for 1 h followed by treatment of 10 μM Aβ42 for 72 h. The known selective p38 inhibitor, SB202190, at 0.05 μM was used as a reference control. ELISA analysis was performed with specific antibody against Tau pS396 to quantify cellular tau phosphorylation levels. Fold changes were calculated relative to the control with ± SEM (n = 6). Data were analyzed by one-way ANOVA with Tukey’s multiple comparison test. ####p < 0.0001 relative to vehicle control; ****p < 0.0001 relative to the 10 μM Aβ42 treatment.
Conversely, pretreatment of TPPU (0.01, 0.1, and 1 μM) in the cells significantly mitigated the Aβ42-induced tau phosphorylation levels at the site pS396 in a dose-dependent manner (p < 0.0001). To determine whether such remedy on tau hyperphosphorylation is due to the p38β inhibition by TPPU, we used a selective p38α/β inhibitor SB202190 as a reference control in the cellular assay.35 The results demonstrated that TPPU is pharmacologically equivalent to SB202190 in terms of the outcome on p-tau alleviation, which implies the p38β inhibition by TPPU in the cells. In addition, the data showed that TPPU (0.01 μM) possesses a stronger alleviative effect on tau hyperphosphorylation than that of SB202190 at a 5-fold higher dosage level (0.05 μM).
TPPU and EETs Prevent Aβ-induced Depolarization of Mitochondrial Membrane Potential and Mitochondrial Dysfunction.
Aβ oligomers induce oxidative stress by producing reactive oxygen species (ROS) and activate proinflammatory genes, which cause mitochondrial dysfunction.2 To assess the protective effects of TPPU on mitochondrial integrity and function against Aβ toxicity, the changes of mitochondrial membrane potential (Δψm) in SH-SY5Y human nerve cells were measured with a JC 10 assay. In this assay, the JC-10 dyes generate red-fluorescent aggregates (Em = 590 nm) within the healthy mitochondria upon membrane polarization. However, if mitochondria dysfunctions, the dyes will be released in a greenfluorescent monomer form (Em = 525 nm) through the collapsed/depolarized mitochondrial membrane.40 Because TPPU protected Aβ42-induced neurotoxicity with an EC50 value of 48.6 nM (Figure 5B–C), we used relevant doses of TPPU (50 nM and 100 nM) to test its effect on Δψm. As shown in Figure 8, 10 μM Aβ42 treatment for 72 h decreased Δψm as the monomer/aggregate ratio (525/590 nm) increased by approximately 140% compared with the control (p < 0.0001). Such a deficit of mitochondrial membrane induced by Aβ42 was similar to that of a reference compound FCCP, a known ionophore capable of depolarizing the mitochondrial membrane. Conversely, cells pretreated with TPPU for 1 h followed by 10 μM Aβ42 treatment for 72 h significantly decreased fluorescence ratios of 525/590 nm in a dose-dependent manner (p < 0.0001), indicating the apparent protection of mitochondrial integrity against Aβ42 in the cells.
Figure 8.

TPPU and EETs prevented Aβ42-induced mitochondrial dysfunction in differentiated SH-SY5Y cells. Cellular mitochondrial membrane potential (Δψm) was evaluated with JC-10 assay. FCCP at 50 μM or Aβ42 at 10 μM depolarized Δψm as indicated by increasing JC-10 monomer/aggregate fluorescence ratio (525/590 nm). Pretreatment with TPPU, EETs, or co-dose for both significantly prevented the depolarized condition of mitochondria in the presence of 10 μM Aβ42 in 72 h. Data were the mean of duplicate of six independent experiments with ± SEM (n = 6). Data were analyzed by one-way ANOVA with Tukey’s multiple comparison test. ####p < 0.0001 relative to the vehicle control; ***p < 0.001, ****p < 0.0001 relative to the 10 μM Aβ42 treatment. FCCP at 50 μM was used as a reference control.
Figure 10.

Proposed neuroprotective mechanisms of TPPU against Alzheimer’s disease.
To determine whether sEH is critical in regulating Δψm for mitochondrial integrity, we assayed the sEH substrate, EETs, in SH-SY5Y cells. The data substantiated the effect that pretreatment with EETs at 0.1 μM attenuated the loss of Δψm induced by Aβ42 (p < 0.001) in SH-SY5Y cells, and cotreatment of 0.1 μM TPPU with 0.1 μM EETs synergistically restored Δψm to normal levels against Aβ42 toxicity (p < 0.0001). Our data agreed with the other studies that EETs protect mitochondrial functions in rat hippocampal astrocytes.41 The results collectively suggest that sEH inhibition by TPPU or treatment with EETs results in prevention from Aβ-induced mitochondrial dysfunction in SH-SY5Y nerve cells.
TPPU Suppresses Activation and Nuclear Translocation of the Transcription Factor NF-κB in SH-SY5Y Cells.
The transcription factor NF-κB is a master regulator of inflammatory responses. Aβ oligomers destabilize beneficial epoxy fatty acids like EETs and activate p38 MAPK/NF-κB pathways in neurons and glial cells,31, 42–43 thereby promoting neuroinflammation and primarily microglial inflammation in AD brains.6–7 NF-κB is inactive in the cytoplasm, bound to the inhibitory IκB proteins. Upon activation by p38 MAPK phosphorylation, the homo- or heterodimeric complexes of NF-κB will release and translocate to the nucleus, where they activate proinflammatory gene expression.6 Given TPPU being a potent inhibitor of both sEH and p38 kinase, we evaluated the effects on perturbation of the p38 MAPK/NF-κB signaling in response to Aβ42 stimuli in SH-SY5Y cells. Aβ42 at 5 μM significantly increased nuclear concentrations of all five subunits of NF-κB (p50, p52, p65, RelB, and c-Rel) in SH-SY5Y cells after 8 h (Figure 9). In particular, the nuclear p50 and p65 quantities were approximately 7.8-fold and 12.8-fold, respectively, higher than those of the untreated controls, while the nuclear p52, RelB, and c-Rel were approximately 3 to 5-fold more concentrated in the Aβ42 treated cells. Conversely, TPPU at 0.05 and 0.1 μM inhibited nuclear translocation of all five subunits (p < 0.0001), and particularly significant for p65. To determine whether TPPU blocking NF-κB activation is due to the p38β kinase inhibition, we used dexamethasone (DEX) as a reference control that has been reported to inhibit both p38 MAPK and NF-κB.44 The effects of TPPU on suppressing activation and nuclear translocation of NF-κB were comparable to DEX at 0.1 μM.
Figure 9.

TPPU suppressed activation and nuclear translocation of the transcription factor NF-κB in differentiated SH-SY5Y cells. NF-κB family members (p50, p52, p65, RelB, and c-Rel) in the nuclear extracts were monitored with TransAM NF-κB assay. 5 μM Aβ42 treatment for 8 h significantly induced translocation of NF-κB to the nucleus. Pretreatment with various concentrations of TPPU (0.05 to 0.1 μM) or 0.1 μM DEX for 1 h followed by treatment of 5 μM Aβ42 for 8 h significantly reduced levels of all five NF-κB subunits in neuronal nuclei. Data were the mean of duplicate of six independent experiments with ± SEM (n = 6). Data were analyzed by one-way ANOVA with Tukey’s multiple comparison test. ####p < 0.0001 relative to vehicle control; ••••p < 0.0001 relative to the 5 μM Aβ42 treatment. Dexamethasone (DEX) known as the NF-κB/p38 MAPK inhibitor for anti-inflammatory activities was used as a reference control.
DISCUSSION
The sEH and p38 MAPKs are key mediators in inflammatory processes. sEH plays a pivotal role in the metabolism of lipid epoxides such as EETs. The cytochrome P450−epoxide hydrolase axis is clinically relevant to inflammatory responses. Therefore, sEH has implicated in many human inflammatory diseases, including cardiovascular diseases, hypertension, ischemia, diabetes, chronic kidney disease, cancer, neuropathic pain, and neurodegenerative disorders.8–9, 11 p38 MAPKs are responsive to cellular stresses such as ROS, LPS, cytokines, UV light, DNA damage, and heat/osmotic shock. In AD, toxic Aβ oligomers bind to certain receptors such as RAGE and nAChR in brain cells, thereby stimulating abnormal activation of p38 MAPK signaling,31, 38–39 causing hyperphosphorylation of tau proteins27 and excessively high activity of transcription factor NF-κB in neuroinflammation.18, 20 These collective pathological events are believed to be the key elements of the amyloid cascade in AD.45 Given that neuroinflammation is a major characteristic in most neurodegenerative diseases, the overall aim of this study was to investigate the neuroprotective effects and mechanistic understanding of a potent sEH inhibitor, TPPU, as a potential intervention for treating AD.
TPPU was originally developed as a urea-based sEH inhibitor to reduce inflammatory and neuropathic pain.23 The pharmacological efficacy of TPPU has been demonstrated in many preclinical animal models, including mice, rats, dogs, and non-human primates with chronic inflammatory conditions.9, 15, 23, 46–49 In a previous study, the brain-to-plasma ratio of TPPU was 0.18, suggesting that it is a brain penetrant.13 TPPU selectively targets primate sEH with a low nanomolar potency (human, IC50 = 45 nM; monkey, IC50 = 16 nM), whereas its potencies in other vertebrate species are relatively weaker (Table 1). The fine-tuned ADMET profiles and the blood-brain barrier permeability of TPPU make it as a suitable CNS drug candidate for neurodegenerative diseases.10, 13–16 Nevertheless, despite extensive pharmacological studies on inflammatory disorders targeting sEH, there is still limited knowledge deeming whether sEH is a viable therapeutic target of AD and the inhibitors such as TPPU may be applicable in AD intervention.
To answer above scientific questions, we used differentiated SH-SY5Y human cells as a defined nerve cell model of AD because they express functional sEH and p38β kinase that are of interest in our study (Figure 3). The results support our hypothesis that TPPU is a potent dual inhibitor of human sEH and human p38β kinase. Treatment of TPPU alone or a drug combination using t-AUCB (sEH inhibitor) plus SB202190 (p38α/β inhibitor) exerts a synergistic neuroprotection against Aβ toxicity (Figure 5 and 6). In addition, TPPU being a p38β kinase inhibitor can alleviate tau hyperphosphorylation and maintain healthy tau−microtubule association for neuronal differentiation and axonogenesis in SH-SY5Y cells. Although both p38 MAPK and GSK-3β are tau protein kinases,19, 50 TPPU selectively inhibits p38β (IC50, 270 nM) over GSK-3β (IC50, > 10 μM) (Figure 2) and alleviates tau hyperphosphorylation at the site pS396 that is found aberrant in AD brains (Figure 7).
Mitochondria are the primary energy source crucial for the viability of human cells. Their dysfunction has been implicated in many disease conditions ranging from cancer, diabetes, cardiovascular diseases to neurodegenerative disorders.2, 51 In AD, toxic Aβ oligomers increase oxidative stress and ER stress, impair mitochondrial function, and trigger programmed cell death of neurons. Particularly in mitochondria, Aβ causes formation of mitochondrial permeability transition pores and disrupts the electron transport chain, consequently leading to the pathological ROS generation.52 Such ROS feeds back to exacerbate mitochondrial dysfunction. As shown in Figure 8, the sEH inhibition by TPPU protects nerve cells against Aβ42 at least in part through maintaining the membrane integrity, intracellular ionic charges, and mitochondrial function, which in turn increase neuronal survival. Additionally, the p38β inhibition by TPPU might contribute to reducing mitochondrial oxidative stress downstream of the AD signaling pathway (Figure 10), thereby resulting in neuroprotection. These data collectively suggest that sEH/p38β dual inhibitors could be a potential therapy in regulating mitochondrial homeostasis and preventing AD. They also support a hypothesis that mitochondrial dysfunction leads to a pathological increase of ROS levels, a feedback loop of ROS on mitochondria, and the shift in the ER stress response from cell homeostasis to inflammatory status.9, 53 Modulating the mitochondrial–ROS–ER stress axis by sEH/p38β dual inhibitors is an attractive strategy for treating AD.
Importantly, the results indicate that TPPU shows an effect to mitigate neuroinflammation. By inhibiting p38 MAPK, TPPU effectively suppresses activation and nuclear translocation of NF-κB responsive to amyloid stimuli (Figure 9), suggesting the downstream repressive effect on proinflammatory genes by blocking NF-κB signaling. Such anti-inflammatory properties could also be reinforced through inhibition of neuronal sEH to stabilize the endogenous EETs level. While all of the primary targets of EETs are still unknown, increasing evidence shows that EETs inhibit ER/oxidative stresses and NF-κB activation to modulate inflammatory responses in cells.9, 12, 54 On the other hand, it has become apparent that toxic p-tau oligomers can spread through extracellular vesicles from neurons to astrocytes and microglia, and in turn trigger release of proinflammatory cytokines such as TNF-α, IL-1β, and IL-6 in gliosis, thereby aggravating chronic neuroinflammation.4, 20, 55 Since elevated p38 MAPK activity is prone to increase tauopathy in neurons, inhibition of p38β kinase by TPPU may help reduce tau-mediated neuroinflammation in the brain.
As illustrated above, TPPU intervenes the canonical amyloid cascade of AD, offering multiple benefits in alleviating neuroinflammation, mitochondrial dysfunction, tau hyperphosphorylation, and eventual death of neurons. A plausible proposal of neuroprotective mechanisms of TPPU against AD is summarized in Figure 10. Since no disease-modifying therapies for AD have been approved by the US-FDA, investigating drug candidates with a dual inhibitory function targeting both sEH and p38 MAPK may offer a great promise to find effective AD treatment. Neuroinflammation is a prominent feature in most neurodegenerative diseases including AD, where sEH and p38 MAPK regulate distinct but separate inflammatory pathways.3, 6–7 Interestingly, TPPU being a urea-based small-molecule inhibitor shows a good selectivity to p38β kinase over the other three isoforms (i.e., p38α, p38δ, and p38γ). The p38 isoform-selectivity of TPPU is particularly advantageous for which it would potentially reduce the risk of off-target effects of a drug. In fact, a p38α kinase selective inhibitor, neflamapimod (VX-745),56 is currently in phase II clinical trials in AD patients, which highlights the therapeutic relevance of p38 MAPKs in AD pathology. While TPPU shows approximately 3-fold selectivity to p38β over p38γ, it serves as a promising drug lead for developing new selective sEH/p38β dual inhibitors through medicinal chemistry approaches in the future.50, 57
CONCLUSIONS
The present study in conjunction with the prior evidence gives new insights into an investigational small-molecule inhibitor, TPPU, for potential AD therapy. We conducted in vitro enzymatic assay screening to identify new targets of TPPU and then functionally validated in a cell model of AD, with a primary aim at understanding the molecular mechanisms and pharmacology of TPPU in human nerve cells. The present investigation supports the argument that sEH plays a key role in AD pathology and is a novel target worth pursuing. The new findings of TPPU regarding the dual inhibitory properties, the molecular and cellular mechanisms, as well as our curated knowledge in sEH biochemistry and pharmacology would pave a road for developing new selective sEH/p38β dual inhibitors with improved blood-brain barrier permeability via structure-based drug design and optimization for potential therapies in AD and related neurodegenerative diseases. Notwithstanding, because different brain cells may have distinct drug responses, it would be interesting to expand the study to microglia and astrocytes as they are the main contributors of neuroinflammation. In addition, it remains essential to conduct in vivo pharmacological studies of TPPU to ascertain its neuroprotective and anti-inflammatory outcomes as well as its target engagement in preclinical animal models of AD in the future.
METHODS
Chemicals and Reagents.
All solvents and reagents were purchased from commercial sources and were used without further purification. TPPU, t-AUCB, and EETs were synthesized in-house as previously described.23, 34 Liver S-9 fractions were acquired from Xenotech-LLC (Kansas City, KS). Staurosporine, 4-(4-fluorophenyl)-2-(4-hydroxyphenyl)-5-(4-pyridyl)-1H-imidazole (SB202190), FCCP, DEX, phenylmethanesulfonyl fluoride (PMSF), protease inhibitor cocktail, retinoic acid, PEG 400, hexafluoroisopropanol, and Nonidet P-40 were from Sigma-Aldrich (Saint Louis, MO). Aβ42 peptide was from AnaSpec (Fremont, CA). Kinase Selectivity Profiling Assay Kit (Cat# V6854, V6856, V6858, V6924, V6918), ADP-Glo Kinase Assay Kit (Cat# V6930), and CellTiter 96 AQueous One Solution Cell Proliferation MTS Assay Kit (Cat# G3580) were from Promega (Madison, WI). Human Tau pS396 ELISA Kit (Cat# KHB7031) and Cell Extraction Buffer were from Invitrogen (Camarillo, CA). Bradford Protein Assay Kit (Cat# 5000002) was from Bio-Rad (Hercules, CA). JC-10 Mitochondrial Membrane Potential Assay Kit (Cat# ab112134) was from Abcam (Burlingame, CA). TransAM NF-κB Family ELISA Assay Kit (Cat# 43296) was from Active Motif (Carlsbad, CA). Primary antibody anti-EPHX2 (Cat# ab155280) was from Abcam (Cambridge, MA), anti-p38β MAPK (Cat# 2339) and anti-β-actin (Cat# 4970) were from Cell Signaling Technology (Danvers, MA). Alkaline phosphataseconjugated secondary antibodies were obtained from Bio-Rad (Hercules, CA).
sEH Enzyme Assay.
Liver S9 fractions were diluted with sodium phosphate buffer (0.1 M, pH 7.4) containing 0.1 mg/mL of bovine serum albumin (BSA) to ensure that the activity was in the linear range of the assay (< 20% hydrolysis of the substrate). The enzyme activity was measured with [3H]-trans-diphenylpropene oxide (t-DPPO) as a substrate as described previously.58 To 100 μL of diluted extract, 1 μL of inhibitor solution in DMSO was added ([I]final = 2.5 to 10000 nM). The mixture was incubated at 37 °C for 10 min. The enzymatic reaction was started by adding 1 μL of t-DPPO ([S]final = 50 μM). The reaction was carried at 37 °C for 15 min. The reaction was stopped by adding 60 μL of methanol and extracted with 200 μL of isooctane. The diol product was measured by quantifying the amount of radioactivity in 40 μL of the water phase using a liquid scintillation counter (TriCarb 2810TR, Perkin Elmer, Shelton, CT). Each assay was done in triplicate (n =3). Data were plotted as a percent of inhibition (100% activity measured in the absence of inhibitor but in the presence of 1% DMSO) as a function of the concentration of inhibitor. The IC50 values were determined by non-linear regression of the data, using a curve fitted onto a sigmoidal, logistic three-parameter equation.
To measure the sEH activities in SH-SY5Y cells, the cells were seeded at a density of 3 × 105 cells/mL. When it reached 80% confluency, cells were treated with different concentrations of TPPU for 24 h. After treatment, the cells were harvested and pelleted by centrifugation at 600 g, 4 °C, for 15 min. The cell pellet was then resuspended in 0.5 mL of sodium phosphate buffer (20 mM, pH 7.4) containing 5 mM of EDTA. In a glass tube containing 10 μg of BSA, 100 μL of the cell suspension was added. The sEH activity was measured using [3H]-t-DPPO as a substrate ([S] = 50 μM). The mixture was incubated at 37 °C for 60 min. The remaining substrate was extracted with 200 μL of isooctane. The diol product was quantified by measuring radioactivity remaining in water phase by liquid scintillation (TriCarb 2810TR, Perkin Elmer, Shelton, CT). The sEH activities were normalized by the total protein concentration using a BCA assay. Each assay was done in triplicate (n =3).
Kinase Luminescent Assay.
Kinase inhibition was assessed with the ADP-Glo Kinase Assay according to the procedure described previously.27–28 For screening, 5 ng/μL of kinase solutions were assayed in a 20 μL reaction mixture containing 50 ng/μL substrate, 40 mM Tris, pH 7.5, 20 mM MgCl2, 0.1 mg/mL BSA, 50 μM dithiothreitol (DTT), 25 μM ATP, varying concentrations of TPPU solutions or 0.2% PEG 400 vehicle in a 384-well microplate. The reaction mixture was incubated for 1 h at room temperature followed by the addition of the ADP-Glo reagents according to the manufacturer’s protocol. The pan-kinase inhibitor staurosporine was used at 1 μM as a reference control. Inhibition was presented as the percentage of the kinase activity relative to the vehicle control. Inhibition curves were analyzed by four-parameter regression. Samples were analyzed in duplicate in six independent experiments (n = 6).
Cell Culture and Differentiation.
An SH-SY5Y human neuroblastoma cell line (ATCC CRL-2266; Sigma-Aldrich, Saint Louis, MO) was cultured in 1:1 (v/v) DMEM/F12 media supplemented with 2 mM glutamine, 10% heat-inactivated fetal bovine serum (FBS) and 1% antibiotics including penicillin and streptomycin. Cell cultures with three to four passages were used for the experiments in compliance with ATCC recommendation. After reaching 70–80% confluence, cells were subcultured on poly-D-lysine plates with 10 μM retinoic acid in a reduced serum media (1% FBS) to induce postmitotic differentiation.27 Differentiation was confirmed by the change to polygonal morphology and extension of long neurites. Cells were cultured at 37 °C in a fully humidified atmosphere containing 5% CO2.
Aβ42 Oligomer Preparation.
The toxic oligomers of Aβ42 were prepared as described previously.27 Briefly, lyophilized Aβ42 peptide was dissolved in hexafluoroisopropanol, dried under vacuum, and stored at −20 °C. Immediately prior to use, the peptide residue was reconstituted in the DMEM/F12 media to make a 0.1 mM stock solution and incubated at 4 °C for 24 h to form diffusible oligomers. The toxicity of Aβ42 oligomers at a final concentration of 5 or 10 μM were confirmed by cell viability assay.
Chemical Treatment in SH-SY5Y Cells.
SH-SY5Y human neuroblastoma cells were seeded at a density of 3 × 105 cells/mL in a 6-well or 96-well poly-D-lysine plate, or a 10 cm poly-D-lysine culture dish in the DMEM/F12 media containing 10 μM retinoic acid and 1% FBS to induce postmitotic differentiation. The cells were incubated under regular culture conditions for attachment. After 24 h of plating, the cells were pretreated with different concentrations of test chemicals or a vehicle control for 1 h following coincubation with 5 or 10 μM Aβ42 during a desired time period.
Anti-Aβ42 Neurotoxicity Assay and Neuronal Morphology Analysis.
The assay procedure was previously described.27–28 SH-SY5Y cells were treated with chemicals in a 96-well plate under the culture condition described above. Staurosporine at 1 μM was used as a reference control for cytotoxicity, while 0.05 μM SB202190 was used as a reference control for p38 inhibition. After experimental treatment for 72 h, the cells were subjected to microscopic analyses of neuronal morphology and a CellTiter 96 AQueous One Solution Cell Proliferation MTS assay according to the manufacturer’s instruction. Cell viability was normalized by comparing the colorimetric intensity of nontreated control cells in the MTS assay (100% viability) with that of the solvent blank without cell culture (0% viability). The neuroprotection curve was normalized as the percentage of the cell viability relative to the nontreated control (100%) and 10 μM Aβ42 treatment (0%) and was analyzed by four-parameter regression. Samples were analyzed in duplicate in six independent experiments (n = 6).
Western Blotting.
The differentiated SH-SY5Y cells were washed with phosphate buffered saline (PBS) and lysed with phosphate protein extraction buffer (20 mM Na4P2O7, pH 7.4, 115 mM NaCl) containing 1 mM PMSF and a protease inhibitor cocktail. Concentrations of the harvested proteins were determined by a BioSpec-nano. Equal amounts of total proteins for each sample (10 μg per lane) were resolved by 10% SDS-PAGE and transferred onto PVDF membranes. The membranes were blocked with 5% BSA overnight and hybridized with primary antibodies against EPHX2, p38β MAPK, or β-actin at room temperature for 2 h. After washes, the membranes were incubated with corresponding alkaline phosphatase-conjugated secondary antibodies at room temperature for 1 h, and the immunoreactive bands were detected by the BCIP/NBT reagent. Densitometric analysis was performed on three independent samples per experiment (n = 3).
Anti-Tau Hyperphosphorylation ELISA Analysis.
The assay procedure was previously described.28 Briefly, SH-SY5Y cells were treated with chemicals in a 6-well plate under the culture condition described above. The known selective p38α/β inhibitor SB202190 at 0.05 μM was used as a reference control. After 72 h, the harvested cells were washed with PBS and lysed with cell extraction buffer containing 10 mM Tris, pH 7.4, 100 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1 mM NaF, 20 mM Na4P2O7, 2 mM Na3VO4, 1% Triton X-100, 10% glycerol, 0.1% sodium dodecyl sulfate (SDS), 0.5% sodium deoxycholate, 1 mM PMSF and a protease inhibitor cocktail (Sigma-Aldrich). Total protein concentrations were determined with the Bradford assay (Bio-Rad). The phosphorylated human tau at pS396 site (a specific site of p38 kinase found in AD)19 in the cell lysate was quantified with the human Tau (Phospho) [pS396] ELISA kit according to the manufacturer’s protocol (Invitrogen, Camarillo, CA). Absorbance at 450 nm was read with a Multiskan Go microplate reader (Thermo Scientific, Waltham, MA). Samples were analyzed in duplicate in six independent experiments (n = 6).
JC-10 Mitochondrial Membrane Potential Assay.
The assay procedure was previously described.40 SH-SY5Y cells were treated with chemicals in a 96-well plate under the culture condition described above. The mitochondrial ionophore, carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone (FCCP), at 50 μM was used as a reference control. After an incubation at 37 °C for 72 h, the cells were subject to a JC-10 mitochondrial membrane potential assay according to the manufacturer’s instruction (Abcam, Burlingame, CA). Briefly, JC-10 dyeloading solution was added to each well and incubated at 37 °C, 5% CO2 for 1 h. Fluorescence intensities (Ex/Em =490/525 nm, and Ex/Em = 540/590 nm) of each well were monitored. The ratio of fluorescence intensity (525/590 nm) was used to determine the mitochondrial membrane potential (Δψm). Increasing ratios indicate mitochondrial membrane depolarization. Samples were analyzed in duplicate in six independent experiments (n = 6).
Nuclear Extraction of Cell Lysate.
The harvested SH-SY5Y cells were washed with ice-cold PBS and incubated with a hypotonic buffer containing 20 mM Tris-HCl, pH 7.4, 10 mM NaCl, 3 mM MgCl2, 0.5% Nonidet P-40, 1 mM PMSF, and a protease inhibitor cocktail on ice for 15 min, and vortexed. The cell homogenate was centrifuged (1000g) for 10 min at 4 ºC. The resulting supernatant containing the cytosolic fraction was removed. The nuclear pellet was lysed with cell extraction buffer containing 10 mM Tris, pH 7.4, 100 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1 mM NaF, 20 mM Na4P2O7, 2 mM Na3VO4, 1% Triton X-100, 10% glycerol, 0.1% SDS, 0.5% sodium deoxycholate, 1 mM PMSF, and a protease inhibitor cocktail followed by centrifugation (14000g) for 30 min at 4 ºC. Nuclear protein concentrations were determined with the Bradford assay.
Nuclear NF-κB DNA-binding ELISA Analysis.
The assay procedure was previously described.59–60 SH-SY5Y cells were treated with chemicals in a 6-well plate under the culture condition described above. Dexamethasone (DEX, 0.1 μM) known as a MAPK/NF-κB inhibitor with anti-inflammatory activities was used as a reference control. After the experimental treatment for 8 h, the nuclear proteins were extracted from harvested cells. TransAM NF-κB Family Assay Kit (Active Motif, Carlsbad, CA) was used to determine concentrations of p50, p52, p65, RelB, and c-Rel in cellular nuclear fractions via a colorimetric DNA-binding ELISA in response to the NF-κB consensus sequence (5’-GGGACTTTCC-3’). Wild-type and mutated consensus oligonucleotides were used to monitor the NF-κB binding specificity. Nuclear NF-κB levels were quantified by measuring the absorbance at 450 nm on a Multiskan Go microplate reader. Samples were analyzed in duplicate in six independent experiments (n = 6).
General Procedures for Instrumental Analysis.
Fluorescent/luminescent measurement was performed on an Agilent Cary Eclipse fluorescence spectrophotometer. Optical absorbance was measured on a Multiskan GO microplate reader. Microscopic images were taken under a Nikon Diaphot inverted tissue culture microscope with Optronics MicroFire microscope camera (Nikon Precision Inc, Belmont, CA).
Statistical Analysis.
The data and statistical analyses were complied with the recommendations on experimental design and analysis in pharmacology. Data were presented as mean ± SEM or ± SD of three to six independent experiments performed in duplicate. The data were analyzed by one-way ANOVA with Tukey’s multiple comparison post-hoc test as well as Student’s t-test. The p values less than 0.05 were considered statistically significant. Analysis was performed using GraphPad Prism 6 and SigmaPlot 13.0.
Acknowledgments
Funding Sources
This work was supported in part by the USDA (Hatch project HAW5032-R), Hawaii Community Foundation (18ADV-90801), NIH-NIMHD grant 8G12MD007601, NIH-NIEHS Superfund Program P42ES04699, and NIH-NINDS grant U54NS079202. B.D.H. is a NIH-NIEHS RIVER Awardee (1R35 ES030443-01).
ABBREVIATIONS
- AD
Alzheimer’s disease
- Aβ42
β-amyloid fragment peptide 1–42
- TPPU
1-trifluoromethoxyphenyl-3-(1-propionylpiperidin-4-yl) urea
- t-AUCB
trans-4-(4-[3-adamantan-1-yl-ureido]-cyclohexyloxy)-benzoic acid
- EETs
epoxyeicosatrienoic acids
- DHETs
dihydroxyeicosatrienoic acids
- [3H]-t-DPPO
[3H]-trans-diphenylpropene oxide
- DEX
dexamethasone
- FCCP
carbonyl cyanide-4-(trifluoromethoxy) phenylhydrazone
- PEG 400
polyethylene glycol 400
- sEH
soluble epoxide hydrolases
- p38 MAPK
p38 mitogen-activated protein kinase
- GSK-3β
glycogen synthase kinase-3β
- ERK2
extracellular signal regulated kinase 2
- JNK1
c-Jun N-terminal kinase 1
- JNK3
c-Jun N-terminal kinase 3
- CDK1/CyclinA
cyclin-dependent kinase 1 with subunit cyclin A
- CDK2/CyclinE
cyclin-dependent kinase 2 with subunit cyclin E
- CDK3/CyclinE
cyclin-dependent kinase 3 with subunit cyclin E
- CDK5/p25
cyclin-dependent kinase 5 with subunit p25
- CDK5/p35
cyclin-dependent kinase 5 with subunit p35
- CDK6/CyclinD
cyclin-dependent kinase 6 with subunit cyclin D
- CDK9/CyclinK
cyclin-dependent kinase 9 with subunit cyclin K
- CLK1
dual specificity protein kinase 1
- AKT1
v-akt murine thymoma viral oncogene homolog 1
- p70S6Kβ
p70 ribosomal protein S6 kinase beta
- PDK1
phosphoinositide-dependent kinase 1
- PKA
protein kinase A
- PKC
protein kinase C
- PRKG1
cGMP-dependent protein kinase 1
- ROCK1
Rho-associated, coiled-coil containing protein kinase 1
- RSK2
ribosomal protein S6 kinase 2
- AMPK
AMP-activated protein kinase with subunits
- CAMKIIα
Ca2+/calmodulin-dependent protein kinase IIα
- CAMKIIγ
Ca2+/calmodulin-dependent protein kinase IIγ
- CAMKIV
Ca2+/calmodulin-dependent protein kinase IV
- DAPK1
death-associated protein kinase 1
- STK33
serine/threonine-protein kinase 33
- CK2α1
casein kinase 2α1
- DNA-PK
DNA-dependent protein kinase
- CK1α1
casein kinase 1α1
- CK1ε
casein kinase 1ε
- CK1γ1
casein kinase 1γ1
- VRK2
vaccinia related kinase 2
- RAGE
receptor for advanced glycation end-products
- nAChR
nicotinic acetylcholine receptor
- NF-κB
nuclear factor-κB
- IκB
inhibitor of κB
- TNF-α
tumor necrosis factor-α
- IL-1β
interleukin-1β
- IL-6
interleukin-6
- ROS
reactive oxygen species
- ER
endoplasmic reticulum
Footnotes
ASSOCIATED CONTENT
Supporting Information. The Supporting Information is available free of charge on the ACS Publications website at DOI: XXX.
Detail of the kinase selectivity screening (PDF).
Conflict of Interest
B.D.H. is a founder of EicOsis Human Health which is developing sEH inhibitors for treatment of inflammatory and neuropathic pain.
Publisher's Disclaimer: “Just Accepted” manuscripts have been peer-reviewed and accepted for publication. They are posted online prior to technical editing, formatting for publication and author proofing. The American Chemical Society provides “Just Accepted” as a service to the research community to expedite the dissemination of scientific material as soon as possible after acceptance. “Just Accepted” manuscripts appear in full in PDF format accompanied by an HTML abstract. “Just Accepted” manuscripts have been fully peer reviewed, but should not be considered the official version of record. They are citable by the Digital Object Identifier (DOI®). “Just Accepted” is an optional service offered to authors. Therefore, the “Just Accepted” Web site may not include all articles that will be published in the journal. After a manuscript is technically edited and formatted, it will be removed from the “Just Accepted” Web site and published as an ASAP article. Note that technical editing may introduce minor changes to the manuscript text and/or graphics which could affect content, and all legal disclaimers and ethical guidelines that apply to the journal pertain. ACS cannot be held responsible for errors or consequences arising from the use of information contained in these “Just Accepted” manuscripts.
REFERENCES
- (1).Becker RE, Greig NH, Giacobini E, Schneider LS, and Ferrucci L. (2014) A new roadmap for drug development for Alzheimer’s disease, Nat. Rev. Drug Discov 13, 156–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Lin MT, and Beal MF (2006) Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases, Nature 443, 787–795. [DOI] [PubMed] [Google Scholar]
- (3).Griffin WST (2013) Neuroinflammatory cytokine signaling and Alzheimer’s disease, N. Engl. J. Med 368, 770–771. [DOI] [PubMed] [Google Scholar]
- (4).Chu D, and Liu F. (2019) Pathological changes of tau related to Alzheimer’s disease, ACS Chem. Neurosci 10, 931–944. [DOI] [PubMed] [Google Scholar]
- (5).Oset-Gasque MJ, and Marco-Contelles J. (2018) Alzheimer’s disease, the “one-molecule, one-target” paradigm, and the multitarget directed ligand approach, ACS Chem. Neurosci 9, 401–403. [DOI] [PubMed] [Google Scholar]
- (6).Glass CK, Saijo K, Winner B, Marchetto MC, and Gage FH (2010) Mechanisms underlying inflammation in neurodegeneration, Cell 140, 918–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Calsolaro V, and Edison P. (2016) Neuroinflammation in Alzheimer’s disease: Current evidence and future directions, Alzheimers Dement. 12, 719–732. [DOI] [PubMed] [Google Scholar]
- (8).Morisseau C, and Hammock BD (2013) Impact of soluble epoxide hydrolase and epoxyeicosanoids on human health, Annu. Rev. Pharmacol. Toxicol 53, 37–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Wagner KM, McReynolds CB, Schmidt WK, and Hammock BD (2017) Soluble epoxide hydrolase as a therapeutic target for pain, inflammatory and neurodegenerative diseases, Pharmacol. Ther 180, 62–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Ren Q, Ma M, Yang J, Nonaka R, Yamaguchi A, Ishikawa K. i., Kobayashi K, Murayama S, Hwang SH, Saiki S, Akamatsu W, Hattori N, Hammock BD, and Hashimoto K. (2018) Soluble epoxide hydrolase plays a key role in the pathogenesis of Parkinson’s disease, Proc. Natl. Acad. Sci. U.S.A 115, E5815-E5823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Imig JD, and Hammock BD (2009) Soluble epoxide hydrolase as a therapeutic target for cardiovascular diseases, Nat. Rev. Drug Discov 8, 794–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Dennis EA, and Norris PC (2015) Eicosanoid storm in infection and inflammation, Nat. Rev. Immunol 15, 511–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Ren Q, Ma M, Ishima T, Morisseau C, Yang J, Wagner KM, Zhang J-C, Yang C, Yao W, Dong C, Han M, Hammock BD, and Hashimoto K. (2016) Gene deficiency and pharmacological inhibition of soluble epoxide hydrolase confers resilience to repeated social defeat stress, Proc. Natl. Acad. Sci. U.S.A 113, E1944-E1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Chen Y, Tian H, Yao E, Tian Y, Zhang H, Xu L, Yu Z, Fang Y, Wang W, Du P, and Xie M. (2017) Soluble epoxide hydrolase inhibition promotes white matter integrity and long-term functional recovery after chronic hypoperfusion in mice, Sci. Rep 7, 7758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Minaz N, Razdan R, Hammock BD, and Goswami SK (2018) An inhibitor of soluble epoxide hydrolase ameliorates diabetes-induced learning and memory impairment in rats, Prostaglandins Other Lipid Mediat. 136, 84–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Ma M, Ren Q, Yang J, Zhang K, Xiong Z, Ishima T, Pu Y, Hwang SH, Toyoshima M, Iwayama Y, Hisano Y, Yoshikawa T, Hammock BD, and Hashimoto K. (2019) Key role of soluble epoxide hydrolase in the neurodevelopmental disorders of offspring after maternal immune activation, Proc. Natl. Acad. Sci. U.S.A 116, 7083–7088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Bodles AM, and Barger SW (2005) Secreted β-amyloid precursor protein activates microglia via JNK and p38-MAPK, Neurobiol. Aging 26, 9–16. [DOI] [PubMed] [Google Scholar]
- (18).Munoz L, and Ammit AJ (2010) Targeting p38 MAPK pathway for the treatment of Alzheimer’s disease, Neuropharmacology 58, 561–568. [DOI] [PubMed] [Google Scholar]
- (19).Martin L, Latypova X, Wilson CM, Magnaudeix A, Perrin M-L, Yardin C, and Terro F. (2013) Tau protein kinases: Involvement in Alzheimer’s disease, Ageing Res. Rev 12, 289–309. [DOI] [PubMed] [Google Scholar]
- (20).Leyns CEG, and Holtzman DM (2017) Glial contributions to neurodegeneration in tauopathies, Mol. Neurodegener 12, 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Giuliani D, Bitto A, Galantucci M, Zaffe D, Ottani A, Irrera N, Neri L, Cavallini GM, Altavilla D, Botticelli AR, Squadrito F, and Guarini S. (2014) Melanocortins protect against progression of Alzheimer’s disease in triple-transgenic mice by targeting multiple pathophysiological pathways, Neurobiol. Aging 35, 537–547. [DOI] [PubMed] [Google Scholar]
- (22).Lee JK, and Kim N-J (2017) Recent advances in the inhibition of p38 MAPK as a potential strategy for the treatment of Alzheimer’s disease, Molecules 22, 1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Rose TE, Morisseau C, Liu J-Y, Inceoglu B, Jones PD, Sanborn JR, and Hammock BD (2010) 1-Aryl-3-(1-acylpiperidin-4-yl)urea inhibitors of human and murine soluble epoxide hydrolase: Structure−activity relationships, pharmacokinetics, and reduction of inflammatory pain, J. Med. Chem 53, 7067–7075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Hwang SH, Wecksler AT, Zhang G, Morisseau C, Nguyen LV, Fu SH, and Hammock BD (2013) Synthesis and biological evaluation of sorafenib- and regorafenib-like sEH inhibitors, Bioorg. Med. Chem. Lett 23, 3732–3737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Dumas J, Smith RA, and Lowinger TB (2004) Recent developments in the discovery of protein kinase inhibitors from the urea class, Curr. Opin. Drug Discov. Devel 7, 600–616. [PubMed] [Google Scholar]
- (26).Noble MEM, Endicott JA, and Johnson LN (2004) Protein kinase inhibitors: Insights into drug design from structure, Science 303, 1800–1805. [DOI] [PubMed] [Google Scholar]
- (27).Liang Z, Zhang B, Su WW, Williams PG, and Li QX (2016) C-Glycosylflavones alleviate tau phosphorylation and amyloid neurotoxicity through GSK3β inhibition, ACS Chem. Neurosci 7, 912–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Liang Z, and Li QX (2018) Discovery of selective, substrate-competitive, and passive membrane permeable glycogen synthase kinase-3β inhibitors: Synthesis, biological evaluation, and molecular modeling of new C-glycosylflavones, ACS Chem. Neurosci 9, 1166–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Agholme L, Lindström T, Kågedal K, Marcusson J, and Hallbeck M. (2010) An in vitro model for neuroscience: Differentiation of SH-SY5Y cells into cells with morphological and biochemical characteristics of mature neurons, J. Alzheimer’s Dis 20, 1069–1082. [DOI] [PubMed] [Google Scholar]
- (30).Jahn K, Wieltsch C, Blumer N, Mehlich M, Pathak H, Khan AQ, Hildebrandt H, and Frieling H. (2017) A cell culture model for investigation of synapse influenceability: Epigenetics, expression and function of gene targets important for synapse formation and preservation in SH-SY5Y neuroblastoma cells differentiated by retinoic acid, J. Neural. Transm 124, 1341–1367. [DOI] [PubMed] [Google Scholar]
- (31).Arora K, Cheng J., and Nichols RA (2015) Nicotinic acetylcholine receptors sensitize a MAPK-linked toxicity pathway on prolonged exposure to β-amyloid, J. Biol. Chem 290, 21409–21420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).del Pino J, Marco-Contelles J, López-Muñoz F, Romero A, and Ramos E. (2018) Neuroinflammation signaling modulated by ASS234, a multitarget small molecule for Alzheimer’s disease therapy, ACS Chem. Neurosci 9, 2880–2885. [DOI] [PubMed] [Google Scholar]
- (33).Abdu E, Bruun DA, Yang D, Yang J, Inceoglu B, Hammock BD, Alkayed NJ, and Lein PJ (2011) Epoxyeicosatrienoic acids enhance axonal growth in primary sensory and cortical neuronal cell cultures, J. Neurochem 117, 632–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Hwang SH, Tsai H-J, Liu J-Y, Morisseau C, and Hammock BD (2007) Orally bioavailable potent soluble epoxide hydrolase inhibitors, J. Med. Chem 50, 3825–3840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Karahashi H, Nagata K, Ishii K, and Amano F. (2000) A selective inhibitor of p38 MAP kinase, SB202190, induced apoptotic cell death of a lipopolysaccharide-treated macrophage-like cell line, J774.1, Biochim. Biophys. Acta, Mol. Basis Dis 1502, 207–223. [DOI] [PubMed] [Google Scholar]
- (36).Bauer D, Redmon N, Mazzio E, Taka E, Reuben JS, Day A, Sadrud-Din S, Flores-Rozas H, Soliman KFA, and Darling-Reed S. (2015) Diallyl disulfide inhibits TNFα induced CCL2 release through MAPK/ERK and NF-Kappa-B signaling, Cytokine 75, 117–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).He L, Kulesskiy E, Saarela J, Turunen L, Wennerberg K, Aittokallio T, and Tang J. (2018) Methods for high-throughput drug combination screening and synergy scoring, In Cancer Systems Biology: Methods and Protocols (von Stechow L, Ed.), pp 351–398, Springer New York, New York, NY. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Origlia N, Righi M, Capsoni S, Cattaneo A, Fang F, Stern DM, Chen JX, Schmidt AM, Arancio O, Yan SD, and Domenici L. (2008) Receptor for advanced glycation end product-dependent activation of p38 mitogen-activated protein kinase contributes to amyloid-β-mediated cortical synaptic dysfunction, J. Neurosci 28, 35213530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Liu R, Wu C-X, Zhou D, Yang F, Tian S, Zhang L, Zhang T-T, and Du G-H (2012) Pinocembrin protects against β-amyloid-induced toxicity in neurons through inhibiting receptor for advanced glycation end products (RAGE)-independent signaling pathways and regulating mitochondrion-mediated apoptosis, BMC Med. 10, 105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Sakamuru S, Li X, Attene-Ramos MS, Huang R, Lu J, Shou L, Shen M, Tice RR, Austin CP, and Xia M. (2012) Application of a homogenous membrane potential assay to assess mitochondrial function, Physiol. Genomics 44, 495–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Sarkar P, Zaja I, Bienengraeber M, Rarick KR, Terashvili M, Canfield S, Falck JR, and Harder DR (2014) Epoxyeicosatrienoic acids pretreatment improves amyloid β-induced mitochondrial dysfunction in cultured rat hippocampal astrocytes, Am. J. Physiol. Heart Circ. Physiol 306, H475–H484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Sarkar P, Narayanan J, and Harder DR (2011) Differential effect of amyloid beta on the cytochrome P450 epoxygenase activity in rat brain, Neuroscience 194, 241–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Valerio A, Boroni F, Benarese M, Sarnico I, Ghisi V, Bresciani LG, Ferrario M, Borsani G, Spano P, and Pizzi M. (2006) NF-κB pathway: A target for preventing β-amyloid (Aβ)-induced neuronal damage and Aβ42 production, Eur. J. Neurosci 23, 1711–1720. [DOI] [PubMed] [Google Scholar]
- (44).Danova K, Klapetkova A, Kayserova J, Sediva A, Spisek R, and Jelinkova LP (2015) NF-kappaB, p38 MAPK, ERK½, mTOR, STAT3 and increased glycolysis regulate stability of paricalcitol/dexamethasone-generated tolerogenic dendritic cells in the inflammatory environment, Oncotarget 6, 14123–14138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Selkoe DJ, and Hardy J. (2016) The amyloid hypothesis of Alzheimer’s disease at 25 years, EMBO Mol. Med 8, 595–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Liu J-Y, Tsai H-J, Hwang SH, Jones PD, Morisseau C, and Hammock BD (2009) Pharmacokinetic optimization of four soluble epoxide hydrolase inhibitors for use in a murine model of inflammation, Br. J. Pharmacol 156, 284–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Tsai H-J, Hwang SH, Morisseau C, Yang J, Jones PD, Kasagami T, Kim I-H, and Hammock BD (2010) Pharmacokinetic screening of soluble epoxide hydrolase inhibitors in dogs, Eur. J. Pharm. Sci 40, 222–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Ulu A, Appt SE, Morisseau C, Hwang SH, Jones PD, Rose TE, Dong H, Lango J, Yang J, Tsai HJ, Miyabe C, Fortenbach C, Adams MR, and Hammock BD (2012) Pharmacokinetics and in vivo potency of soluble epoxide hydrolase inhibitors in cynomolgus monkeys, Br. J. Pharmacol 165, 1401–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Sasso O, Wagner K, Morisseau C, Inceoglu B, Hammock BD, and Piomelli D. (2015) Peripheral FAAH and soluble epoxide hydrolase inhibitors are synergistically antinociceptive, Pharmacol. Res 97, 7–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Liang Z, and Li QX (2018) Harnessing the π-cation interaction in rational drug design: Discovery of potent and isoform-specific GSK-3β inhibitors for Alzheimer’s disease, Alzheimers Dement. 14, P301. [Google Scholar]
- (51).Murray M, Dyari HRE, Allison SE, and Rawling T. (2014) Lipid analogues as potential drugs for the regulation of mitochondrial cell death, Br. J. Pharmacol 171, 2051–2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Pérez MJ, Ponce DP, Aranguiz A, Behrens MI, and Quintanilla RA (2018) Mitochondrial permeability transition pore contributes to mitochondrial dysfunction in fibroblasts of patients with sporadic Alzheimer’s disease, Redox Biol. 19, 290–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Inceoglu B, Bettaieb A, Haj FG, Gomes AV, and Hammock BD (2017) Modulation of mitochondrial dysfunction and endoplasmic reticulum stress are key mechanisms for the wide-ranging actions of epoxy fatty acids and soluble epoxide hydrolase inhibitors, Prostaglandins Other Lipid Mediat. 133, 68–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Terashvili M, Sarkar P, Nostrand MV, Falck JR, and Harder DR (2012) The protective effect of astrocyte-derived 14,15-epoxyeicosatrienoic acid on hydrogen peroxide-induced cell injury in astrocyte-dopaminergic neuronal cell line co-culture, Neuroscience 223, 68–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Nilson AN, English KC, Gerson JE, Barton Whittle T, Nicolas Crain C, Xue J, Sengupta U, Castillo-Carranza DL, Zhang W, Gupta P, and Kayed R. (2016) Tau oligomers associate with inflammation in the brain and retina of tauopathy mice and in neurodegenerative diseases, J. Alzheimer’s Dis 55, 1083–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Duffy JP, Harrington EM, Salituro FG, Cochran JE, Green J, Gao H, Bemis GW, Evindar G, Galullo VP, Ford PJ, Germann UA, Wilson KP, Bellon SF, Chen G, Taslimi P, Jones P, Huang C, Pazhanisamy S, Wang Y-M, Murcko MA, and Su MSS (2011) The discovery of VX-745: A novel and selective p38α kinase inhibitor, ACS Med. Chem. Lett 2, 758–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Liang Z, and Li QX (2018) π–Cation interactions in molecular recognition: Perspectives on pharmaceuticals and pesticides, J. Agric. Food Chem 66, 3315–3323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Borhan B, Mebrahtu T, Nazarian S, Kurth MJ, and Hammock BD (1995) Improved radiolabeled substrates for soluble epoxide hydrolase, Anal. Biochem 231, 188–200. [DOI] [PubMed] [Google Scholar]
- (59).Renard P, Ernest I, Houbion A, Art M, Le Calvez H, Raes M, and Remacle J. (2001) Development of a sensitive multi-well colorimetric assay for active NFκB, Nucleic Acids Res. 29, e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Higa JK, Liang Z, Williams PG, and Panee J. (2012) Phyllostachys edulis compounds inhibit palmitic acid-induced monocyte chemoattractant protein 1 (MCP-1) production, PLoS ONE 7, e45082. [DOI] [PMC free article] [PubMed] [Google Scholar]