

Abstract
Von Willebrand factor (VWF), an exceptionally large multimeric plasma glycoprotein, functions to initiate coagulation by agglutinating platelets in the blood stream to sites of vascular injury. This primary hemostatic function is perturbed in type 2 dysfunctional subtypes of von Willebrand disease (VWD) by mutations that alter the structure and function of the platelet GPIbα adhesive VWF A1 domains. The resulting amino acid substitutions cause local disorder and misfolding of the native structure of the isolated platelet GPIbα adhesive A1 domain of VWF in both gain-of-function (type 2B) and loss-of-function (type 2M) phenotypes. These structural effects have not been explicitly observed in A1 domains of VWF multimers native to blood plasma. New mass spectrometry strategies are applied to resolve the structural effects of 2B and 2M mutations in VWF to verify the presence of A1 domain structural disorder in multimeric VWF harboring type 2 VWD mutations. Limited trypsinolysis (LTMS) and hydrogen deuterium exchange (HXMS) are applied to wild type and VWD variants of the single A1, A2 and A3-domains, an A1A2A3 tridomain fragment of VWF, plasmin-cleaved dimers of VWF, multimeric recombinant VWF, and normal VWF plasma concentrates. Comparatively, these methods show that mutations known to misfold the isolated A1-domain increase the rate of trypsinolysis and the extent of hydrogen deuterium exchange in local secondary structures of A1 within multimeric VWF. VWD mutation effects are localized to the A1 domain without appreciably affecting the structure and dynamics of other VWF domains. The intrinsic dynamics of A1 observed in recombinant fragments of VWF are conserved in plasma derived VWF. These studies reveal that structural disorder does occur in VWD variants of the A1 domain within multimeric VWF and provides strong support for VWF misfolding as a result of some, but not all, type 2 VWD variants.
Keywords: von Willebrand factor
Graphical Abstract

3. Introduction.
The von Willebrand Factor (VWF) is the largest blood protein and arguably the most sophisticated protein in human plasma [1]. Consisting of more than 2000 amino acid residues, the mature monomer unit of VWF (~ 250kDa) is a complex architecture of repeat A, C and D domains (D’D3 A1A2A3 D4 C1-C6 CK) that is assembled tail-to-tail at the C-terminal cysteine knot (CK) domains [2–4] as disulfide-linked dimers (~ 500kDa) in the endoplasmic reticulum which then multimerize head-to-head via disulfide bonds at the N-terminal D’ domain in the Golgi apparatus to MDa high molecular weight multimers. The N-terminal VWF propeptide (D1D2, 763 amino acids) facilitates multimerization and is severed in the process [5,6]. VWF secretion into the blood from vascular endothelial cells is highly regulated [7] occurring via several modes of exocytosis involving the post trans-Golgi network storage granules [8], Weibel-Palade bodies (WPB), which fuse with the plasma membrane of vascular endothelial cells. The VWF propeptide and the multimeric VWF are secreted into the vasculature where the rheological shear of blood flow unravels the dense packing of VWF multimers into strings with exposed A1 domains, free to bind platelet GPIbα receptors and begin primary hemostasis.
Primary hemostasis is entirely dependent on the proper function of the VWF A1 domains which contribute to the arrest of bleeding through binding platelet GPIbα receptors under shear stress, aggregating the firm platelet adhesion to exposed subendothelial vascular connective tissue, and establishing the formation of a platelet plug necessary for coagulation. Like a fishing line with multiple hooks, the multimeric structure of VWF facilitates the cooperative binding of platelet GPIbα receptors to multiple A1 domain sites which become exposed under shear stress. Mutations associated with the inherited autosomal dominant bleeding disorder, von Willebrand disease (VWD) within the A1 domains of VWF, can enhance, diminish or abolish the binding affinity of A1 for platelet GPIbα [9]. In any case, the gain- or loss-of-function caused by these mutations leads to a bleeding phenotype of variable severity [10] with high efficiency binding promoting platelet clumping, subsequent thrombocytopenia, and VWF depletion [11] at one extreme and inefficient binding [12] leading to deficient or a lack of platelet agglutination at the other extreme.
Much of the literature has focused on the intramolecular interactions involved in rheological exposure of the A1 domain to platelet GPIbα [13]. Glycosylation [14,15], intra-VWF interdomain interactions [16–18], N-terminal flanking peptide interactions with A1 [16,19–24], and even the physical spacing between D’D3 and A1 domains [25] have been implicated to contribute to the autoinhibition and regulation of VWF-mediated platelet adhesion. All are mechanisms that shield the A1 domain, either through spacial exclusion via bulky glycosyl groups [14,26] or by the association of A1 with adjacent VWF domains [16,18,27]. Some of these properties have been implicated to have a potential role in von Willebrand Disease [16,23,25], particularly type 2B VWD, since these gain-of-function mutations cluster [28] in a region spatially close to the N- and C-terminal flanking sequences which connect A1 to adjacent domains of VWF.
While quaternary structural interactions within VWF are responsive to rheological shear and VWD mutations [27], mutations in both type 2B gain-of-function and type 2M loss-of-function VWD phenotypes have substantial effects on the structure [10,11,29,30] and stability [10,12,31,32] of the isolated A1 domain. Relative to the wild-type native conformation, an increase in the stability dampens the structural dynamics resulting in a more rigid tertiary structure and loss-of-function [12]. By contrast, a decrease in stability has been observed to result in enhanced function, but depending on the structural location and type of mutation, substantial destabilization can result in disordered conformations in both VWD functional phenotypes [10,11]. Monoclonal antibody studies [33] indicate that mutations may alter the folding of the A1 domain within multimeric VWF, but there is no concrete biophysical evidence that the structural abnormalities caused by mutations actually occur within the A1 domains of plasma VWF multimers.
In this study, two proteolytic assays coupled with mass spectrometry are presented that provide evidence supporting A1 misfolding within VWD multimers. The strategy utilizes limited trypsinolysis (LTMS) and hydrogen-deuterium exchange mass spectrometry (HXMS) to assess the conformational dynamics of plasma-derived VWF and a collection of recombinant wild-type and VWD variants known to disorder the A1 domain as a single domain, an A1A2A3 tridomain, in multimeric VWF, and in plasmin-cleaved dimers of VWF. The structural effects of VWD mutations in A1 are observed to have minimal effect on structural dynamics of neighboring domains, exerting their conformational consequence primarily on the A1 domain itself. Mutations affect secondary structures both local to and C-terminally distant from the mutation site within the globular A1 domain indicating a potential for allostery. The structural dynamic properties of the wild-type A1 are conserved in each of the recombinant expression constructs and, within experimental error, are identical to the A1 dynamics in plasma VWF, indicating that the single A1 domain and A1A2A3 tridomain constructs used here, and in our prior publications [10–12,20,21,27,29–32,34–37], are accurate models for structural and functional studies of VWD. Overall, the results indicate that the intrinsic conformational dynamics observed in recombinant domain fragments of VWF are preserved in the biologically-relevant multimeric forms of VWF present in blood plasma.
4. Results.
4.1. Mutation effects on secondary structure in the single A1 domain are less discernible in A1A2A3 tridomain.
Illustrated in Fig.1A, we propose that VWD mutations variably affect the stability and structure of A1 through changes in the local dynamics of the domain within distinct structural regions. Stabilization of A1 by the type 2M G1324S variant suppresses local dynamics and diminishes GPIbα binding function [12] whereas destabilization increases function [31, 32]. Greatly enhanced dynamics leads to local unfolding in both gain and loss-of-function, depending on the type and structural location of amino acid substitutions involved [10]. The type 2B variants, P1337L, V1314D, V1316M, and reduced A1 (RCAM), and the type 2M variants, S1285F and F1369I, shown in Fig.1B are extreme cases that result in a detectable loss of circular dichroic (CD) ellipticity and a loss of the urea-induced native unfolding transition characteristic of the wild-type A1 domain.
Figure 1:
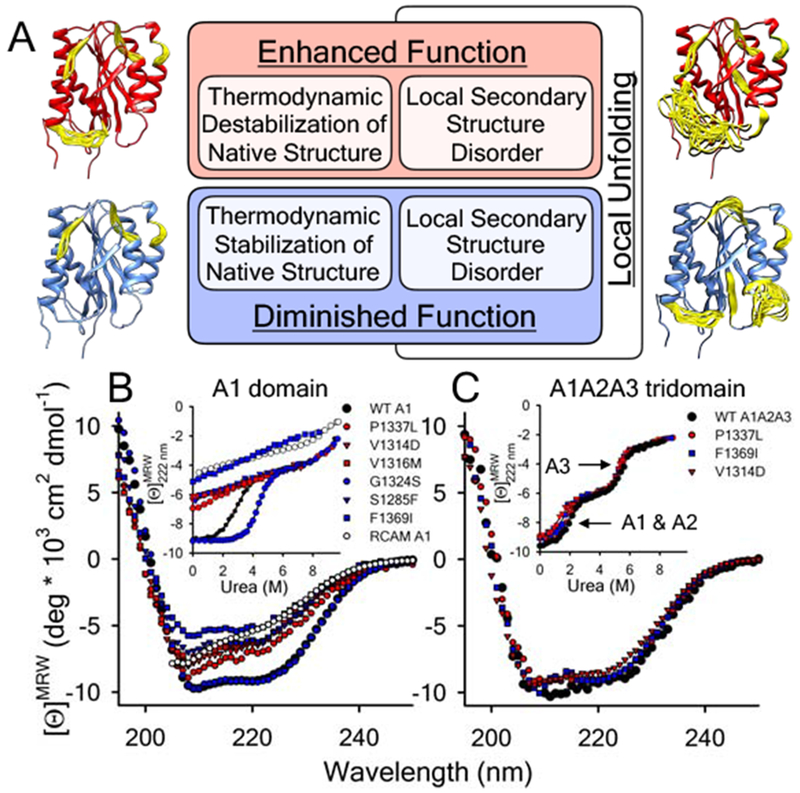
A) Proposed local secondary structure dynamics driven affinity regulation base on what is currently understood about how VWD mutations alter A1 domain structure and function [10–12,27,29–32,34]. Models of the A1 domain (PDB ID = 1AUQ [61]), generated with Mini-Protein MODelling (MPMOD) [62] and rendered using UCSF Chimera [63]. B & C) Far-UV CD spectra and urea denaturations (Insets) of the A1 (panel B) and A1A2A3 (panel C) VWD variants studied by LTMS and HXMS. B) wild-type A1 domain and VWD variants, P1337L, V1314D, V1316M, G1324S, S1285F, F1369I, and reduced A1. Inset: urea-induced unfolding of the proteins monitored at λ = 222nm. C) wild-type, P1337L, V1314D and F1369I A1A2A3 tridomains. Inset: urea-induced unfolding of the tridomains monitored at λ = 222nm. A1 and A2 domain unfolding precedes unfolding of the A3 domain (arrows). Black = wild-type, Red = type 2B VWD gain-of-function (GOF), Blue = type 2M VWD loss-of-function (LOF). Far-UV CD data for the A1 domain in panel B was originally published in [10].
The effect of P1337L, F1369I and V1314D on the CD ellipticity of the A1A2A3 tridomain construct of VWF is, however, smaller relative to wild-type (Fig.1C). Urea denaturation of A1A2A3 also shows only small decreases in the midpoint of the first (low urea) unfolding transition [35]. Far-UV CD spectroscopy is therefore less proficient at resolving local effects in larger multi-domain proteins because the total amount of secondary structure observable to the spectrometer is greater, therefore, a smaller fraction of the total observable secondary structure content of A1A2A3 is altered by the mutation.
4.2. Mutation effects on multimerization and intracellular localization are variable.
Fig.2A compares the multimerization pattern of three recombinant VWD variants of VWF known to disorder the A1 domain; P1337L (type 2B), S1285F (type 2M), and V1316M (type 2B). P1337L has normal multimerization consistent with prior observations [38]. Compared to wild-type VWF, the multimer bands of V1316M and S1285F are shifted in running distance, more diffuse with less intensity, and are quantitatively deficient in the high molecular weight (HMW) multimers. Quantification of fluorescence band intensities corresponding to the number of dimers per multimer (n) show that (In/n)/I1 for V1316M and S1285F is decreased relative to normal for all multimer bands. S1285F has previously been observed to have a loss of high molecular weight multimers, but high molecular weight multimers were restored when co-expressed with normal VWF as a hybrid [33]. These comparative differences indicate that structural effects of mutations in the A1 domain may affect the quality of multimerization, but is likely to depend on the extent of structural damage caused by the mutation since P1337L multimers are not different from normal.
Figure 2:

A) Top: Multimer gels of recombinant VWF. Lanes from left to right = (1) HEK293 (negative) control, (2) wild-type rVWF, (3) P1337L (2B, GOF) rVWF, (4) S1285F (2M, LOF) rVWF, and (5) V1316M (2B, GOF) rVWF. Horizontal lines on the right indicate the number of VWF dimers. Middle: Multimer gels are digitized (band intensity relative to background control as a function of gel running distance) for wild-type, P1337L, S1285F and V1316M rVWF. Bottom: The normalized band intensity ratio relative to the initial band intensity (In/I0) is plotted as a function of the number of dimers for each VWF variant. Inset: the difference (Δ(In/I0) wild-type-Mutant) between each mutant relative to wild-type rVWF. B-E) Intracellular immunofluorescence of VWF expression in HEK293 cells. B) wild-type VWF and VWD variants C) P1337L, D) S1285F, and E) V1316M were transiently expressed in HEK293 cells. 48hrs post transfection, cells were fixed and VWF proteins (left) and the ER marker protein PDI (middle) were detected by indirect immunofluorescence. Antibodies employed were rabbit anti-VWF and mouse anti-PDI detected by goat anti-rabbit AF488 and goat anti-mouse AF546, respectively. Colocalization appears in yellow in the overlays (right). Z-stacks were recorded at room temperature with a confocal microscope (TCS SP5, Leica, Wetzlar, Germany) using an HC PL APO CS2 63.0x1.40 OIL UV objective and the following settings: an image size of 512 x 512 pixels, laser power of the 543 and 488 lasers was set to 9% and 20%, respectively. All plains of the respective Z-stacks were merged using the Z project function of the ImageJ software [59].
To determine how these mutations affect intracellular localization, wild-type VWF and variants P1337L, S1285F, and V1316M were recombinantly expressed in HEK293 cells for 48h. VWF and the Endoplasmic Reticulum (ER)-specific marker protein, Protein Disulfide Isomerase (PDI), were detected by immunofluorescence [2]. S1285F (Fig.2D) showed increased co-localization with the PDI as compared to wild-type VWF, P1337L, and V1316M (Fig.2B, C, & E). This observation that S1285F lacks formation of Weibel-Palade bodies (WPBs) may indicate altered folding of A1 domains in S1285F VWF, but some S1285F fluorescence did not co-localize with PDI indicating residual secretion. V1316M and P1337L did not exhibit a significant difference in the intracellular localization compared to wild-type VWF, all of which were able to get packaged into WPBs.
The observations in Figs.1 & 2 establish that while local secondary structure effects of mutations are readily distinguished by far-UV CD in the single A1 domain, they are are more difficult to observe in VWF and the consequences of local disorder (if present at all in VWD variants of multimeric VWF) to the extent of multimerization and intracellular localization appear to be variable.
4.3. New strategies to observe mutation-induced structural disorder in VWF.
Two mass spectrometry methods, limited trypsinolysis (LTMS) and hydrogen deuterium exchange (HXMS), are developed to determine whether mutations induce structural disorder in A1 domains of multimeric VWF. Fig.3A illustrates models of the A1 domain dynamics consistent with published spectroscopic [10,30], thermodynamic [12,32], and HXMS data [11], where stabilization suppresses native conformational dynamics and structural disorder can occur in different secondary elements of the domain.
Figure 3:
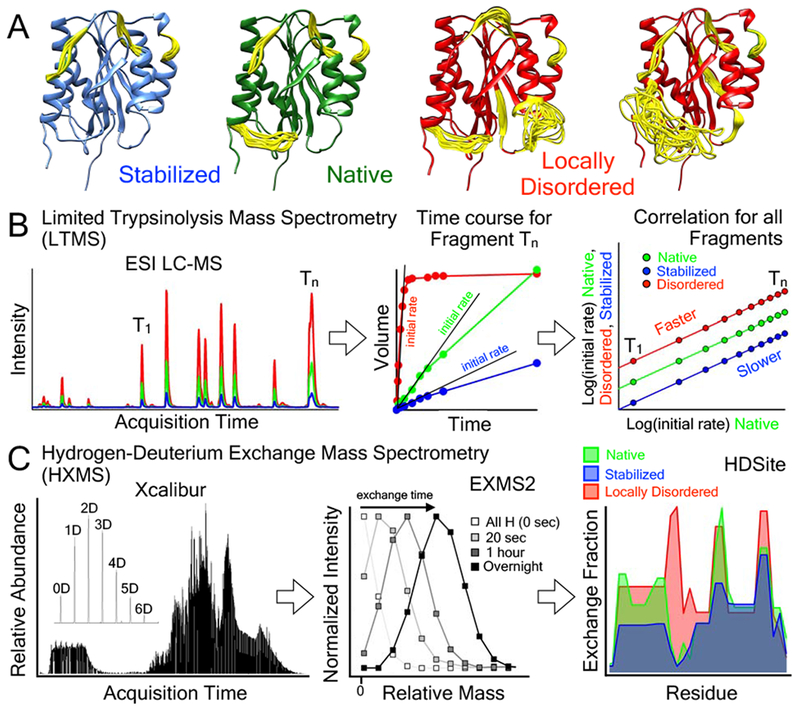
Mass Spectrometry methods to identify local disorder in VWF. A) Schematic models of the A1 domain (PDB ID = 1AUQ [61]), generated with Mini-Protein MODelling MPMOD [62] and rendered using UCSF Chimera [63]. Yellow loops represent dynamic or disordered regions of the A1 domain experimentally measured by HXMS. B) Schematic representation of the LTMS procedure used to evaluate the local flexibility of the A1 domain containing various mutations. Native, stabilized or locally disordered VWF is incubated with a defined amount of trypsin. Aliquots were taken after certain time points, and after quenching of the proteolysis, peptides were analyzed via RP-HPLC followed by injection into an ESI Mass spectrometer. Accumulation of peptides over time was recorded and initial slopes were determined. A double logarithmic correlation of the slopes of the stabilized and misfolded proteins with the slopes of the native protein provides an estimate for the susceptibility to proteolysis and hence yields insight into structural alterations and local flexibility. C) Procedure for HXMS. Fully hydrogenated protein was incubated in 80% 2H2O to allow the secondary structure dependent exchange of backbone amide hydrogens with solvent deuterium. At various time points, exchange is stopped by a drop in pH. Proteins are cleaved with pepsin and proteolysis peptides are separated and injected into the mass spectrometer. EXMS2 [42,43] is used to identify deuterated peptides and HDSite is used for residue deconvolution [44].
The LTMS strategy is to determine the initial proteolysis rate by trypsin, Fig.3B. Trypsinolysis is performed at various defined time intervals and the initial rate is determined from the peak volume of trypsin fragments (T1–Tn) acquired in the mass spectrometer as a function of time. Initial rates are increased for destabilized or locally disordered variants [39], decreased for stabilized variants [12] relative to the wild-type variant, and are either shifted up or down, respectively, on a double log plot of the VWD rates versus the wild-type rates.
The HXMS strategy is to determine steady-state time-dependent exchange of amide hydrogens with solvent deuterium when diluted into 2H2O buffer, Fig.3C [40–45]. Depending on local conformational dynamics of the peptide backbone, structurally protected amide hydrogens exchange slowly and amides in dynamic or disordered regions of structure exchange quickly. Following time dependent hydrogen-exchange (HX) and acidic quenching, on-column pepsinolysis is performed and the shift in mass of proteolytic fragments acquired by the mass spectrometer due to deuterium incorporation into the backbone amides is measured [42, 43] and converted to exchange fraction [44] throughout the residue sequence. In stabilized variants, the exchange fraction is reduced relative to wild-type variants without altering the residue dependent pattern of exchange. Local disorder is observed as an enhanced exchange fraction in destabilized regions and in new structural regions where the exchange either does not occur or is very low in the wild-type variant.
4.4. LTMS.
Fig.4 illustrates that the global effect of VWD mutations that cause disorder in A1 is to enhance the rate of trypsinolysis of the single A1 domain and A1 in the A1A2A3 tridomain or in multimeric VWF. Panels A-E correlate trypsinolysis rates of VWD variants (y-coordinate) to that of the wild-type construct (x-coordinate). The slope of these plots represent a global increase or decrease of the rates as a consequence of the mutation. Each data point represents a different trypsin proteolytic fragment of VWF. Relative to wild-type A1, the type 2B VWD variants, P1337L and reduced and carboxyamidated A1 (RCAM A1, disulfide is chemically blocked from reforming) [11], enhance proteolysis rates 4.0 ± 0.2 and 3.2 ± 0.3 fold, respectively, Fig.4A. Conversely, the stabilizing type 2M VWD variant, G1324S, decreases the proteolysis rates to 0.064 ± 0.005 [12]. Similarly within A1 of the A1A2A3 tridomain, trypsinolysis rates are enhanced 2.0 ± 0.2 fold for P1337L, 10.4 ± 0.7 fold for the type 2M VWD variant, F1369I, and 7.4 ± 0.8 fold for the type 2B VWD variant, V1314D, Fig.4B. The enhanced proteolytic rates of A1 are counter balanced by a small reduction of trypsinolysis rates of A2 and A3 domains, Fig.4C. The stabilization of A2 and A3 against trypsin cleavage in the context of a mutation in A1 is in agreement with prior thermodynamic observations that A3 domain imparts some of its stability to the A1A2A3 complex [31]. When the A1 domain is disordered, the stability of the A3 domain is restored due to perturbed domain interactions that are coupled to the stability of A1. These new observations indicate that A2 behaves in a similar manner in the context of the tridomain. In multimeric VWF, trypsinolysis rates of the A1 domain are enhanced 2.6 ± 0.2 fold for P1337L, 3.3 ± 0.6 fold for the type 2M VWD variant, S1285F, and 3.0 ± 0.6 fold for the type 2B variant, V1316M, Fig.4D. Mutations in A1 do not affect the proteolytic rates of C and D domains of VWF outside of the A1A2A3 tridomain region Fig.4E. Fig.4F demonstrates that while trypsinolysis rates of A1 within the wild-type A1A2A3 tridomain are similar to wild-type VWF, those for the single A1 domain are enhanced 3.2 ± 0.3 fold, as expected due to the lack of glycosylation and other VWF domains.
Figure 4:

LTMS correlations between trypsinolysis rates (hr−1) of wild-type (x-coordinates) and mutant A1, A1A2A3, and multimeric VWF constructs (y-coordinates). A) Initial rates of wild-type, P1337L, G1324S and for RCAM A1 relative to wild-type A1. B) Initial rates of the A1 domain within the wild-type, P1337L, F1369I, and V1314D A1A2A3 tridomain relative to wild-type A1A2A3. C) Initial rates of A2 and A3 domains within the wild-type, P1337L, F1369I, and V1314D A1A2A3 tridomain relative to wild-type A1A2A3. D) Initial rates of the A1 domain within the wild-type, P1337L, S1285F, and V1316M VWF relative to the wild-type VWF. E) Initial rates of all other domains outside of the A1A2A3 domain region of VWF in wild-type, P1337L, S1285F, and V1316M VWF relative to the wild-type VWF. F) Control: Initial rates of the wild-type A1, A1A2A3, and VWF constructs relative to wild-type VWF. Panels A-F) Trypsinolysis rates for all data points for every construct are available in the LTMS Supporting Information. Lines represent linear fits with a zero intercept. Linear fits and rates are translated to Log:Log scale. Rates above wild-type indicate faster proteolysis. Those below wild-type represent a slower cleavage of the protein. Error bars on each datapoint represent fitting errors of the initial slope of tryptic peptide peak volume with respect to time (see LTMS Supporting Information Figures S3–S10).
The observed enhanced trypsinolysis rates in each of the VWF constructs containing mutations known to cause secondary structure disorder in A1 (Fig.1B) confirm that the structure of A1 within multimeric VWF is altered because the proteolytic trend in the rates is consistent across all VWF constructs. The scatter in the data points indicates the possibility for local effects corresponding to specific secondary structure regions, but proteolysis alone, however, cannot directly decipher residual structure in partially-folded states since the first cleavage alters the covalent linkage and the conformation of the protein. Substrate specificity [46] and/or incomplete peptide fragmentation resulting in product inhibition of the proteolysis [47] could outweigh the intrinsic dynamic fluctuation kinetics of the protein leading to potential false positives or negatives regarding the location and identification of disordered secondary structure regions. Some of the tryptic fragments were cleaved multiple times resulting in the loss of these peptides at longer incubation times following their initial accumulation (see LTMS Supporting Information Fig.S3–S10). To obviate these complications, HXMS was used to identify local disorder in the A1 domains of VWF.
4.5. HXMS.
Fig.5A compares a 1hr hydrogen exchange (HX) of the wild-type single domains, A1, A2, and A3 overlaid on the wild-type A1A2A3 tridomain as a function of the residue numbered sequence. Two constructs of the single A1 domain were expressed in E.coli and HEK293 cell culture, respectively, for comparison with the A1A2A3 tridomain also expressed in HEK293 cell culture. Within the A1 domain, the HX is similarly distributed throughout the secondary structures regardless of whether A1 is expressed as a single domain in E.coli or HEK293 cells or in the context of the A1A2A3 tridomain fragment of VWF. To corroborate this observation, peptide envelopes of specific peptides (Fig.5B) located throughout the A1 domain are comparatively similar and/or identical within experimental error for each of the expression constructs.
Figure 5:
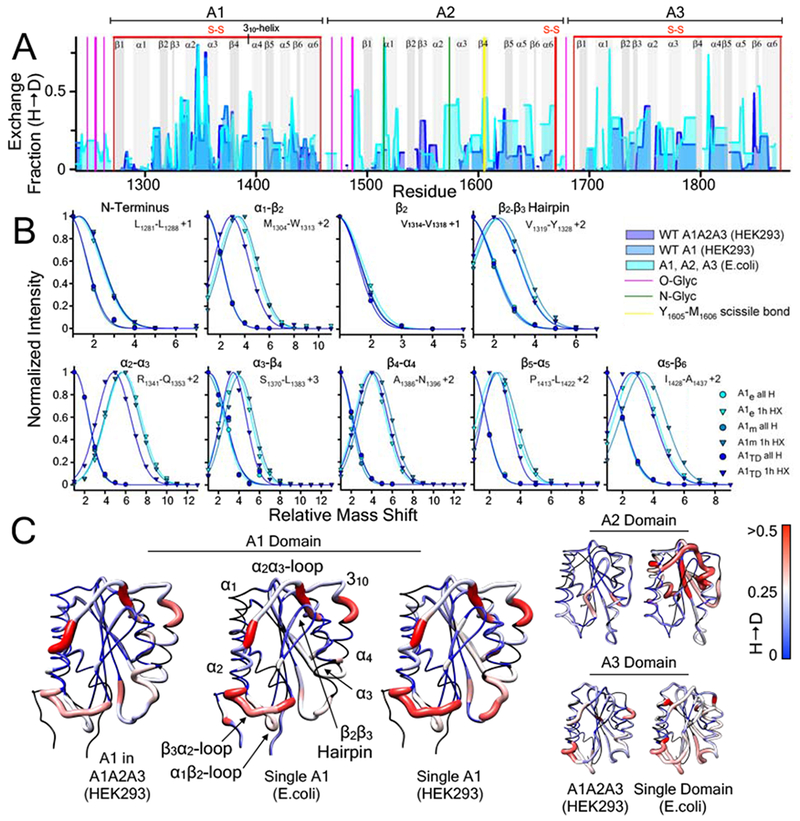
A) 1 hour hydrogen deuterium exchange (H → D) of wild-type A1, A2, and A3 domains (E. coli, cyan area), wild-type A1 domain (HEK293, blue area), and A1 domain within the wild-type A1A2A3 tridomain (HEK293, purple area) as a function of residue position. HXMS of A1, A2, and A3 (from E.coli) and A1A2A3 (from HEK293) were performed in triplicate at 1, 5, 10, 20 and 30 minutes, 1, 3 and 6 hours, and overnight time points. HXMS of A1 (from HEK293) was performed in triplicate at 1 minute, 1 hour and overnight (HXMS Supporting Information). Gray vertical areas represent the indicated secondary structure elements (top). Red vertical lines represent disulfide bonds. Magenta and green vertical lines indicate residues that are O-glycosylated and N-glycosylated respectively. Yellow vertical line represents the Y1605-M1606 ADAMTS13 scissile bond. B) Peptide envelopes (normalized intensity versus mass shift relative to the ”all H” peak) of eight peptides covering various secondary structure regions of the A1 domain expressed in E.coli, HEK293, and within A1A2A3 (HEK293). Relative to the ”all H” peptide at zero time, 1hour HX of peptides in each secondary structure region is observed to be the same regardless of the expression construct. C) HX fraction mapped onto the structures of the A1, A2, A3 domains (pdb IDs 1AUQ [61], 3GXB [64], and 1AO3 [65], respectively). Black = not resolved, blue = 0, white = 0.25, red ≥ 0.5. C) All structures are rendered using UCSF Chimera [63].
Mapped onto the A1, A2, and A3 crystal structures in Fig.5C, HX occurs throughout the solvent exposed secondary structures to different extents. HX is greatest in the loops between α1 and β2, β3 and α2, following α2 and prior to α3, and the 310-helix. These regions of A1 secondary structure are intrinsically dynamic since their fraction of deuteration accumulates quickly and/or is fairly constant throughout the time range in which the kinetics were determined (HXMS Supporting Information). Given small differences in residue resolution of the three A1 expression constructs, Fig.5 illustrates that the intrinsic dynamics of the A1 domain are independent of the expression construct.
By contrast, the A2 domain is significantly less dynamic in the A1A2A3 tridomain than in the E.coli derived single domain. The single A2 exchanges more rapidly than A2 in the tridomain where N-glycosylation occurs at the beginning of α1 and in the loop between α2 through the beginning of α3 and in the β4-strand where the ADAMTS13 Y1605-M1606 scissile bond is located. These observations indicate that A2 is much more stable in the of A1A2A3 tridomain and the presence of N-glycosylation. They also support prior thermodynamic studies that N-glycosylation stabilizes A2 against proteolysis by ADAMTS13 [48] as the single E.coli-derived A2 domain is reported to be a very efficient substrate for ADAMTS13 activity in plasma [49].
Similar to A1, HX in the A3 domain occurs more prominently in the loops between α1 and β2 and between β3 and α2. HX in these regions and in the loop between α3 and β4 are enhanced in the single A3 domain relative to that in the A1A2A3 tridomain. HX throughout the sequence of the A3 domain in A1A2A3 are moderately diminished relative to the single A3 domain indicating that the intrinsic dynamics of A3 contribute to the stabilization of the A1A2A3 complex, an observation in agreement with the above LTMS results (Fig.4C) and prior thermodynamic studies [31] that indicate domain interactions stabilize the tridomain complex.
Comparison of the HX dynamics of the three VWD variants of A1, type 2M G1324S, and type 2B P1337L and reduced A1 (RCAM), with wild-type A1, Fig.6A, demonstrate that mutations affect local secondary structure elements of the A1 domain. While each of the VWD variants share much of the intrinsic dynamic structural regions in common with wild-type A1, HX differences throughout the domain demonstrate that conformational changes occur in the immediate structural vicinity of the mutation and allosteric to the mutation site.
Figure 6:
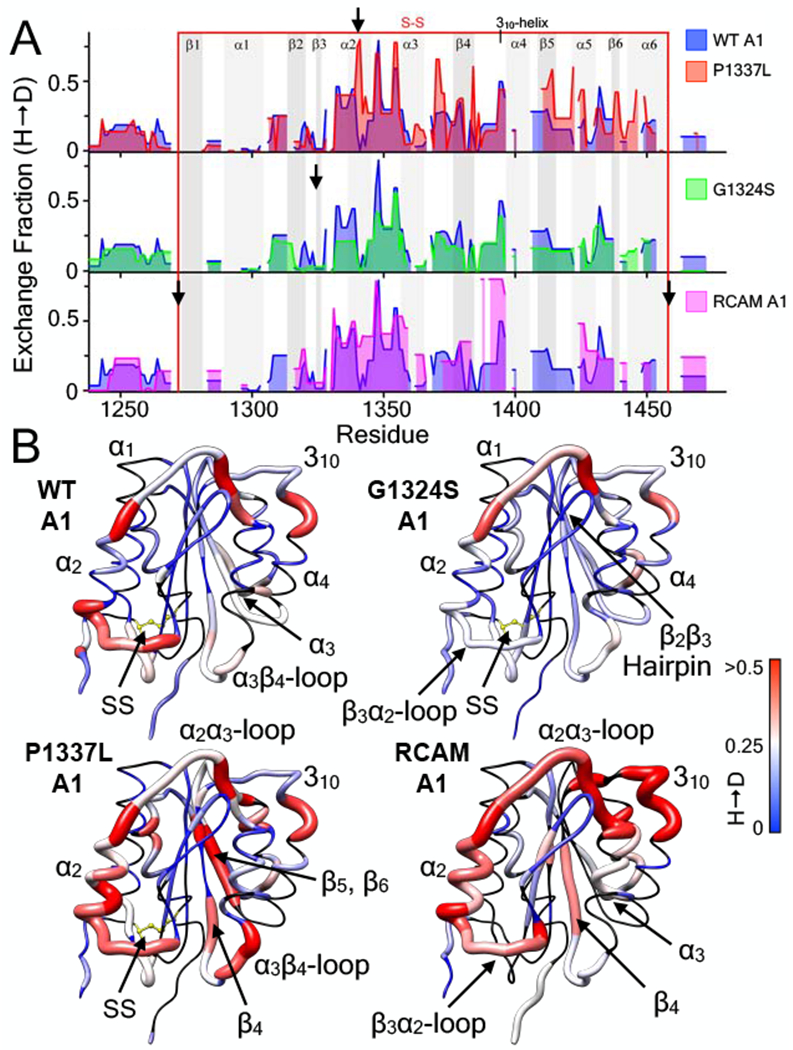
Effect of VWD mutations on the hydrogen deuterium exchange (H → D) of the A1 domain (E. coli). A) 1 hour hydrogen deuterium exchange (H → D) of wild-type A1 (all panels, blue area), P1337L A1 (type 2B GOF VWD, top panel, red area), G1324S A1 (type 2M LOF VWD, middle panel, green area), and RCAM A1 (type 2B GOF VWD where cysteines are reduced and carboxyamidated [11, 30], bottom panel, magenta area) as a function of residue position. HXMS of wild-type and RCAM A1 were previously performed in triplicate at 1, 5, 10, 20 and 30 minutes, 1, 3 and 6 hours, and overnight time points (see [11]). HXMS of P1337L A1 and G1324S A1 were performed in triplicate at 1 minute, 1 hour and overnight (HXMS Supporting Information). Heavy black arrows indicate the location of the mutation in the sequence. Gray vertical areas represent the indicated secondary structure elements (top). Red vertical lines represent the disulfide bond. B) HX fraction mapped onto the structure of the A1 domain (pdb ID = 1AUQ [61]). Black = not resolved, blue = 0, white = 0.25, red ≥ 0.5. All structures are rendered using UCSF Chimera [63].
Mapped onto the A1 crystal structures in Fig.6B, HX is locally enhanced in the α2-helix immediately C-terminal to the P1337L mutation site. HX is also allosterically enhanced in the loop connecting α3 and β4 and on the opposite face of the protein in β5 and β6. Stabilization by G1324S [12] is observed as a reduced HX in the hairpin, the loop between β3 and α2, and the loop following α2. Reduction in exchange is also observed throughout the remaining sequence of the G1324S A1 domain. All of the intrinsic exchangeable regions in wild-type A1 are enhanced in RCAM A1 and local disorder is observed in α2 and the loops prior to α2 and α3. HX is also enhanced in the central β-sheet in parts of β2 and β4. The similarities between P1337L and RCAM A1 are enhanced exchange in α2 and in the various regions of the central β-sheet indicating that variable extents of disorder in α2 may regulate the affinity of A1 for GPIbα.
Gain and loss-of-function VWD mutations that disorder the A1 domain, V1314D (type 2B) and F1369I (type 2M), respectively, were also investigated by HXMS in the A1A2A3 tridomain, Fig.7A. The gain-of-function of V1314D is so severe that platelets become immobilized under rheological shear flow without any observable translocation kinetics. By contrast, F1369I does not bind platelets, Fig.7B. The HX is mapped to the single domain crystal structures (Fig.7C).
Figure 7:

Effect of VWD mutations on the hydrogen deuterium exchange (H → D) of the A1A2A3 domain (HEK293). A) 1 hour hydrogen deuterium exchange (H → D) of wild-type A1A2A3 (top and bottom panels, blue area), V1314D A1 (type 2B GOF VWD, top panel, red area), and F1369I A1 (type 2M LOF VWD, bottom panel, green area) as a function of residue position. HXMS was performed in triplicate at 1, 5, 10, 20 and 30 minutes, 1, 3 and 6 hours, and overnight time points (HXMS Supporting Information). Heavy black arrows indicate the location of the mutation in the sequence. Gray vertical areas represent the indicated secondary structure elements (top). Red vertical lines represent disulfide bonds. Magenta and green vertical lines indicate residues that are O-glycosylated and N-glycosylated respectively. Yellow vertical line represents the Y1605-M1606 ADAMTS13 scissile bond. B) Platelet adhesion to V1314D A1A2A3 (top) and lack of adhesion to F1369I (bottom). Platelets adhered to V1314D do not translocate (see supplemental movies). Shear rate is 1500 s−1. C) HX fraction mapped onto the structures of the A1, A2, A3 domains (pdb IDs 1AUQ [61], 3GXB [64], and 1AO3 [65], respectively). Black = not resolved, blue = 0, white = 0.25, red ≥ 0.5. All structures are rendered using UCSF Chimera [63].
Consistent with the other gain-of-function variants in the single A1 domain expressed in E.coli, V1314D in A1A2A3 results in enhanced HX in the α2-helix. It also enhances HX in β2, the C-terminal part of α3, the loop into β4, and in β5 and α5 allosteric to the mutation site. The HX of F1369I is near identical to wild-type except for the local enhanced exchange around the mutation in the C-terminal part of α3, the center of the α2-α3 loop (residues G1351 - A1355), and a stabilization and diminished HX of the C-terminal part of the α2-helix, Fig.7A.
The differences observed in the A2 and A3 domains are small compared to those in the A1 domain. Although mutations in A1 can dissociate of the A1A2A3 tridomain complex [27], the HX is not appreciably altered in either A2 or A3 except for a moderate enhancement in α5 and β6 of A3, whose hydrogen bonding may be stabilized by interdomain association.
The primary difference in the conformational dynamics between gain and loss-of-function appears to be the HX changes in the α2-helix because this is enhanced relative to wild-type for V1314D and diminished relative to wild-type for F1369I. Thus α2 is likely to be a stable helix in type 2M VWD variants and a more dynamic or disordered helix in type 2B VWD variants [11,50]. Another difference is the additional enhanced exchange in the C-terminal regions of the A1 domain resulting from gain-of-function mutations. This is evident for the type 2B V1314D variant of the A1A2A3 tridomain (Fig.7A) and the P1337L and RCAM A1 variants of the single A1 domain (Fig.6A). This is not evident for the type 2M F1369I variant of A1A2A3 nor the G1324S variant of the single A1 domain. In these cases, gain-of-function appears to destabilize C-terminal secondary structures allosteric to and concomitant with destabilization of α2. By contrast, the loss-of-function F1369I variant stabilizes α2 with little effect on the C-terminal secondary structures even though it introduces local disorder in the α3-helix. The loss-of-function G1324S variant stabilizes the entire A1 structure.
4.6. HXMS of recombinant and plasma-derived VWF confirms the observed mutation-induced local disorder of A1 in single and tridomains.
HXMS was also performed on VWF obtained from commercial plasma concentrates to validate that the intrinsic exchange dynamics observed in A1 and in A1A2A3 are also observed in VWF from its native environment, blood plasma. This experimental endeavor was extremely difficult and a number of measures were taken to ensure its success (see HXMS Supporting Information). To avoid practical experiment problems with the liquid chromatography due to the very large size of VWF multimers, plasmin was used to cleave multimeric VWF (obtained from Wilate, Alphanate, Humate P and Koãte DVI plasma VWF concentrates) to N-terminal dimers with A1 at the C-termini and C-terminal dimers with A2 and A3 at the N-termini. Plasmin cleaves the linker between A1 and A2 [51]. Fig.8A shows the exchange fraction as a function of residue number in the mature VWF monomeric unit. Extensive disulfide bonding outside of the A domains limits the resolution, but coverage within the A1, A2 and A3 domains was quite good. Twelve individual 1 hr HX measurements (2 on Alphanate, 4 on humate P, 1 on Koãte DVI, and 5 on Wilate) were determined to obtain this resolution. HX in a few fragments of the VWF propeptide (D1D2) within these plasma concentrates were also observed.
Figure 8:
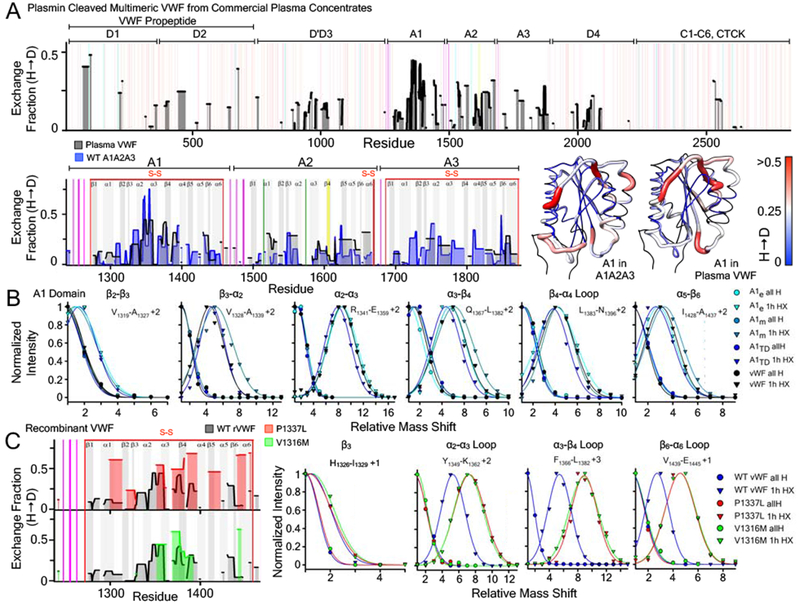
A) Top: 1 hour hydrogen deuterium exchange (H → D) of multimeric VWF isolated from plasma concentrates (black area) as a function of residue position. Red vertical lines represent disulfide bonds. Magenta and green vertical lines indicate residues that are O-glycosylated and N-glycosylated respectively. Yellow vertical line represents the Y1605-M1606 ADAMTS13 scissile bond. Bottom Left: Comparison of the HX obtained from the A1A2A3 tridomain (blue area) with that of plasma VWF (black area) within the A1, A2, and A3 domains. Bottom Right: HX fraction mapped onto the structures of the A1, A2, A3 domains (pdb ID 1AUQ [61]) is remarkable similar despite the lower resolution of the HX obtained from plasma VWF. B) Peptide envelopes (normalized intensity versus mass shift relative to the ”all H” peak) of six peptides covering various secondary structure regions of the A1 domain expressed in E.coli, HEK293, within A1A2A3 (HEK293), and in VWF isolated from plasma concentrates. Relative to the ”all H” peptide at zero time, 1hour HX of peptides in each secondary structure region is observed to be the same regardless of the recombinant expression construct or the plasma VWF. C) Left: Effect of VWD mutations (P1337L (red), V1316M (green)) on the hydrogen deuterium exchange (H → D) of the A1 domain within recombinant multimeric VWF (HEK293). Right: Peptide envelopes (normalized intensity versus mass shift relative to the ”all H” peak) of four peptides covering various secondary structure regions of the A1 domain in recombinant VWF. Variants are observed to increase the exchange in peptide envelopes indicating an enhanced local disorder of A1 in these structural regions.
The exchange pattern observed in the A domains obtained from the plasma VWF concentrates is remarkably similar to the A1A2A3 tridomain and the single A1 domain Figs.5 & 8. Mapped onto the structure of the A1 domain, HX within the A1 domain β3-α2 loop is slightly more protected in VWF relative to A1A2A3. Within experimental error, however, comparisons of the HX illustrate that the intrinsic structural dynamics of A1 are very similar regardless of its quaternary structural environment. A quantitative comparison of specific peptide envelopes, Fig.8B, confirms that the exchange dynamics are similar, if not identical, throughout the secondary structure elements of the A1 domain in each VWF construct. Relative to A1A2A3, the HX of A2 in VWF increased, likely because of the plasmin cleavage of the A1A2 linker in VWF, but HX in the A3 domain was similar though less resolved in VWF than in A1A2A3.
In Fig.8C, HX of plasmin-cleaved recombinant P1337L and V1316M VWF dimers (HEK293) were compared with that of wild-type VWF obtained from plasma concentrates within the A1 domain. The resolution of the mutant VWF is lower due to issues of expression and purification yield and on column pepsin digestion, however, a number of specific peptides were identified in each variant to illustrate that an enhanced exchange occurs in the P1337L and V1316M disease variants relative to wild-type VWF. This observation indicates that these mutations also cause local disorder within the A1 domains of multimeric VWF, consistent with that of the recombinant single A1 domain and A1A2A3 tridomain fragments in Figs.6 & 7.
5. Discussion.
Prior structure function studies in our lab have demonstrated that mutations causing type 2B and type 2M von Willebrand disease can alter the proper folding of the A1 domain. Of the nineteen type 2 VWD variants characterized, twelve have been identified that misfold the A1 domain, six in each phenotype [10, 34]. Here, we show that the far-UV CD spectroscopy used to characterize single A1 domain becomes less sensitive to conformational changes in the larger A1A2A3 recombinant fragments of VWF, thereby eliminating its utility in providing proof that VWD mutations misfold A1 in multimeric VWF. Furthermore, the multimer patterns and the intracellular localization of recombinant VWD variants of VWF suggest there may be problems with the proper folding, but also do not provide direct evidence of it. The two mass spectrometry methods developed here were designed to test the misfolding hypothesis inherent to prior observations of VWD mutation effects on the structure of the single A1 domain and provide evidence for the occurrence of local secondary structure disorder caused by VWD mutations in the A1 domains of full-length VWF.
LTMS and HXMS complement each other’s weaknesses to demonstrate that VWD mutations cause misfolding of A1 in multimeric VWF. The advantage of LTMS is that it can be performed on the full-length multimeric state of VWF, but its weakness is low resolution of local structural dynamics. While many trypsin fragments are produced, their rates of accumulation are uniformly increased or decreased indicating that the A1 domain is either destabilized or stabilized, respectively. LTMS is less certain about whether local disorder occurs in the absence of prior knowledge of the single domain, but the increased rates are consistent with the presence of local disorder. The advantage of HXMS is that it provides higher resolution for deciphering local disorder, but experimental limitations on the HPLC require the use of plasmin to digest VWF prior to the exchange reactions to enable online pepsinolysis and separation of the resulting peptides. Plasmin digestions of VWF generate N-terminal dimers of D’D3-A1 and C-terminal dimers of A2A3-D4-C1-C6-CTCK that break interactions between the A domains which may stabilize A1 in VWD variants of VWF. The very large molecular weight of these fragments also limits the resolution of HXMS on VWF. Nevertheless, HX of C-terminally exposed A1 domains in the D’D3-A1 dimers illustrates that the intrinsic dynamics of A1 in plasma VWF are, within experimental error, identical to the HX of A1 in the recombinant proteins.
Setting these weaknesses aside, both methods produce conclusions consistent with what is already known about A1 from the more traditional biophysical methods. 1) Mutations that misfold A1 increase the rate of trypsinolysis and the extent of hydrogen deuterium exchange in local secondary structures of A1 in VWF. 2) Mutation effects are localized to the A1 domain without appreciably affecting other domains. 3) The intrinsic dynamics of A1 observed in recombinant fragments of VWF are conserved in plasma derived VWF. These observations support the hypothesis that VWD mutations do misfold A1 in the context of the multimeric plasma VWF.
The HXMS data reported here do not support other mass spectrometry studies which claim an autoinhibitory mechanism mediated by a direct interaction of the N- and C-terminal flanking sequences of A1 with its α1 and α2-helices [23, 24]. Studies by Deng et al. show that the α1 and α2 helices exchange much faster in an N-terminally truncated A1 than in A1 with its N-terminus intact suggesting an interaction that protects the amide hydrogens from exchanging within these helices. In order for HXMS to detect such an interaction, either the intramolecular hydrogen bonds within these helices are stabilized or hydrogen bonds are formed between residue amides of these helices and amides in the N- or C-terminal flanking sequences. First, the experiments reported here indicate that α1-helix does not appreciably exchange in either wild-type or in any of the VWD variants (Figs.5–8). The amide backbone of α1 is protected due to the hydrogen bonded helical structure which is stable even as an isolated peptide (see CD spectra of β1-α1, residues S1273-W1313, Fig.S29 of the HXMS Supporting Information). Second, HX of residues in the α2-helix, which is claimed to be suppressed by the hypothetical interaction, do exchange in A1, A1A2A3, and plasmin-cleaved VWF dimers. Furthermore, the N-terminal residues in A1 and VWD variants of A1 also exchange (Fig.6A) indicating that amides in the N-terminal flanking region do not participate in any hydrogen bonds that might form in the putative autoinhibitory interaction. Although it could be argued that the lack of HX in the α1 helix suggests autoinhibition, each of the recombinant proteins studied here have been shown to bind platelet GPIbα and platelets both in the presence and absence of shear flow according to their phenotype determined affinity [10–12,20, 21, 27, 31], excepting severe disordered type 2M VWD variants.
Removing the N-terminal flanking sequence potentiates platelet activation when A1A2A3 is bound to fibrinogen [20]. This suggests that the presence of glycosylation stemming from this sequence in A1A2A3 is inhibitory to platelet adhesion and activation, as indicated by Nowak et al. [15]. Although glycosylation is known to stabilize proteins [52, 53], its stabilizing effect is predominantly a result of destabilizing the unfolded state with little effect on the native state stability. As a result, the equilibrium stability of the wild-type A1 domain and the conformational dynamics of its native structure are unaltered by the presence of glycosylation [26]. Platelet adhesion is therefore sterically inhibited by glycosylation resulting in loss of GPIbα affinity as a result of volume exclusion [26] rather than a direct stabilizing interaction between the A1 domain and its flanking sequences [23, 24]. Compared to the functional changes caused by VWD mutations, functional suppression by the steric exclusion of glycosylated flanking peptide regions is likely a common denominator in health and disease and, therefore, irrelevant because the structural effects of VWD mutations can increase platelet adhesion times many fold over wild-type A1 or abolish adhesion all together [10].
When considering disease mechanisms, it is important to remember that VWD is a genetic disorder, the cause of which are mutations that result in amino acid changes or deletions in the VWF protein. In the vasculature, everyone’s VWF is shear-stressed [54] because everyone’s VWF feels the force [1] of rheological blood flow regardless of whether they have type 2 VWD. While glycosylation and intra-VWF or inter-VWF molecular interactions do affect VWF activity, the intrinsic conformational dynamics of A1 domains in VWD must outweigh these contributions, particularly in cases where mutations misfold A1 and consequently abolish the native interactions that facilitate the von Willebrand factor’s resistance to rheological activation.
6. Conclusions.
Due to the large size and molecular weight of VWF, new mass spectrometry methods were developed to detect mutation-induced local disorder in the A1 domains of multimeric VWF. The LTMS and HXMS results described demonstrate a consistent agreement between the observed conformational dynamics of A1 as a single domain or part of the larger more biologically relevant fragments of VWF. HXMS shows a consistent pattern of hydrogen-deuterium exchange throughout the wild-type A1 sequence regardless of whether it is expressed by itself, together with A2 and A3, or in plasmin-cleaved dimers of multimeric VWF. Relative to wild-type, mutations that disorder the single A1 domain enhance rates of tryptic proteolysis in A1 as well as in A1A2A3 and full-length multimeric VWF. Furthermore, HXMS demonstrates that the A1 domain becomes locally disordered to different extents and in different structural regions of A1 consistently across all the recombinant fragments of VWF studied.
Despite limitations of each of the methods taken individually, together they support the principle of local disorder in disease states of VWF and provide common regional hotspots associated with gain- and loss-of-function phenotypes of VWD. Specifically, enhanced dynamics of the α2-helix are associated with a gain-of-function as observed in each of the three type 2B VWD variants investigated. Conversely, suppressed dynamics of the α2-helix are observed in each of the loss-of-function type 2M VWD variants. The dynamics of α2 are stabilized in both the natively folded G1324S variant and the partially disordered F1369I variant indicating that, although other structural regions can become disordered in type 2M variants, a stable α2-helix constitutes at least part of the mechanism for loss-of-function. Additional studies on the dynamics of hydrogen deuterium exchange in misfolded type 2M variants will further elucidate the role of conformational disorder in loss-of-function phenotypes.
7. Methods.
7.1. Protein expression and purification.
Wild type VWF A1 (Q1238-P1471) and its mutants G1324S and P1337L were expressed in E. coli M15 cells as fusion constructs containing an N-terminal His6 tag. Inclusion body preparation, refolding and purification of the proteins were performed as previously described [12, 55]. RCAM A1 was prepared in 2 M GdnHCl by reduction of the single disulfide bond connecting N- and C-termini of A1 with 6 mM DTT, followed by blocking with 12 mM iodoacetamide as previously described [27, 30]. A2 and A3 domains were expressed and purified from E. coli M15 cells as fusion constructs containing an N-terminal His6 tag. A2 was purified in the presences of CaCl2. Wild type A1A2A3 tridomains (amino acids Q1238 - G1874) and the mutants P1337L, V1314D and F1369I in A1A2A3 were expressed in HEK293 cells as fusion proteins containing a C-terminal His6 tag and purified via Ni2+ affinity chromatography as described previously [34, 35]. The purity of the A1 and of A1A2A3 domains was confirmed via Analytical Gel filtration and/or RP-HPLC as recently described [55]. A1 and A1A2A3 concentrations were measured on a Shimadzu UV2101PC spectrophotometer using extinction coefficients of 15350 L/mol/cm for the A1 domain and 74497 L/mol/cm for A1A2A3.
7.2. Recombinant VWF and VWF Plasma Concentrates.
Wild type full-length VWF (amino acids M1-K2813) was expressed in HEK293 cells and cultured as described previously [56, 57]. P1337L, V1316M and S1285F were inserted into full-length cDNA of wild-type VWF within the mammalian expression vector pcDNA3 [57] by site-directed mutagenesis employing the QuickChange kit (Stratagene). The VWF concentrates Wilate (Octapharma), Koãte DVI (Bayer), Humate-P (CSL Behring) and Alphanate (Grifols) were provided by the Mayo Clinic Special Coagulation Laboratory. All concentrates were excessively dialyzed into PBS prior to usage. An SDS-PAGE gel of all four concentrates can be found in the HXMS Supporting Information (Fig.S27). VWF was quantified by fluorescence and the commercial REAADS antigen activity assay (Diapharma1) as described in the LTMS Supporting Information (Fig.S11).
7.3. Multimer gel analysis.
Separation of VWF multimers was performed in the Mayo Clinic Special Coagulation Laboratory via horizontal slab gel electrophoresis at 12.5°C using 50mM TrisHCl, 0.1% SDS, 0.38M glycine, pH 8.3 as running buffer. The electrophoresis and the subsequent in-gel staining of the VWF multimers was performed as previously described [58]. Gels were scanned at 700nm on an infrared imager (Odyssey, Li-Cor Biosciences) and converted into x,y-coordinates where the x-axis represents the running distance and the y-axis the relative band intensity. Multimer gels were quantitatively compared by normalizing the peak intensities of each by the number of dimers, N, in each multimer band and calculating the ratio of the dimer normalized intensities relative to that of the lowest molecular weight band intensity, , where N1=1. This normalized intensity per dimer ratio is plotted as a function of the number of dimers in the multimer bands.
7.4. Immunofluorescence and confocal microscopy.
HEK293 cells were cultured in Dulbecco Modified Eagle Medium (DMEM, Invitrogen) with 10% [v/v] fetal bovine serum (Invitrogen) and 1% penicillin/streptavidin at 37°C and 5% CO2. For immunofluorescence HEK293 cells were seeded in Ibidi treat 8-well μ-slides (Ibidi, Martinsried, Germany) 24hrs prior transfection with Lipofectamine LTX (Invitrogen) according to the manufacturers instructions. 48hrs after transfection cells were washed twice with 1x PBS and fixed with 3% paraformaldehyde (PFA) in PBS for 10 min at 37°C, then the cells were permeabilized using 0.3% Triton in PBS for 3min at 37°C and finally blocked with 1% BSA in PBS for 20min at 37°C. After each step cells were washed twice with PBS. Antibodies used were: rabbit anti-VWF (DAKO; 1:1,000), mouse anti-PDI (abcam, ab2792, 1:400), goat anti-rabbit AF488 (Invitrogen, 1:5,000), goat anti-mouse AF546 (Invitrogen, 1:5,000). Z-stacks were recorded at RT with a confocal microscope (TCS SP5, Leica, Wetzlar, Germany) using an HC PL APO CS2 63.0 x 1.40 OIL UV objective and the following settings: an image size of 512 x 512 pixels, laser power of the 543 nm and 488 nm lasers was set to 9% and 20%, respectively. 3D reconstruction was performed using the ImageJ software [59]. Three independent experiments were performed.
7.5. Circular dichroism spectroscopy.
All Circular Dichroism measurements were performed on an Aviv Biomedical Model 420SF circular dichroism spectrometer. Far UV CD spectra of A1 and of A1A2A3 were recorded at 20°C between 195 and 260nm using a 0.1 or 1.0mm quartz cell. The integration time for all spectra was 60s, the bandwidth and step width 1nm. Isothermal urea induced unfolding was followed at 20° C at a wavelength of 222nm using quartz cells with a path length of either 1.0 or 2.0mm. All samples were allowed to equilibrate overnight at 20°C. The CD signal of each sample was collected and averaged for 5 and 10min with an integration time of 1s. All CD spectra and single wavelength measurements were corrected for the signal of the corresponding buffer and converted to mean ellipticity per amino acid residue.
7.6. Platelet adhesion to A1A2A3 tridomains under rheological shear flow.
The parallel plate flow assay on immobilized C-terminal 6xHis tagged wild-type, V1314D, and F1369I A1A2A3 tridomains was performed as previously described [10–12, 26] using Cellix Vena8 CGS biochips on a Zeiss Axio Observer-A1 microscope operated by Zen2012 software. Citrated whole blood was perfused over the Cu2+-chelated proteins at a shear rate of 1500s−1. After 3-4 minutes TBS was perfused through the channel to remove red blood cells. A 60s video was recorded in phase contrast mode using a PCO.edge camera operated at 25 frames per second. No platelet translocation was observed on V1314D A1A2A3 as platelet adhesion was irreversible. Platelets translocated on wild-type A1A2A3 as previously described [34]. Platelets did not adhere to F1369I A1A2A3.
7.7. LTMS and HXMS.
Limited trypsinolysis and hydrogen deuterium exchange mass spectrometry were performed as previously described [11, 12, 39] in the Supporting Information. The LTMS Supporting Information contains detailed experimental methods for the time dependent limited trypsinolysis data on VWF and its domains, including plots of trypsin peptide mapping, representative chromatographs, kinetics of peptide peak volume as a function of time, tables of tryptic peptides identified, and methods for quantifying VWF concentrations. The HXMS Supporting Information contains a table of metrics for statistical reproducibility of HXMS data on VWF and its domains as recommended by Masson et al. [60] as well as detailed experimental methods, plots of pepsin peptide mapping, and time dependent resolution of deuterium incorporation into VWF structures.
Supplementary Material
Research Highlights.
Structure-function studies of the isolated recombinant A1 domain of von Willebrand factor (VWF) show that type 2 von Willebrand disease (VWD) mutations misfold A1 in both gain-of-function and loss-of function phenotypes, but their structural effects have not been explicitly observed in multimeric VWF native to blood plasma.
Since the large multimeric structure of VWF limits the utility of traditional biophysical spectroscopy, two complementary mass spectrometry methods are developed with the resolution to detect structural disorder within the A1 domains of recombinant multimeric VWD variants of VWF and normal plasma VWF.
These studies reveal that type 2 VWD mutations can and do alter the structure of VWF A1 domains without appreciably affecting the structure of other domains within VWF, the multimerization of VWF, or its cellular localization.
8. Acknowledgements.
This study was supported, in part, by research funding from the National Institutes of Health Grant HL109109 from NHLBI (M.A.), the Mayo Clinic Division of Hematology Small Grants Program (CCaTS UL1TR000135 M.A.), the Mayo Clinic Department of Laboratory Medicine and Pathology Collaborative Research Funds (M.A., D.C.), the Mayo Clinic Center for Biomedical Discovery (M.A.), the Great Lakes Hemophilia Foundation and Health Resources and Services Administration through the Mayo Clinic Comprehensive Hemophilia Treatment Center (M.A., R.K.P.), and the German Research Foundation (DFG) to the Research Group FOR1543: Shear flow regulation of hemostasis - bridging the gap between nanomechanics and clinical presentation (R.S., M.A.B., T.O.). We thank the technical support from staff of the Mayo Clinic Special Coagulation Laboratory, the Mayo Clinic Proteomics Core for technical support with limited trypsinolysis mass spectrometry and analysis, and the UKE Microscopy Imaging Facility (UMIF) for technical support and providing the Leica SP5 microscope. We also gratefully acknowledge Drs. S. Walter Englander and Leland Mayne for very helpful scientific discussions regarding optimization of HXMS and the establishment of HXMS technology in our lab. In addition, we also acknowledge charitable contributions from Mark Davies’ Cycle von Willebrand Disease, which have defrayed, in part, publication costs.
Abbreviations Used:
- LTMS
limited trypsinolysis mass spectrometry
- HXMS
hydrogen deuterium exchange mass spectrometry
- RCAM
reduced and carboxyamidated
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure of conflicts of interest.
The authors state that they have no conflict of interest.
References
- [1].Springer T, “von willebrand factor, jedi knight of the bloodstream.,” Blood, vol. 124, no. 9, pp. 1412–1425, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Lippok S, Kolsek K, Löf A, Eggert D, Vanderlinden W, Müller J, König G, Obser T, Röhrs K, Schneppenheim S, Budde U, Baldauf C, Aponte-Santamaría C, Gräter F, Schneppenheim R, Rädler J, and Brehm M, “von willebrand factor is dimerized by protein disulfide isomerase.,” Blood, vol. 127, no. 9, pp. 1183–1191, 2016. [DOI] [PubMed] [Google Scholar]
- [3].Zhou Y and Springer T, “Highly reinforced structure of a c-terminal dimerization domain in von willebrand factor.,” Blood, vol. 123, no. 12, pp. 1785–1793, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Zhou Y, Eng E, Zhu J, Lu C, Walz T, and Springer T, “Sequence and structure relationships within von willebrand factor.,” Blood, vol. 120, no. 2, pp. 449–458, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Purvis A and Sadler J, “A covalent oxidoreductase intermediate in propeptide-dependent von willebrand factor multimerization.,” J Biol Chem, vol. 279, no. 48, pp. 49982–49988, 2004. [DOI] [PubMed] [Google Scholar]
- [6].Sadler J, “von willebrand factor: two sides of a coin.,” J Thromb Haemost, vol. 3, no. 8, pp. 1702–1709, 2005. [DOI] [PubMed] [Google Scholar]
- [7].Nightingale T and Cutler D, “The secretion of von willebrand factor from endothelial cells; an increasingly complicated story.,” J Thromb Haemost, vol. 11 Suppl 1, pp. 192–201, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Valentijn K and Eikenboom J, “Weibel-palade bodies: a window to von willebrand disease.,” J Thromb Haemost, vol. 11, no. 4, pp. 581–592, 2013. [DOI] [PubMed] [Google Scholar]
- [9].Keeney S and Cumming A, “The molecular biology of von willebrand disease.,” Clin Lab Haematol, vol. 23, no. 4, pp. 209–230, 2001. [DOI] [PubMed] [Google Scholar]
- [10].Tischer A, Madde P, Moon-Tasson L, and Auton M, “Misfolding of vwf to pathologically disordered conformations impacts the severity of von willebrand disease.,” Biophys J, vol. 107, no. 5, pp. 1185–1195, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Tischer A, Machha V, Frontroth J, Brehm M, Obser T, Schneppenheim R, Mayne L, Walter Englander S, and Auton M, “Enhanced local disorder in a clinically elusive von willebrand factor provokes high-affinity platelet clumping.,” J Mol Biol, vol. 429, no. 14, pp. 2161–2177, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Tischer A, Campbell J, Machha V, Moon-Tasson L, Benson L, Sankaran B, Kim C, and Auton M, “Mutational constraints on local unfolding inhibit the rheological adaptation of von willebrand factor.,” J Biol Chem, vol. 291, no. 8, pp. 3848–3859, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Fu H, Jiang Y, Yang D, Scheiflinger F, Wong W, and Springer T, “Flow-induced elongation of von willebrand factor precedes tension-dependent activation.,” Nat Commun, vol. 8, no. 1, p. 324, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Schulte am Esch J, Robson S, Knoefel W, Eisenberger C, Peiper M, and Rogiers X, “Impact of o-linked glycosylation of the vwf-a1-domain flanking regions on platelet interaction.,” Br J Haematol, vol. 128, no. 1, pp. 82–90, 2005. [DOI] [PubMed] [Google Scholar]
- [15].Nowak A, Canis K, Riddell A, Laffan M, and McKinnon T, “O-linked glycosylation of von willebrand factor modulates the interaction with platelet receptor glycoprotein ib under static and shear stress conditions.,” Blood, vol. 120, no. 1, pp. 214–222, 2012. [DOI] [PubMed] [Google Scholar]
- [16].Madabhushi S, Zhang C, Kelkar A, Dayananda K, and Neelamegham S, “Platelet gpiba binding to von willebrand factor under fluid shear:contributions of the d’ d3-domain, a1-domain flanking peptide and o-linked glycans.,” J Am Heart Assoc, vol. 3, no. 5, p. e001420, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Aponte-Santamaría C, Huck V, Posch S, Bronowska A, Grässle S, Brehm M, Obser T, Schneppenheim R, Hinterdorfer P, Schneider S, Baldauf C, and Gräter F, “Force-sensitive autoinhibition of the von willebrand factor is mediated by interdomain interactions.,” Biophys J, vol. 108, no. 9, pp. 2312–2321, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Posch S, Aponte-Santamaría C, Schwarzl R, Karner A, Radtke M, Gräter F, Obser T, König G, Brehm M, Gruber H, Netz R, Baldauf C, Schneppenheim R, Tampé R, and Hinterdorfer P, “Single molecule force spectroscopy data and bd- and md simulations on the blood protein von willebrand factor.,” Data Brief, vol. 8, pp. 1080–1087, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Nakayama T, Matsushita T, Dong Z, Sadler J, Jorieux S, Mazurier C, Meyer D, Kojima T, and Saito H, “Identification of the regulatory elements of the human von willebrand factor for binding to platelet gpib. importance of structural integrity of the regions flanked by the cys1272-cys1458 disulfide bond.,” J Biol Chem, vol. 277, no. 24, pp. 22063–22072, 2002. [DOI] [PubMed] [Google Scholar]
- [20].Auton M, Sowa K, Behymer M, and Cruz M, “N-terminal flanking region of a1 domain in von willebrand factor stabilizes structure of a1a2a3 complex and modulates platelet activation under shear stress.,” J Biol Chem, vol. 287, no. 18, pp. 14579–14585, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Tischer A, Cruz M, and Auton M, “The linker between the d3 and a1 domains of vwf suppresses a1-gpibalpha catch bonds by site-specific binding to the a1 domain.,” Protein Sci, vol. 22, no. 8, pp. 1049–1059, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Ju L, Dong J, Cruz M, and Zhu C, “The n-terminal flanking region of the a1 domain regulates the force-dependent binding of von willebrand factor to platelet glycoprotein ib?.,” J Biol Chem, vol. 288, no. 45, pp. 32289–32301, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Deng W, Wang Y, Druzak S, Healey J, Syed A, Lollar P, and Li R, “A discontinuous autoinhibitory module masks the a1 domain of von willebrand factor.,” J Thromb Haemost, vol. 15, no. 9, pp. 1867–1877, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Deng W, Voos KM, Colucci JK, Legan ER, Ortlund EA, Lollar P, and Li R, “Delimiting the autoinhibitory module of von willebrand factor,” J Thromb Haemost, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Zhang C, Kelkar A, Nasirikenari M, Lau J, Sveinsson M, Sharma U, Pokharel S, and Neelamegham S, “The physical spacing between the von willebrand factor d’d3 and a1 domains regulates platelet adhesion in vitro and in vivo.,” J Thromb Haemost, vol. 16, no. 3, pp. 571–582, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Tischer A, Machha V, Moon-Tasson L, Benson L, and Auton M, “Glycosylation sterically inhibits platelet adhesion to von willebrand factor without altering intrinsic conformational dynamics.,” J Thromb Haemost, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Auton M, Sowa K, Smith S, Sedlák E, Vijayan K, and Cruz M, “Destabilization of the a1 domain in von willebrand factor dissociates the a1a2a3 tri-domain and provokes spontaneous binding to glycoprotein ibalpha and platelet activation under shear stress.,” J Biol Chem, vol. 285, no. 30, pp. 22831–22839, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Jong A. d. and Eikenboom J, “Von willebrand disease mutation spectrum and associated mutation mechanisms,” Thromb Res, 2017. [DOI] [PubMed] [Google Scholar]
- [29].Zimmermann M, Tischer A, Whitten S, and Auton M, “Structural origins of misfolding propensity in the platelet adhesive von willebrand factor a1 domain.,” Biophys J, vol. 109, no. 2, pp. 398–406, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Tischer A, Madde P, Blancas-Mejia L, and Auton M, “A molten globule intermediate of the von willebrand factor a1 domain firmly tethers platelets under shear flow.,” Proteins, vol. 82, no. 5, pp. 867–878, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Auton M, Zhu C, and Cruz M, “The mechanism of vwf-mediated platelet gpibalpha binding.,” Biophys J, vol. 99, no. 4, pp. 1192–1201, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Auton M, Sedlák E, Marek J, Wu T, Zhu C, and Cruz M, “Changes in thermodynamic stability of von willebrand factor differentially affect the force-dependent binding to platelet gpibalpha.,” Biophys J, vol. 97, no. 2, pp. 618–627, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Stepanian A, Ribba A, Lavergne J, Fressinaud E, Juhan-Vague I, Mazurier C, Girma J, and Meyer D, “A new mutation, s1285f, within the a1 loop of von willebrand factor induces a conformational change in a1 loop with abnormal binding to platelet gpib and botrocetin causing type 2m von willebrand disease.,” Br J Haematol, vol. 120, no. 4, pp. 643–651, 2003. [DOI] [PubMed] [Google Scholar]
- [34].Machha V, Tischer A, Moon-Tasson L, and Auton M, “The von willebrand factor a1-collagen iii interaction is independent of conformation and type 2 von willebrand disease phenotype.,” J Mol Biol, vol. 429, no. 1, pp. 32–47, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Auton M, Cruz M, and Moake J, “Conformational stability and domain unfolding of the von willebrand factor a domains.,” J Mol Biol, vol. 366, no. 3, pp. 986–1000, 2007. [DOI] [PubMed] [Google Scholar]
- [36].Cruz M, Diacovo T, Emsley J, Liddington R, and Handin R, “Mapping the glycoprotein ib-binding site in the von willebrand factor a1 domain.,” J Biol Chem, vol. 275, no. 25, pp. 19098–19105, 2000. [DOI] [PubMed] [Google Scholar]
- [37].Cruz M, Yuan H, Lee J, Wise R, and Handin R, “Interaction of the von willebrand factor (vwf) with collagen. localization of the primary collagen-binding site by analysis of recombinant vwf a domain polypeptides.,” J Biol Chem, vol. 270, no. 18, pp. 10822–10827, 1995. [DOI] [PubMed] [Google Scholar]
- [38].Baronciani L, Federici A, Cozzi G, Canciani M, and Mannucci P, “Biochemical characterization of a recombinant von willebrand factor (vwf) with combined type 2b and type 1 defects in the vwf gene in two patients with a type 2a phenotype of von willebrand disease.,” J Thromb Haemost, vol. 5, no. 2, pp. 282–288, 2007. [DOI] [PubMed] [Google Scholar]
- [39].Tischer A, Machha V, Rösgen J, and Auton M, “cooperative collapse” of the denatured state revealed through clausius-clapeyron analysis of protein denaturation phase diagrams.,” Biopolymers, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Wang L, Pan H, and Smith D, “Hydrogen exchange-mass spectrometry: optimization of digestion conditions.,” Mol Cell Proteomics, vol. 1, no. 2, pp. 132–138, 2002. [DOI] [PubMed] [Google Scholar]
- [41].Mayne L, Kan Z, Chetty P, Ricciuti A, Walters B, and Englander S, “Many overlapping peptides for protein hydrogen exchange experiments by the fragment separation-mass spectrometry method.,” J Am Soc Mass Spectrom, vol. 22, no. 11, pp. 1898–1905, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Kan Z, Mayne L, Chetty P, and Englander S, “Exms: data analysis for hx-ms experiments.,” J Am Soc Mass Spectrom, vol. 22, no. 11, pp. 1906–1915, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Kan Z, Ye X, Skinner J, Mayne L, and Englander S, “Exms2: An integrated solution for hydrogen-deuterium exchange mass spectrometry data analysis.,” Anal Chem, 2019. [DOI] [PubMed] [Google Scholar]
- [44].Kan Z, Walters B, Mayne L, and Englander S, “Protein hydrogen exchange at residue resolution by proteolytic fragmentation mass spectrometry analysis.,” Proc Natl Acad Sci U S A, vol. 110, no. 41, pp. 16438–16443, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Mayne L, “Hydrogen exchange mass spectrometry.,” Methods Enzymol, vol. 566, pp. 335–356, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Park C and Marqusee S, “Probing the high energy states in proteins by proteolysis.,” J Mol Biol, vol. 343, no. 5, pp. 1467–1476, 2004. [DOI] [PubMed] [Google Scholar]
- [47].Kasper J, Andrews E, and Park C, “Product inhibition in native-state proteolysis.,” PLoS One, vol. 9, no. 10, p. e111416, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Lynch C and Lane D, “N-linked glycan stabilization of the vwf a2 domain.,” Blood, vol. 127, no. 13, pp. 1711–1718, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Cruz M, Whitelock J, and Dong J, “Evaluation of adamts-13 activity in plasma using recombinant von willebrand factor a2 domain polypeptide as substrate.,” Thromb Haemost, vol. 90, no. 6, pp. 1204–1209, 2003. [DOI] [PubMed] [Google Scholar]
- [50].Liu G, Fang Y, and Wu J, “A mechanism for localized dynamics-driven affinity regulation of the binding of von willebrand factor to platelet glycoprotein ibalpha,” J Biol Chem, vol. 288, no. 37, pp. 26658–26667, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Brophy T, Ward S, McGimsey T, Schneppenheim S, Drakeford C, O’Sullivan J, Chion A, Budde U, and O’Donnell J, “Plasmin cleaves von willebrand factor at k1491-r1492 in the a1-a2 linker region in a shear- and glycan-dependent manner in vitro.,” Arterioscler Thromb Vasc Biol, vol. 37, no. 5, pp. 845–855, 2017. [DOI] [PubMed] [Google Scholar]
- [52].Solá R and Griebenow K, “Effects of glycosylation on the stability of protein pharmaceuticals.,” J Pharm Sci, vol. 98, no. 4, pp. 1223–1245, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Shental-Bechor D and Levy Y, “Effect of glycosylation on protein folding: a close look at thermodynamic stabilization,” Proc Natl Acad Sci U S A, vol. 105, no. 24, pp. 8256–8261, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Brehm M, Huck V, Aponte-Santamaría C, Obser T, Grässle S, Oyen F, Budde U, Schneppenheim S, Baldauf C, Gräter F, Schneider S, and Schneppenheim R, “von willebrand disease type 2a phenotypes iic, iid and iie: A day in the life of shear-stressed mutant von willebrand factor.,” Thromb Haemost, vol. 112, no. 1, pp. 96–108, 2014. [DOI] [PubMed] [Google Scholar]
- [55].Campbell J, Tischer A, Machha V, Moon-Tasson L, Sankaran B, Kim C, and Auton M, “Data on the purification and crystallization of the loss-of-function von willebrand disease variant (p.gly1324ser) of the von willebrand factor a1 domain.,” Data Brief, vol. 7, pp. 1700–1706, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Schneppenheim R, Budde U, Obser T, Brassard J, Mainusch K, Ruggeri Z, Schneppenheim S, Schwaab R, and Oldenburg J, “Expression and characterization of von willebrand factor dimerization defects in different types of von willebrand disease.,” Blood, vol. 97, no. 7, pp. 2059–2066, 2001. [DOI] [PubMed] [Google Scholar]
- [57].Schneppenheim R, Michiels J, Obser T, Oyen F, Pieconka A, Schneppenheim S, Will K, Zieger B, and Budde U, “A cluster of mutations in the d3 domain of von willebrand factor correlates with a distinct subgroup of von willebrand disease: type 2a/iie.,” Blood, vol. 115, no. 23, pp. 4894–4901, 2010. [DOI] [PubMed] [Google Scholar]
- [58].Pruthi R, Daniels T, Heit J, Chen D, Owen W, and Nichols W, “Plasma von willebrand factor multimer quantitative analysis by in-gel immunostaining and infrared fluorescent imaging.,” Thromb Res, vol. 126, no. 6, pp. 543–549, 2010. [DOI] [PubMed] [Google Scholar]
- [59].Schneider C, Rasband W, and Eliceiri K, “Nih image to imagej: 25 years of image analysis.,” Nat Methods, vol. 9, no. 7, pp. 671–675, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Masson G, Burke J, Ahn N, Anand G, Borchers C, Brier S, Bou-Assaf G, Engen J, Englander S, Faber J, Garlish R, Griffin P, Gross M, Guttman M, Hamuro Y, Heck A, Houde D, Iacob R, Jørgensen T, Kaltashov I, Klinman J, Konermann L, Man P, Mayne L, Pascal B, Reichmann D, Skehel M, Snijder J, Strutzenberg T, Underbakke E, Wagner C, Wales T, Walters B, Weis D, D Wilson, Wintrode P, Zhang Z, Zheng J, Schriemer D, and Rand K, “Recommendations for performing, interpreting and reporting hydrogen deuterium exchange mass spectrometry (hdx-ms) experiments.,” Nat Methods, vol. 16, no. 7, pp. 595–602, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Emsley J, Cruz M, Handin R, and Liddington R, “Crystal structure of the von willebrand factor a1 domain and implications for the binding of platelet glycoprotein ib.,” J Biol Chem, vol. 273, no. 17, pp. 10396–10401, 1998. [DOI] [PubMed] [Google Scholar]
- [62].Whitten S, García-Moreno B, and Hilser V, “Ligand effects on the protein ensemble: unifying the descriptions of ligand binding, local conformational fluctuations, and protein stability.,” Methods Cell Biol, vol. 84, pp. 871–891, 2008. [DOI] [PubMed] [Google Scholar]
- [63].Pettersen E, Goddard T, Huang C, Couch G, Greenblatt D, Meng E, and Ferrin T, “Ucsf chimera–a visualization system for exploratory research and analysis.,” J Comput Chem, vol. 25, no. 13, pp. 1605–1612, 2004. [DOI] [PubMed] [Google Scholar]
- [64].Zhang Q, Zhou Y-F, Zhang C-Z, Zhang X, Lu C, and Springer TA, “Structural specializations of a2, a force-sensing domain in the ultralarge vascular protein von willebrand factor,” Proc Natl Acad Sci U S A, vol. 106, no. 23, pp. 9226–9231, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Bienkowska J, Cruz M, Atiemo A, Handin R, and Liddington R, “The von willebrand factor a3 domain does not contain a metal ion-dependent adhesion site motif,” J Biol Chem, vol. 272, no. 40, pp. 25162–25167, 1997. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.