

ABSTRACT
Oncolytic viruses represent a promising therapeutic modality, but they have yet to live up to their therapeutic potential. Safety and efficacy concerns impel us to identify least toxic oncolytic agents that would generate durable and multifaceted anti-tumor immune responses to disrupt the tumors. Here we describe a rational engineered oncolytic herpes virus (OVH) that is a selective killer for targeting tumors, has strong safety records, induces complete regression of tumors in multiple tumor models, and elicits potent antitumor immunity. By far, the potential of OVs in promoting the tumor antigen-specific humoral immune responses remains obscure. In this study, we found that effective treatment by OVH induced immunogenic cell death, which facilitates to elicit humoral immune responses. Depletion experiments revealed that B cells were required for maximal antitumor efficacy of oncolytic immunotherapy. Both serum transfer and antibody treatment experiments revealed that endogenous oncolysis-induced antigen-targeting therapeutic antibodies can lead to systemic tumor regression. Our data demonstrate that tumor-targeting immune modulatory properties confer oncolytic OVH virotherapy as potent immunotherapeutic cancer vaccines that can generate specific and efficacious antitumor humoral responses by eliciting endogenous tumor antigen-targeting therapeutic antibodies in situ, resulting in an efficacious and tumor-specific therapeutic effect.
KEYWORDS: Oncolytic virus, tumor-targeting, therapeutic antibodies, cancer vaccine, anti-tumor effect
Introduction
Although great advances in cancer immunotherapy have been achieved over the past few years, cancer is still a major problem with high morbidity and mortality rates, and novel active immunotherapies are still needed.1,2
Oncolytic viruses (OVs) can infect, specifically replicate within and directly kill tumor cells, as well as induce systemic antitumor immunity.3 OVs have emerged as promising anticancer agents, with several generations of viruses having been evaluated in clinical trials for efficacy testing.4 The effectiveness of an oncolytic herpes simplex virus containing GM-CSF (talimogene laherparepvec, T-VEC) has been proven with a 26% objective response rate in patients with advanced melanoma in a randomized phase III clinical trial, and this virus received approval by the FDA as the first OV in 2015.5 More recent studies suggest that T-VEC can also sensitize tumor cells to immune checkpoint therapy, resulting in significantly improved therapeutic outcomes.6 Although oncolytic virotherapy has vast potential, there are numerous challenges, including tumor selectivity, safety, immunogenicity and magnitude of boosting antitumor immune response, regarding what it can achieve as a monotherapy.7–9 As such, great efforts are now being made to develop a newer generation of OV therapies that can be used as standalone anticancer agents with least toxic and durable anti-tumor effect.
Although the importance of OVs in antitumor activity has been demonstrated in many types of solid cancers, their roles and potential in antitumor immunity have not been fully investigated, which is an obvious impediment to oncolytic virus drug discovery and clinical investigation.10 Many OVs can elicit antitumor immune responses directly or indirectly by inducing immunogenic cell death in infected tumor cells.11,12 This immunogenic oncolysis helps to sensitize host innate and adaptive immune responses by releasing pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs), which in turn facilitate dendritic cell infiltration and cross-presentation of tumor-associated antigens (TAAs) that promote phagocytosis of dead or injured virus-infected tumor cells and lead to antigen-agnostic boosting of neoantigen-specific cytotoxic T lymphocyte (CTL) responses, making OVs highly complementary to other immunotherapies that only target one or a few immunosuppressive pathways.13,14 However, the potential of OVs in modulating both antigenicity and adjuvanticity of tumor cells and promoting the tumor antigen-specific humoral immune response remain obscure, and the underlying mechanism of generating endogenous oncolysis-induced antigen-targeting therapeutic antibodies has not been clearly illustrated.
Herpes Simplex Virus type I (HSV-1) offers particular advantages for use as an oncolytic vector in cancer therapy.15 We therefore undertook a stepwise design and development strategy to create a novel effective HSV-1 agent. First, ICP0 and ICP34.5, which are essential for HSV-1 replication in nondividing cells (normal cells) but are dispensable in rapidly dividing cells (tumor cells), were both deleted to make a better replication-selective and attenuated oncolytic virus. ICP0 is needed to stimulate translation of viral mRNA in quiescent cells and plays a key role in blocking IFN-induced inhibition of viral infection,16 so ICP0-null HSV-1 replicates more efficiently in tumor cells than in normal cells. ICP34.5 intercepts the interferon-induced PKR-mediated block to virus replication, which is usually disabled in tumor cells, thereby allowing ICP34.5-null HSV-1 mutants to proceed with viral infection.17 The deletion of ICP34.5 gene also can significantly reduce the neurotoxicity of virus to host.18 Second, the telomerase reverse transcriptase (hTERT) promoter, which is transcriptionally active in tumor cells, has been utilized to drive the exogenous gene expression.19 Approaches to develop a replication-selective oncolytic adenovirus by replacing the promoters of essential genes with the hTERT promoter, were well-documented.20–23 We thus proceeded to select the hTERT promoter for regulating the expression of viral essential gene that further optimized the safety of the virus.
In this study, we developed a rational engineered hTERT promoter-regulated ICP0 and ICP34.5-null oncolytic HSV-1 virus (OVH) and explored its safety, antitumor activity and mechanism of action in multiple tumor models. OVH was proved to be an excellent immunotherapeutic OV with strong safety records, good oncolytic properties and multiple immune boosting effects, suggesting extensive application prospects for OVH. To reveal the antitumor mechanisms with the goal of illustrating the unique immunotherapeutic potential of OVH, we found that OVH virotherapy cured the majority of mice with established tumors by eliciting efficacious and tumor antigen-targeting therapeutic antibody-mediated humoral responses to disrupt the tumors. These results demonstrate that the humoral arm of antitumor immunity contributes to the maximal therapeutic effect of OVH against different tumors. This suggests that the vaccine response elicited by OVs is potent and durable, which may provide novel insights into potentially immunotherapeutic cancer vaccine using oncolytic virus.
Materials & methods
Mice
C57BL/6, BALB/c mice and BALB/c nu/nu mice were purchased from the Shanghai Slack Laboratory Animal Co., Ltd. B6.129P2-Igh-Jtm1Cgn/J B cell-deficient mice (JHT) were kindly provided by C.C. Xiao (Xiamen University), which were originally purchased from the Jackson Laboratory. Mice were used in studies when 6–8 weeks old unless otherwise indicated. All animal work was conducted under the approval of the Institutional Animal Care and Use Committee at Xiamen University (XMULAC20150016).
Cells
Hepa1-6, Renca and A20 cells were purchased from the American Type Culture Collection (ATCC). L-02, Huh7, HCCLM3, MHHC97H, Cal-27, FaDu and Detroit 562 were purchased from the China Infrastructure of Cell Line Resources (Beijing, China). CEN1 and CEN2 were kindly provided by H.L. Chen (The University of Hong Kong). The other human cell lines were purchased from the ATCC. Tumor cell lines and normal cell lines were maintained in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal bovine serum (FBS), 100 U/ml penicillin, and 100 µg/ml streptomycin. Primary human umbilical vein endothelial cells (HUVECs) were purchased from ScienCell and cultured according to the manufacturer’s instructions. Primary mouse hepatocytes were isolated by the two-step collagenase perfusion technique and cultured in plates pre-coated with 0.1% rat tail collagen (SIGMA-ALDRICH).24 Hybridomas were cultured in RPMI 1640 containing 10% fetal bovine serum (FBS), 100 U/ml penicillin, and 100 µg/ml streptomycin. All cell lines and assay cultures were maintained at 37°C and 5% CO2.
Antibodies
Polyclonal antibodies including anti-ICP27 antibody VP-20, anti-β-Actin antibody C4 and anti-VP5 antibody 6F10 were from Santa Cruz. Polyclonal anti-ICP34.5 antibodies were raised against synthetic peptides synthesized from Shanghai Sangon Biotech (the epitope sequence is listed in Table S3). Anti-ICP0 mAb 5H7, anti-gD mAb DL6 and anti-ICP4 mAb H943 were from Santa Cruz. Polyclonal cleaved caspase-3 (Asp175) antibodies, cleaved PARP (Asp214) antibodies, PARP antibodies and anti-caspase-3 mAb 8G10, anti-caspase-8 mAb 1C12, anti-cleaved Caspase-8 (Asp391) mAb 18C8 were from Cell Signaling Technology (CST). Polyclonal anti-Ki67 antibodies were from Novus. Annexin V-PE antibody was from BEYOTIME. Antibodies used for flow cytometry were purchased from Bioss (calreticulin, ALEXA FLUOR® 647). Antibodies used for immunofluorescence were purchased from Invitrogen (goat anti-mouse IgG PE).
Tissue samples
Human glioma and ovarian tumor tissues from cancer patients were obtained from The First Affiliated Hospital of Xiamen University and Zhongshan Hospital Xiamen University and the quality of cancers were further verified by histopathological examination of the samples. The study was conducted in accordance with the ethical principles of the Declaration of Helsinki and was approved by the Human Ethics Committee of the Xiamen University. Informed consent was obtained from each patient before collection. The information for cancer patients involved in the study was summarized in Table S4. The isolation of tumor cells was following the protocol reported previously with minor modifications.25
Virus and plasmids
HSV-1 strain KOS virus was purchased from ATCC and propagated in U-2 OS cells. The viral titer was determined in U-2 OS cells as previously described.26 For the construction of the donor plasmids, different strategies were used. To generate the 27p plasmid, the 5ʹ and 3ʹ regions flanking the genomic regions of core ICP27 promoter (from nt113422 to nt113590) were amplified with the primer pair #1 and #2, and then sequentially cloned into BamHI and PstI digested PUC57 vector with a linker containing the PmeI and SpeI sites. To generate the 27p-hTERT plasmid, the region for core ICP27 promoter in 27p was replaced with DNA fragments containing the core hTERT promoter,27 using the restriction enzymes PmeI and SpeI. To generate the fICP0 plasmid, the 5ʹ and 3ʹ regions flanking the genomic regions of ICP0 were amplified with the primer pair #3 and cloned into HindIII and EcoRI digested PUC57 vector. To generate the fICP0-GFP plasmid, the ICP0 region of picp0 was replaced with DNA fragments encoding eGFP using the restriction enzymes NcoI and SalI. To generate the PF0 plasmid, the DNA fragment covered 478 bp upstream of the ICP34.5 initiation codon to 409 bp downstream of the ICP0 stop codon was amplified with the primer pair #4 and ligated into PstI and SacI digested pMD18-T vector. The d34.5/0GFP plasmid was derived from PF0 that the ICP34.5 and ICP0 coding region was replaced by a DNA fragment encoding eGFP at the NcoI and SalI site. All primers used in this study are listed in Table S5.
Virus construction
Generation of recombinant virus was using a cell-based recombination method previously described.26 dICP0 was an ICP0-null mutant derived from KOS, in which both copies of the ICP0 coding sequence were replaced by the eGFP gene by homologous recombination using donor plasmid fICP0-GFP. OVN was an ICP34.5 and ICP0-null mutant derived from KOS, in which both copies of the ICP34.5 and ICP0 coding sequence were replaced by the eGFP gene by homologous recombination using donor plasmid d34.5/0GFP. OVH was derived from OVN in which both copies of the ICP34.5 and ICP0 coding sequence were replaced as described in constructing OVN and the ICP27 core promoter was replaced with the core hTERT promoter by homologous recombination using donor plasmid p27p-hTERT. All three recombinant viruses were selected and purified repeatedly in U-2 OS cells. All three recombinants were propagated and purified repeatedly in U-2 OS cells. After serial passage, a HSV-1 variant named OVH was isolated and verified with higher cell-killing ability relative to its parent virus. All viruses were propagated and assayed in U-2 OS cells.
Virus verification
Virus genomic DNA was extracted by using the QIAamp DNA Blood MiniKit (Qiagen). The genomic region of each gene was amplified using KOD-plus high-fidelity DNA polymerase (Toyobo) and given primer sets (#5-8). The identities of PCR products were confirmed by direct DNA sequencing. Total RNA was extracted from virus-infected cells using TRIzol (Life Technologies), and reverse transcription was performed with 1 μg total RNA using random primers and AMV Reverse Transcriptase (Promega) according to the manufacturer’s instructions. Quantitative PCR was performed with SsoAdvanced™ Universal SYBR® Green (Promega) and given primer sets (#9-11) using an Applied Biosystems 7500 Real-Time PCR System (Life Technologies).
Genome sequencing
Genomic DNA was isolated from infected U-2 OS cells using standard protocols.28 An unpaired 350-bp Illumina library was generated and double-end sequenced using the HiSeq sequencing platform (Novogene). The resulting reads were assembled initially into large contigs. Viral genomes were compared using KOS genome sequence (Genbank: JQ673480) as a reference.
Virus replication assay and titration
Cells were seeded in 6 cm plates at 106 cells/dish and infected with KOS, dICP0, OVN, OVH (1 PFU/cell) or mock-infected, separately. After 48 h of infection, the infected cells were either harvested and thereafter subjected to virus titration or examined by fluorescence microscopy and thereafter subjected to western blot. The titers of the amplified viruses were determined on U-2 OS monolayers.
Cell viability assay
To evaluate the cell-killing ability of KOS, dICP0, OVN and OVH, cells were seeded in 6 cm plates at 106 cells/well and infected with virus (1 PFU/cell) or mock-infected. Remaining cells were harvested from individual virus infected cells at 72 h post-infection, and counted by trypan blue exclusion method. For each time point, cell viability was expressed as the means of percentage reduction in infected versus uninfected cells. To evaluate the cell-killing ability of OVH, cells were seeded in 6 cm dishes at 3 × 106 cells/dish and infected with OVH (1 PFU/cell) or mock-infected. After 72 h infection, the number of viable cells were counted by trypan blue exclusion method. The percentage of viable cells was calculated using the formula: Percent viable cells (%) = (Viable cells in OVH infected wells)/(Viable cells in mock-infected control wells × 100%.
Apoptosis assay
Cells were infected with OVH at an MOI of 0.5 PFU/cell. After 48 h of infection, the cells were harvested and stained with the Pacific Blue-labeled annexin V (Invitrogen) and PI. Apoptotic cell death was determined by flow cytometric analysis using the BD FACSDiva Software on a FACSAria II cell sorter (BD).
Assays for detecting the immunogenic cell death (ICD) determinants
Cells were infected with OVH at an MOI of 1 PFU/cell. After 48 h of infection, the cells were harvested and stained with the anti-calreticulin (Bioss), and flow cytometric analysis was used to determine expression of calreticulin on the surfaces of treated cells. The released ATP in the supernatant was measured by an ATPlite Luminescence kit (PerkinElmer), and the HMGB1 in the supernatant was measured by an HMGB1 ELISA kit (TECAN).
Western blot analysis
Cells were lysed in RIPA lysis buffer containing protease inhibitor cocktail (Roche), and the generated cell lysates were separated by SDS-PAGE and transferred onto a nitrocellulose membrane. After membranes were washed and blocked, they were probed with indicated primary antibody overnight. After rigorous washing, they were probed with horseradish-peroxidase (HRP) conjugated secondary antibodies and finally visualized with the Lumi-LightPLUS Western blotting Substrate (Roche).
Immunostaining
Hepa1-6 cells and mouse primary hepatocytes were grown on CellCarrier-96 microplate overnight. Primary mAbs to Hepa1-6 cells were added to the culture media and incubated for 48 h at 4°C. Wells were then washed and stained with PE conjugated goat anti mouse IgG and DAPI for 1 h at 37°C. Imaging was performed on Operetta® High Content Imaging System (PerkinElmer).
Neurovirulence study
Four-week-old female BALB/c mice were anesthetized with sodium thiopental (60 mg/kg) and placed in a stereotaxic instrument (Stoelting). Intracerebral inoculation was performed with a 281/2 gauge needle between the outer canthus of the eye, the front of the pinna, and midline of the head.29 Vehicle (PBS), 1 × 104 PFU of KOS, 1 × 105 PFU, 1 × 106 PFU and 1 × 107 PFU of dICP0, OVN or OVH, and 4 × 107 PFU of OVH were used in this study. 10 μl of virus was slowly injected at a depth of 4.5 mm from the skull surface over a period of 10 min. After another 3 min, the needle was slowly withdrawn. Mice were monitored for 30 days following inoculation.
Blood cell analysis
OVH was injected intravenously (i.v.) at a dose of 5 × 107 PFU in 0.5 ml in female BALB/c mice. Mice were bled on day 1 and day 30 post-first treatment for a total of two blood draws using plastic K2EDTA tubes. Determination of white blood cells (WBC), platelet (PLT), lymphocytes, red blood cells (RBC) and neutrophil granulocytes, were performed on Mindray Hematology Analyzer BC-5000 according to manufacturer’s instructions.
Serum chemistry
BALB/c mice were injected intravenously (i.v.) with OVH as previously described. Mice were bled on the day of first treatment with PBS or OVH and bled every five days thereafter until the 40th day after treatment for a total of six blood draws. The sera were isolated via centrifugation, aliquot, and then immediately stored at −80°C to allow for subsequent batch processing. Determination of the albumin (ALB), total protein (TP), creatinine (CREA), uric acid (UA), UREA and total bilirubin (TBil) levels as well in the aspartate aminotransferase (AST) and alanine aminotransferase (ALT) activity were performed on Mindray BS-200 Analyzer, according to manufacturer’s instructions.
Mouse models and treatment protocols
For establishment of human xenograft tumors, BALB/c nu/nu mice were subcutaneously (s.c.) implanted with seven human tumor cell lines respectively, including 7.5 × 106 human lung tumor cells (H1299), 5 × 106 human liver tumor cells (Hep3B), 1 × 106 human breast tumor cells (MCF-7), 5 × 106 human nasopharynx tumor cells (CEN2), 5 × 106 human colon tumor cells (HCT116), 1 × 106 human glioma cells (Glioma), 1 × 106 human ovarian tumor cells (Ovarian cancer) in 100 μl of PBS. Once the tumors reached 100 ~ 200 mm3, OVH was intratumorally injected at a dose of 107 PFU, at day 0 (relative to treatment) and every 3 days thereafter until the total three dosages were finished.
For establishment of Hepa1-6 tumors, 5 × 106 cells in 100 μl of PBS were s.c. injected into the both flanks of C57BL/6 mice or JHT mice and allowed to establish for 10 days. For establishment of Renca or A20 tumors, 5 × 106 Renca cells or 1 × 106 A20 cells in 100 μl of PBS were injected subcutaneously into the both flanks of BALB/c mice and allowed to establish for 10 or 12 days. Once the tumors reached indicated volume, OVH was intratumorally injected at indicated dosages. Tumor volumes were measured every 3 days for 30 days using calipers and calculated by using the formula Volume = (L×(W)2)/2, where L and W represent the largest and the smallest diameters, respectively. Mice were weighed every 3 days for 30 days using precision electronic balance. The tumor-free incidence was presented as the percentage of tumor-free mice in total treated mice.
For syngeneic tumor re-challenging experiments, naive or tumor-free mice cured by OVH therapy, which survived for 80 days, were s.c. re-challenged with 1 × 107 corresponding tumor cells. For heterologous tumor re-challenging experiments, naive or Renca tumor-free BALB/c mice by OVH therapy, which survived for 80 days, were s.c. re-challenged with 1 × 107 A20 tumor cells. The incidence of rejecting secondary challenge was presented as the percentage of tumor-free mice in total re-challenged mice.
Serum transfusion experiments
Transfer of serum from PBS-treated mice or OVH-cured mice to newly established tumor-bearing C57BL/6 mice were performed on all three syngeneic tumor models. For each experiment, mice were bled at 21 days post-first treatment with PBS or OVH, serum was harvested from each mouse and incubated at 56°C for 0.5 h to inactivate complement. The serum from OVH-cured mice was either untreated or preprocessed by removing the IgG antibodies using Protein A/G beads (GE Health). 7 days prior to serum transfusion, Hepa1-6 tumors were s.c. pre-implanted in naïve mice. Once the tumors reached 100 mm3, the mice were i.v. treated with 0.3 ml of sera diluted in 1 ml saline every 3 days until the total three dosages were finished. Mice were weighed every 3 days from transfer treatment initiation until end of follow-up. Tumor volumes were measured every 3 days for 30 days and tumors were dissected for weight measurement and photographing at the end of follow-up.
Antibody production
OVH-cured Hepa1-6 mice, which did not relapse and survived for more than 45 days, were s.c. re-challenged with 1 × 107 Hepa1-6 cells, and subjected to hybridoma generation process following the protocol reported previously with minor modifications.30 In brief, splenocytes from OVH-cured Hepa1-6 mice and mouse SP2/0 myeloma cells were fused at a ratio of 5:1 using PEG 1450 (SIGMA-ALDRICH). The resulting hybridomas were plated over 20 96-well plates and initially grown in RPMI 1640 medium supplemented with 20% fetal bovine serum for 24 h. Then the hybridoma cells were selected with HAT conditioned medium for 2 weeks. The positive clones were screened and selected by indirect cell-based ELISPOT assay. The Hepa1-6 cells and mouse hepatocytes were fixed with 1% paraformaldehyde in PBS for 5 min and followed by permeabilization with 0.1% Triton X-100 in PBS at room temperature for 5 min.100 µl supernatant was added to the monolayer cells and incubated at 37°C for 1 h. After washing, HRP-conjugated secondary antibody was added and incubated 37°C for 1 h. After reaction with TMB substrates (SIGMA-ALDRICH), the spots were developed within 15 min. Last, the plates were spin dried and counted using ImmunSpot@S5 UV Analyzer (Cellular Technology). mAbs were prepared by ascites production and purified by using protein G chromatography according to the manufacturer’s instructions.
Antibody reactivity analysis
1 µg/ml diluted mAbs were incubated with Hepa1-6 cells at 37°C for 1 h, respectively. After stringent washing, PE conjugated anti-mouse IgG (Fc specific) antibody was added and incubated for 0.5 h. The positive stained cells were analyzed by flow cytometry. For the competitive binding assay, 3F11 F(ab)2 fragments were prepared using a F(ab)2 Fragmentation Kit (Geno). Hepa1-6 cells were incubated with the same amount of 2G2 and PBS or 3F11 F(ab)2 (1 µg/ml), then stained with PE conjugated anti-mouse IgG (Fc specific) antibody, and finally subjected to flow cytometric analysis.
Antibody therapy
To test the therapeutic effect of antibodies against Hepa1-6 tumors, once tumors reached indicated sizes, antibodies were intraperitoneally (i.p.) injected at a dose of 200 μg, at day 0 and every 3 days thereafter until the total three dosages were finished. The tumor size and weight change were monitored for 24 days. To test the therapeutic effect of 3F11 in BALB/c nu/nu mice, once tumors reached 150 mm3, 3F11 were i.p. injected at a dose of 400 μg, at day 0 and every 3 days thereafter until the total three dosages were finished. The tumor size was monitored for 18 days.
Depletions
Depletion of immune cells was performed with corresponding depleting rat mAbs against different immune markers. When the tumors reached 100 mm3, all depleting antibodies (anti-NK1.1 (clone PK136), anti-macrophage (F4/80) and rat IgG isotype control antibodies) were i.p. administered beginning 3 days before initiation of 3F11 therapy, at a dose of 400 μg per antibody for a total three dosages. 3F11 were i.p. injected at a dose of 200 μg, at day 0 and every 3 days thereafter until the total three dosages were finished. The tumor size was monitored for 24 days.
Histology
Vital tissues (including lung, liver, brain, heart, kidney, spleen, muscle, intestine) were collected at sacrifice and kept in 10% buffered formalin. The fixed tissues were histologically analyzed by standard hematoxylin-eosin (H&E) staining. For immunohistochemistry (IHC) staining of proliferation marker (Ki67) and apoptosis marker (cleaved caspase3), animals were sacrificed at 24 h after receiving treatment of PBS or OVH, and HeLa, CEN2 and Hepa1-6 tumors were fixed in 10% formalin, embedded in paraffin. IHC staining was performed using an Ultrasensitive SP kit (Maxim) and a DAB detection kit (Maxim) according to the manufacturer’s instructions. 5 μm sectioned specimens were stained with indicated antibodies and subsequently incubated with HRP conjugated secondary antibodies (Maxim). Then the sections were washed, counterstained with hematoxylin (SIGMA-ALDRICH) and mounted with cover slips. Images were captured with a research-level upright microscope (Olympus).
Statistical analysis
Statistical analyzes were performed using GraphPad Prism 7 software (GraphPad Software). Statistical parameters and methods are reported in the Figures and the Figure Legends. For all statistical analyzes, differences were considered significant when a P value was below or equal to 0.05. Data for survival was evaluated using log-rank test.
Results
Selective killing of tumor cells by a rational engineered HSV-1 virus, OVH
To generate an oncolytic HSV-1 virus with good tumor selectivity and oncolytic properties, we first rationally designed three generations of HSV-1 recombinant constructs (dICP0, OVN and OVH) for parallel comparison, each of which contained different genetic modifications (Figure 1A). dICP0 is an ICP0-null, attenuated HSV-1 virus with a certain degree of tumor selectivity as previously described.26,31 OVN is an ICP0- and ICP34.5-null HSV-1 virus with reduced neurovirulence due to the additional deletions of ICP34.5. OVH is an OVN derivative, in which the essential gene ICP27 is under the regulation of the tumor-specific hTERT promoter. All these recombinant viruses were verified by sequencing the PCR products (Fig. S1A), whole genome sequencing and observing gene expression (Figure 1B and Fig. S1B). Then, we examined the expression of immediate early genes and late genes in various infected human normal cell lines and human tumor cell lines. In the three normal cell lines, HUVECs, L-02 and HEL299, the ICP27 expression of OVH was significantly reduced at 3 ~ 9 h after exposure to 0.5 PFU/cell compared to that of other recombinant viruses (Figure 1C). However, in the three tumor cell lines, MCF-7, Hep3B and H1299, the ICP27 expression of OVH was expressed in a time-dependent manner, showing a similar expression pattern to the other HSV-1 recombinant viruses (Figure 1D). The expression of late genes (gD and vp5) showed similar results (Fig. S1C), which further support the selectivity of the hTERT promoter to tumors in regulating ICP27 expression of OVH. Next, we compared the replication efficiency of these viruses. We infected the cells at an MOI of 1 and then measured the viral titers. OVH showed a significantly reduced replication efficiency in the human normal cell lines but not in human tumor cell lines (Figure 1E). Compared to OVN, OVH showed a further reduction of its replication capability only in the human normal cell lines, which suggests that OVH had greater tumor selectivity. In addition, the cell-killing potency of OVH in the human normal cell lines was significantly decreased compared to that of the other HSV-1 recombinant viruses, while their oncolytic potency of all three viruses was similar in the human tumor cell lines (Figure 1F). All these data indicate that tumor-selective replication contributes to the tumor-targeting property of OVH.
Figure 1.
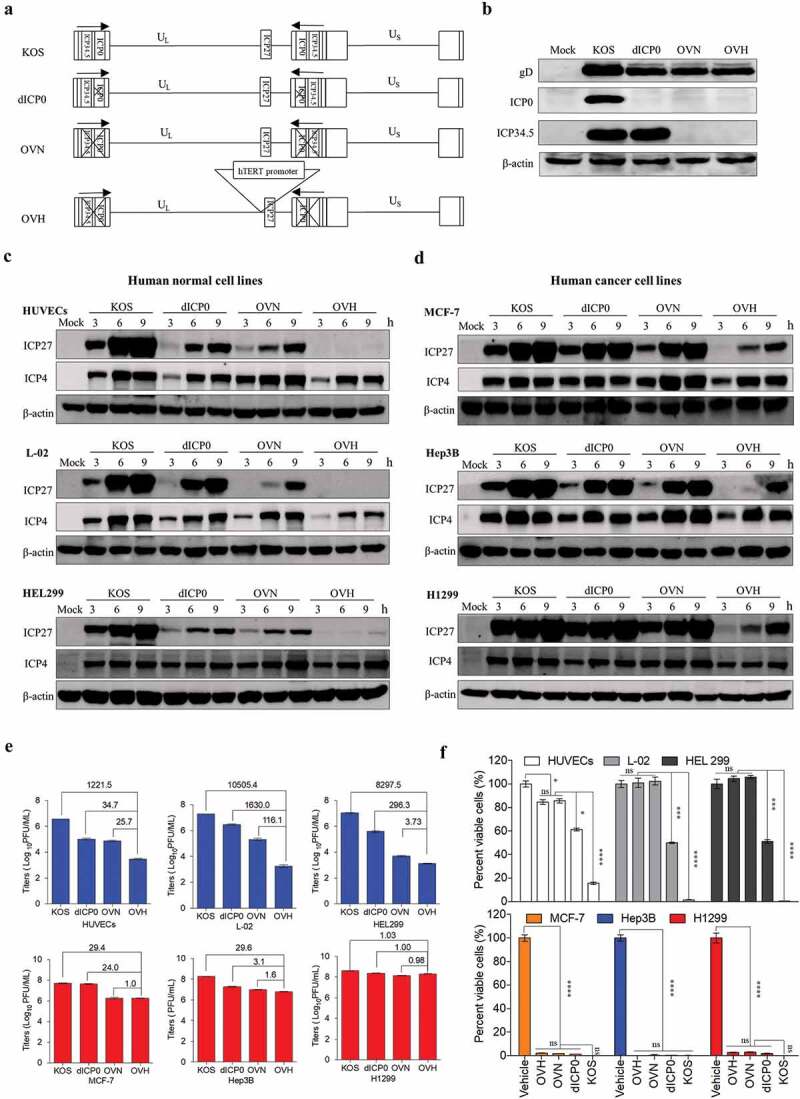
Development of a novel hTERT promoter-regulated oncolytic HSV-1 virus (OVH) with selective oncolytic capability. (A) Schematic diagram of KOS and KOS-derived HSV-1 recombinant constructs (dICP0, OVN and OVH) used in this study. (B) Western blot analysis of ICP0 and ICP34.5 expression in various infected U-2 OS cells 48 h after virus infection. (C-D) Western blot analysis of ICP27 and ICP4 expression in various infected human normal cell lines (HUVECs, L-02 and HEL299) (C) and human tumor cell lines (MCF-7, Hep3B and H1299) (D) 3 h, 6 h, and 9 h after virus infection. (E) Viral replication assays were performed on various infected cell lines (MOI = 1 PFU/cell). Viruses harvested from infected cells 48 h after virus infection were titrated. Fold changes between groups were calculated and shown. (F) Cell viability was measured in various infected cell lines 72 h after virus infection (MOI = 1 PFU/cell). Remaining cells harvested from individual virus infected cells were measured by trypan blue exclusion method. Values are means of three independent experiments, data are shown as means ± SEM. *P < .05, ***P < .001, ****P < .0001, ns, not significant by one-way ANOVA test for F.
Safety profile of OVH in mice
Two different toxicity evaluation models, the murine lethal challenge model and systemic challenge model, were established to evaluate the safety and potential toxicity of OVH (Figure 2A and D). To test safety, we first evaluated the neurovirulence of intracerebrally injected OVH in BALB/c mice. The mice were challenged with a single inoculation of PBS, KOS, dICP0, OVN and OVH with indicated dosages (1 × 104 PFU per dose to 4 × 107 PFU per dose). Approximately 90% of mice in the OVN-challenged group receiving 1 × 107 PFU per dose survived compared to 100% of mice survived in the OVH-challenged group receiving either 1 × 107 PFU per dose or 4 × 107 PFU per dose, while all the mice in the KOS-challenged group receiving 1 × 104 PFU per dose died (Figure 2B). The results showed that, in vivo, OVH exhibited over 4000-fold reduced neurovirulence compared to KOS, over 400-fold reduced neurovirulence compared to dICP0 and over 4-fold reduced neurovirulence compared to OVN. OVH showed increased sensitivity to anti-herpes drug (ganciclovir, GCV) compared to KOS (Figure 2C), thus suggesting that OVH could be easily controlled if unexpected viral replication was observed. Moreover, we established a systemic challenge model to evaluate the systemic toxicity of OVH in BALB/c mice by a single intravenous injection of virus (5 × 107 PFU per dose) (Figure 2D). On day 30, histological analysis of vital tissues of OVH-injected mice and vehicle-injected mice, including lung, liver, brain, heart, kidney, spleen, muscle and intestine, was performed by H&E staining. No obvious pathological abnormalities were observed (Figure 2E). Importantly, OVH did not cause histological changes in brain tissues since encephalitis caused by HSV-1 can result in significant brain damage. Some pathology signs, including inflammatory infiltrates and signs of apoptosis, were not observed in either lymphoid or non-lymphoid tissues. Taken together, we found that OVH-injected mice showed least tissue toxicity. Hematology studies demonstrated no overt leukocytopenia, leukocytosis or anemia and no abnormalities in the platelet and neutrophil counts (Figure 2G–J). We observed slightly decreased white blood cell counts 1 day after OVH injection, but they returned to normal after several days (Figure 2F). We also observed no significant difference in body weight between the OVH-injected group and the vehicle-injected group during the course of the study (Figure 2K). Serum chemistry studies demonstrated no significant difference in the ALB, TP, CREA, UA, urea and T-Bil levels as well in the AST and ALT activity between the OVH-injected group and the vehicle-injected group during the course of the study (Figure 2L–T). All the results support the conclusion that OVH is relatively safe and exhibits low toxicity in mice, thus providing important safety evidence for the clinical use of OVH therapy in the future.
Figure 2.
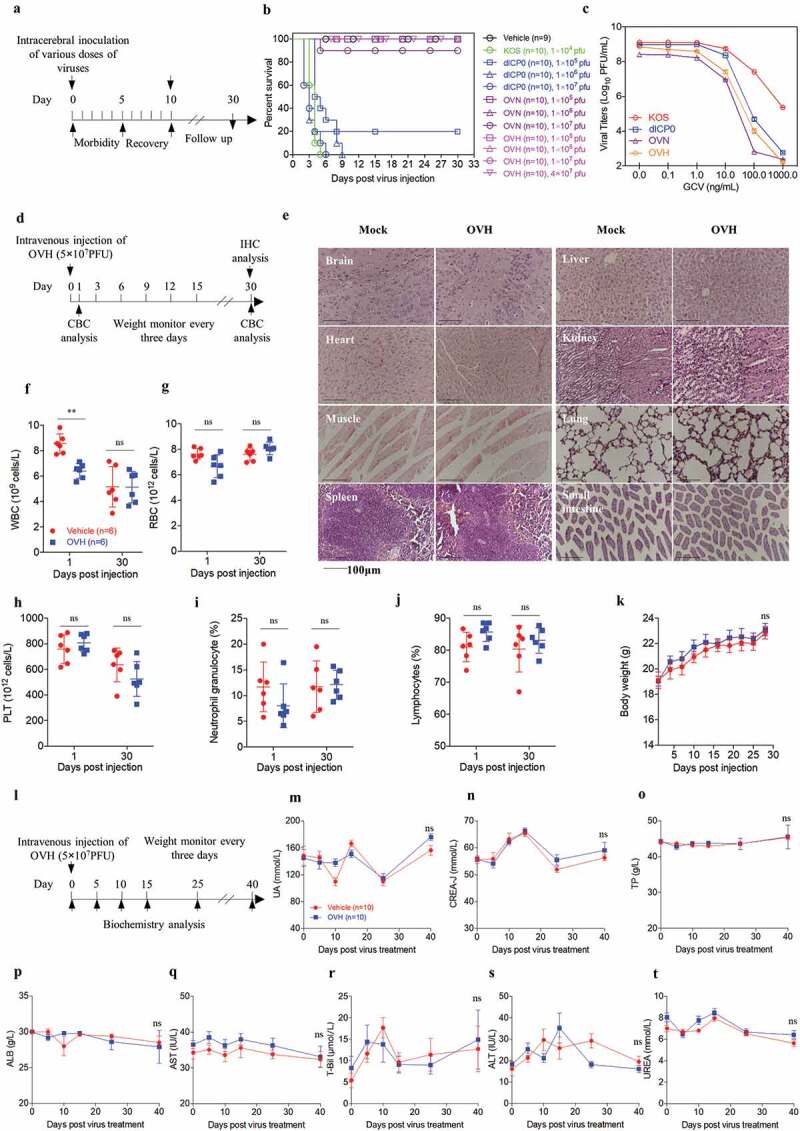
Safety evaluation of OVH in vivo. (A) Murine lethal challenge model and timeline of challenge and follow-up. (B) Survival over time for mice intracerebrally (i.c.) injected with indicated dosages of viruses or PBS in 10 μl. (C) The replication efficiency of KOS, dICP0, OVN and OVH in U-2 OS cells exposed to different doses of ganciclovir (0 ng/mL~1000 ng/mL, MOI = 0.05 PFU/cell). (D) Timeline of the toxicity study. Complete blood count (CBC) analysis and immunohistochemistry (IHC) analysis were performed at indicated time points. (E-J) Mice intravenously injected with a single dose of OVH (5 × 107 PFU) were bled on day 1 and day 30 post-injection, and WBC, PLT, lymphocytes, RBC and neutrophil granulocytes were measured, n = 6 BALB/c mice per group. H&E staining of representative tissue sections from vehicle- and OVH-injected mice on day 30 following virus injection (E). H&E images were obtained using a microscope (OLYMPLUS), with a 20 × objective. (K) The body weight of the treated mice was monitored over a 30-day period. (l) Timeline of biochemistry analysis after mice were intravenously challenged with 5 × 107 PFU OVH or PBS. (M-T) ALB, TP, CREA, UA, UREA, and T-Bil levels as well as AST and ALT activity were measured, n = 10 BALB/c mice per group. Arrows indicate corresponding time points. Data are shown as means ± SEM. **P < .01, ns, not significant by unpaired two-tailed Student’s t tests for E, F, G, I, J or repeated measure ANOVA for K, M-T.
Oncolytic efficacy of OVH in vitro and in vivo
To explore the oncolytic efficacy of OVH in vitro, we first examined the tumor cell-killing effects of OVH on various cultured human tumor cell lines. All tested cell lines are listed and categorized in Table S1. OVH remarkably induced cell death in tumor cells (Figure 3A). Of the 49 tumor cell lines that we tested, at 72 h after OVH infection at an MOI of 1, 17 lines showed a more than 90% decrease in cell viability, and 38 lines showed a more than 50% decrease in cell viability. To test the hypothesis that the tumor cell-killing capability of OVH correlated with virus growth, we tested the replication titers of OVH in different cell line sets based on their sensitivity to OVH (high sensitivity (HS), sensitivity (S) and refractory to OVH (R)); these cell line sets were grouped by the degree of OVH-induced cell death (Table S2). A positive correlation between viral titer and tumor cell-killing capability was observed, and the replication of OVH was better in OVH-sensitive tumor cells than in refractory cells (Figure 3B).
Figure 3.
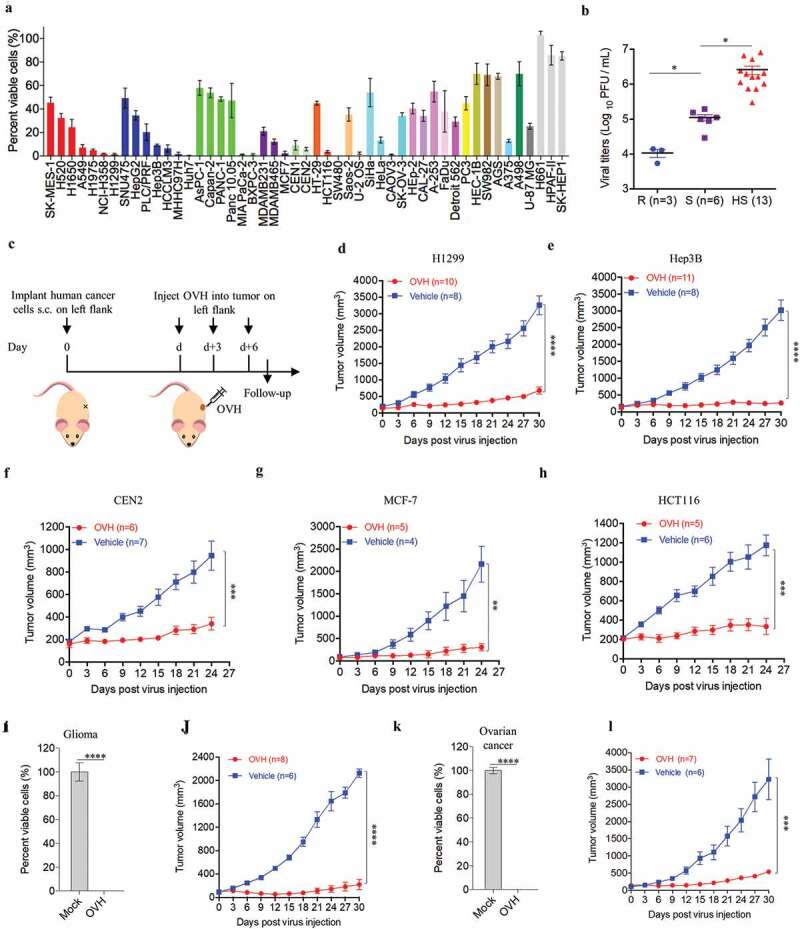
Oncolytic efficacy of OVH in vitro and in vivo. (A) Cell viability assay was performed on a panel of human tumor cell lines 72 h after virus infection (MOI = 1 PFU/cell). (B) Viral titers in selected infected cell lines exhibiting high sensitivity (HS, n = 13), sensitivity (S, n = 6) and refractory (R, n = 3) to OVH. (C) Timeline of treatment in BALB/c nu/nu mice. Arrows indicate corresponding time points. (D-H) Growth of vehicle- or OVH-treated tumor xenografts in nude mice. Mice were subcutaneously (s.c.) inoculated with H1299 (D), Hep3B (E), CEN2 (F), MCF-7 (G) or HCT116 (H) cells in the left flank and treated d days later with OVH. Tumor growth was monitored over a 24 ~ 30-day period. In D, E, d = 30; in F, G, H, d = 14. (I-L) Two human primary tumor cell lines were isolated from surgical tumor specimens of glioma and ovarian cancer patients and infected with OVH (MOI = 1) or vehicle for 72 h. Oncolytic efficacy of OVH against glioma in vitro (I) and in vivo (J) was observed. Oncolytic efficacy of OVH against ovarian cancer in vitro (K) and in vivo (L) was observed. In J, L, d = 14. The number of mice (N) used in the experiments is shown in the individual figures. All values are presented as the mean ± SEM. *P < .05, **P < .01, ***P < .001, ****P < .0001, ns, not significant by one-way ANOVA test for B, unpaired two-tailed Student’s t tests for I, K or repeated measure ANOVA for D-H, J, L.
To further evaluate the in vivo tumor cell-killing potential of OVH, we established a subcutaneous xenograft nude mouse model implanted with different human tumor cell lines (H1299, Hep3B, CEN2, MCF-7 and HCT116) (Figure 3C). Once the tumors reached 100 mm3, mice xenografted with each tumor were randomized to receive intratumoral injections of three doses of OVH (1 × 107 PFU per dose) or vehicle. Consistent with the in vitro experiments, significant tumor inhibition was observed in the OVH-treated mice (Figure 3D–H). Additionally, we isolated and established two human primary tumor cell lines from surgically dissected tumor specimens of glioma and ovarian cancer patients. OVH remarkably induced cell death in glioma and ovarian tumor cells in vitro (Figure 3I and K). Tumor growth was significantly inhibited in the OVH-treated group compared to that in the vehicle-treated group (Figure 3J and L). We proposed that OVH-induced oncolysis was effective at promoting tumor regression in the subcutaneous xenograft models.
OVH therapy eradicates the injected and distant large tumors in syngeneic mice models
Because of the limited viral spread in local tumors, it is important for oncolytic virotherapy to induce sufficient tumor lysis and a systemic tumor response, especially in syngeneic mice models bearing large established tumors. We explored intratumoral OVH injection as an attempt to elicit a systemic antitumor immune response that would be potentially active against metastatic tumors. Thus, we established bilateral flank tumor models in immunocompetent mice. OVH administration into the right flank tumor resulted in viral replication within the injected tumors, but no virus was detected in the distant (left flank) tumors based on virus titration (Fig. S2). We first evaluated the antitumor efficacy of OVH in three immunogenic tumor models: the Hepa1-6 mouse liver tumor model, Renca mouse kidney tumor model and A20 mouse B cell lymphoma model (Figure 4A, D and G). Once the tumors reached 250 mm3 for the Hepa1-6 and A20 tumors or 100 mm3 for the Renca tumors, the mice were randomized to receive intratumoral injection of three doses of OVH (1 × 107 PFU per dose) or vehicle in the right flank. OVH therapy induced robust tumor eradication and durable cures in 100% of the mice implanted with Hepa1-6 tumors (Figure 4B and C), 66.7% of the mice implanted with Renca tumors (Figure 4E and F) and 75% of the mice implanted A20 tumors (Figure 4H and I). OVH treatment resulted in tumor rejection not only in virus-injected tumors but also in the distant flank tumors (Figure 4B–I). Although each of the three tumor models showed a slightly different efficacy, OVH therapy exhibited substantially high efficacy in tumor eradication of the injected and distant large established tumors. To determine whether this effect was durable, a re-challenge with the corresponding tumor cells was administered on day 80 post-first treatment, and 100% of tumor-free long-term survivors cured by OVH therapy rejected these cells, suggesting that intratumoral OVH therapy can induce durable and protective antitumor memory (Figure 4J–M). However, the antitumor memory was only restricted to the injected tumor type, and long-term survivors cured from Renca tumors could not reject a re-challenge with the A20 tumor cells (Figure 4N), suggesting that the observed antitumor effect in the distant tumors was dependent on the injected tumor type and not a result of nonspecific inflammation generated by OVH infection. A number of previous studies have demonstrated that neoantigen-specific cytotoxic T lymphocyte (CTL) responses played vital role in OV-induced systemic antitumor effect.7,32 However, little is known about the potential of OVs in promoting the tumor antigen-specific humoral immune responses. In our study, we evaluated endogenous antibody responses in OVH-cured mice. In two syngeneic tumor models, an immunoblot of serum from OVH-treated C57BL/6 mice bearing Hepa1-6 tumors against Hepa1-6 lysate but not against Renca lysate revealed that these endogenous antibodies recognized numerous antigens of the treated tumors and the induced antibody response was also tumor-specific (Figure 4O and P). Since Hepa1-6, A20 and Renca tumor cells were highly susceptible to OVH infection and regarded as high immunogenic tumor cells (Fig. S3, A and B). We also evaluated the oncolytic efficacy of OVH in poorly immunogenic tumor models, such as murine Lewis lung carcinoma tumor model (LL/2 tumor model, Fig. S3, B and C) and murine prostate cancer model (TRAMP-C2 tumor model, Fig. S3, B and F). The results showed that OVH can significantly inhibit both injected and distant flank tumor growth in these two tumor models, but no complete regression of injected and distant tumors was observed. (Fig. S3, C-H). More obvious tumor growth inhibition was observed in OVH-injected tumors compared to distant tumors (Fig. S3, F and H). Taken together, these results indicated that the anti-tumor response induced by oncolytic virotherapy, especially for the distant tumors, was associated with the immunogenicity of tumor cells as well as the tumor cell susceptibility to OVH infection.
Figure 4.
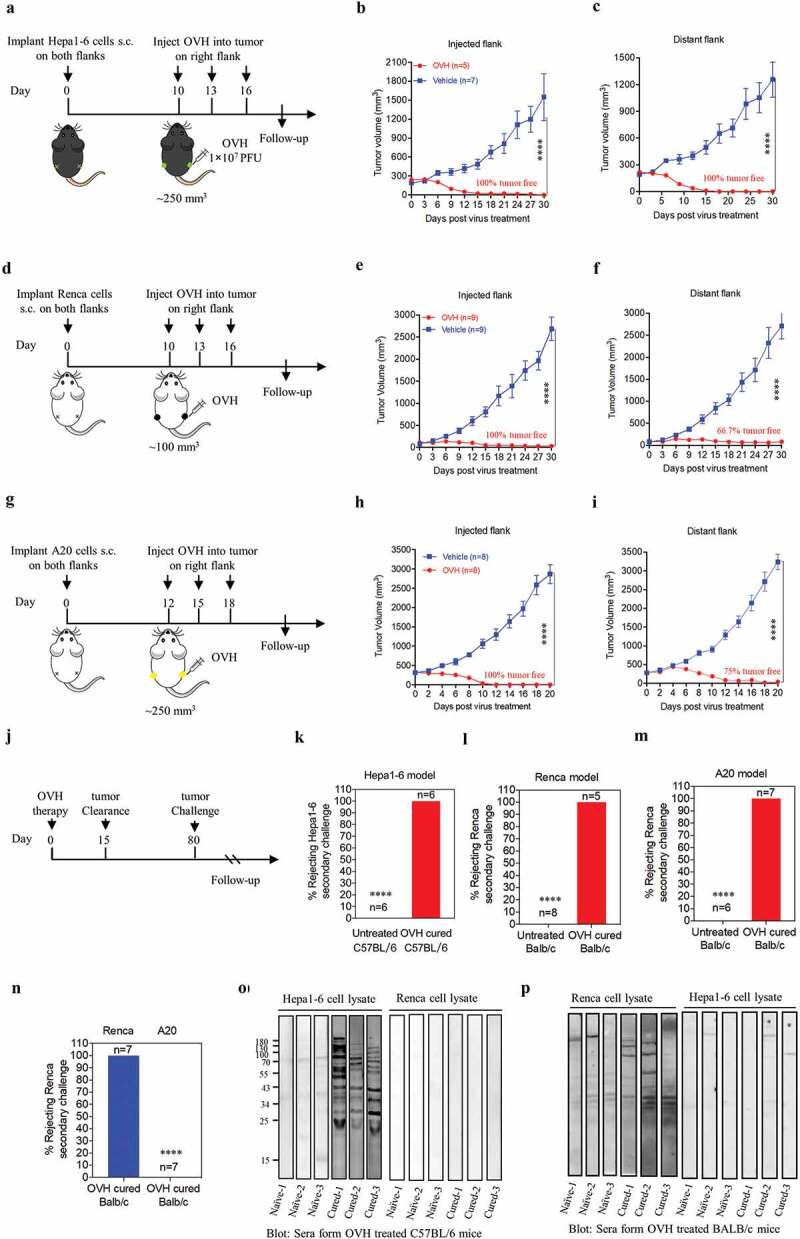
OVH therapy can eradicate local and distant tumors in the immunogenic tumor models. (A, D, G) Timeline of treatment in C57BL/6 and BALB/c mice. Mice were s.c. inoculated with Hepa1-6 (A), Renca (D) or A20 cells (G) in both flanks, and treated 10 or 12 days later with OVH or vehicle. (B) Tumor growth of virus-injected (right flank) Hepa1-6 tumors. (C) Growth of distant (left flank) Hepa1-6 tumors. (E) Tumor growth of virus-injected Renca tumors. (F) Tumor growth of distant Renca tumors. (H) Tumor growth of virus-injected A20 tumors. (I) Tumor growth of distant A20 tumors. (J-M) The subset of cured long-term survivors from the Hepa1-6 model (K), Renca model (l), and A20 model (M) groups, together with naïve mice, were re-challenged with 1 × 107 of the corresponding tumor cells. The percentage of long-term survivors that rejected the secondary challenge was calculated. (N) A subset of cured long-term survivors from the Renca model and A20 model groups were re-challenged with 1 × 107 Renca tumor cells. The percentage of long-term survivors that rejected secondary challenge was calculated. (O) Western blot analysis of Hepa1-6 cell lysate and Renca cell lysate with serum from OVH-treated C57BL/6 mice bearing Hepa1-6 tumors or naïve mice. (P) Western blot analysis of Hepa1-6 cell lysate and Renca cell lysate with serum from OVH-treated BALB/c mice bearing Renca tumors or naïve mice. The number of mice (N) used in the experiments is shown in individual figures. All values are presented as the mean ± SEM. **P < .01, ***P < .001, ****P < .0001 by repeated measure ANOVA for B, C, E, F, H, I or unpaired two-tailed Student’s t tests for K-N.
OVH induces immunogenic cell apoptosis
Understanding the immunogenicity of dying or dead tumor cells induced by OVs is important when considering the potency of these viruses to sensitize host innate and humoral immune responses. FACS analysis showed significant upregulation of annexin V staining at 48 h after OVH infection in four tested tumor cell lines (Figure 5A). Our study revealed that early and late apoptosis were involved in OVH-induced oncolysis in four different tumor cell lines. We next tested the downstream caspase cascades in OVH-treated tumor cells by detecting the activity of the apoptotic initiator caspase-8, apoptotic executioner caspase-3 and apoptotic marker PARP. The cleaved forms of all these markers were increased after OVH infection (Figure 5B), indicating that the OVH-induced oncolysis can lead to evident apoptotic cell death. To determine the immunogenic profile of OVH-infected tumor cell lines, tumor cells were infected with OVH for 48 h at an MOI of 1 PFU/cell. The infected cells and supernatants were harvested and analyzed for expression of the ICD associated DAMPs. The levels of secreted ATP, HMBG1 and surface expression of calreticulin were evidently upregulated in OVH-infected tumor cells (Figure 5C–E). Moreover, we employed IHC staining to analyze the profile of OVH-induced oncolysis in tumors. In xenograft nude mouse model, significant upregulation of apoptotic signals (cleaved caspase3) and a reduction in proliferation signals (Ki67) were observed in OVH-injected HeLa tumors and OVH-injected CEN2 tumors (Figure 5F and G), suggesting OVH treatment can induce robust tumor cell apoptosis in situ. In the Hepa1-6 mouse liver cancer model, OVH therapy induced robust tumor cell apoptosis not only in injected tumors but also in distant tumors (Figure 5H). These data suggested that OVH had greater potential to induce immunogenic cell-mediated apoptosis by releasing DAMPs both in vitro and in vivo, leading to systemic anti-tumor immune responses.
Figure 5.
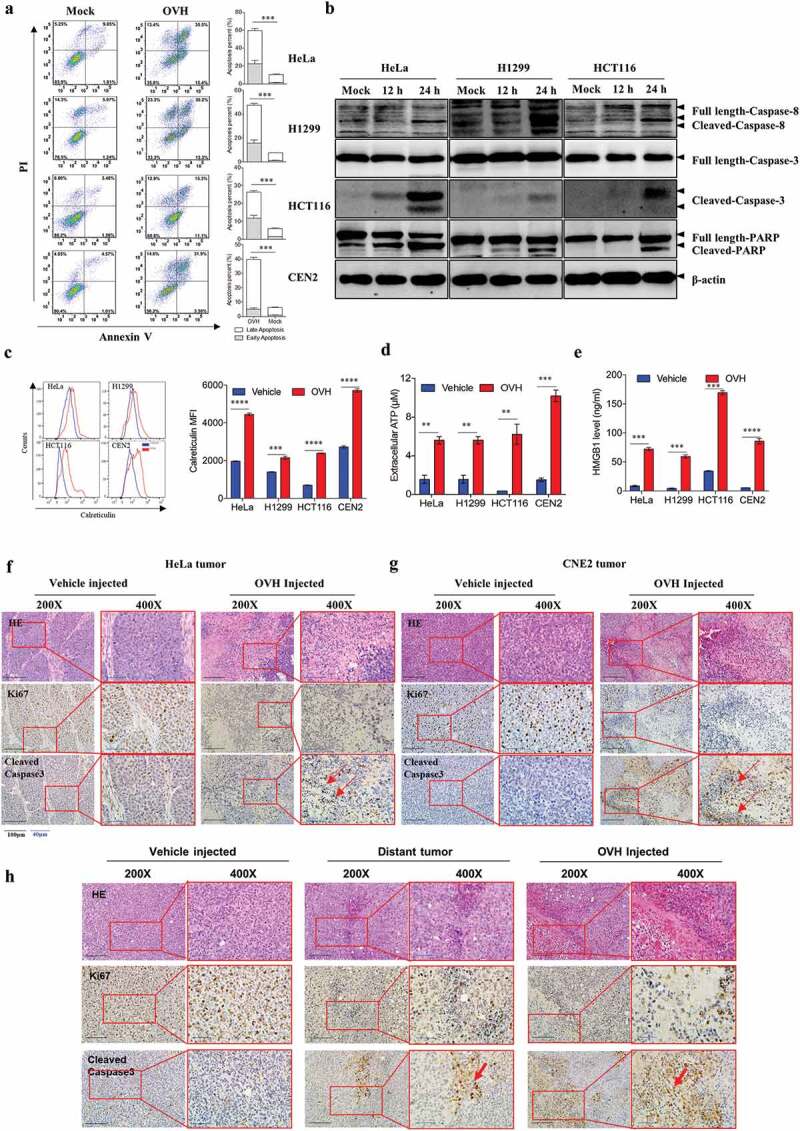
OVH triggers immunogenic cell apoptosis in susceptible tumor cells. (A) Determination of the levels of apoptosis in HeLa, H1299, HCT116 and CEN2 cell lines left uninfected or infected with OVH at an MOI of 0.5 PFU/cell for 48 h using annexin-V/PI-labeled flow cytometry. Graphs represent pooled data from three independent experiments. (B) Western blot analysis of the expression of full-length and cleaved caspase-8, caspase-3 and PARP in infected HeLa, H1299, HCT116 cell lines at 48 h after virus infection. (C-E) HeLa, H1299, HCT116 and CEN2 cell lines were either left uninfected or infected with OVH at an MOI of 1 PFU/cell. Cells and supernatants were harvested at 48 h post-infection. The surface expression of calreticulin was determined by flow cytometric analysis of the cells (C), and the levels of released ATP (D) and HMGB1 (E) were determined by analysis of the supernatants. (F-G) H&E and IHC analysis of an apoptotic marker (cleaved-caspase3) and proliferation marker (Ki67) in human xenograft HeLa tumors (F) and CEN2 tumors (G) at 24 h after receiving intratumoral injection of two doses of OVH (1 × 107 PFU per dose) or vehicle. (H) Mice were s.c. inoculated with Hepa1-6 cells in both flanks and received intratumoral injection of a single dose of OVH on the right flank (1 × 107 PFU per dose) or vehicle. H&E and IHC analysis of an apoptotic marker (cleaved-caspase3) and proliferation marker (Ki67) in both flanks of Hepa1-6 xenografts (virus-injected tumor and distant tumor) at 5 days after treatment. H&E and IHC images were obtained using a microscope (OLYMPLUS), with indicated objectives. Values are the means of three independent experiments, data are shown as means ± SEM. **P < .01, ***P < .001, ****P < .0001, ns, not significant by unpaired two-tailed Student’s t tests.
Tumor regression by oncolysis-induced endogenous antibody responses
To understand the cellular mechanisms underlying the observed durable and protective antitumor effect of OVH therapy. First, we analyzed the endogenous antibody responses induced by OVH therapy in all three syngeneic tumor models (Figure 6A). To evaluate the functions of these endogenous antibodies, we developed an approach to deplete all the antibodies from the serum of OVH-cured mice and then carried out the serum transfusion experiments (Figure 6B). Using this strategy, we found that OVH-elicited antibodies had therapeutic potential, as only untreated sera obtained from mice successfully cured of Hepa1-6 tumors by OVH treatment can reject the Hepa1-6 tumor growth of naïve recipients (Figure 6C, E and F). The results from three syngeneic tumor models unanimously demonstrated that OVH therapy-induced antibodies were functional and tumor-specific (Figure 6C–N). We also observed no significant difference in body weight among the treated groups during the course of the study (Figure 6D, H and L). Moreover, administration of OVH therapy to Hepa1-6 tumor-bearing JHT mice (300 mm3), which fail to produce functional B cells as they lack the gene for the heavy chain joining region (Fig. S4), led to a significantly reduced frequency of durable cures as that observed in wild-type B6 mice. OVH therapy induced robust tumor eradication and durable cures in 50% of B6 mice implanted with Hepa1-6 tumors, but 25% of the JHT mice implanted with Hepa1-6 tumors (Figure 7A–C). In JHT mice, OVH treatment induced direct virus-induced oncolysis within injected tumors but not in distant tumors (Fig. S5) and led to better tumor rejection in virus-injected tumors (Figure 7E–G), resulting in a decreased ratio of tumor-free mice. Despite the prominent tumor rejection observed in the injected tumors, complete distant tumor rejection with long-term survival was not observed in treated JHT mice, which suggests that an endogenous antibody response dependent on B cells was required for maximal antitumor efficacy of OVH therapy.
Figure 6.
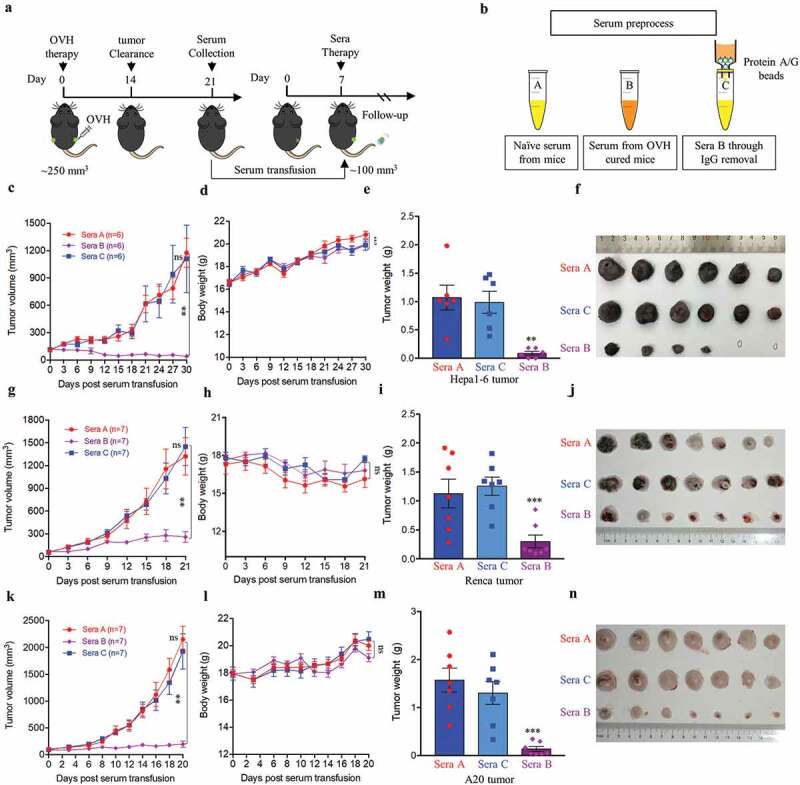
OVH-induced oncolysis elicits efficacious humoral response against tumors. (A) Timeline of the serum transfusion experiment. Arrows indicate corresponding time points. (B) Overview of serum used for antibody depletion experiments. Sera A or B were collected 21 days after vehicle or OVH treatment, separately. Sera C was generated by filtering sera B through an IgG removal protein A/G beads. (C-F) Sera A, B and C (0.3 ml) from Hepa1-6 model mice were transferred into naïve recipients pre-implanted with Hepa1-6 tumors. Tumor growth (C) and body weight (D) of the treated mice were monitored over a 30-day period. (G-J) Sera A, B and C (0.3 ml) from Renca model mice were transferred into naïve recipients pre-implanted with Renca tumors. Tumor growth (G) and body weight (H) of the treated mice were monitored over a 21-day period. (K-N) Sera A, B and C (0.3 ml) from A20 model mice were transferred into naïve recipients pre-implanted with A20 tumors. Tumor growth (K), and body weight (L) of the treated mice were monitored over a 20-day period. Tumors from each tumor model were dissected and weighed at experiment termination (E, F, I, J, M, N). All values are presented as the mean ± SEM. **P < .01, ns, not significant by repeated measure ANOVA (C, G, K) or by one-way ANOVA (E, I, M).
Figure 7.
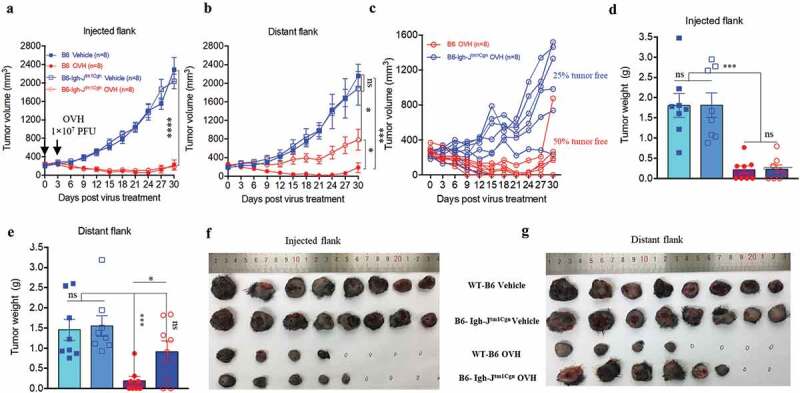
B cells were required for maximal antitumor efficacy of oncolytic virotherapy. (A, B) Evaluation of the antitumor effects of OVH therapy was carried out in wild-type C57BL/6 (B6) and JHT mice. Mice were s.c. inoculated with Hepa1-6 cells in both flanks and treated 10 days later and every 3 days thereafter until the total two dosages were finished, with OVH or vehicle. Black arrow indicates virus injection. Tumor growth of virus-injected (A) and distant (B) Hepa1-6 tumors. (C) Tumor growth of distant Hepa1-6 tumors in OVH-treated B6 and JHT mice. (D-G) The tumors in both flanks from each group were dissected and weighed at experiment termination (F, G). Tumor weight of virus-injected tumors (D) and distant tumors (E). All values are presented as the mean ± SEM. *P < .05, ***P < .001, ****P < .0001, ns, not significant by repeated measure ANOVA (A, B) or by one-way ANOVA (D, E).
Oncolytic OVH can elicit endogenous tumor antigen-targeting therapeutic antibodies
Because an in situ-elicited humoral response was observed in OVH-treated mice, we next assessed whether OVH-induced antibodies were tumor-specific. We attempted to isolate the monoclonal antibodies (mAbs) from OVH-cured mice and test their in vivo functions in the absence of OVH (Figure 8A). Sixteen mAbs, which specifically reacted with mouse liver tumor cells but not with mouse primary liver cells, were isolated and characterized (Figure 8B and Fig. S5). Two mAbs (3F11 and 2G2) with greater reactivity were used for functional studies. 3F11 and 2G2 specifically recognized the tumor cells and reacted with an unknown antigen on the surface of Hepa1-6 cells (Figure 8C). In Hepa1-6 mouse liver tumor model, mAb therapy induced robust tumor eradication and durable cures in 100% of the mice when tumor reached 50 mm3 (Figure 8D) and no obvious toxicity was observed in the mAb-treated group during the treatment (Figure 8E). Due to higher reactivity of 3F11 against the same antigen of Hepa1-6 cells compared to 2G2 as well as other antibodies, 3F11 treatment showed best tumor inhibition when tumor reached 100 mm3 (Fig. S7). Interestingly, the binding activity of 2G2 to Hepa1-6 cells was reduced by the addition of 3F11 F (ab’)2, suggesting that 3F11 and 2G2 may recognize the same antigen epitope (Figure 8F). We then assessed the cellular mechanisms underlying the observed antitumor effect of mAb therapy. Antibody depletion experiments showed that macrophages and natural killer (NK) cells were critical to mAb-mediated tumor regression (Figure 8G–H). Tumor growth was also significantly inhibited by mAb therapy in BALB/c nu/nu mice implanted with Hepa1-6 tumors (Figure 8I). Taken together, the results of these experiments indicate that OVH therapy elicited therapeutic antibody responses to tumor antigens that were not directly recognized by the host immunity.
Figure 8.
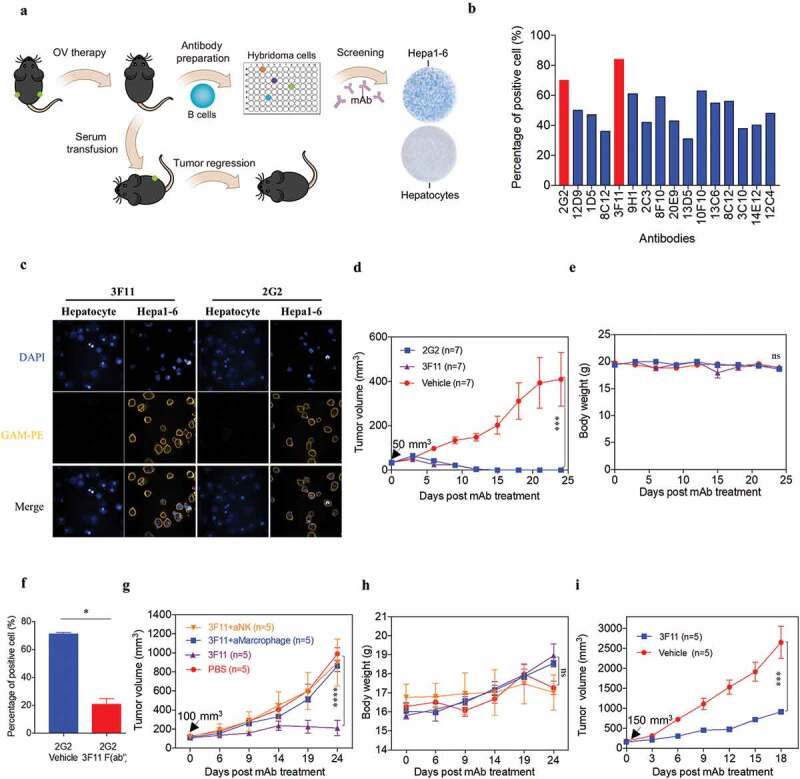
OVH therapy induces endogenous tumor-specific antibody responses. (A) Schematic diagram of Hepa1-6-specific mAb generation from OVH-cured Hepa1-6 mice. (B) Sixteen mAbs were selected from C57BL/6 spleen×sp2/0 hybridomas, which specifically recognized Hepa1-6 tumor cells and did not cross-react with mouse primary hepatocytes. (C) Hepa1-6 cells and mouse primary hepatocytes were stained with each mAb (3F11 or 2G2) and PE conjugated anti-mouse IgG (Fc specific) antibody, then analyzed by flow cytometry. Representative images of cells stained with each mAb were shown. (D, E) Evaluation of the antitumor effects of 3F11 and 2G2. Tumor growth (D) and body weight (E) were monitored over a 24-day period. (F) Competitive binding assay between 3F11 and 2G2 against Hepa1-6 cells. Hepa1-6 cells were incubated with 2G2 and 3F11 F (ab’)2, then stained with PE conjugated anti-mouse IgG (Fc specific) antibody and analyzed by flow cytometry. (G) Tumor growth was monitored after depleting antibodies specific for the indicated surface markers (n = 5 mice per group). (H) Body weight was monitored over a 24-day period. (I) Evaluation of the antitumor effects of 3F11. Tumor growth was monitored over an 18-day period. Black arrow indicates initial tumor volume for treatment. All values are presented as the mean ± SEM. *P < .05, ***P < .001, ****P < .0001, ns, not significant by repeated measure ANOVA (D, E, G, H, I) or by unpaired two-tailed Student’s t tests (F).
Discussion
Oncolytic virotherapy has been classified as a target therapy and a novel type of immunotherapy. The selective destruction of tumors by OVs leads to systemic antitumor immunity, such as innate anti-viral immune responses and antigen-specific CTL responses.33 Advances in oncolytic virotherapy have shown exciting promise for the treatment of solid cancers and great potential for boosting tumor immunity to control distant metastases, making OVs an attractive immunotherapy for treating advanced cancer.33–35 In this study, we introduced OVH as an improved immunotherapeutic OV with strong safety records, good oncolytic properties and cancer vaccine boosting effects.
The balance between tumor selectivity and cell-killing ability is the core determinant for the therapeutic potent of OVs.36,37 To achieve good tumor selectivity and oncolytic properties, an ICP0 and ICP34.5 null oncolytic HSV-1 virus regulated by the human hTERT promoter (OVH) was designed and developed. OVH efficiently and specifically lysed tumor cell lines in vitro and significantly reduced tumor size and increased survival of xenograft mice implanted with multiple human tumor cells in vivo. There are two properties of OVH that are important because they are responsible for its high tumor selectivity: (i) HSV-1 ICP0 and ICP34.5 double deletions make HSV-1 an attenuated virus, which allows it to efficiently kill tumor cells without harming healthy non-cancerous cells and possess remarkably reduced neurovirulence and systemic toxicity;17,31 and (ii) the introduction of the hTERT promoter to regulate ICP27 protein expression further attenuates OVH activity in healthy non-cancerous cells and normal tissues, which have very low hTERT promoter activity.38–40 It is worth noting that the neurovirulence of OVH is significantly reduced by over 4,000-fold compared to that of KOS, and by over 4,00-fold compared to that of ICP34.5-null virus or ICP0-null virus,41 which indicates OVH as an improved, least toxic agent for long-term tumor control.
OVs appear to be effective antitumor agents upon intratumoral administration, most studies focus on the cellular arm of antitumor immunity and demonstrated virus-mediated tumor inhibition. An increasing body of evidence suggests that the cytotoxic T cells may critically influence the outcome of oncolytic virotherapy.42–47 Several OVs have been demonstrated to increase the number of monocyte-derived DCs and thereafter to increase the activity of cytotoxic CD8+ lymphocytes by promoting antigen presentation.48,49 Additionally, NK cells play a dual role in oncolytic virotherapy, driving both antitumor and anti-OV responses.50 On one hand, NK cells may adversely affect oncolytic virotherapy by preventing the intratumoral spread of OVs and thereby limiting the extent of virus-mediated oncolysis.51,52 On the other hand, NK cells have also been found to support OV-mediated tumor elimination by enhancing NK-cell-dependent killing of tumor cells.53,54 However, very few studies have demonstrated virus-mediated immune clearance of established tumors.55 OVH had a significant antitumor effect in immunocompromised mice and tumor clearance propensity in syngeneic mice bearing large established tumors. To date, complete eradication of immunogenic tumors in immunocompetent mouse cancer models bearing large tumors has only been reliably achieved by adoptively transferred T cells, and many immunotherapy and cancer vaccine studies focus on CD8+ T cell responses to tumors, but the important roles of humoral immune effector cells in this process have not been described.1,55 It has remained unclear whether the tumor antigen-specific antibody response can be stimulated by OVs to overcome a diverse range of advanced tumors in mice.56 Here, for the first time, we demonstrate the capacity of endogenous tumor antigen-specific antibodies elicited by OVH immunotherapy can eliminate large established tumors in several syngeneic tumor models, including poorly immunogenic tumors (LL/2 and TRAMP-C2). Endogenous antibody-mediated humoral responses were primed by ICD and oncolysis-released antigens, and OVH treatment induced the creation of antibodies that exhibited protective functions when transferred to naïve recipients. In view of the importance of the endogenous antibody response, we supposed that B cells may play a major role in producing protective antibodies against tumors. We found the systemic antitumor activity (distant flank tumor inhibition) mediated by virotherapy was dampened in B cell-deficient (JHT) mice, which suggests B cells were required for the maximal efficacy of virotherapy. Furthermore, tumor antigen-specific therapeutic mAbs selected from tumor-free syngeneic mice, which were boosted by OV-mediated oncolysis and specifically targeted tumor cells, demonstrated promising antitumor activity. Our data obtained from depletion experiments suggest antitumor activity of those endogenous antibodies (such as 3F11) may be dependent on macrophages and NK cells by direct antibody-dependent phagocytosis and/or cell cytotoxicity against tumors. The importance of this mechanism of action versus the viral oncolytic effect may likely vary depending on the tumor types, but this novel type of oncolytic cancer vaccine provides a powerful strategy to identify effective B cell target antigens that are shared by tumors of a given type.
Although oncolytic virotherapy has vast potential, there are hurdles as to what it can achieve as a standalone therapy because of the complexity of the tumor microenvironment (TME), the tumor cell susceptibility to OV infection and the limits of the drug delivery route.8–10 As such, it is worth noting that enormous efforts are now being made to find rational combination therapies that can further enhance the antitumor efficacy of OVs. One of the good examples is the combination of T-VEC with anti-PD1 checkpoint blockade therapy, which has achieved great success in treating advanced melanoma.6 Checkpoint blockade therapies, aiming to overcome the immunosuppressive TME and enhance antitumor immune responses, have been considered as the most successful approaches to fight cancer by far, but only a small group of patients with high tumor mutation burdens benefit from this treatment. These findings call for the development of combinatorial strategies that could make a larger patient population benefit from therapy. The importance of ICD for initiating an antitumor response has previously been demonstrated.11,12,57 PAMPs, released ATP, HMGB1 and ecto-calreticulin drive adjuvanticity and effective antigen-presenting cell (APC) engagement in TME. Our studies identify OVH is an ideal vector to initiate TAAs-specific immune responses induced by ICD. As expected, the therapeutic efficacy of OVH was better in highly immunogenic tumor model than in poorly immunogenic tumor model. Our results also shed the light on the importance of the immunogenicity of tumor cells in oncolytic virotherapy.
Several questions remain and should be addressed in future studies. First, the mechanisms by which OVH induces endogenous tumor-antigen-specific humoral immunity should be elucidated in the future. In addition, analyzes of the binding targets for these selected therapeutic mAbs may result in the identification of antibody therapeutics, novel targets for therapeutic mAbs and other active immunotherapeutics. Next, to prevent the early shutdown of oncolytic virotherapy–induced antitumor immunity, immune signature of OV inflamed TME must be determined in future studies. Finally, the optimal combination strategy for virotherapy to overcome the resistance to immunotherapy, especially for non-inflamed cold tumor, should be further investigated.
In summary, we have demonstrated that a novel oncolytic and immunotherapeutic herpes simplex virus can lead to robust humoral immune responses in the setting of various tumors, in addition to the direct cell-killing effect of OVH. By combining in situ antibody immune responses with CD8+ T cell-mediated immune responses, OVH may have the potential to be a powerful cancer therapy and combination partner for other immunotherapies. Insights from this novel OV also provide a provoking thought regarding the expansion of the power of OVs as immunotherapeutic cancer vaccines that can generate specific and efficacious antitumor immune responses by eliciting endogenous tumor antigen-targeting therapeutic antibodies in situ to disrupt tumors,58,59 resulting in an efficacious and tumor-specific therapeutic effect.
Funding Statement
This work was supported by grant [2018ZX10301404-001-002] from the National Science and Technology Major Project of the Ministry of Science and Technology of the People's Republic of China (MOST) and [grant 81571990 and grant 31730029] from the National Natural Science Foundation of China.
Acknowledgments
We thank H.L. Chen (The University of Hong Kong) for kindly providing the CEN1 and CEN2 cell lines.
Disclosure of Potential Conflict of Interest
No potential conflicts of interest were disclosed.
Supplementary Material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A.. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell. 2017;168:707–18. doi: 10.1016/j.cell.2017.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Khalil DN, Smith EL, Brentjens RJ, Wolchok JD. The future of cancer treatment: immunomodulation, cars and combination immunotherapy. Nat Rev Clin Oncol. 2016;13:273–290. doi: 10.1038/nrclinonc.2016.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lichty BD, Breitbach CJ, Stojdl DF, Bell JC. Going viral with cancer immunotherapy. Nat Rev Cancer. 2014;14:559–567. doi: 10.1038/nrc3770. [DOI] [PubMed] [Google Scholar]
- 4.Russell SJ, Peng KW, Bell JC. Oncolytic virotherapy. Nat Biotechnol. 2012;30:658–670. doi: 10.1038/nbt.2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Andtbacka RH, Kaufman HL, Collichio F, Amatruda T, Senzer N, Chesney J, Delman KA, Spitler LE, Puzanov I, Agarwala SS, et al. Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J Clin Oncol. 2015;33:2780–2788. doi: 10.1200/JCO.2014.58.3377. [DOI] [PubMed] [Google Scholar]
- 6.Ribas A, Dummer R, Puzanov I, VanderWalde A, Andtbacka RHI, Michielin O, Olszanski AJ, Malvehy J, Cebon J, Fernandez E, et al. Oncolytic virotherapy promotes intratumoral T cell infiltration and improves anti-PD-1 immunotherapy. Cell. 2017;170:1109–1119 e1110. doi: 10.1016/j.cell.2017.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Russell SJ, Barber GN. Oncolytic viruses as antigen-agnostic cancer vaccines. Cancer Cell. 2018;33:599–605. doi: 10.1016/j.ccell.2018.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fountzilas C, Patel S, Mahalingam D. Oncolytic virotherapy, updates and future directions. Oncotarget. 2017;8:102617–102639. doi: 10.18632/oncotarget.18309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ferguson MS, Lemoine NR, Wang Y. Systemic delivery of oncolytic viruses: hopes and hurdles. Adv Virol. 2012;2012:805629. doi: 10.1155/2012/805629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lawler SE, Speranza MC, Cho CF, Chiocca EA. Oncolytic viruses in cancer treatment: a review. JAMA Oncol. 2017;3:841–849. doi: 10.1001/jamaoncol.2016.2064. [DOI] [PubMed] [Google Scholar]
- 11.Kepp O, Senovilla L, Vitale I, Vacchelli E, Adjemian S, Agostinis P, Apetoh L, Aranda F, Barnaba V, Bloy N, et al. Consensus guidelines for the detection of immunogenic cell death. Oncoimmunology. 2014;3:e955691. doi: 10.4161/21624011.2014.955691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bartlett DL, Liu Z, Sathaiah M, Ravindranathan R, Guo Z, He Y, Guo ZS. Oncolytic viruses as therapeutic cancer vaccines. Mol Cancer. 2013;12:103. doi: 10.1186/1476-4598-12-103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yin J, Markert JM, Leavenworth JW. Modulation of the intratumoral immune landscape by oncolytic herpes simplex virus virotherapy. Front Oncol. 2017;7:136. doi: 10.3389/fonc.2017.00136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Robert C. Checkpoint blockade plus oncolytic virus: a hot therapeutic cancer strategy. Trends Mol Med. 2017;23:983–985. doi: 10.1016/j.molmed.2017.09.008. [DOI] [PubMed] [Google Scholar]
- 15.Varghese S, Rabkin SD. Oncolytic herpes simplex virus vectors for cancer virotherapy. Cancer Gene Ther. 2002;9:967–978. doi: 10.1038/sj.cgt.7700537. [DOI] [PubMed] [Google Scholar]
- 16.Walsh D, Mohr I. Phosphorylation of eIF4E by Mnk-1 enhances HSV-1 translation and replication in quiescent cells. Genes Dev. 2004;18:660–672. doi: 10.1101/gad.1185304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu BL, Robinson M, Han ZQ, Branston RH, English C, Reay P, McGrath Y, Thomas SK, Thornton M, Bullock P, et al. ICP34.5 deleted herpes simplex virus with enhanced oncolytic, immune stimulating, and anti-tumour properties. Gene Ther. 2003;10:292–303. doi: 10.1038/sj.gt.3301885. [DOI] [PubMed] [Google Scholar]
- 18.MacLean AR, ul-Fareed M, Robertson L, Harland J, Brown SM. Herpes simplex virus type 1 deletion variants 1714 and 1716 pinpoint neurovirulence-related sequences in glasgow strain 17+ between immediate early gene 1 and the ‘a’ sequence. J Gen Virol. 1991;72(Pt 3):631–639. doi: 10.1099/0022-1317-72-3-631. [DOI] [PubMed] [Google Scholar]
- 19.Kim NW, Piatyszek MA, Prowse KR, Harley CB, West MD, Ho PL, Coviello G, Wright W, Weinrich S, Shay J, et al. Specific association of human telomerase activity with immortal cells and cancer. Science. 1994;266:2011–2015. doi: 10.1126/science.7605428. [DOI] [PubMed] [Google Scholar]
- 20.Wirth T, Zender L, Schulte B, Mundt B, Plentz R, Rudolph KL, Manns M, Kubicka S, Kühnel F, et al. A telomerase-dependent conditionally replicating adenovirus for selective treatment of cancer. Cancer Res. 2003;63:3181–3188. [PubMed] [Google Scholar]
- 21.Huang TG, Savontaus MJ, Shinozaki K, Sauter BV, Woo SL. Telomerase-dependent oncolytic adenovirus for cancer treatment. Gene Ther. 2003;10:1241–1247. doi: 10.1038/sj.gt.3301987. [DOI] [PubMed] [Google Scholar]
- 22.Kawashima T, Kagawa S, Kobayashi N, Shirakiya Y, Umeoka T, Teraishi F, Taki M, Kyo S, Tanaka N, Fujiwara T, et al. Telomerase-specific replication-selective virotherapy for human cancer. Clin Cancer Res. 2004;10:285–292. doi: 10.1158/1078-0432.CCR-1075-3. [DOI] [PubMed] [Google Scholar]
- 23.Huang Q, Zhang X, Wang H, Yan B, Kirkpatrick J, Dewhrist MW, Li CY. A novel conditionally replicative adenovirus vector targeting telomerase-positive tumor cells. Clin Cancer Res. 2004;10:1439–1445. doi: 10.1158/1078-0432.CCR-03-0122. [DOI] [PubMed] [Google Scholar]
- 24.Li WC, Ralphs KL, Tosh D. Isolation and culture of adult mouse hepatocytes. Methods Mol Biol. 2010;633:185–196. [DOI] [PubMed] [Google Scholar]
- 25.Zhao W, Wang L, Han H, Jin K, Lin N, Guo T, Chen Y, Cheng H, Lu F, Fang W, et al. 1B50-1, a mAb raised against recurrent tumor cells, targets liver tumor-initiating cells by binding to the calcium channel alpha2delta1 subunit. Cancer Cell. 2013;23:541–556. doi: 10.1016/j.ccr.2013.02.025. [DOI] [PubMed] [Google Scholar]
- 26.Lin C, Li H, Hao M, Xiong D, Luo Y, Huang C, Yuan Q, Zhang J, Xia N, et al. Increasing the efficiency of CRISPR/Cas9-mediated precise genome editing of HSV-1 virus in human cells. Sci Rep. 2016;6:34531. doi: 10.1038/srep34531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Takakura M, Kyo S, Kanaya T, Hirano H, Takeda J, Yutsudo M, Inoue M. Cloning of human telomerase catalytic subunit (hTERT) gene promoter and identification of proximal core promoter sequences essential for transcriptional activation in immortalized and cancer cells. Cancer Res. 1999;59:551–557. [PubMed] [Google Scholar]
- 28.Maclean AR. Preparation of HSV-DNA and production of infectious virus. Methods Mol Med. 1998;10:19–25. doi: 10.1385/0-89603-347-3:19. [DOI] [PubMed] [Google Scholar]
- 29.Lynas C, Hill TJ, Maitland NJ, Love S. Latent infection with the MS strain of herpes simplex virus type 2 in the mouse following intracerebral inoculation. J Neurol Sci. 1993;120:107–114. doi: 10.1016/0022-510X(93)90033-U. [DOI] [PubMed] [Google Scholar]
- 30.Huang CH, Yuan Q, Chen PJ, Zhang YL, Chen CR, Zheng QB, Yeh S-H, Yu H, Xue Y, Chen Y-X, et al. Influence of mutations in hepatitis B virus surface protein on viral antigenicity and phenotype in occult HBV strains from blood donors. J Hepatol. 2012;57:720–729. doi: 10.1016/j.jhep.2012.05.009. [DOI] [PubMed] [Google Scholar]
- 31.Hummel JL, Safroneeva E, Mossman KL. The role of ICP0-Null HSV-1 and interferon signaling defects in the effective treatment of breast adenocarcinoma. Mol Ther. 2005;12:1101–1110. doi: 10.1016/j.ymthe.2005.07.533. [DOI] [PubMed] [Google Scholar]
- 32.Holay N, Kim Y, Lee P, Gujar S. Sharpening the edge for precision cancer immunotherapy: targeting tumor antigens through oncolytic vaccines. Front Immunol. 2017;8:800. doi: 10.3389/fimmu.2017.00800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Romero D. Immunotherapy: oncolytic viruses prime antitumour immunity. Nat Rev Clin Oncol. 2018;15:135. doi: 10.1038/nrclinonc.2018.15. [DOI] [PubMed] [Google Scholar]
- 34.Samson A, Scott KJ, Taggart D, West EJ, Wilson E, Nuovo GJ, Thomson S, Corns R, Mathew RK, Fuller MJ, et al. Intravenous delivery of oncolytic reovirus to brain tumor patients immunologically primes for subsequent checkpoint blockade. Sci Transl Med. 2018;10:eaam7577. doi: 10.1126/scitranslmed.aam7577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Desjardins A, Gromeier M, Herndon JE 2nd, Beaubier N, Bolognesi DP, Friedman AH, Friedman HS, McSherry F, Muscat AM, Nair S, et al. Recurrent Glioblastoma treated with recombinant poliovirus. N Engl J Med. 2018;379:150–161. doi: 10.1056/NEJMoa1716435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Russell SJ, Peng KW. Oncolytic virotherapy: a contest between apples and oranges. Mol Ther. 2017;25:1107–1116. doi: 10.1016/j.ymthe.2017.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Le Boeuf F, Batenchuk C, Vaha-Koskela M, Breton S, Roy D, Lemay C, Cox J, Abdelbary H, Falls T, Waghray G, et al. Model-based rational design of an oncolytic virus with improved therapeutic potential. Nat Commun. 2013;4:1974. doi: 10.1038/ncomms2974. [DOI] [PubMed] [Google Scholar]
- 38.Kyo S, Inoue M. Complex regulatory mechanisms of telomerase activity in normal and cancer cells: how can we apply them for cancer therapy? Oncogene. 2002;21:688–697. doi: 10.1038/sj.onc.1205163. [DOI] [PubMed] [Google Scholar]
- 39.Huang FW, Hodis E, Xu MJ, Kryukov GV, Chin L, Garraway LA. Highly recurrent TERT promoter mutations in human melanoma. Science. 2013;339:957–959. doi: 10.1126/science.1229259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang W, Ge K, Zhao Q, Zhuang X, Deng Z, Liu L, Li J, Zhang Y, Dong Y, Zhang Y, et al. A novel oHSV-1 targeting telomerase reverse transcriptase-positive cancer cells via tumor-specific promoters regulating the expression of ICP4. Oncotarget. 2015;6:20345–20355. doi: 10.18632/oncotarget.3884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sundaresan P, Hunter WD, Martuza RL, Rabkin SD. Attenuated, replication-competent herpes simplex virus type 1 mutant G207: safety evaluation in mice. J Virol. 2000;74:3832–3841. doi: 10.1128/JVI.74.8.3832-3841.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kowalsky SJ, Liu Z, Feist M, Berkey SE, Ma C, Ravindranathan R, Dai E, Roy EJ, Guo ZS, Bartlett DL, et al. Superagonist IL-15-armed oncolytic virus elicits potent antitumor immunity and therapy that are enhanced with PD-1 blockade. Mol Ther. 2018;26:2476–2486. doi: 10.1016/j.ymthe.2018.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zamarin D, Ricca JM, Sadekova S, Oseledchyk A, Yu Y, Blumenschein WM, Wong J, Gigoux M, Merghoub T, Wolchok JD, et al. PD-L1 in tumor microenvironment mediates resistance to oncolytic immunotherapy. J Clin Invest. 2018;128:1413–1428. doi: 10.1172/JCI98047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zamarin D, Holmgaard RB, Subudhi SK, Park JS, Mansour M, Palese P, Merghoub T, Wolchok JD, Allison JP, et al. Localized oncolytic virotherapy overcomes systemic tumor resistance to immune checkpoint blockade immunotherapy. Sci Transl Med. 2014;6:226ra232. doi: 10.1126/scitranslmed.3008095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Freedman JD, Duffy MR, Lei-Rossmann J, Muntzer A, Scott EM, Hagel J, Campo L, Bryant RJ, Verrill C, Lambert A, et al. An oncolytic virus expressing a T-cell engager simultaneously targets cancer and immunosuppressive stromal cells. Cancer Res. 2018;78:6852–6865. doi: 10.1158/0008-5472.CAN-18-1750. [DOI] [PubMed] [Google Scholar]
- 46.Li X, Wang P, Li H, Du X, Liu M, Huang Q, Wang Y, Wang S. The efficacy of oncolytic adenovirus is mediated by T-cell responses against virus and tumor in syrian hamster model. Clin Cancer Res. 2017;23:239–249. doi: 10.1158/1078-0432.CCR-16-0477. [DOI] [PubMed] [Google Scholar]
- 47.Zamarin D, Holmgaard RB, Ricca J, Plitt T, Palese P, Sharma P, Merghoub T, JD Wolchok, Allison JP. Intratumoral modulation of the inducible co-stimulator ICOS by recombinant oncolytic virus promotes systemic anti-tumour immunity. Nat Commun. 2017;8:14340. doi: 10.1038/ncomms14340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Marelli G, Howells A, Lemoine NR, Wang Y. Oncolytic viral therapy and the immune system: a double-edged sword against cancer. Front Immunol. 2018;9:866. doi: 10.3389/fimmu.2018.00866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Errington F, Steele L, Prestwich R, Harrington KJ, Pandha HS, Vidal L, de Bono J, Selby P, Coffey M, Vile R, et al. Reovirus activates human dendritic cells to promote innate antitumor immunity. J Immunol. 2008;180:6018–6026. doi: 10.4049/jimmunol.180.9.6018. [DOI] [PubMed] [Google Scholar]
- 50.Bhat R, Rommelaere J. Emerging role of natural killer cells in oncolytic virotherapy. Immunotargets Ther. 2015;4:65–77. doi: 10.2147/ITT.S55549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Alvarez-Breckenridge CA, Yu J, Price R, Wojton J, Pradarelli J, Mao H, Wei M, Wang Y, He S, Hardcastle J, et al. NK cells impede glioblastoma virotherapy through NKp30 and NKp46 natural cytotoxicity receptors. Nat Med. 2012;18:1827–1834. doi: 10.1038/nm.3013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Altomonte J, Wu L, Chen L, Meseck M, Ebert O, Garcia-Sastre A, Fallon J, Woo SL. Exponential enhancement of oncolytic vesicular stomatitis virus potency by vector-mediated suppression of inflammatory responses in vivo. Mol Ther. 2008;16:146–153. doi: 10.1038/sj.mt.6300343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Miller CG, Fraser NW. Requirement of an integrated immune response for successful neuroattenuated HSV-1 therapy in an intracranial metastatic melanoma model. Mol Ther. 2003;7:741–747. doi: 10.1016/S1525-0016(03)00120-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bhat R, Dempe S, Dinsart C, Rommelaere J. Enhancement of NK cell antitumor responses using an oncolytic parvovirus. Int J Cancer. 2011;128:908–919. doi: 10.1002/ijc.25415. [DOI] [PubMed] [Google Scholar]
- 55.Moynihan KD, Opel CF, Szeto GL, Tzeng A, Zhu EF, Engreitz JM, Williams RT, Rakhra K, Zhang MH, Rothschilds AM, et al. Eradication of large established tumors in mice by combination immunotherapy that engages innate and adaptive immune responses. Nat Med. 2016;22:1402–1410. doi: 10.1038/nm.4200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kim MK, Breitbach CJ, Moon A, Heo J, Lee YK, Cho M, Lee JW, Kim S-G, Kang DH, Bell JC, et al. Oncolytic and immunotherapeutic vaccinia induces antibody-mediated complement-dependent cancer cell lysis in humans. Sci Transl Med. 2013;5:185ra163. doi: 10.1126/scitranslmed.3005361. [DOI] [PubMed] [Google Scholar]
- 57.Sharma P, Allison JP. Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell. 2015;161:205–214. doi: 10.1016/j.cell.2015.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yarchoan M, Johnson BA 3rd, Lutz ER, Laheru DA, Jaffee EM. Targeting neoantigens to augment antitumour immunity. Nat Rev Cancer. 2017;17:209–222. doi: 10.1038/nrc.2016.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.van der Burg SH, Arens R, Ossendorp F, van Hall T, Melief CJ. Vaccines for established cancer: overcoming the challenges posed by immune evasion. Nat Rev Cancer. 2016;16:219–233. doi: 10.1038/nrc.2016.16. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.