

ABSTRACT
Mutations in the enzyme isocitrate dehydrogenase 1 (IDH1) lead to metabolic alterations and a sustained formation of 2-hydroxyglutarate (2-HG). 2-HG is an oncometabolite as it inhibits the activity of α-ketoglutarate-dependent dioxygenases such as ten-eleven translocation (TET) enzymes. Inhibitors of mutant IDH enzymes, like ML309, are currently tested in order to lower the levels of 2-HG. Vitamin C (VC) is an inducer of TET enzymes. To test a new therapeutic avenue of synergistic effects, the anti-neoplastic activity of inhibition of mutant IDH1 via ML309 in the presence of VC was investigated in the colon cancer cell line HCT116 IDH1R132H/+ (harbouring a mutated IDH1 allele) and the parental cells HCT116 IDH1+/+ (wild type IDH1). Measurement of the oncometabolite indicated a 56-fold higher content of 2-HG in mutated cells compared to wild type cells. A significant reduction of 2-HG was observed in mutated cells after treatment with ML 309, whereas VC produced only minimally changes of the oncometabolite. However, combinatorial treatment with both, ML309 and VC, in mutated cells induced pronounced reduction of 2-HG leading to levels comparable to those in wild type cells. The decreased level of 2-HG in mutated cells after combinatorial treatment was accompanied by an enhanced global DNA hydroxymethylation and an increased gene expression of certain tumour suppressors. Moreover, mutated cells showed an increased percentage of apoptotic cells after treatment with non-cytotoxic concentrations of ML309 and VC. These results suggest that combinatorial therapy is of interest for further investigation to rescue TET activity and treatment of IDH1/2 mutated cancers.
KEYWORDS: Vitamin C, epigenetics, IDH1, TET, cancer cells
Introduction
Initiation and progression of cancerous lesions arise not only from genetic mutations but also from aberrant epigenetic mechanisms such as DNA methylation. Here, failure to maintain a balance between DNA hypomethylation and hypermethylation gives rise to subsequent pathological processes in the cell. While genome-wide DNA hypomethylation is associated with chromosomal instability, aberrant DNA hypermethylation in CpG islands of gene promoter regions can lead to gene silencing [1–3]. This, in turn, can facilitate tumorigenesis and cancer progression due to decreased expression of, e.g., tumour suppressor genes and disruption of cell cycle regulation, apoptosis, and/or DNA repair.
Aberrant DNA hypermethylation has been associated with several types of cancer, including gliomas, acute myeloid leukaemia (AML) and cancers of the lung, breast, ovaries, and colon [4–6]. Members of the ten-eleven translocation (TET) dioxygenase family are capable of oxidizing methylated 2 ´-deoxycytidines to hydroxymethylated 2´-deoxycytidines – e.g., 5-methyl-2´-deoxycytidine (5-mdC) to 5-hydroxymethyl-2´-deoxycytidine (5-hmdC) [7] – a known mechanism of active DNA demethylation. TETs can further oxidize 5-hmdC to 5-formyl-2´-deoxycytidine (5-fdC) and 5-carboxy-2´-deoxycytidine (5-cadC), which are eventually replaced by unmodified 2´-deoxycytidines as a result of thymine-DNA glycosylase (TDG)-mediated base excision repair [8–10]. This cascade of 5-mdC oxidation by TETs might represent an active gene demethylation mechanism leading to transcriptional reactivation. In addition to its role as a demethylation intermediate, 5-hmdC has unique epigenetic properties that result in specific gene expression profiles [11,12]. Indeed, a genome-wide decrease in 5-hmdC levels is considered an epigenetic hallmark in many cancers [13,14] and several studies have highlighted the diagnostic and prognostic value of this mechanism.
From a therapeutic point of view, a limited number of known TET enzyme activators can significantly increase genome-wide 5-hmdC levels. Vitamin C (VC) is one of the best-known substrates of the TET enzymes as well as a potent antioxidant and reducing agent [15]. It has been proposed that VC is responsible for the restoration of TET enzyme catalytic activity through the reduction of Fe(III) to Fe(II) [16,17]. This hypothesis has, however, been contradicted by other studies in which other strong reducing agents were unable to enhance the TET-mediated hydroxylation of 5-mdC [18–20]. The mechanism by which VC contributes to increased levels of TET-mediated hydroxylation of 5-mdC is, therefore, presently unclear.
Aberrant DNA hypermethylation has been identified in AML and gliomas, as well as in colorectal cancer and was linked to decreased TET enzyme activity due to mutations in the isocitrate dehydrogenase (IDH) family, specifically IDH1 or IDH2 [4,21,22]. Mutations at the codon 132 of IDH1 result in replacement of an arginine by a histidine (IDH1R132H) and are present in >70% of adult WHO grade II/III gliomas and in >80% of secondary glioblastoma multiforme (GBM), a highly aggressive brain tumour [23–25]. In gliomas, the IDH1R132H mutation is the most frequent, though other types of cancer causing base substitutions in IDH1 and IDH2 have also been reported [25,26]. Colorectal cancers harbour an IDH1 mutation exclusively accompanied by the CpG island methylator phenotype (CIMP), which is characterized by aberrantly coordinated DNA hypermethylation of CpG dinucleotides in a subset of promoters of genes silenced during tumorigenesis [27].
The switch of amino acids at the active site of IDH enzymes is crucial for the enzyme’s function. The IDH1R132H substitution not only disrupts physiological oxidative decarboxylation of isocitrate to α-ketoglutarate as performed by the wild type enzyme, it can also irreversibly convert α-ketoglutarate to the structurally similar oncometabolite 2-hydroxyglutarate (2-HG) in large amounts and as a consequence inhibit α-ketoglutarate-dependent dioxygenases such as the TET enzymes [28–31]. Indeed, a specific CpG island methylator phenotype (CIMP) in gliomas and colorectal cancers, that is characterized by DNA hypermethylation, has been associated with IDH mutations, which has led to several speculations that IDH mutations contribute to tumorigenesis via altered epigenetic regulation [27,32].
Additionally, a previous study demonstrated ‘rewiring’ of metabolic profiles in IDH1 mutant (mutated IDH1) gliomas: the production of pyruvate from lactate, along with glutamate anaplerosis, was used to replenish the tricarboxylic acid (TCA) cycle and produce isocitrate and α-ketoglutarate, the latter of which serves as a substrate for mutant IDH enzymes to further generate 2-HG [33]. Concordantly, other work has shown >80% of 2-HG produced by mutant IDH enzymes in colorectal carcinoma cells derives from glutamine, which is converted to glutamate [34,35]. This is in contrast to IDH1 wild type gliomas, which predominantly use pyruvate for lactate production and predominantly depend on glycolysis and acetate anaplerosis. The clear metabolic ‘rewiring’ exhibited by IDH1 mutated gliomas has several other implications such as enzyme inhibition and epigenetically mediated silencing of genes encoding for metabolic enzymes [33].
For these tumours, IDH mutations have become a target for novel therapeutic approaches including the use of hypomethylating agents – i.e., DNMT inhibitors or enhancers of TET enzyme activity – and inhibitors of mutant IDH enzymes [36,37]. Although the hypomethylating agents like decitabine or 5-azacytidine were originally developed as cytostatic agents for leukaemia chemotherapy, their epigenetic properties have since been revealed to play an important role in their anti-cancer activity [38–42]. A multitude of new compounds for the treatment of tumours bearing mutated IDH1 and IDH2 are presently in clinical trials [25]. Innovative combinations of epigenetically active substances and chemotherapeutics have been shown to improve actions against solid tumours and leukaemia. However, combinations of mutant IDH inhibitors and compounds capable of reactivating DNA hydroxymethylation, such as VC (ascorbate), have yet to be investigated as a novel therapeutic avenue in IDH mutated cancers. This newly discovered function of VC prompted us to determine whether additive or synergistic effects can be achieved when sub-toxic concentrations of the mutant IDH1 inhibitor ML309 are used in combination with high-doses of VC. Investigations were conducted in mutated IDH1 (HCT116 IDH1R132H/+) and wild type colon cancer cells (HCT116 IDH1+/+) and to assess whether such an epigenetic therapy has therapeutic application in cancers.
Due to the excessive generation of the oncometabolite 2-HG, mutant IDH1 drives tumorigenesis and cancer progression by impairing the efficacy of epigenetically active TET enzymes, promoting aberrant DNA methylation patterns. Targeting one of the most frequent cancer-associated mutant alleles, IDH1R132H, may be a promising therapeutic approach. The aim of this work was, therefore, to investigate TET-mediated DNA hydroxymethylation and how it is affected by the repercussions of mutated IDH1. As such, this work aims to contribute to the identification of mechanisms capable of mitigating the carcinogenic effects of the IDH mutations frequently found in a variety of cancer types, including colorectal cancer and brain tumours.
Results
2-HG levels in IDH1R132H/+ cells decrease upon treatment with ML309 and VC
IDH1 mutations are associated with altered IDH1 enzyme function which induces the overproduction of the neomorphic metabolite 2-HG. In order to validate the increased abundance of 2-HG in HCT116 IDH1R132H/+ cells compared to HCT116 IDH1+/+ cells, 2-HG content was analysed by LC-MS/MS analysis (Figure 1(a)). Indeed, the intracellular concentration of 2-HG was 56-fold higher in IDH1R132H/+ cells (8.5 nmol/mg protein) compared to that in parental wild type cells (0.15 nmol/mg protein). Mutant IDH1 inhibition by 10 µM ML309 significantly reduced 2-HG levels in HCT116 IDH1R132H/+ cells. However, the level of 2-HG in wild type cells was not achieved (Figure 1(b)). VC (1 mM) on its own reduced 2-HG levels only marginally in IDH1 mutated cells, but combinational treatment with ML309 and VC decreased 2-HG levels to those found in IDH1+/+cells.
Figure 1.
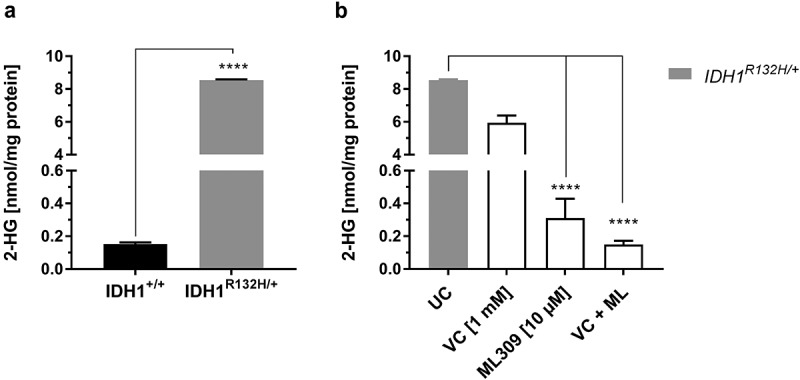
Measurement of 2-HG levels in HCT116 wild type IDH1 (IDH1+/+) and mutant IDH1 (IDH1R132H/+) cells.
(a) Comparison of 2-HG levels in untreated IDH1+/+ and IDH1R132H/+ (mean ± SD; **** p < 0.0001; n = 3), (b) Influence of VC (1 mM) and ML309 (10 µM) treatment on 2-HG levels in IDH1R132H/+ cells after 48 h (mean ± SD; **** p < 0.0001; n = 3)
Genome-wide DNA hydroxymethylation is decreased in IDH1R132H/+cells
As 2-HG inhibits TET activity, we aimed to assess whether elevated 2-HG levels in mutant cells are accompanied by a lower DNA hydroxymethylation status. Indeed, the 5-hmdC/dC levels were more than 50% lower in IDH1R132H/+ cells as compared to IDH1+/+ cells (0.004% hmdC/dC and 0.008% hmdC/dC, respectively) (Figure 2(a)). It should be mentioned that the genome wide DNA methylation was not affected in IDH1R132H/+ cells. As shown in Figure 2(b), 5 -mdC/dC levels were similar in IDH1+/+ and IDH1R132H/+ cells, both cell types contained approximately 5% mdC/dC.
Figure 2.
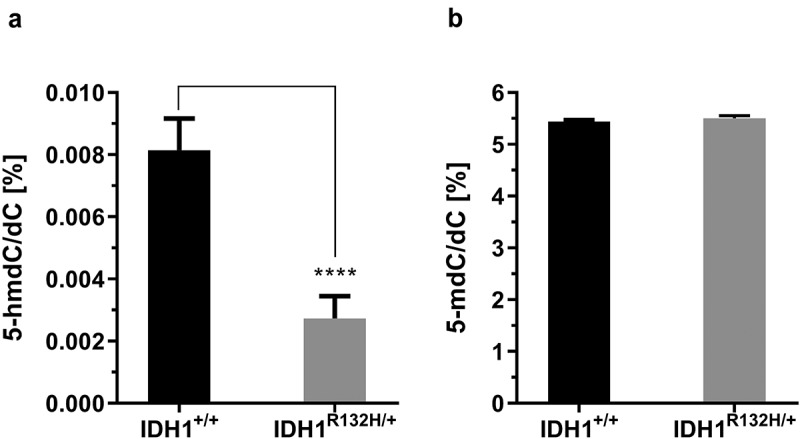
Genomic 5-hydroxymethyl-2´-deoxycytidine (5-hmdC) and 5-methyl-2´-deoxycytidine (5-mdC) levels in HCT116 wild type IDH1 (IDH1+/+) and mutant IDH1 (IDH1R132H/+) cells.
(a) 5-hmdC/dC levels in untreated IDH1+/+ and IDH1R132H/+ (mean ± SD; ****p < 0.0001; n = 3), (b) The 5-mdC/dC levels in IDH1+/+ and IDH1R132H/+ (mean ± SD; n = 3)
VC increases genome-wide DNA hydro-xymethylation in IDH1+/+ and IDH1r132h/+cells
VC is known to activate TET dioxygenases and increase global DNA hydroxymethylation. Therefore, 5-hmdC/dC levels were measured in HCT116 IDH1+/+ and IDH1R132H/+ cells in response to VC via isotope-dilution LC-MS/MS. The IDH1+/+ cells exhibited increased 5-hmdC/dC levels after 48 h treatment with VC in a concentration-dependent manner. Differences were significant at 10 µM VC and reached maximal at 1 mM VC (Figure 3(a)), which is consistent with a previous report [36]. However, in IDH1R132H/+ cells VC incubation only slightly increased 5-hmdC/dC levels, but not in a significant manner. To evaluate whether VC has an impact on the DNA methylation status of HCT116 IDH1+/+ or IDH1R132H/+ cells, 5-mdC/dC ratios were plotted against increasing concentrations of VC. As it is evident from supplementary Figure S2, VC did not contribute to an altered DNA methylation status neither in mutated nor in wild type cells.
Figure 3.
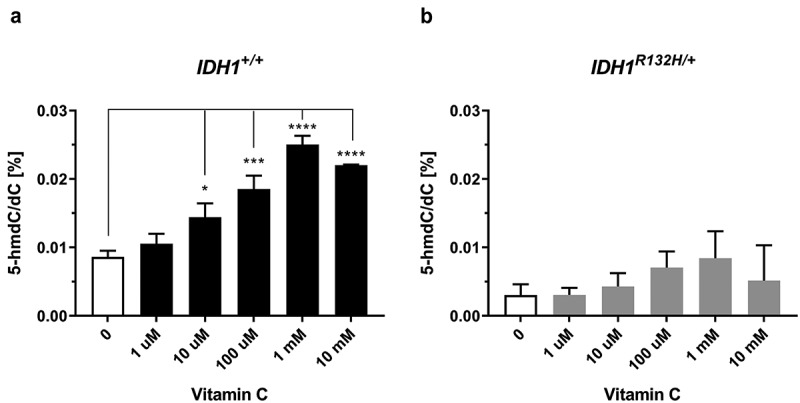
Impact of VC on genomic 5-hmdC/dC levels in IDH1+/+ and IDH1R132H/+ cells.
(a,b) Quantification of 5-hmdC/dC levels in IDH1+/+ and IDH1R132H/+ treated for 48 h with increasing concentrations of VC as indicated (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 – significant results; error bars = SD; n = 3).
Combinatorial treatment of ML309 and VC increases the genome-wide DNA hydroxymethylation
As 2-HG is highly overproduced in IDH1 mutated cells, which is accompanied by an inhibition of TET enzymes and reduced 5-hmdC/dC levels, it can be assumed that inhibition of mutant IDH1 will restore TET dioxygenase activity and increase 5-hmdC/dC levels. Surprisingly however, treatment of IDH1R132H/+ cells with the mutant IDH1 inhibitor ML309, although reducing 2-HG contents, but not to the level of wild type cells, did not significantly alter 5-hmdC/dC levels (Figure 4). Therefore, it was of major interest to determine whether co-incubation of both compounds, VC and ML309, improved the epigenetic outcome with regards to genomic 5-hmdC/dC levels. Remarkably, the combination of both compounds synergistically increased the DNA hydroxymethylation in IDH1R132H/+ cells. After co-incubation of 1 mM VC with 1 or 10 µM ML309, we found significantly enhanced 5-hmdC/dC levels. In the presence of 1 mM VC a significant increase relative to matched ML309 only treated cells was seen (p < 0.001 for 1 µM ML309 and p < 0.01 for 10 µM ML309) (Figure 4(b)).
Figure 4.
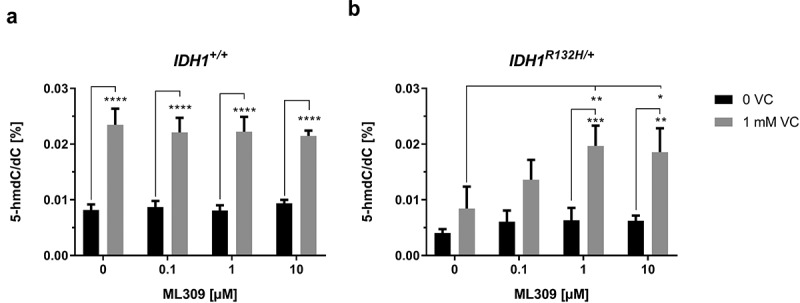
Impact of ML309 and VC on genomic 5-hmdC/dC levels in IDH1+/+ and IDH1R132H/+ cells.
(a,b) Quantification of 5-hmdC/dC levels in IDH1+/+ and IDH1R132H/+ treated for 48 h with increasing concentrations of ML309 alone or in combination with vitamin C (1 mM) as indicated (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 – significant results; error bars = SD; n = 3).
In contrast, VC and ML309 combinations had no impact on the DNA methylation status of HCT116 IDH1+/+ or IDH1R132H/+ cells. As shown in Figure S2, Figure 5.5-mdC/dC ratios were plotted against increasing concentrations of ML309, but did not contribute to an altered DNA methylation status in wild type or mutant IDH1 harbouring cells.
Figure 5.
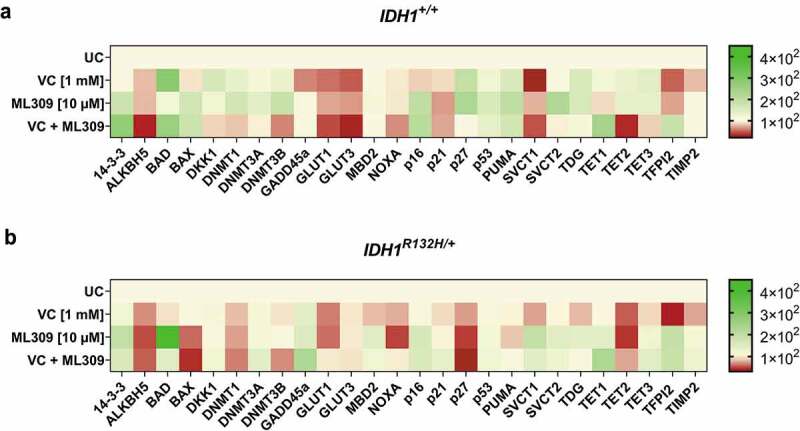
Gene expression profile of 25 genes after single or combinatorial treatment with VC and ML309 for 48 h in IDH1+/+ and IDH1R132H/+ cells.
Heat map of relative mRNA-levels of the differential expressed genes in (a) IDH1+/+ and (b) IDH1R132H/+ cells after treatment with VC (1 mM), ML309 (10 µM) and combinatorial treatment. (relative to the HMBS housekeeper gene, using the 2ΔΔCT method)
VC and ML309 induce significant changes in mRNA profile of IDH1+/+ and IDH1r132h/+ cells
To examine whether the increased genome-wide 5-hmdC levels in response to VC and ML309 led to a direct impact on the mRNA expression of epigenetically regulated genes, differential gene expression of 25 specific target genes was analysed. Indeed, the treatment with VC and ML309 alone or in combination had a significant impact on the expression of the analysed genes in both, mutated and wild type cells. Apparently, the treatment modulated the expression of genes involved in glucose uptake, apoptotic pathways and vitamin C uptake. Though we found certain similar expression changes, there were significant differences between HCT116 IDH1+/+ or IDH1R132H/+ cells (Figure 5).
Both cell lines showed a significant decrease of gene expression in ALKBH5, DKK1, DNMT1, DNMT3B, GLUT1, GLUT3, NOXA and TET2 after combined treatment with VC and ML309. Interestingly, in IDH1+/+ and IDH1R132H/+ cells, the treatment with VC and ML309 led to an increased expression of BAD, which is part of the p53 signalling pathway. This gene can be activated in part by the 14-3-3 protein, whose gene expression was also up-regulated in both cell types. Accordingly, we found an increase in transcripts of down-stream gene products like p16, as well. This leads to the assumption that in IDH1R132H/+ cells, at least in part, the induction of apoptosis and cell cycle arrest may be associated to the combinatorial treatment of ML309 and VC via the GADD45a pathway. Indeed, mutated cells showed an increased GADD45a mRNA expression. Frequently, cancer cells show a high expression of glucose transporters GLUT1 and GLUT3 due to the metabolic changes and high demands of energy through neoplastic transformation [43–45]. In IDH1+/+ cells, GLUT1 and GLUT3 were significantly reduced upon treatment with VC, whereas in IDH1R132H/+ cells also a reduction, though to a smaller extent, was detected. As overexpression of these transporters is associated with poor survival in several cancers [46], we suggest a positive outcome of high-dose VC treatment as therapeutically avenue in IDH1+/+ and IDH1R132H/+ cancer cells. Further, several genes involved in epigenetic regulations showed a different mRNA profile after treatment with VC and ML309. The combinatorial treatment led to a DNMT1 and DNMT3B down-regulation in both cell types. As one could speculate, that the increase of 5-hmdC/dC levels after treatment with VC was due to an increase of TET gene expression, we measured the differential expression, as well. The combinatorial treatment led to an increase of TET1 mRNA expression in mutated and wild type cells. In contrast, TET2 mRNA expression was down-regulated after treatment with VC and ML309. The controversial regulation does not explain the increase of 5-hmdC/dC levels in the cells. As the expression analysis of the selected genes indicated a differential regulation in response to VC and ML309, it was of interest to examine whether this gene regulation influences cell growth and apoptosis.
Enhanced cell death by VC in pharmacological concentrations in combination with ML309
The cytotoxic potential of increasing concentrations of mutant IDH1 inhibitor ML309 with or without VC (1 mM) on HCT116 IDH1+/+ or IDH1R132H/+ was evaluated by means of MTT reduction assay (Figure 6). Upon the 24 h treatment with increasing concentrations of ML309, mutated and wild type IDH1 cells became less viable. Mutant cells exhibited a higher response rate with a calculated IC50 of approximately 45 µM and wild type cells did not have a relative viability lower than 55% after treatment with the highest tested concentration of 50 µM ML309. The co-incubation with 1 mM VC and ML309 led to a significant decrease of cell viability in mutant cells (IC50: 19 µM) as well as in wild type cells (IC50: 29 µM). These data indicate that co-administration of 1 mM VC was detrimental to cell viability at all ML309 concentrations for both tested cell lines. Overall, cell toxicity was more pronounced for IDH1R132H/+ compared to IDH1+/+ cells with all tested treatment strategies.
Figure 6.
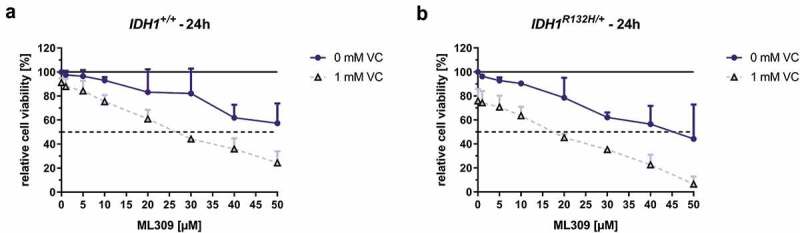
Effect of mutant IDH1 inhibitor ML309 and VC on cell viability of HCT116 IDH1+/+ or IDH1R132H/+.
HCT116 cells were exposed to increasing concentrations of ML309, as indicated. The incubation was carried out for 24 h in HCT116 IDH1+/+ (a) or IDH1R132H/+ (b) with ML309 alone or in combination with VC (1 mM), and cell viabilities assessed by MTT assay. Data are expressed as a percentage of the untreated control (100% are indicated as black line), viable cell levels <75% were taken to indicate cytotoxic induction (error bars = SD; n = 3), dotted line shows 50% viability for determination of IC50 values.
Apoptosis induction in IDH1R132H/+ colon cancer cells upon treatment with ML309 and VC
For further elucidation, whether the single or combinatorial treatment with ML309 and VC was able to induce apoptosis in HCT116 IDH1+/+ or IDH1R132H/+ cells, selected treatments were analysed by flow cytometry after staining with Annexin V and propidium iodide (Figure 7). A significant induction of total apoptosis relative to untreated cells was only evident in HCT116 IDH1R132H/+ cells. Stimulation with ML309 (10µM) led to a significant increase of the apoptotic rate, which was further enhanced after combinatorial treatment with 1 mM VC (Figure 7). Interestingly, the increased rate of apoptosis after combinatorial treatment was due to an enhanced amount of early apoptotic cells.
Figure 7.
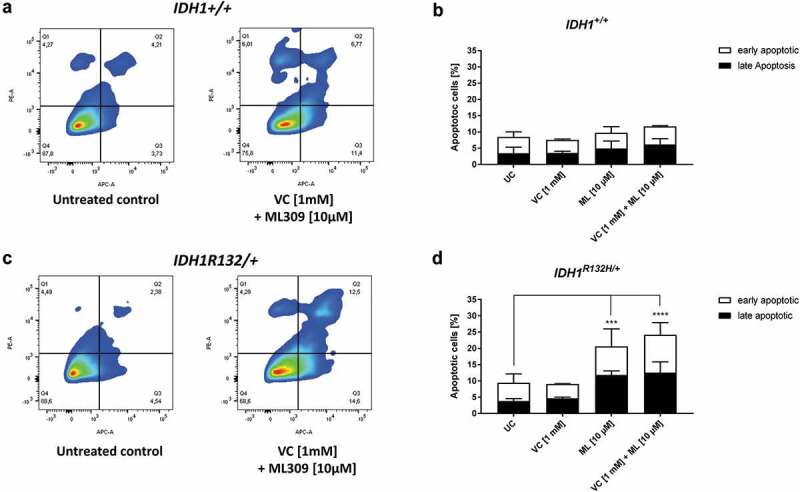
Measurement of apoptotic cells upon VC and ML309 incubation.
(a, c) Representative contour plots of Annexin V/propidium iodide staining in cells treated for 48 h with combination of VC and ML309, as indicated. Q1 – gates for necrotic cells, Q2 – for late apoptosis, Q3 – for early apoptosis and Q4 – for healthy cells. (b, d) Quantitative representation of early and late apoptotic cells after treatment with VC, ML309 or combined with indicated concentrations. (error bars = SD; n = 3; Statistical significance of the treated groups to the untreated control was calculated using 2-way ANOVA and Tukey post-test: * = p < 0.05, ** = p < 0.01, *** = p < 0.001, **** = p < 0.0001).
Discussion
In this study, we present the first attempt to investigate a synergistic effect of VC and mutant IDH1 inhibitors in IDH1 mutated cancer cells. By combining the compounds, we aimed to reverse the epigenetic aberrations of mutated IDH1 cancer cells by activating the TET enzymes with VC and simultaneously inhibit the neomorphic action of mutated IDH1 enzymes via the mutant IDH1 inhibitor ML309.
Aberrant epigenetic modifications are a hallmark of cancer progression, and restoring a balance between hyper- and hypomethylating events has emerged as a promising counteraction against neoplasia for therapeutic use. Originally, VC has attracted attention as a potential cancer therapy through potential induction of oxidative stress that selectively kills cancer cells [47,48]. VC has also been shown to act as a potent activator and possible co-factor of the TET dioxygenases, which mediate the oxidation of 5-mdC to 5-hmdC in genomic DNA, a possible therapeutic avenue for active demethylation in aberrantly hypermethylated cancer loci [18,49]. Notably, there is an underestimated prevalence of VC deficiencies in cancer patients [50–52], and pharmacologically active concentrations of up to 1 g/kg bw VC have been shown to significantly improve the outcomes of the epigenetic cancer therapies [50].
Cells harbouring IDH1 mutations show a significant overproduction of the oncometabolite 2-HG, which is able to promote cellular transformation towards cancer. As this effect can be reversed by inhibition of the production of 2-HG, the development of specific compounds able to repress IDH activity is of great interest. Here, we investigated whether VC can enhance the anti-carcinogenic actions of the mutant IDH1 inhibitor ML309 in IDH1 mutated cells, and whether this innovative combination of these compounds can be used as a model therapy option for cancers harbouring IDH1 mutations. Although our studies were only performed with the human colorectal carcinoma cell line HCT116 with a heterozygous knock-in of IDH1 dominant-negative (R132H) point mutation, a similar outcome can be speculated in other IDH1 mutated cancer cells.
Mutations in IDH1 and IDH2 are frequently observed in various cancers ranging from solid cancers like glioblastomas, chondrosarcoma or colorectal cancers and in different forms of leukaemia like AML [25]. Unusually to other cancer-related aberrations, the most common mutation in IDH1 and the less common mutation in IDH2 confer a neomorphic gain-of-function. One prevalent IDH1 mutation involves a histidine substitution for an arginine (IDH1R132H/+), which leads to an aberrantly working IDH1 enzyme and to the overproduction of the oncometabolite 2-HG. Especially high-grade gliomas show a high prevalence of mutated IDH1 or IDH2 genes of up to 90%, whereas IDH1 is most often mutated. Even in up to 30% of AML leukaemia IDH1/2 mutations are found [29]. Certain subclasses of colorectal cancers with a CpG island show IDH1 mutations, whereas 2-HG can effectively inhibit the function of Fe(II)-dioxygenases like TET enzymes. One aim of the present study was to determine whether 2-HG production in IDH1R132H/+ HCT116 cells could be reduced by application of the mutant IDH1 inhibitor ML309. In addition, it was of interest whether TET reactivation mediated by VC could effectively rescue 5-hmdC/dC levels in IDH1R132H/+ HCT116 cells.
Firstly, the excessive production of 2-HG in IDH1R132H/+ cells was confirmed by a highly sensitive LC-ESI-MS/MS approach, revealing up 56-fold 2-HG content relative to HCT116 IDH1+/+ cells. As expected, this was also accompanied by a significant reduction of the 5-hmdC/dC levels in line with the binding of 2-HG to numerous class of Fe(II)-dependent dioxygenases, including the TET enzymes, and their subsequent inhibition. However, genome-wide DNA hypermethylation in IDH1R132H/+ cells was not observed. Similarly, treatment with ML309 or VC failed to evoke marked changes 5-mdC/dC levels. By contrast, the treatment with ML309 significantly reduced the 2-HG levels in the IDH1R132H/+ cells, where treatment with VC alone did not. VC is a potent activator of TET activity, as seen by the increased 5-hmdC/dC levels in IDH1+/+ cells. Although in IDH1 mutant cells, the VC mediated increase of 5-hmdC/dC was not significant, we found a synergistical activation of TET activity after co-incubation with VC and ML309. In detail, the combinatorial treatment significantly put the 5-hmdC/dC levels up to 0.02%, around 4 times higher than in untreated IDH1R132H/+ cells. This was surprising with regard to the currently presumed model of VC action on TET enzymes. Until now, it was not clear that VC can reactivate TET-enzymes in IDH1R132H/+ gain-of-function cells. Due to its structure, VC itself should be unsuitable to efficiently rescue 2-HG-mediated inhibition of TET activity. In contrast, 2-HG exhibits structural similarities to the TET cofactor α-ketoglutarate and thereby performs competitive inhibition of α-ketoglutarate-dependent oxygenases [53,54], including TET enzymes. Yet, it is unlikely that competitive displacement of 2-HG can be exerted by VC itself, the detailed mode of action needs to be further explored. The previously proposed idea by Monfort [17] and others was that VC exerts its positive effects on DNA hydroxymethylation by recycling Fe(II) for TET enzyme activity. In that case, VC might not be able to rescue DNA hydroxymethylation in cells that produce excessive amounts of 2-HG, which is a competitive antagonist of the TET cofactor α-ketoglutarate. Based on our findings, we suggest that in some way VC and ML309 act synergistically in order to mediate a displacement of 2-HG at the catalytically active site of TET and to reactivate its full function. This disparity of proposed scientific approaches punctuates the requirement for further investigation. In contrast to the combinatorial treatment, we found that incubation with increasing concentrations of mutant IDH1 inhibitor ML309 alone led to no significant increase of 5-hmdC/dC levels neither in IDH1R132H/+ mutant cells nor IDH1+/+ wild type cells. Here, the reduction of 2-HG levels was successful; however without increase of 5-hmdC/dC levels. This discrepancy can be explained by the fact that the mutant IDH1 inhibitor ML309 was not able to diminish 2-HG to those contents found in wild type cells. Further studies to investigate the mode of action of the mutant IDH1 inhibitor on the reactivation of TETs will be needed here.
In addition, we show that VC and ML309 alone, but especially the combination, induced a significant increase of apoptotic cells in IDH1R132H/+ cells. However, in IDH1+/+ cells, the treatment did not lead to any detectable apoptosis induction. This finding underlines the IDH1 mutant-specific mode of action of the combinatorial treatment of these two compounds, which was evident for the TET activity and 5-hmdC/dC increase, as well. Most interestingly, apoptosis induction was accompanied by significant gene activation of a subset of analysed genes. Here, we found that GADD45a was one of the highest re-expressed genes after combinatorial treatment. The GADD45a gene product is increased following stressful growth arrest conditions. The DNA damage-induced transcription of this gene is mediated by both p53-dependent and -independent mechanisms. Interestingly, in IDH1+/+ and IDH1R132H/+ cells, the treatment with VC and ML309 led to increases in the expression of BAD, which is part of the p53 signalling pathway. This gene can be activated in part by the 14-3-3 protein, whose gene expression was up-regulated in both cell types, as well. Accordingly, we found an increase in transcripts of down-stream gene products like p16, as well. This leads to the assumption that in IDH1R132H/+ cells, at least partly, the induction of apoptosis and cell cycle arrest may be associated to the combinatorial treatment of ML309 and VC via the GADD45a pathway.
Frequently, cancer cells show a high expression of glucose transporters GLUT1 and GLUT3 due to the metabolic changes and high demands of energy through neoplastic transformation [43,55,56]. In IDH1+/+ and IDH1R132H/+ cells, GLUT1 and GLUT3 levels were reduced upon treatment with VC. As overexpression of these transporters is associated with poor survival in several cancers [44], we purpose a positive outcome of high-dose VC treatment as therapeutical avenue in IDH1+/+ and IDH1R132H/+ cancer cells.
Further, genes involved in epigenetic regulations show a different mRNA profile between IDH1+/+ and IDH1R132H/+cells. The combinatorial treatment led to a DNMT1 and DNMT3B down-regulation in both cell types. As one could speculate that the increase of 5-hmdC/dC levels after treatment with VC was due to increase of TET gene expression, we measured the differential expression, as well. Surprisingly, TET1 and TET2 gene expressions were regulated in an opposite manner after treatment with VC and ML309. Therefore, the synergistic effect on the 5-hmdC/dC levels after combinatorial treatment in IDH1R132H/+cells cannot be explained solely by TET expression. Further studies for the exact mode of action by VC and ML309 on TET activity are needed.
It has been shown that IDH1 mutated cancer cells confer certain liabilities of other enzymes, which may open different avenues of treatment [25,29,45]. This can lead to a decrease of resistance to specific therapies during the cancer treatment. As shown in this study, we found that the combinatorial treatment with VC and ML309, as one mutant IDH1 inhibitor, can effectively induce apoptosis and gene expression of certain tumour suppressors. Therefore, it would be of great interest to further investigate the beneficial outcome of this treatment routine in mutated IDH1 cancers in vivo.
Several studies have underlined the clinical potential of high-dose ascorbate in anticancer therapies [46]. However, due to further insights into the physiological mechanisms of VC, a combinatorial treatment with other epigenetic [36,57] or immunomodulatory [58,59] compounds can be recommended [60,61]. The benefit of co-treatment with VC and decitabine in cancer cells or with checkpoint inhibitors, such as CTLA4 or PD-L1 has been proved and even, radiotherapy [62] and chemotherapy [63,64] can be supported by high-dose VC co-treatment.
Conclusion
This study is the first to investigate the combination of the mutant IDH1 inhibitor ML309 and high VC doses as a potential anti-cancer therapy. In summary, combinatorial treatment showed an important impact on the epigenetic landscape of IDH1 mutant cancer cells that corresponded to significant increases of apoptotic cell population within these. Furthermore, the combination of both compounds led to significant increases in genome-wide 5-hmdC/dC levels and expression of previously down-regulated tumour suppressor genes, as well as a shift towards a more apoptotic state without significant cytotoxic induction. To the best of our knowledge, this is the first time a direct link between VC treatment and a decrease in genome-wide 2-HG levels has been observed. Although this study mainly concentrated on the HCT116 cell line with an introduced IDH1 mutation, we can conclude that our results indicate anticancer therapies in IDH1R132H/+ mediated by non-toxic concentrations of ML309 are capable of enhancing anti-cancer activities when administered in combination with high-dose VC. As such, our data suggest that therapeutic responses to low, non-toxic doses of ML309 can be improved by additional VC supplementation.
Materials and methods
Cell culture and treatment
For cell culture experiments the human colorectal carcinoma cell line HCT116 with a heterozygous knock-in of IDH1 dominant-negative (R132H) point mutation was used. HCT116 IDH1+/+ (parental) and HCT116 IDH1R132H/+ knock-in human colorectal cell lines were generated by rAAV targeting technology GENESIS [65] and obtained from Horizon Discovery (Cambridge, United Kingdom) as frozen cultures.
For the cultivation of cells pre-warmed RPMI 1640 medium with GlutaMAX supplement (Thermo Fisher, Darmstadt, Germany), containing 10% foetal bovine serum (FBS) and 1% penicillin/streptomycin (growth medium) was used. The cells were maintained in 5% CO2 at 37°C. The cells were allowed to reach 90% confluency before subcultivation. The cell lines were confirmed as negative for mycoplasma infection within 6 months prior to use.
VC and ML309 [66] were purchased from Sigma (Schnelldorf, Germany). HCT116 IDH1+/+ and HCT116 IDH1R132H/+ cells were treated, if not other stated, with 1 mM VC and increasing concentrations of ML309 (1–50 µM) for 24 h within the scope of a cell viability assay. The maximal tolerable doses of VC was determined in a previous study [36]. Based on the results of the cell viability assay the concentrations of ML309 for further experiments were chosen. For the apoptosis assay, the cells were treated with 10 µM ML309 and 1 mM VC. The gene expression analysis was conducted after treatment of the cells with selected concentrations of 1 mM VC and 10 µM ML309. In order to analyse the genome-wide 5-mdC/dC and 5-hmdC/dC levels, the cells were treated with increasing concentrations of VC (1 µM – 10 mM) and 0.1, 1 and 10 µM ML309 alone or in combination with 1 mM VC.
For all cell-based assessments, test compounds were dissolved and diluted in RPMI 1640 medium supplemented with 1% FBS and 1% penicillin/streptomycin (test medium). Cells were treated with the respective test compounds in test medium 24 h after seeding. To ensure continuous stability of the test compounds throughout the experiments, the test medium was changed every 24 h with freshly dissolved or diluted test compounds.
Cytotoxicity assessment via MTT assay
A 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reduction assay was used to indirectly determine the cytotoxic potential of VC (1 mM) with increasing concentrations of ML309 (1 µM to 50 µM) in HCT116 IDH1+/+ and HCT116 IDH1R132H/+ cells. The reduction of yellow MTT to blue formazan via mitochondrial reductase is an effective indicator for metabolic activity. As this reaction can only proceed in metabolically active cells, the amount of generated blue dye correlates with the amount of viable cells. By exploiting this process, one can indirectly measure cell viability of proliferating cells via colorimetric measurement [67].
For the assessment of the cytotoxic potential of the test compounds, HCT116 IDH1+/+ or IDH1R132H/+ cells were seeded in 96-well plates and incubated in test medium for 24 h at 37°C. Cells in unamended medium were used as vehicle control, and cells treated with 0.01% or 0.005% SDS were used as positive control. After the respective incubation times, test media were substituted for medium containing 0.5 mg/ml MTT for 4 h at 37°C. Following the substitution of MTT medium for DMSO, the plates were shaken at 350 rpm in darkness at room temperature for 20 min. Measurement of the samples’ respective optical densities was performed at λ = 540 nm by means of a FLUOstar OPTIMA plate reader. The biologically relevant threshold of 80% cell viability was defined as the onset of cytotoxicity upon administration of test compounds.
Quantification of 2-hydroxyglutarate using liquid chromatography tandem-mass spectrometry (LC-MS/MS)
The quantification of 2-HG levels in HCT116 IDH1+/+ or IDH1R132H/+ cells was conducted according to a previously described method [68]. Cell pellets were lysed in deionized water using a Bead Ruptor 12 (Omni International, Kennesaw, USA). Protein content (by Bradford assay) and 2-HG were determined in the cell lysates. For 2-HG quantification, 5 nmol of 3-amino-4-hydroxybenzoic acid (AHBA) were added to lysate aliquots (50 µl) as internal standard. Analyses were conducted with an Agilent 1260 Infinity LC system coupled to an Agilent 6490 triple quadrupole-mass spectrometer (both from Waldbronn, Germany) interfaced with an Agilent Jet Stream (AJS) electrospray ion source operating in the negative ion mode (ESI-). Chromatographic separation was carried out using an YMC-Triart C18 plus column (3 μm, 3 × 100 mm) equipped with a guard column of the same material. Water and methanol, both supplemented with 10 mM ammonium formate and 0.02% formic acid, were used as eluents. Cell lysate supernatants were injected into a mobile phase consisting of 100% water. 2-HG and AHBA were eluted from the column with an 8-min linear gradient to 70% methanol at a flow rate of 0.25 ml/min. 2-HG and AHBA eluted from the separation column at 3.8 and 6.5 min, respectively. The total run time for one analysis was 14 min, including re-equilibration of the LC system. The following ion source parameters were used: drying gas temperature = 225°C, drying gas flow = 15 l/min of nitrogen, sheath gas temperature = 350°C, sheath gas flow = 12 l/min of nitrogen, nebulizer pressure = 40 psi, capillary voltage = 4000 V, nozzle voltage = 500 V. The optimized ion funnel parameters were: high pressure RF voltage = 90 V and low pressure RF voltage = 60 V. 2-HG was quantified by external calibration in relation to the internal standard AHBA (2-HG range: 0–500 µM, fixed AHBA: 50 µM) using the multiple reaction monitoring (MRM) approach. The following mass transitions were recorded (optimized collision energies in parentheses): 2-HG: m/z 147.0 → 129.1 (8 eV), m/z 147.0 → 85.1 (12 eV), m/z 147.0 → 56.9 (20 eV); AHBA: m/z 152.0 → 107.9 (12 eV). The water loss, represented by m/z 147.0 → 129.1 for 2-HG, was used for quantification. The dwell time for each of the four mass transitions recorded was 175 ms. The determined 2-HG amounts were normalized to the actual protein content of the cell pellet used for lysis.
Determination of genome-wide DNA methylation and hydroxymethylation using isotope-dilution liquid chromatography tandem-mass spectrometry (LC-MS/MS)
HCT116 IDH1+/+ and HCT116 IDH1R132H/+ cells were treated with VC and ML309 in different concentrations as indicated for 24 h. The genomic DNA was then isolated with the QIAamp DNA Mini Kit (Qiagen, Hilden, Germany). Samples of genomic DNA (ranging between 10 and 20 µg) were hydrolysed to 2´-deoxynucleosides following a protocol previously described [69]. [15N2,13C1]dC, 5-mdC-d3 and 5-hmdC-d3 (all from Toronto Research Chemicals, Toronto, Canada) were used as internal standards for quantification of dC, 5-mdC and 5-hmdC in DNA hydrolyzates by isotope-dilution LC-MS/MS as reported recently [36]. Briefly, analytes were chromatographically separated with an Agilent 1260 Infinity LC system coupled to an Agilent 6490 triple quadrupole mass spectrometer (both from Agilent Technologies, Waldbronn, Germany) that was operated in multiple reaction monitoring (MRM) mode for detection of 2´-deoxynucleoside analytes. Precursor ions were generated by positive mode electrospray ionization (ESI+). The following mass transitions were used for quantification (accordant internal standard in parentheses): dC: m/z 228.1 (231.1) → 112.0 (114.9); 5-mdC: m/z 242.1 (245.1) → 126.0 (129.0) and 5-hmdC: m/z 258.1 (261.1) → 142.0 (145.0).
Apoptosis assessment via flow cytometric analysis
Levels of apoptotic and dead cells were determined by flow cytometry using eBioscience™ Annexin V Apoptosis Detection Kit APC (Thermo Fisher, Darmstadt, Germany). The differentiation between late-apoptotic or necrotic and early-apoptotic cells was facilitated by staining the cells with the fluorescent dye propidium iodide and allophycocyanin (APC)-coupled Annexin V prior to flow cytometric analysis [70]. For this purpose, HCT116 IDH1+/+ or IDHR132H/+ cells were seeded and treated with VC (1 mM) or ML309 (10 µM) alone and with combination of both for 48 h. Cells grown in unamended medium were used as vehicle control, and cells treated with TNFα and cycloheximide (20 ng/ml, 25 µg/ml, respectively) 24 h prior to cell harvest were used as positive control for the induction of apoptosis. The protein synthesis inhibitor cycloheximide was included in the positive control to induce pro-apoptotic effects of TNFα. Per run 10,000 events were counted and analysed on a FACSCanto II (BD Biosciences, Heidelberg, Germany). FlowJo software (Treestar, Ashland, USA) was used for data analysis.
RNA extraction and quantitative real-time PCR
Altered gene expression in treated or untreated HCT116 IDH1+/+ and HCT116 IDH1R132H/+ cells was analysed by means of quantitative real-time polymerase chain reaction (qPCR). The procedure included the treatment of cells, isolation of RNA, cDNA synthesis and qPCR, as previously described [36]. RNA extraction was performed with the RNA High Pure RNA Kit (Roche, Mannheim, Germany) according to the manufacturer’s instructions. The cDNA synthesis was then conducted using the RevertAid reverse transcriptase (Thermo Fisher, Darmstadt, Germany). Therefore, the synthesis of cDNA, 3 µg of isolated total RNA from treated and untreated HCT116 IDH1+/+ and HCT116 IDH1R132H/+ cells was brought to a volume of 11 µl with nuclease-free water, mixed with 1 µl oligo (dT)18 primers and incubated at 65°C for 5 min. Nuclease-free water (1 µl), 5X Reaction Buffer (4 µl), 10 mM dNTP mix (2 µl) and RevertAid M-MuLV Reverse Transcriptase (1 µl) were added to each sample and gently mixed. Samples were incubated at 45°C for 1 h, followed by a 5-min heating period at 70°C and cooling off at 4°C. The qRT-PCR was performed using the Maxima SYBR Green qPCR Mix (ThermoFisher, Darmstadt, Germany) on a LightCycler 480 II Real-Time PCR system (Roche, Mannheim, Germany). Quantification was performed using the ΔΔ Ct method with hHMBS expression as an internal reference. Melt curve analysis confirmed that all the qRT-PCR products were generated in the form of double-stranded DNA. An overview of the analysed target genes is given in Table 1. Forward and reverse primers were optimized or verified for optimization and obtained from Eurofins Genomics (Hamburg, Germany).
Table 1.
Primer sequences for the RT-qPCR.
| Target gene | Primer sequence (5´ – 3´) |
|---|---|
| 14-3-3s | forward: TAG GCG CTG TTC TTG CTC CAA reverse: ACC AGT GGT TAG GTG CGC TCA |
| BAD | forward: GAT CGG GCT TGG GGT GAG AC reverse: TCA TCT GTC TGC CGG GTC TG |
| BAX | forward: GAC ATT GGA CTT CCT CCG GG reverse: ACA GGG ACA TCA GTC GCT TC |
| DKK1 | forward: GGG TCT TTG TCG CGA TGG TA reverse: CGG GTA CGG CTG GTA GTT G |
| DNMT1 | forward: ACC TGG CTA AAG TCA AAT CC reverse: ATT CAC TTC CCG GTT GTA AG |
| DNMT3A | forward: ACT ACA TCA GCA AGC GCA AG reverse: CAT CCA CCA AGA CAC AAT GC |
| DNMT3B | forward: CCA GCT CTT ACC TTA CCA TC reverse: CAG ACA TAG CCT GTC GCT TG |
| GADD45a | forward: ACG ATC ACT GTC GGG GTG TA reverse: AAT GTG GAT TCG TCA CCA GCA |
| GLUT1 | forward: CTG CTC ATC AAC CGC AAC reverse: CTT CTT CTC CCG CAT CAT CT |
| GLUT3 | forward: CAG CGA GAC CCA GAG ATG reverse: TTG GAA AGA GCC GAT TGT AG |
| HMBS | forward: ACC AAG GAG CTT GAA CAT GC reverse: GAA AGA CAA CAG CAT CAT GAG |
| ITGA4 | forward: GTT TTC CAG AGC CAA ATC CA reverse: GCC AGC CTT CCA CAT AAC AT |
| NOXA | forward: GCT GGA AGT CGA GTG TGC TA reverse: GGA GTC CCC TCA TGC AAG TT |
| p16 | forward: GAG CAG CAT GGA GCC TTC reverse: CCT CCG ACC GTA ACT ATT CG |
| p21 | forward: AGT GGA CAG CGA GCA GCT GA reverse: TAG AAA TCT GTC ATG CTG GTC TG |
| p27 | forward: AAA CGT GCG AGT GTC TAA CGG GA reverse: CGC TTC CTT ATT CCT GCG CAT TG |
| p53 | forward: TGT GGG ATG GGG TGA GAT TTC Reverse: CTG TTG GTC GGT GGG TTG |
| PUMA | forward: GAC CTC AAC GCA CAG TAC GA Reverse: TAA GGG CAG GAG TCC CAT GA |
| SVCT1 | forward: TCA TCC TCC TCT CCC AGT ACC T reverse: AGA GCA GCC ACA CGG TCA |
| SVCT2 | forward: TCT TTG TGC TTG GAT TTT CGA T reverse: ACG TTC AAC ACT TGA TCG ATT C |
| TET1 | forward: GCT GCT GTC AGG GAA ATC AT reverse: ACC ATC ACA GCA GTT GGA CA |
| TET2 | forward: CCA ATA GGA CAT GAT CCA GG reverse: TCT GGA TGA GCT CTC TCA GG |
| TET3 | forward: TCG GAG ACA CCC TCT ACC AG reverse: CTT GCA GCC GTT GAA GTA CA |
| TFPI2 | Forward: GCA CAT GCA CGT TTG CAA TC Reverse: CCA GAT GAA GCT ACT TGT ATG |
| TIMP2 | forward: CCA GAA GAA GAG CCT GAA CCA reverse: GTC CAT CCA GAG GCA CTC ATC |
| TRAIL | forward: TGC GTG CTG ATC GTG ATC TT reverse: TCT TGG AGT CTT TCT AAC GAG C |
Statistical analysis
Statistical differences were determined by multiple t-tests between untreated and treated groups, where p-values ≤ 0.05 were considered as significant. Differences in apoptosis induction were calculated by 2 way ANOVA analysis, where p-values ≤ 0.05 were considered as significant. All statistical analyses were conducted using the software GraphPad Prism (GraphPad Software, Inc., La Jolla, USA).
Funding Statement
This work was supported by NutriAct – Competence Cluster Nutrition Research Berlin-Potsdam funded by the Federal Ministry of Education and Research (FKZ: 01EA1408A-B); Bundesministerium für Bildung und Forschung [01EA1408A-B].
Acknowledgments
This work was supported by NutriAct – Competence Cluster Nutrition Research Berlin-Potsdam funded by the Federal Ministry of Education and Research (FKZ: 01EA1408A-B). Special thanks go to Dr. Guy Yealland for language editing of the manuscript. We would like to thank Monika Haseloff and Caue Egea Rodrigues for the excellent technical assistance in conducting the experiments.
Disclosure statement
No potential conflict of interest was reported by the authors.
Author Contributions
CG and BK designed experiments; CG, FS and AB performed experiments; CG, FS, TH and AB analysed data; CG, FS, AB, TH and BK wrote the manuscript, CG and BK advised on experimental design and provided critical feedback, all authors reviewed the manuscript.
Supplementary material
Supplementary data for this article can be accessed here.
References
- [1].Baylin SB, Herman JG.. DNA hypermethylation in tumorigenesis: epigenetics joins genetics. Trends Genet. 2000. April;16(4):168–174. [DOI] [PubMed] [Google Scholar]
- [2].Eden A, Gaudet F, Waghmare A, et al. Chromosomal instability and tumors promoted by DNA hypomethylation. Science. 2003. April 18;300(5618):455. [DOI] [PubMed] [Google Scholar]
- [3].Baylin SB, Jones PA. Epigenetic determinants of cancer. Cold Spring Harb Perspect Biol. 2016. September 1;8(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Figueroa ME, Abdel-Wahab O, Lu C, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 2010. December 14;18(6):553–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Lu C, Ward PS, Kapoor GS, et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature. 2012. February 15;483(7390):474–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Zheng S, Houseman EA, Morrison Z, et al. DNA hypermethylation profiles associated with glioma subtypes and EZH2 and IGFBP2 mRNA expression. Neuro Oncol. 2011. March;13(3):280–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Tahiliani M, Koh KP, Shen Y, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009. May 15;324(5929):930–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Ito S, D’Alessio AC, Taranova OV, et al. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature. 2010. August 26;466(7310):1129–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Ito S, Shen L, Dai Q, et al. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science. 2011. September 2;333(6047):1300–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].He YF, Li BZ, Li Z, et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science. 2011. September 2;333(6047):1303–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Hu L, Li Z, Cheng J, et al. Crystal structure of TET2-DNA complex: insight into TET-mediated 5mC oxidation. Cell. 2013. December 19;155(7):1545–1555. [DOI] [PubMed] [Google Scholar]
- [12].Pastor WA, Aravind L, Rao A. TETonic shift: biological roles of TET proteins in DNA demethylation and transcription. Nat Rev Mol Cell Biol. 2013. June;14(6):341–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Lian CG, Xu Y, Ceol C, et al. Loss of 5-hydroxymethylcytosine is an epigenetic hallmark of melanoma. Cell. 2012. September 14;150(6):1135–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Delatte B, Deplus R, Fuks F. Playing TETris with DNA modifications. Embo J. 2014. June 2;33(11):1198–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Hore TA, von Meyenn F, Ravichandran M, et al. Retinol and ascorbate drive erasure of epigenetic memory and enhance reprogramming to naive pluripotency by complementary mechanisms. Proc Natl Acad Sci U S A. 2016. October 25;113(43):12202–12207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Minor EA, Court BL, Young JI, et al. Ascorbate induces ten-eleven translocation (Tet) methylcytosine dioxygenase-mediated generation of 5-hydroxymethylcytosine. J Biol Chem. 2013. May 10;288(19):13669–13674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Monfort A, Wutz A. Breathing-in epigenetic change with vitamin C. EMBO Rep. 2013. April;14(4):337–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Blaschke K, Ebata KT, Karimi MM, et al. Vitamin C induces Tet-dependent DNA demethylation and a blastocyst-like state in ES cells. Nature. 2013. August 8;500(7461):222–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Coulter JB, O’Driscoll CM, Bressler JP. Hydroquinone increases 5-hydroxymethylcytosine formation through ten eleven translocation 1 (TET1) 5-methylcytosine dioxygenase. J Biol Chem. 2013. October 4;288(40):28792–28800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Yin R, Mao SQ, Zhao B, et al. Ascorbic acid enhances Tet-mediated 5-methylcytosine oxidation and promotes DNA demethylation in mammals. J Am Chem Soc. 2013. July 17;135(28):10396–10403. [DOI] [PubMed] [Google Scholar]
- [21].Ko M, An J, Pastor WA, et al. TET proteins and 5-methylcytosine oxidation in hematological cancers. Immunol Rev. 2015. January;263(1):6–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Turcan S, Makarov V, Taranda J, et al. Mutant-IDH1-dependent chromatin state reprogramming, reversibility, and persistence. Nat Genet. 2018. January 01;50(1):62–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Balss J, Meyer J, Mueller W, et al. Analysis of the IDH1 codon 132 mutation in brain tumors. Acta Neuropathol. 2008. December;116(6):597–602. [DOI] [PubMed] [Google Scholar]
- [24].Bleeker FE, Lamba S, Leenstra S, et al. IDH1 mutations at residue p.R132 (IDH1(R132)) occur frequently in high-grade gliomas but not in other solid tumors. Hum Mutat. 2009. January;30(1):7–11. [DOI] [PubMed] [Google Scholar]
- [25].Losman JA, Kaelin WG Jr.. What a difference a hydroxyl makes: mutant IDH, (R)-2-hydroxyglutarate, and cancer. Genes Dev. 2013. April 15;27(8):836–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Miller JJ, Shih HA, Andronesi OC, et al. Isocitrate dehydrogenase-mutant glioma: evolving clinical and therapeutic implications. Cancer. 2017;123(23):4535–4546. [DOI] [PubMed] [Google Scholar]
- [27].Whitehall VL, Dumenil TD, McKeone DM, et al. Isocitrate dehydrogenase 1 R132C mutation occurs exclusively in microsatellite stable colorectal cancers with the CpG island methylator phenotype. Epigenetics. 2014. November;9(11):1454–1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Chowdhury R, Yeoh KK, Tian YM, et al. The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep. 2011. May;12(5):463–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Dang L, White DW, Gross S, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009. December 10;462(7274):739–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Koivunen P, Lee S, Duncan CG, et al. Transformation by the (R)-enantiomer of 2-hydroxyglutarate linked to EGLN activation. Nature. 2012. February 15;483(7390):484–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Xu W, Yang H, Liu Y, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell. 2011. January 18;19(1):17–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Noushmehr H, Weisenberger DJ, Diefes K, et al. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell. 2010. May 18;17(5):510–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Khurshed M, Molenaar RJ, Lenting K, et al. In silico gene expression analysis reveals glycolysis and acetate anaplerosis in IDH1 wild-type glioma and lactate and glutamate anaplerosis in IDH1-mutated glioma. Oncotarget. 2017. July 25;8(30):49165–49177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Grassian AR, Parker SJ, Davidson SM, et al. IDH1 mutations alter citric acid cycle metabolism and increase dependence on oxidative mitochondrial metabolism. Cancer Res. 2014. June 15;74(12):3317–3331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Izquierdo-Garcia JL, Viswanath P, Eriksson P, et al. Metabolic reprogramming in mutant IDH1 glioma cells. PLoS One. 2015;10(2):e0118781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Gerecke C, Schumacher F, Edlich A, et al. Vitamin C promotes decitabine or azacytidine induced DNA hydroxymethylation and subsequent reactivation of the epigenetically silenced tumour suppressor CDKN1A in colon cancer cells. Oncotarget. 2018. August 28;9(67):32822–32840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Mondesir J, Willekens C, Touat M, et al. IDH1 and IDH2 mutations as novel therapeutic targets: current perspectives. J Blood Med. 2016;7:171–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Saba HI. Decitabine in the treatment of myelodysplastic syndromes. Ther Clin Risk Manag. 2007. October;3(5):807–817. [PMC free article] [PubMed] [Google Scholar]
- [39].Seymour JF, Dohner H, Butrym A, et al. Azacitidine improves clinical outcomes in older patients with acute myeloid leukaemia with myelodysplasia-related changes compared with conventional care regimens. BMC Cancer. 2017. December 14;17(1):852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Schuh AC, Döhner H, Pleyer L, et al. Azacitidine in adult patients with acute myeloid leukemia. Crit Rev Oncol Hematol. 2017. August;116:159–177. [DOI] [PubMed] [Google Scholar]
- [41].Pleyer L, Dohner H, Dombret H, et al. Azacitidine for front-line therapy of patients with AML: reproducible efficacy established by direct comparison of international phase 3 trial data with registry data from the Austrian Azacitidine registry of the AGMT study group. Int J Mol Sci. 2017. February 15;18(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Pleyer L, Greil R. Digging deep into “dirty” drugs - modulation of the methylation machinery. Drug Metab Rev. 2015. May;47(2):252–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Younes M, Brown RW, Stephenson M, et al. Overexpression of Glut1 and Glut3 in stage I nonsmall cell lung carcinoma is associated with poor survival. Cancer. 1997. September 15;80(6):1046–1051. [DOI] [PubMed] [Google Scholar]
- [44].Chen X, Lu P, Zhou S, et al. Predictive value of glucose transporter-1 and glucose transporter-3 for survival of cancer patients: A meta-analysis. Oncotarget. 2017. February 21;8(8):13206–13213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Molenaar RJ, Maciejewski JP, Wilmink JW, et al. Wild-type and mutated IDH1/2 enzymes and therapy responses. Oncogene. 2018. April;37(15):1949–1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Ngo B, Van Riper JM, Cantley LC, et al. Targeting cancer vulnerabilities with high-dose vitamin C. Nat Rev Cancer. 2019. May;19(5):271–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Yun J, Mullarky E, Lu C, et al. Vitamin C selectively kills KRAS/BRAF mutant colorectal cancer cells by targeting GAPDH [10.1126/science.aaa5004]. Science. 2015;350(6266):1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Sant DW, Mustafi S, Gustafson CB, et al. Vitamin C promotes apoptosis in breast cancer cells by increasing TRAIL expression. Sci Rep. 2018. March 28;8(1):5306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Chen J, Guo L, Zhang L, et al. Vitamin C modulates TET1 function during somatic cell reprogramming. Nat Genet. 2013. December;45(12):1504–1509. [DOI] [PubMed] [Google Scholar]
- [50].Liu M, Ohtani H, Zhou W, et al. Vitamin C increases viral mimicry induced by 5-aza-2′-deoxycytidine. Proc Nat Acad Sci. 2016;113(37):10238–10244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Schleicher RL, Carroll MD, Ford ES, et al. Serum vitamin C and the prevalence of vitamin C deficiency in the United States: 2003–2004 National Health and Nutrition Examination Survey (NHANES). Am J Clin Nutr. 2009. November;90(5):1252–1263. [DOI] [PubMed] [Google Scholar]
- [52].Mayland CR, Bennett MI, Allan K. Vitamin C deficiency in cancer patients. Palliat Med. 2005. January;19(1):17–20. [DOI] [PubMed] [Google Scholar]
- [53].Gross S, Cairns RA, Minden MD, et al. Cancer-associated metabolite 2-hydroxyglutarate accumulates in acute myelogenous leukemia with isocitrate dehydrogenase 1 and 2 mutations. J Exp Med. 2010. February 15;207(2):339–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Yen KE, Bittinger MA, Su SM, et al. Cancer-associated IDH mutations: biomarker and therapeutic opportunities. Oncogene. 2010. December 9;29(49):6409–6417. [DOI] [PubMed] [Google Scholar]
- [55].Sharen G, Peng Y, Cheng H, et al. Prognostic value of GLUT-1 expression in pancreatic cancer: results from 538 patients. Oncotarget. 2017. March 21;8(12):19760–19767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Gonzalez-Menendez P, Hevia D, Mayo JC, et al. The dark side of glucose transporters in prostate cancer: are they a new feature to characterize carcinomas? Int J Cancer. 2018. June 15;142(12):2414–2424. [DOI] [PubMed] [Google Scholar]
- [57].Zhao H, Zhu H, Huang J, et al. The synergy of Vitamin C with decitabine activates TET2 in leukemic cells and significantly improves overall survival in elderly patients with acute myeloid leukemia. Leuk Res. 2018;66:1–7. [DOI] [PubMed] [Google Scholar]
- [58].Carr AC, Maggini S. Vitamin C and immune function. Nutrients. 2017. November 3;9(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Ang A, Pullar JM, Currie MJ, et al. Vitamin C and immune cell function in inflammation and cancer. Biochem Soc Trans. 2018. October 19;46(5):1147–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Nauman G, Gray JC, Parkinson R, et al. Systematic review of intravenous ascorbate in cancer clinical trials. Antioxidants (Basel). 2018. July 12;7(7):89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Kuiper C, Vissers MC. Ascorbate as a co-factor for fe- and 2-oxoglutarate dependent dioxygenases: physiological activity in tumor growth and progression. Front Oncol. 2014;4:359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Schoenfeld JD, Sibenaller ZA, Mapuskar KA, et al. O2(-) and H2O2-mediated disruption of Fe metabolism causes the differential susceptibility of NSCLC and GBM cancer cells to pharmacological ascorbate. Cancer Cell. 2017. April 10;31(4):487–500 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Mustafi S, Camarena V, Volmar CH, et al. Vitamin C sensitizes melanoma to BET inhibitors. Cancer Res. 2018. January 15;78(2):572–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Ma Y, Chapman J, Levine M, et al. High-dose parenteral ascorbate enhanced chemosensitivity of ovarian cancer and reduced toxicity of chemotherapy. Sci Transl Med. 2014. February 5;6(222):222ra18. [DOI] [PubMed] [Google Scholar]
- [65].Fernandez SL, Russell DW, Hurlin PJ. Development of human gene reporter cell lines using rAAV mediated homologous recombination. Biol Proced Online. 2007. December;24(9):84–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Davis M, Pragani R, Popovici-Muller J, et al. ML309: A potent inhibitor of R132H mutant IDH1 capable of reducing 2-hydroxyglutarate production in U87 MG glioblastoma cells. Probe Reports from the NIH Molecular Libraries Program. Bethesda (MD). 2010. [PubMed] [Google Scholar]
- [67].Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983. December 16;65(1–2):55–63. [DOI] [PubMed] [Google Scholar]
- [68].Berger RS, Ellmann L, Reinders J, et al. Degradation of D-2-hydroxyglutarate in the presence of isocitrate dehydrogenase mutations. Sci Rep. 2019. May 15;9(1):7436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Schumacher F, Herrmann K, Florian S, et al. Optimized enzymatic hydrolysis of DNA for LC-MS/MS analyses of adducts of 1-methoxy-3-indolylmethyl glucosinolate and methyleugenol. Anal Biochem. 2013. March 1;434(1):4–11. [DOI] [PubMed] [Google Scholar]
- [70].Vermes I, Haanen C, Steffens-Nakken H, et al. A novel assay for apoptosis. Flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labelled Annexin V. J Immunol Methods. 1995. July 17;184(1):39–51. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.