

Abstract
The limited therapeutic options and increasing drug-resistance call for next-generation of influenza antivirals. Due to the essential function in viral replication and high sequence conservation among influenza viruses, influenza polymerase PA-PB1 protein-protein interaction becomes an attractive drug target. Here, we developed an in vitro split luciferase complementation-based assay to speed up screening of PA-PB1 interaction inhibitors. By screening 10,000 compounds, we identified two PA-PB1 interaction inhibitors, R160792 and R151785, with potent and broad-spectrum antiviral activity against a panel of influenza A and B viruses, including amantadine -, oseltamivir-, or dual resistant strains. Further mechanistic study reveals that R151785 inhibits PA nuclear localization, reduces the levels of viral RNAs and proteins, and inhibits viral replication at intermediate stage, all of which are in line with its antiviral mechanism of action. Overall, we developed a robust high throughput-screening assay for screening broad-spectrum influenza antivirals targeting PA-PB1 interaction and identified R151785 as a promising antiviral drug candidate.
Graphical Abstract
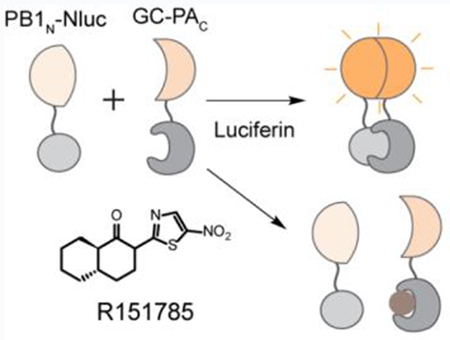
INTRODUCTION
Influenza viruses are negative-sense, segmented single-strand RNA viruses and belong to Orthomyxoviruses family.1 Influenza viruses are responsible for seasonal influenza as well as sporadic influenza pandemic that cause high morbidity and mortality worldwide. Seasonal influenza is estimated to result in about 3 to 5 million cases of severe illness, and about 290 000 to 650 000 respiratory deaths worldwide [https://www.who.int/news-room/fact-sheets/detail/influenza-(seasonal)]. In United States, it is estimated that the seasonal influenza has resulted in 9.3 million to 49.0 million illnesses, 140,000 to 960,000 hospitalizations and 12,000 to 79,000 deaths annually since 2010 (https://www.cdc.gov/flu/about/burden/index.html). In addition, the past pandemics of 1918 (H1N1), 1957-1958 (H2N2), and 1968 (H3N2) caused millions of deaths worldwide.2 In particular, the 1918 influenza pandemic was responsible for the deaths of approximately 40 million people worldwide,3 and the most recent 2009 H1N1 pandemic caused an estimated 201,200 deaths globally.4
Currently, influenza infections can be controlled by vaccination and antiviral drugs.5 Vaccination is the most common prophylaxis with an overall 60% effectiveness. However, vaccines have limited efficacy in immune-compromised patients and have to be re-formulated annually due to the changes of the circulating influenza viruses in each season. By now, there are three classes of FDA-approved anti-influenza drugs: two M2 ion channel blockers, amantadine and rimantadine,6 three neuraminidase (NA) inhibitors, oseltamivir, zanamivir, and peramivir , and one PA cap-dependent endonuclease inhibitor, baloxavir marboxil, which was approved for use in the United States in October of 2018.7 Due to the high mutation rate of influenza virus, the emergence of drug resistant influenza virus strain is common and also difficult to predict. In reality, the use of M2 inhibitors amantadine and rimantadine is no longer recommended because of the widespread of M2-S31N mutation among circulating influenza A viruses.8,9 Moreover, resistance to oseltamivir has been continuously reported and the 2008-2009 seasonal influenza H1N1 was nearly completely resistant to oseltamivir due to the NA-H274Y mutation.10 For the most recent approved drug, baloxavir marboxil, resistance has been reported in both clinical trials and patients treated with this drug.11 Thus, next generation of broad-spectrum antiviral drugs with a novel mechanism of action and a high genetic barrier to drug resistance is urgently needed for the prevention and treatment of drug-resistant or newly emerging influenza viruses.12
Influenza polymerase plays a central role in virus replication: it is responsible for the synthesis of viral mRNAs and viral genomic RNA (vRNA) via an intermediate complementary RNA (cRNA). 13 Influenza polymerase is a heterotrimeric complex consisting of three subunits, PA, PB1, and PB2 (Figure 1A). The three subunits forms a non-covalent assembly to exert the polymerase function. Due to the multiple enzymatic functions as well as highly conserved protein sequence and 3D structure among influenza A and B stains, influenza polymerase has become as an attractive antiviral drug target.14–17 Specifically, the PA endonuclease, PB1 polymerase, and PB2 cap-binding domains are all valid antiviral targets and have been actively pursued for developing new antiviral therapies.18 Of note, the recently approved drug, baloxavir marboxil, is a PA endonuclease inhibitor. T-705 targets the RNA-dependent RNA polymerase PB1. The PB2 cap-binding inhibitor, JNJ-63623872, inhibits the cap-snatching function of PB2. Both T-705 and JNJ-63623872 are currently in human clinical trials.
Figure 1. X-ray crystal structures of influenza polymerase complex.

(A) X-ray crystal structure of the bat influenza A/H17N10 virus polymerase complex (PDB: 4WSB).15 PA: lightblue, PB1: green, PB2: yellow. (B) X-ray crystal structure of PAC-PB1N (PDB: 3CM8).19 PAC: lightblue, PB1N: green.
In addition, protein-protein interactions play pivotal roles in the formation of influenza virus polymerase complex. Therefore, inhibition of influenza polymerase’s function through disruption of its subunit protein-protein interactions, such as the PA-PB1 interaction and PB1-PB2 interaction, has been proved to be an attractive strategy for developing broad-spectrum antiviral drugs against different influenza virus strains.18, 20, 21 Among the drugs in development targeting influenza virus polymerase subunit interactions, PA-PB1 inhibitors were the first-in-class and were actively pursued. Aided by the crystal structures of PA and PB1 (Figure 1B), structure-based virtual screening has been performed, leading to the discovery a number of hit compounds.22–25 In addition, enzyme-linked immunosorbent assay (ELISA) and cell-based Split Luciferase Complementation (SLC) assay have been successfully applied to screen influenza polymerase subunit interactions inhibitors.26 However, both ELISA and cell-based SLC assays suffer intrinsic disadvantages. ELISA requires expensive antibodies and time-consuming coating, blocking, and washing steps, thus not suitable for high-throughput screening (HTS). Whereas the cell-based SLC assay might identify false positives that inhibit protein synthesis or false negatives that are cell cytotoxic. To overcome these limitations and speed up drug discovery, we developed an in vitro SLC-based HTS assay to identify inhibitors that could directly inhibit the PA-PB1 interactions. Despite numerous SLC systems have been used in cell-based assay, there are only a few studies on in vitro SLC system using firefly luciferase due to the issue of protein purification or stability,27,28,29,30. Therefore, firefly luciferase was chosen for our in vitro SLC-based HTS assay. Using this assay, we identified two potent PA-PB1 interaction inhibitors with broad-spectrum antiviral activity against both influenza A and B viruses. The mechanism of action of the most potent hit, R151785, was further confirmed in ELISA assay as well as cellular PA localization assay, time-of-addition, western blot and RT-qPCR assays. Collectively, the in vitro SLC assay appears to be suitable for HTS of influenza polymerase PA-PB1 inhibitors, and targeting influenza polymerase PA-PB1 subunit interaction is a promising strategy in developing broad-spectrum antiviral drugs.
RESULTS AND DISCUSSION
Development of an in vitro SLC-based PA-PB1 interaction assay.
To explore small molecules that could abolish the interaction between influenza PA and PB1, we aimed to develop an in vitro SLC-based PA-PB1 interaction assay (Figure 2A). The principle of SLC assay is that the luciferase enzymatic activity of the firefly luciferase will only be restored if the N-terminal luciferase fragment (Nfluc) and C-terminal luciferase fragment (Cfluc) assemble in a native form. The driving force of this assembly is the interaction between the fusion partner proteins PAC and PB1N (Figure 2A). Key parameter to the success of this assay is the direction of the fusion proteins, meaning that both the Nfluc-Cfluc and the PAC-PB1N interactions should not be affected in the fusion constructs. After examining the X-ray crystal structure of PAC-PB1N (PDB: 3CM8)31 (Figure 1B), we decided to construct and express three fusion partners: GST-Cfluc-PAC (GC-PAC), PB1N-Nfluc (PB1N-Nluc), and Nfluc-PB1N (Nluc-PB1N) (Figure 2B). The design was based on the X-ray crystal structure of PA-PB1 (Figure 1B) and it is important to make sure Nfluc-Cfluc interactions will not interfere with PA-PB1 interactions. Therefore, we chose to attach the fusion proteins far away from the PA-PB1 interaction site. In general, SLC assay is used in the cellular assay or in cell lysates without purification, probably due to the instability of the Nfluc or Cfluc. Nevertheless, we overcame this difficulty and were able to successfully express and purify all three fusion proteins (Figure 2C). In the next step, we set to test either GC-PAC/PB1N-Nluc pair or the GC-PAC/Nluc-PB1N pair will restore the luciferase activity of the firefly luciferase. As shown in Figure 2D, none of these fusion proteins alone produced detectable luminescence. Gratifyingly, GC-PAC/PB1N-Nluc pair gave the highest luminescence signal. The GC-PAC/Nluc-PB1N pair also restored the firefly luciferase activity, but the signal was much weaker, suggesting this is not an ideal combination. Given these results, GC-PAC/PB1N-Nluc pair was chosen for the following experiments. The next step is to establish the assay condition for the HTS. When the GC-PAC was fixed at a concentration of 60 nM, we observed a dose-dependent SLC signal increase with continuous titration of PB1N-Nluc (Figure 2E). As such, the final concentrations of GC-PAC and PB1N-Nluc were both set as 60 nM. Finally, to verify the specificity of the assay, which is an important factor for any HTS assay, we tested the inhibitory effect of a known inhibitor of PAC-PB1N interaction, the PB1N peptide (PB11-15), in the established in vitro SLC-based PA-PB1 interaction assay. As expected, PB11-15 reduced the SLC signals in a dose-dependent manner with an IC50 value of 0.14 ± 0.08 μM (Figure 2F). Taken together, our results indicate that the luminescence signal from GC-PAC/ PB1N-Nluc pair is specifically due to the interaction between PAC and PB1N, and this assay could be used to screen small molecule inhibitors of PAC and PB1N protein-protein interaction.
Figure 2. The in vitro PA-PB1 SLC assay.

(A) Cartoon representation of the assay principle for SLC assay. (B) The influenza PAC/PN1BN SLC constructs. GST-Cfluc (aa 398-550)-PAC aa 239-716 (named as GC-PAC), PB1N (aa 1-25)-Nfluc (aa 2-415) (named as PB1N-Nluc), and Nfluc (aa 2-415)- PB1N (aa 1-25) (named as Nluc-PB1N). (C) SDS-PAGE gel showing purified GC-PAC, PB1N-Nluc and Nluc-PB1N. (D) The positions of Nluc and Cluc affect luciferase activity. Equal concentrations (100 nM) of each pair of PAC-PB1N constructs were mixed (or alone) and incubated with luciferin substrate. (E) Dose-response of GC-PAC/PB1N-Nluc pair. GC-PAC at 60 nM was included in each experiment. Concentration of PB1N-Nluc was varied as indicated. (F) The influenza PB1 aa 1-15 peptide, PB11-15, inhibited the SLC in a dose-dependent manner. In all graphs, means and SD from triplicate experimental data were shown.
Discovery of potent PA-PB1 interaction inhibitors by HTS.
Next, we optimized the in vitro SLC-based PAC-PB1N interaction assay in 96-well plate format for HTS (Figure 3A). To evaluate the assay, PB11-15 peptide and DMSO was used as positive control and negative control, respectively. As shown in Figure 3B, 10 μM of PB11-15 significantly and robustly inhibited SLC with a significant signal/background (S/B) ratio of 159 ± 20. The calculated Z-factor is 0.81, which is far more than 0.5, indicating this is an excellent HTS assay. Upon completion of optimization, we performed an HTS of 10,000 of compounds from the MyriaScreen Diversity Collection. Along with the screening, 11 control plates were included to monitor the quality of screening. All the values of Z’-factor are > 0.5 with an average of 0.76 ± 0.03, indicating a high quality of HTS (Figure 3C). In the primary screening, a total of 105 compounds were identified that had more than 95% inhibition at 20 μM. These compounds were subsequently tested for cellular antiviral activity in virus-induced cytopathic effect (CPE) assay. The purpose was to rule out compounds that are either cell membrane impermeable or are cytotoxic. Two compounds, R160792 and R151785 (Figure 3D), which shared similar structure, showed potent antiviral activity against A/WSN/33 (H1N1) virus at 10 μM (> 70% protection, data not shown). To verify the screening results, we performed dose-response analyses of R160792 and R151785 in the in vitro SLC-based PAC-PB1N interaction assay. As shown in Figure 3E, both R160792 and R151785 showed inhibitory activity in a dose-dependent manner with IC50 values of 12.15 and 4.78 μM, respectively. To further confirm the direct inhibition of PAC-PB1N interaction by R160792 and R151785, we tested these two compounds in the ELISA assay (Figure 3F), which has been widely used to identify the PA-PB1 interaction inhibitors. Similarly, both R160792 and R151785 shows inhibitory activity in a dose-dependent manner with IC50 values of 20.47 and 18.54 μM, respectively. As a positive control, the PB11-15 peptide had an IC50 value of 0.26 μM in the ELISA assay. Taken together, these in vitro assay results confirmed the inhibitory activity of R160792 and R151785 in PA-PB1 interaction.
Figure 3. Screening of potent PA-PB1 interaction inhibitors by HTS.

(A) Illustration of HTS with in vitro PA-PB1 SLC assay. The procedure was detailed in METHODS. Left and right panels represent the compounds without and with inhibitory activity, respectively. (B) Evaluation of HTS using PB11-15 (10 μM) and DMSO as positive and negative controls. Means and SD, Z’ factor and S/B (signal/background) from two experiments with 16 repeats were shown. Asterisks indicate statically significant difference compared to DMSO (***, p < 0.001; student’s t-test). (C) Z’ factors of 11 control plates. (D) Chemical structures of R160792 and R151785. (E) IC50 curves of R160792 and R151785 in the SLC assay. (F) IC50 curves of R160792 and R151785 in the PA-PB1 interaction ELISA assay. PB11-15 was served as a positive control. In E and F, means and SD from triplicate experimental data were shown.
R160792 and R151785 had potent antiviral activity against both influenza A and B viruses.
To investigate the antiviral effects of compounds, we first tested the cytotoxicity of R160792 and R151785 in MDCK cells. The calculated CC50 values of R160792 and R151785 were 132 ± 32 μM and > 300 μM, respectively (Table 1). We then tested the antiviral activities of R160792 and R151785 against a panel of human influenza A and B viruses, including multiple drug-resistant strains, such as the amantadine-resistant strains, A/WSN/1933, A/California/07/2009 (H1N1), A/Switzerland/9715293/2013 (H3N2), B/Wisconsin/1/2010 (Yamagata), and B/Brisbane/60/2008 (Victoria), and the oseltamivir and amantadine dual resistant strains, A/North Carolina/39/2009 (H1N1), A/Washington/29/2009 (H1N1), A/Texas/4/2009 (H1N1), and A/Denmark/528/2009 (H1N1) (Figure 4 A and Table 1), which pose greater threat to public health.32 Despite the diverse genetic background and drug sensitivity, the replication of all the tested viruses was inhibited by R160792 and R151785 in a dose-dependent manner, suggesting a broad-spectrum antiviral activity of R160792 and R151785. Although R160792 and R151785 share similar structure, R151785 had better antiviral activity, less cellular cytotoxicity, and, thus, a higher SI (selection index; ratio of CC50/EC50) than that of R160792 (Table 1), therefore, R151785 was selected for the following studies
Table 1.
EC50 values of R160792 and R151785 against influenza A or B virus.
| Influenza Strains | Drug sensitivity | EC50 (μM)a | SIb | ||
|---|---|---|---|---|---|
| R160792 | R151785 | R160792 | R151785 | ||
| A/Solomon Islands/03/2006 (H1N1) | Amantadine Sensitive Oseltamivir Sensitive | 2.89 ± 0.25 | 0.93 ± 0.11 | 45.7 | > 322.6 |
| A/WSN/1933 (H1N1) | Amantadine Resistant Oseltamivir Sensitive | 6.65 ± 0.48 | 4.66 ± 0.22 | 19.9 | > 64.4 |
| A/California/07/2009 (H1N1) | 2.45 ± 0.15 | 1.43 ± 0.18 | 53.9 | > 209.8 | |
| A/Switzerland/9715293/2013 (H3N2) | 3.09 ± 0.13 | 2.71 ± 0.32 | 42.8 | > 110.1 | |
| A/North Carolina/39/2009 (H1N1) | Amantadine Resistant Oseltamivir Resistant | 4.63 ± 0.53 | 1.59 ± 0.07 | 28.6 | > 188.7 |
| A/Washington/29/2009 (H1N1) | 5.34 ± 0.26 | 2.26 ± 0.37 | 24.8 | > 132.7 | |
| A/Texas/04/2009 (H1N1) | 7.54 ± 0.41 | 2.27 ± 0.32 | 17.5 | > 132.1 | |
| A/Denmark/528/2009 (H1N1) | 5.09 ± 0.81 | 1.75 ± 0.10 | 25.9 | > 171.4 | |
| B/Wisconsin/01/2010 (Yamagata) | Amantadine Resistant Oseltamivir Sensitive | 3.81 ± 0.33 | 3.68 ± 0.31 | 34.7 | > 81.5 |
| B/Brisbane/60/2008 (Victoria) | 2.91 ± 0.11 | 1.33 ± 0.07 | 45.4 | > 225.6 | |
EC50 was determined in the plaque assay using MDCK or AX-4 cells. Plaque areas were quantified for calculation of EC50 using Image J and Prism 7;
CC50 values of R160792 and R151785 were 132 ± 32 μM and > 300 μM, respectively, in MDCK cells. SI is selectivity index, which was calculated by CC50/EC50. Means and SD from two experiments in duplicate or triplicate were shown.
Figure 4. R151785 inhibits both influenza A and B viruses.

(A) Antiviral activity of R151785 against representative influenza A and B viruses. The antiviral activity of R151785 was determined in plaque reduction assay. Monolayers of MDCK or AX4 cells were infected with indicated viruses and incubated with overlays containing a serial of concentrations of R151785, 1 μM of amantadine or 0.2 μM of oseltamivir carboxylate. (B and C) Antiviral activity of R151785 against influenza virus at low to high MOIs. MDCK cells were infected A/California/ 07/2009 (H1N1) (B) and A/Switzerland/9715293/2013 (H3N2) (C) at different MOIs. The MOI of virus and concentration of compound were shown. The images shown were representative results from two repeats.
Next, we further validated the antiviral activity of R151785 in MDCK cells infected with either the A/California/07/2009 (H1N1) virus or the A/Switzerland/9715293/2013 (H3N2) virus at low to high multiplicity of infections (MOIs). Infection with high MOIs mimics delayed treatment when the virus was already amplified in the host. It was found that R151785 significantly suppressed the viral replication even when MOI was as high as 4.9 × 10−1 or 1.8 × 10−1 for both influenza strains (Figure 4B and 4C). In contrast, oseltamivir carboxylate only showed antiviral activity at low MOIs (1.8 × 10−3 or less) but not at high MOIs (1.8 × 10−2 or higher). Overall, R151785 showed potent inhibition against human H1N1 and H3N2 influenza A strains at both low and high MOIs and had superior antiviral efficacy than oseltamivir at higher MOIs.
Antiviral mechanism of R151785.
To gain insights into the cellular antiviral mechanism of R151785, we first confirmed the inhibitory activity of R151785 on PA-PB1 interaction in a cellular context. It is known that transportation of PA and PB1 from cytosol into nucleus requires formation of PA-PB1 complex.33 Blockade of PA-PB1 interaction leads to cytoplasmic accumulation of PA.22 Thus, we transfected HEK 293T cells with a plasmid expressing PA-GFP fusion protein alone or along with a plasmid expressing PB1 and analyzed the subcellular localization of PA-GFP in the presence or absence of R151785. As shown in Figure 5A, PA-GFP largely accumulated in cytosol when it was expressed alone (first panel), whereas PA-GFP was mainly accumulated in nucleus when PA and PB1 were co-expressed in cells (second panel), consistent with previous report.33 Similarly, DMSO treatment had no effect on PA nuclear localization (third panel), whereas treatment with R151785 largely reduced the PA nuclear localization in PA-PB1 co-expressing cells (fourth panel), indicating an inhibitory activity of R151785 against PA-PB1 interaction in the cellular context.
Figure 5. Cellular antiviral mechanism of R151785.

(A) R151785 blocks intracellular PA-PB1 interaction. HEK 293T cells were transfected with the PA-GFP-expressing plasmid alone or along with PB1-expressing plasmid. DMSO or R151785 (30 μM) was added into the cell culture at 6 h post-transfection. At 30 h post-transfection, cells were fixed and stained with DAPI for imaging. Bar = 10 μm. (B-E) Time-of-addition experiment. MDCK cells were infected with A/California/ 07/2009 (H1N1) (MOI = 0.01) at −2 h. After 1-hour incubation at 4 °C for attachment and another 1 h at 37 °C for viral entry, cells were washed with PBS buffer and incubated with DMEM at 37 °C for 12 h, then viruses in culture supernatant were harvested for titration by plaque assay. Oseltamivir carboxylate (1 μM) (C), Baloxavir (30 nM) (D), or R151785 (15 μM) (E) was present during the time as illustrated in (B). ND means No Drug. Data represents mean ± SE from three independent experiments. Asterisks indicate statistically significant difference in comparison with ND (**, p < 0.01, ***, p < 0.001; student’s t-test). (F and G) R151785 reduces viral RNA (F) and protein (G) levels. MDCK cells were infected with A/WSN/33 (H1N1) virus (MOI = 1) and followed by treatment with DMSO, 3, 10 or 30 μM of R151785 for 6 h. Total RNA or protein was extracted for detection by RT-qPCR or western blot. Nucleozin (10 μM) was included as a control. Data represents mean ± SE from three independent experiments. Asterisks indicate statistically significant difference in comparison with DMSO control. *, p < 0.05; **, p < 0.01; ***, p < 0.001 (student’s t-test). (H) R151785 inhibits influenza replication. MDCK cells were infected with A/WSN/33 (H1N1) virus at MOI of 1. Cells were fixed at 6 h p. i. and stained with mouse anti-influenza A NP antibody, rabbit anti-M1 and DAPI to determine the NP, M1, and nucleus, respectively. Bar = 25 μm.
As R151785 inhibits the influenza polymerase PA-PB1 interaction, it is expected to inhibit the intermediate stage of viral replication post viral fusion in the time-of-addition experiment. Furthermore, when the viral polymerase is affected, viral RNA transcription and replication should be reduced, and thus result in reduced expression levels of viral proteins.25 Based on this, we first investigate which stage(s) of viral replication was affected by R151785 treatment. We performed a time-of-addition experiment, in which R151785 was added into culture medium at different time points during the viral replication (Figure 5B–5E). It was found that the efficacy of R151785 gradually decreased when it was added at later stages of viral replication (Figure 5E). Although the PA endonuclease inhibitor, baloxavir, completely inhibited virus replication when it was added at early time points, its efficacy was also gradually reduced when it was added at later time points, suggesting both compounds take effect at similar stage (Figure 5D). In contrast, oseltamivir carboxylate retained potent antiviral activity even when it was added 8 h post viral infection (Figure 5C), consistent with its known mechanism of inhibiting viral egress. Next, to test the effect of R151785 on viral RNA transcription and replication, we performed RT-qPCR assay. Quantification of the viral nucleoprotein (NP) RNA expression levels by RT-qPCR revealed that the levels of vRNA, cRNA, and mRNA were all inhibited in a dose-dependent manner by R151785 (Figure 5F). Furthermore, to test the effect of R151785 on viral protein level, we analyzed the expression levels of several viral proteins by western blot and immunofluorescence assays (Figure 5G and 5H). Compared to DMSO control, R151785 significantly reduced the protein expression levels of HA, M1 and NP in a dose dependent manner (Figure 5G). Similar results were also observed in immunofluorescence assay (Figure 5H). R151785 significantly reduced NP and M1 protein expression levels in a dose dependent manner. Taken together, the results of PA subcellular localization, time-of-addition experiment, RT-qPCR, western blot and immunofluorescence assays are consistent with the antiviral mechanism of R151785 by inhibiting the viral polymerase PA-PB1 interactions.
Docking model of compound R151785 in PAC.
To gain molecular details on how R151785 inhibits the PA-PB1 interaction, we performed molecular docking using Schroödinger Glide software. In the docking model of R151785 in PA, several key interactions were observed (Figure 6): the ketone from R151785 formed a hydrogen bond with the K643 side chain ammonium, the nitro group from R151785 formed another hydrogen bond with the Q408 side chain amide, the thiazole from R151785 formed π-π interaction with the W706 indole ring, and the hydrophobic decalin ring from R151785 fitted into a hydrophobic pocket formed by F658, F707, and F710. Overall, the docking model suggests the high affinity binding of R151785 to the PAC.
Figure 6. Docking model of R151785.

X-ray crystal structure of PA-PB1 (PDB: 3CM8) was used for the docking. (A) Best docking pose of R151785 in PAC. (B) Surface view of the docking model of R151785 in PAC. (C) Ligand interaction diagram of R151785 with residues in the binding site of PAC.
CONCLUSION
Despite the availability of antiviral medications and influenza vaccines, influenza virus infection still causes substantial morbidity and mortality worldwide each year. Due to the reassortment of the virus genome and the error-prone influenza polymerase, the rapid emergence of new influenza virus or drug resistant strains is common. Thus, there is an urgent and continuous need for next generation of antiviral drugs with novel mechanisms of action and a high genetic barrier to drug resistance. Inhibitors targeting polymerase subunit PA-PB1 interaction have shown to have potent and broad-spectrum antiviral activity. Moreover, compared to current FDA approved drugs, this class of inhibitors have no cross resistance with existing influenza antivirals, high genetic barrier to drug resistance, and broad-spectrum antiviral activity against different types and subtypes of influenza A and B viruses. Therefore, PA-PB1 interaction inhibitors have been actively pursued as next generation of antiviral drugs.22–25, 34–38
Drug discovery is a time-consuming and expensive endeavor. To speed up screening new PA-PB1 interaction inhibitors and reduce the cost, we developed an in vitro SLC-based assay for robust HTS (Figure 2 and 3). The SLC-based assay could also be applied for testing other influenza viral PPIs, such as PB1-PB2 interaction and PA-PB2 interaction. Currently there are several other approaches that are available to detect the viral polymerase subunit interactions, such as two-hybrid 39–42, co-immunoprecipitation,43–45 pull-down assay22, 46, Bimolecular Fluorescence Complementation (BiFC),47 SLC,48 and ELISA20, 22, 35, 49. However, pull-down assay and co-IP assay are not suitable for HTS. Although ELISA and the cell-based assays such as the two-hybrid, BiFC and SLC could be adapted to or have been used for HTS, they all have intrinsic limitations: ELISA is time-consuming and expansive, while the above mentioned cell-based assays might select false positives that are toxic to cells or false negatives that are not cell membrane permeable. In contrast, the in vitro SLC-based assay described in this study has overcome above laminations. Since the fusion proteins can be individually expressed and purified, their concentrations can be accurately quantified, therefore ensuring consistent results between different batches of assays. This simple “mix and measure” assay is easy to preform and highly reproducible (Figure 3). Once the proteins were obtained, one assay could be finished in several hours. In addition, it can be adapted in 384-well or 1,536-well plate format to further increase its robustness and throughput. Therefore, this cost-effective and robust assay is ideal for HTS.
As a proof-of concept, a pilot screening of a library of 10,000 compounds using our assay yielded two PA-PB1 interaction inhibitors, R160792 and R151785, with potent cellular antiviral activity. Their inhibitory activity against PA-PB1 interactions has been confirmed by ELISA and cellular mechanistic study (Figures 3 and 5). Both R160792 and R151785 shows potent antiviral activity against amantadine-, oseltamivir-, or double resistant strains. Since both PA and PB1 interaction domains are highly conserved among different subtypes of influenza A and B viruses, both R160792 and R151785 have shown broad-spectrum antiviral activity against different subtypes of influenza A and B viruses (Figure 4A and Table 1).
In conclusion, we have developed a robust HTS method specifically for screening inhibitors of influenza polymerase PA-PB1 interaction. One of the PA-PB1 interaction inhibitor, R151785, with low-micromolar efficacy, broad-spectrum antiviral activity and a high selection index could be further developed to a next generation of therapeutic agent.
METHODS
Detailed information for all methods used can be found in the supporting information.
Supplementary Material
ACKNOWLEDGEMNTS
We thank I. Ghosh for providing pcDNA3.1-Kz-FKBP-27aa-Nfluc and pCDNA3.1-Kz-CFluc-25aa-FRB plasmids, H. Li for providing pGEX-6P-1-GST-Cluc398-NS3 and pET28a-NLuc416-NS2B-E66_stop plasmids, E. Fodor for providing pcDNA-PA-GFP and pcDNA-PB1 plasmids. This work was supported by the University of Arizona startup fund, the Arizona Biomedical Research Centre Young Investigator grant, and the NIH AI 119187 and AI 144887 to J.W.
Footnotes
Supporting Information: This material is available free of charge via the internet at http://pubs.acs.org.
Methods used in this study
REFERENCE
- 1.Braciale TJ, and Webster RG (2013) Textbook of Influenza, Textbook of Influenza, 2nd Edition, 479–482. [Google Scholar]
- 2.Cox NJ, and Subbarao K (2000) Global epidemiology of influenza: past and present, Annu Red Med 51, 407–421. [DOI] [PubMed] [Google Scholar]
- 3.Johnson NP, and Mueller J (2002) Updating the accounts: global mortality of the 1918-1920 “Spanish” influenza pandemic, Bull Hist Med 76, 105–115. [DOI] [PubMed] [Google Scholar]
- 4.Dawood FS, Iuliano AD, Reed C, Meltzer MI, Shay DK, Cheng PY, Bandaranayake D, Breiman RF, Brooks WA, Buchy P, Feikin DR, Fowler KB, Gordon A, Hien NT, Horby P, Huang QS, Katz MA, Krishnan A, Lal R, Montgomery JM, Molbak K, Pebody R, Presanis AM, Razuri H, Steens A, Tinoco YO, Wallinga J, Yu H, Vong S, Bresee J, and Widdowson MA (2012) Estimated global mortality associated with the first 12 months of 2009 pandemic influenza A H1N1 virus circulation: a modelling study, Lancet Infect Dis 12, 687–695. [DOI] [PubMed] [Google Scholar]
- 5.Loregian A, Mercorelli B, Nannetti G, Compagnin C, and Palu G (2014) Antiviral strategies against influenza virus: towards new therapeutic approaches, Cell Mol Life Sci 71, 3659–3683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang J, Li F, and Ma C (2015) Recent progress in designing inhibitors that target the drug-resistant M2 proton channels from the influenza A viruses, Biopolymers 104, 291–309. [DOI] [PubMed] [Google Scholar]
- 7.Zhang J, Hu Y, Hau R, Musharrafieh R, Ma C, Zhou X, Chen Y, and Wang J (2019) Identification of NMS-873, an allosteric and specific p97 inhibitor, as a broad antiviral against both influenza A and B viruses, Eur J Pharm Sci 133, 86–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bloom JD, Gong LI, and Baltimore D (2010) Permissive secondary mutations enable the evolution of influenza oseltamivir resistance, Science 328, 1272–1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bright RA, Shay DK, Shu B, Cox NJ, and Klimov AI (2006) Adamantane resistance among influenza A viruses isolated early during the 2005-2006 influenza season in the United States, JAMA-J Am Med Assoc 295, 891–894. [DOI] [PubMed] [Google Scholar]
- 10.Gubareva LV, Besselaar TG, Daniels RS, Fry A, Gregory V, Huang W, Hurt AC, Jorquera PA, Lackenby A, Leang SK, Lo J, Pereyaslov D, Rebelo-de-Andrade H, Siqueira MM, Takashita E, Odagiri T, Wang D, Zhang W, and Meijer A (2017) Global update on the susceptibility of human influenza viruses to neuraminidase inhibitors, 2015-2016, Antiviral Res 146, 12–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hayden FG, Sugaya N, Hirotsu N, Lee N, de Jong MD, Hurt AC, Ishida T, Sekino H, Yamada K, Portsmouth S, Kawaguchi K, Shishido T, Arai M, Tsuchiya K, Uehara T, Watanabe A, and Baloxavir Marboxil Investigators G (2018) Baloxavir Marboxil for Uncomplicated Influenza in Adults and Adolescents, N Engl J Med 379, 913–923. [DOI] [PubMed] [Google Scholar]
- 12.Shaw ML (2017) The Next Wave of Influenza Drugs, ACS Infect Dis, 3, 691–694. [DOI] [PubMed] [Google Scholar]
- 13.Stevaert A, and Naesens L (2016) The Influenza Virus Polymerase Complex: An Update on Its Structure, Functions, and Significance for Antiviral Drug Design, Med Res Rev 36, 1127–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Das K, Aramini JM, Ma LC, Krug RM, and Arnold E (2010) Structures of influenza A proteins and insights into antiviral drug targets, Nat Struct Mol Biol 17, 530–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reich S, Guilligay D, Pflug A, Malet H, Berger I, Crepin T, Hart D, Lunardi T, Nanao M, Ruigrok RW, and Cusack S (2014) Structural insight into cap-snatching and RNA synthesis by influenza polymerase, Nature 516, 361–366. [DOI] [PubMed] [Google Scholar]
- 16.Yamashita M, Krystal M, and Palese P (1989) Comparison of the three large polymerase proteins of influenza A, B, and C viruses, Virology 171, 458–466. [DOI] [PubMed] [Google Scholar]
- 17.Yuan S, Wen L, and Zhou J (2018) Inhibitors of Influenza A Virus Polymerase, ACS Infect Dis 4, 218–223. [DOI] [PubMed] [Google Scholar]
- 18.Zhang J, Hu Y, Musharrafieh RG, Yin H, and Wang J (2019) Focusing on the influenza virus polymerase complex: recent progress in drug discovery and assay development, Curr Med Chem 26, 2243–2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.He X, Zhou J, Bartlam M, Zhang R, Ma J, Lou Z, Li X, Li J, Joachimiak A, Zeng Z, Ge R, Rao Z, and Liu Y (2008) Crystal structure of the polymerase PAC-PB1N complex from an avian influenza H5N1 virus, Nature 454, 1123–1126. [DOI] [PubMed] [Google Scholar]
- 20.Chase G, Wunderlich K, Reuther P, and Schwemmle M (2011) Identification of influenza virus inhibitors which disrupt of viral polymerase protein-protein interactions, Methods 55, 188–191. [DOI] [PubMed] [Google Scholar]
- 21.Yuan S, Chu H, Ye J, Singh K, Ye Z, Zhao H, Kao RY, Chow BK, Zhou J, and Zheng BJ (2017) Identification of a novel small-molecule compound targeting the influenza A virus polymerase PB1-PB2 interface, Antiviral Res 137, 58–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Muratore G, Goracci L, Mercorelli B, Foeglein A, Digard P, Cruciani G, Palu G, and Loregian A (2012) Small molecule inhibitors of influenza A and B viruses that act by disrupting subunit interactions of the viral polymerase, Proc. Natl. Acad. Sci. U. S. A 109, 6247–6252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tintori C, Laurenzana I, Fallacara AL, Kessler U, Pilger B, Stergiou L, and Botta M (2014) High-throughput docking for the identification of new influenza A virus polymerase inhibitors targeting the PA-PB1 protein-protein interaction, Bioorg Med Chem Lett 24, 280–282. [DOI] [PubMed] [Google Scholar]
- 24.Watanabe K, Ishikawa T, Otaki H, Mizuta S, Hamada T, Nakagaki T, Ishibashi D, Urata S, Yasuda J, Tanaka Y, and Nishida N (2017) Structure-based drug discovery for combating influenza virus by targeting the PA-PB1 interaction, Sci Rep 7, 9500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang J, Hu Y, Foley C, Wang Y, Musharrafieh R, Xu S, Zhang Y, Ma C, Hulme C, and Wang J (2018) Exploring Ugi-Azide Four-Component Reaction Products for Broad-Spectrum Influenza Antivirals with a High Genetic Barrier to Drug Resistance, Sci Rep 8, 4653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li C, Wang Z, Cao Y, Wang L, Ji J, Chen Z, Deng T, Jiang T, Cheng G, and Qin FX (2017) Screening for Novel Small-Molecule Inhibitors Targeting the Assembly of Influenza Virus Polymerase Complex by a Bimolecular Luminescence Complementation-Based Reporter System, J Virol 91, e02282–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shekhawat SS, and Ghosh I (2011) Split-protein systems: beyond binary protein-protein interactions, Curr Opin Chem Biol 15, 789–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bosmans RP, Briels JM, Milroy LG, de Greef TF, Merkx M, and Brunsveld L (2016) Supramolecular Control over Split-Luciferase Complementation, Angew Chem Int Ed Engl 55, 8899–8903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brecher M, Li Z, Liu B, Zhang J, Koetzner CA, Alifarag A, Jones SA, Lin Q, Kramer LD, and Li H (2017) A conformational switch high-throughput screening assay and allosteric inhibition of the flavivirus NS2B-NS3 protease, Plos Pathog 13, e1006411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li Z, Brecher M, Deng YQ, Zhang J, Sakamuru S, Liu B, Huang R, Koetzner CA, Allen CA, Jones SA, Chen H, Zhang NN, Tian M, Gao F, Lin Q, Banavali N, Zhou J, Boles N, Xia M, Kramer LD, Qin CF, and Li H (2017) Existing drugs as broad-spectrum and potent inhibitors for Zika virus by targeting NS2B-NS3 interaction, Cell Res 27, 1046–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yuan P, Bartlam M, Lou Z, Chen S, Zhou J, He X, Lv Z, Ge R, Li X, Deng T, Fodor E, Rao Z, and Liu Y (2009) Crystral structure of an avian influenza polymerase PAN reveals an endonuclease active site, Nature 458, 909–913. [DOI] [PubMed] [Google Scholar]
- 32.Zhang W, and Webster RG (2017) Can we beat influenza?, Science 357, 111. [DOI] [PubMed] [Google Scholar]
- 33.Fodor E, and Smith M (2004) The PA subunit is required for efficient nuclear accumulation of the PB1 subunit of the influenza A virus RNA polymerase complex, J Virol 78, 9144–9153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Trist IM, Nannetti G, Tintori C, Fallacara AL, Deodato D, Mercorelli B, Palu G, Wijtmans M, Gospodova T, Edink E, Verheij M, de Esch I, Viteva L, Loregian A, and Botta M (2016) 4,6-Diphenylpyridines as Promising Novel Anti-Influenza Agents Targeting the PA-PB1 Protein-Protein Interaction: Structure-Activity Relationships Exploration with the Aid of Molecular Modeling, J Med Chem 59, 2688–2703. [DOI] [PubMed] [Google Scholar]
- 35.Yuan S, Chu H, Zhao H, Zhang K, Singh K, Chow BK, Kao RY, Zhou J, and Zheng BJ (2016) Identification of a small-molecule inhibitor of influenza virus via disrupting the subunits interaction of the viral polymerase, Antiviral Res 125, 34–42. [DOI] [PubMed] [Google Scholar]
- 36.Desantis J, Nannetti G, Massari S, Barreca ML, Manfroni G, Cecchetti V, Palu G, Goracci L, Loregian A, and Tabarrini O (2017) Exploring the cycloheptathiophene-3-carboxamide scaffold to disrupt the interactions of the influenza polymerase subunits and obtain potent anti-influenza activity, Eur J Med Chem 138, 128–139. [DOI] [PubMed] [Google Scholar]
- 37.D’Agostino I, Giacchello I, Nannetti G, Fallacara AL, Deodato D, Musumeci F, Grossi G, Palu G, Cau Y, Trist IM, Loregian A, Schenone S, and Botta M (2018) Synthesis and biological evaluation of a library of hybrid derivatives as inhibitors of influenza virus PA-PB1 interaction, Eur J Med Chem 157, 743–758. [DOI] [PubMed] [Google Scholar]
- 38.Nannetti G, Massari S, Mercorelli B, Bertagnin C, Desantis J, Palu G, Tabarrini O, and Loregian A (2019) Potent and broad-spectrum cycloheptathiophene-3-carboxamide compounds that target the PA-PB1 interaction of influenza virus RNA polymerase and possess a high barrier to drug resistance, Antiviral Res, 165, 55–64. [DOI] [PubMed] [Google Scholar]
- 39.Perez DR, and Donis RO (1995) A 48-amino-acid region of influenza A virus PB1 protein is sufficient for complex formation with PA, J Virol 69, 6932–6939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gonzalez S, Zurcher T, and Ortin J (1996) Identification of two separate domains in the influenza virus PB1 protein involved in the interaction with the PB2 and PA subunits: a model for the viral RNA polymerase structure, Nucleic Acids Res 24, 4456–4463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zurcher T, de la Luna S, Sanz-Ezquerro JJ, Nieto A, and Ortin J (1996) Mutational analysis of the influenza virus A/Victoria/3/75 PA protein: studies of interaction with PB1 protein and identification of a dominant negative mutant, J Gen Virol 77 ( Pt 8), 1745–1749. [DOI] [PubMed] [Google Scholar]
- 42.Perez DR, and Donis RO (2001) Functional analysis of PA binding by influenza a virus PB1: effects on polymerase activity and viral infectivity, J Virol 75, 8127–8136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Toyoda T, Adyshev DM, Kobayashi M, Iwata A, and Ishihama A (1996) Molecular assembly of the influenza virus RNA polymerase: determination of the subunit-subunit contact sites, J Gen Virol 77 ( Pt 9), 2149–2157. [DOI] [PubMed] [Google Scholar]
- 44.Ghanem A, Mayer D, Chase G, Tegge W, Frank R, Kochs G, Garcia-Sastre A, and Schwemmle M (2007) Peptide-mediated interference with influenza A virus polymerase, J Virol 81, 7801–7804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ohtsu Y, Honda Y, Sakata Y, Kato H, and Toyoda T (2002) Fine mapping of the subunit binding sites of influenza virus RNA polymerase, Microbiol Immunol 46, 167–175. [DOI] [PubMed] [Google Scholar]
- 46.Naito T, Momose F, Kawaguchi A, and Nagata K (2007) Involvement of Hsp90 in assembly and nuclear import of influenza virus RNA polymerase subunits, J Virol 81, 1339–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hemerka JN, Wang D, Weng Y, Lu W, Kaushik RS, Jin J, Harmon AF, and Li F (2009) Detection and characterization of influenza A virus PA-PB2 interaction through a bimolecular fluorescence complementation assay, J Virol 83, 3944–3955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Deng Q, Wang D, Xiang X, Gao X, Hardwidge PR, Kaushik RS, Wolff T, Chakravarty S, and Li F (2011) Application of a split luciferase complementation assay for the detection of viral protein-protein interactions, J Virol Methods 176, 108–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wunderlich K, Juozapaitis M, Ranadheera C, Kessler U, Martin A, Eisel J, Beutling U, Frank R, and Schwemmle M (2011) Identification of High-Affinity PB1-Derived Peptides with Enhanced Affinity to the PA Protein of Influenza A Virus Polymerase, Antimicrob Agents Chemother 55, 696–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.