

SUMMARY:
High-grade gliomas in patients with neurofibromatosis type 1 are rare and may therefore not be considered in the differential of brain lesions. Here, we describe 5 children with neurofibromatosis type 1; four of them developed various types of high-grade gliomas. The fifth patient had imaging features concerning for a high-grade lesion, but tissue diagnosis showed a low-grade glioma. The cases and literature summary provided here are to raise awareness for the occurrence of high-grade gliomas in children with neurofibromatosis type 1 and the limited ability of imaging features alone to predict a high-grade malignancy.
Brain tumors associated with neurofibromatosis type 1 (NF1) are typically low-grade gliomas (LGGs), such as pilocytic astrocytomas.1 The mutation of neurofibromin, a protein that regulates growth and the proto-oncogene Ras, causes unregulated cell growth and can result in formation of either LGGs or high-grade gliomas (HGGs).2 The incidence of HGG in children with NF1 is low and ranges from 0.28% to 5%.3–5
Using MR imaging for differentiating LGG from HGG can be problematic. While typical imaging features of LGG include lack of contrast enhancement, lack of restricted diffusion, and lack of hyperperfusion,6–9 the lack of MR imaging contrast enhancement in supratentorial brain tumors does not always equate with low-grade malignancy.10–12 In addition, contrast enhancement is a typical imaging feature of pilocytic astrocytoma, a common low-grade tumor in NF1.10,13 Thus, tissue sampling may become necessary for some patients.14
Here, we present several pediatric patients with NF1with suspicion for HGG based on imaging, along with clinical features and neuropathology outcomes. Our goal was to raise awareness of HGG in children with NF1 and to facilitate appropriate management, including tissue sampling.
Materials and Methods
The study received an exempt status by the institutional review board at Children's Healthcare of Atlanta and was Health Insurance Portability and Accountability Act–compliant. mPower™ (Nuance, Burlington, Massachusetts) was used to identify brain imaging studies performed during an 18-month period (January 1, 2015, to June 30, 2016) at our institution. Of 176 unique patients with NF1, five had concerning imaging findings.
Case Series
Patient 1.
A 7-year-old boy with no known medical history presented to an outside institution with a 1-month history of roving eye movements with upgaze palsy, ataxia, and headaches. An outside MR imaging at baseline demonstrated a 2.6 × 2.2 × 2.5 cm pineal region mass (Fig 1) with restricted diffusion (Fig 1C, -D), loss of the NAA peak, and elevated choline on MR spectroscopy. Tissue sampling at an outside facility showed a low-grade pineal parenchymal tumor. A 6-week MR imaging follow-up showed lesion growth to 2.9 × 2.5 × 2.9 cm, which was localized to the tectal plate rather than the pineal gland, and interval development of an irregular and centrally necrotic–appearing area of contrast enhancement (Fig 1B). The patient was diagnosed with NF1 on admission in accordance with diagnostic criteria.15 Subtotal resection of the mass was performed, and pathology was now read as consistent with a midline, H3 K27M–mutant glioblastoma multiforme (On-line Table). When we compared it with the initial pathology, the current specimen displayed a different histomorphology with necrosis and vascular proliferation, which could have been related to intratumoral heterogeneity or possibly secondary to malignant transformation of the tumor. After a 6-week course of focal radiation therapy, the patient is currently stable clinically and by imaging surveillance.
Fig 1.
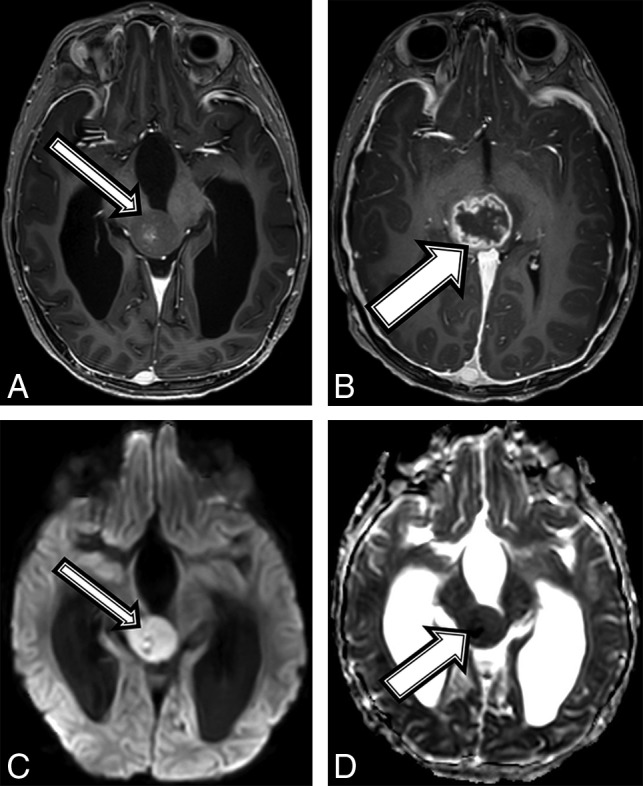
A 7-year-old boy with NF1 and a pathologic diagnosis of a midline, H3 K27M–mutant glioblastoma (patient 1). Axial T1 postcontrast at baseline (A) and after 6 weeks of follow-up (B). Baseline (A, arrow) smoothly marginated T1 hypointense mass is shown centered within the midbrain and tectal plate with mild central enhancement; 6 weeks later, the mass increased in size (B, arrow) with new peripheral enhancement. Imaging at baseline shows a high diffusion signal on DWI (C) and a low signal on ADC (D), suggestive of high cellularity.
Patient 2.
An 11-year-old girl with no known medical history presented with a 1-month history of left upper extremity weakness, left foot drop, facial droop, mild dysarthria, headache, and intermittent nausea and vomiting. MR imaging demonstrated a peripherally enhancing heterogeneous mass containing blood products centered within the right thalamus (Fig 2A). The patient was diagnosed with NF1 on admission in accordance with diagnostic criteria.15 Tissue sampling showed histomorphology consistent with a grade II–III astrocytoma (On-line Table). However, the amount of Ki-67/MIB1 staining was elevated at 10%, and H3 K27M immunostaining was strongly positive, consistent with a diffuse midline, H3 K27M–mutated glioma (World Health Organization grade IV). After chemotherapy induction, follow-up MR imaging demonstrated tumor progression and aggressive features consistent with a high-grade malignancy (Fig 2B). Despite aggressive treatment, the patient's tumor progressed and the patient died 17 months after the initial diagnosis.
Fig 2.
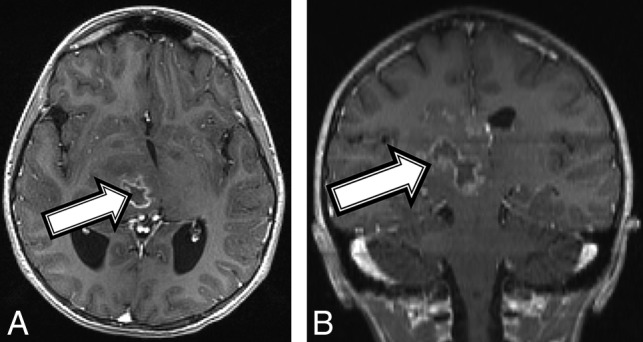
An 11-year-old girl with a final diagnosis of a grade II astrocytoma by histomorphology and a midline, diffuse HGG with H3 K27M mutation based on immunostaining (patient 2). Baseline axial T1 (A) and coronal (B) T1 postcontrast MR imaging shows a heterogeneous mass (A and B, arrows) centered in the right thalamus with a peripherally enhancing central cyst or area of necrosis.
Patient 3.
An asymptomatic 10-year-old boy with a diagnosis of NF1 in accordance with diagnostic criteria15 and previously treated optic glioma underwent a routine brain MR imaging surveillance scan, and a tiny T2 hyperintense focus along the posterior aspect of the right hippocampus was found (Fig 3A). Follow-up MRIs performed 6 and 12 months later demonstrated a progressive increase in size, new heterogeneous contrast enhancement (Fig 3B), and new restricted diffusion (Fig 3C, -D). After gross total tumor resection, neuropathology revealed a high-grade pleomorphic glioma, with imaging and histologic features consistent with a pleomorphic xanthoastrocytoma (On-line Table). Due to the high-grade features of this tumor, he was treated with focal proton radiation therapy. The patient is currently active and asymptomatic 18 months after completion of proton radiation.
Fig 3.
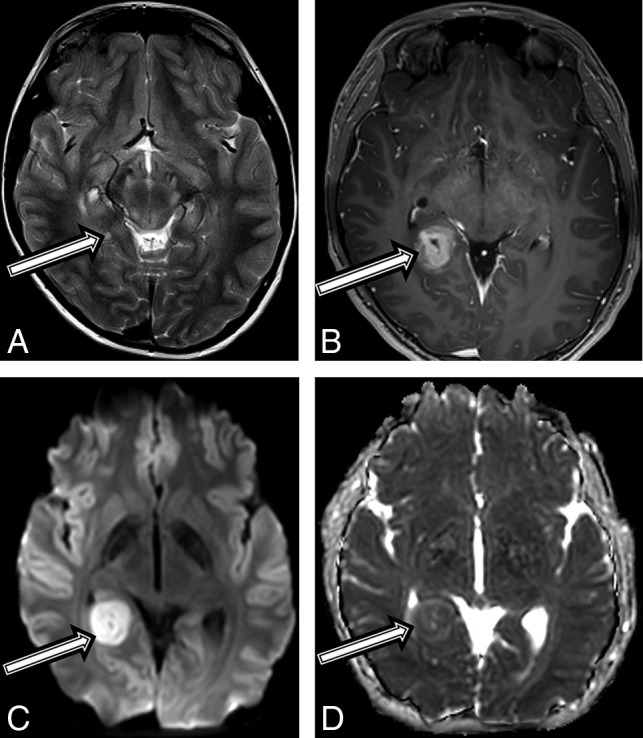
A 10-year-old boy with a final diagnosis of an anaplastic pleomorphic xanthoastrocytoma (patient 3). Axial T2-weighted images at baseline (A) and postcontrast axial T1 at 21 months (B) after diagnosis show a growing, well-defined mass in the right posterior mesial temporal lobe (A, arrow), which developed new inhomogeneous enhancement (B, arrow). MR imaging at 21 months shows new high-diffusion signal on DWI (C) and low signal on ADC (D), suggestive of high cellularity.
Patient 4.
A 4-year-old boy with a history of “skin spots” presented to our institution with headache, nausea and vomiting, weight loss, fatigue, and forehead swelling for 1 month. A brain MR imaging revealed a heterogeneous and partially enhancing mass measuring 3.5 × 2.3 × 2.1 cm in the midbrain tectum with compression of the cerebral aqueduct leading to severe obstructive hydrocephalus (Fig 4A). The patient was diagnosed with NF1 on admission in accordance with diagnostic criteria.15 Tissue biopsy revealed a diffuse, midline World Health Organization grade IV glioma with immunohistochemistry positive for nuclear H3 K27M–mutant protein, α-thalassemia/mental retardation syndrome X-linked (ATRX), and wild type p53, with an MIB-1/Ki-67 index of 10%. However, OncoScan (https://www.thermofisher.com/order/catalog/product/902695) copy-number microarray only revealed a PIK3CA p.H1047R point mutation, but no copy number changes. Given that the molecular findings were inconsistent with a high-grade malignancy, the initial plan was to follow the patient clinically and by MR imaging. On follow-up MR imaging at 3 and 6 months, the lesion remained relatively stable in size but developed multiple peripherally enhancing components within the mass (Fig 4B). The patient experienced intratumoral hemorrhage, and MR imaging at that time showed evidence of tumor dissemination. The patient was started on chemotherapy with carboplatin and vincristine with a plan for re-evaluation for evidence of tumor progression by MR imaging after the induction phase of chemotherapy.
Fig 4.
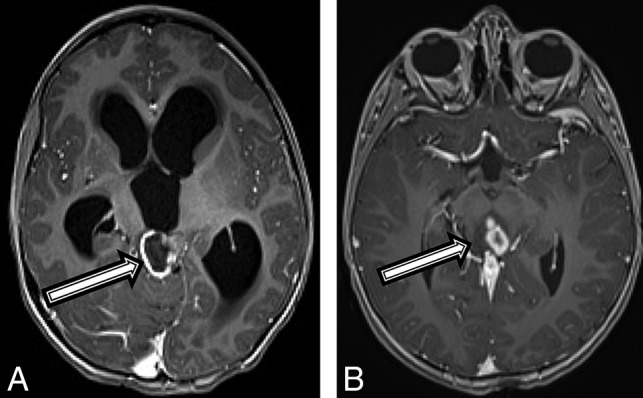
A 4-year-old boy with a diffuse HGG with H3 K27M mutation (patient 4). Axial T1 postcontrast MR imaging (A) at the initial presentation and on 6-month follow-up (B). There was initially a peripherally enhancing T2 hyperintense mass centered within the tectum (A, arrow) causing hydrocephalus. After 6 months, the lesion has increased in size, peripheral enhancement is no longer seen, but multiple centrally enhancing lesions are seen (B, arrow).
Patient 5.
A 7-year-old boy with a medical history of migraines and slowed speech presented to an outside institution with a 2-month history of clumsiness, truncal ataxia, worsening headache, nausea and vomiting, somnolence, and worsened slowed/slurred speech. MR imaging demonstrated a robustly enhancing suprasellar mass and a peripherally enhancing mass centered within the left globus pallidus (Fig 5A), an infiltrating T2 hyperintense signal with punctuate foci of enhancement in the right basal ganglia, and enlargement of the optic nerves, chiasm, and optic radiations. The constellation of findings was most compatible with an infiltrating optic pathway/hypothalamic glioma. The patient was diagnosed with NF1 on admission in accordance with diagnostic criteria.15 Tissue sampling showed a high-grade infiltrating astrocytoma, but without morphologic criteria diagnostic for glioblastoma multiforme, with staining negative for p53 and a mutation of the BRAF gene (V600E) mutant protein (On-line Table). A second biopsy of both the suprasellar mass and the left globus pallidus revealed findings more compatible with a pilocytic astrocytoma with low Ki-67/MIB-1 immunostaining. The patient was intolerant of initial chemotherapy with carboplatin and vincristine, so he was treated with vinblastine for 8 months. Follow-up MR imaging 2 months later showed an enlarging, enhancing mass lesion in the inferior right basal ganglia (Fig 5B). He began treatment with 5′-Inosinic acid, polymers, complex with 5′-cytidylic acid, polymers and L-lysine, homopolymer, compound with cellulose carboxymethyl ether (cancer drug) (Poly-ICLC) at that time and has experienced improvement of neurologic symptoms; his MRI findings have remained stable on immunotherapy with Poly-ICLC for 24 months.
Fig 5.
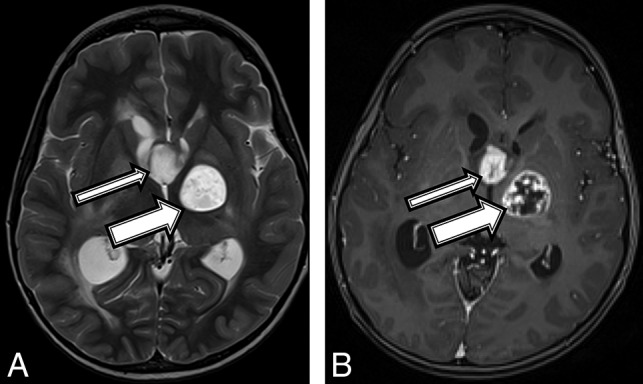
A 7-year-old boy with a final diagnosis of a pilocytic astrocytoma (patient 5). Axial T2 (A) and T1 postcontrast (B) MR images demonstrate a robustly enhancing T2 hyperintense suprasellar mass (thin arrows) and a T1 hypointense, T2 hyperintense mass centered within the left globus pallidus (thick arrows) with robust peripheral and patchy central enhancement.
Discussion
In our case series, new enhancement on MR imaging or a change in the contrast-enhancement pattern in a pre-existing presumed low-grade lesion occurred in 3 of our patients (patients 1–3) and correctly indicated a higher grade malignancy.16 Only 2 patients (patients 1 and 4) had restricted diffusion on conventional DWI and also had a high-grade glioma.7 The use of kurtosis diffusion imaging may have demonstrated superiority to conventional diffusion metrics in distinguishing various cerebral glioma grades.17 Most interesting, patient 5 had concerning clinical and MR imaging signs but turned out to have a low-grade histopathology.
There are 2 scenarios by which an HGG may occur in patients with NF1: 1) Lesions presumed to be pilocytic astrocytoma convert to HGG at that site years later18; or 2) patients with presumed low-grade lesions subsequently develop a high-grade glioma at a different location.19 In our study, 1 patient (patient 3) developed a new lesion at a different site that grew slowly during a 16-month period; the other 4 patients did not have prior neuroimaging. Recent retrospective analyses found that pediatric LGGs rarely transform to HGGs. In 1 retrospective analysis of 886 pediatric LGGs, only 2.9% of pediatric LGGs transformed into HGGs.20
Approximately half of biopsied intracranial tumors in patients with NF1 are pilocytic or pilomyxoid astrocytomas, and the remainder are mostly diffuse/infiltrating gliomas, of which some are high-grade (anaplastic astrocytoma or glioblastoma).1 Lesions of the 3 patients with HGGs in our series were diffuse and contained an H3 K27M point mutation. NF1 mutations have been demonstrated in a subset of H3 K27M–mutant HGGs.21 Another HGG in our series was an anaplastic pleomorphic xanthoastrocytoma, a rare tumor that has been reported several times in the literature in the context of NF1.22–25 Recently, the molecular genetic definition of HGG and apparent LGG has gained more importance, emphasizing the need to obtain tissue surgically.26 The 2-year survival in patients with NF1 with HGG is higher than in patients without NF1.27
Despite the small number of cases, we believe that raising awareness of an HGG in patients with NF1 among neuroradiologists is important. We were unable to evaluate the relative specificity of MR spectroscopy, perfusion, and kurtosis imaging, which may have prevented the tissue biopsy in the patient without HGG.
In conclusion, HGG in children with NF1 is rare, but concerning imaging signs should be taken seriously. We pursue tissue sampling when imaging findings are highly concerning for an HGG, though one of our patients in this series had a low-grade tumor on surgical pathology.
ABBREVIATIONS:
- HGG
high-grade glioma
- LGG
low-grade glioma
- NF1
neurofibromatosis type 1
Footnotes
Paper previously presented as an oral Excerpta at: Annual Meeting of the American Society of Neuroradiology and the Foundation of the ASNR Symposium, April 22–27, 2017; Long Beach, California.
References
- 1. Rodriguez FJ, Perry A, Gutmann DH, et al. Gliomas in neurofibromatosis type 1: a clinicopathologic study of 100 patients. J Neuropathol Exp Neurol 2008;67:240–49 10.1097/NEN.0b013e318165eb75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Fangusaro J. Pediatric high grade glioma: a review and update on tumor clinical characteristics and biology. Front Oncol 2012;2:105 10.3389/fonc.2012.00105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Byrne S, Connor S, Lascelles K, et al. Clinical presentation and prognostic indicators in 100 adults and children with neurofibromatosis 1 associated non-optic pathway brain gliomas. J Neurooncol 2017;133:609–14 10.1007/s11060-017-2475-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Blatt J, Jaffe R, Deutsch M, et al. Neurofibromatosis and childhood tumors. Cancer 1986;57:1225–29 10.1002/1097-0142(19860315)57:6<1225::AID-CNCR2820570627>3.0.CO;2-P [DOI] [PubMed] [Google Scholar]
- 5. Gutmann DH, Rasmussen SA, Wolkenstein P, et al. Gliomas presenting after age 10 in individuals with neurofibromatosis type 1 (NF1). Neurology 2002;59:759–61 10.1212/WNL.59.5.759 [DOI] [PubMed] [Google Scholar]
- 6. Upadhyay N, Waldman AD. Conventional MRI evaluation of gliomas. Br J Radiol 2011;84(Spec No 2):S107–11 10.1259/bjr/65711810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hilario A, Ramos A, Perez-Nuñez A, et al. The added value of apparent diffusion coefficient to cerebral blood volume in the preoperative grading of diffuse gliomas. AJNR Am J Neuroradiol 2012;33:701–07 10.3174/ajnr.A2846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kao HW, Chiang SW, Chung HW, et al. Advanced MR imaging of gliomas: an update. Biomed Res Int 2013;2013:970586 10.1155/2013/970586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sugahara T, Korogi Y, Kochi M, et al. Usefulness of diffusion-weighted MRI with echo-planar technique in the evaluation of cellularity in gliomas. J Magn Reson Imaging 1999;9:53–60 10.1002/(SICI)1522-2586(199901)9:1<53::AID-JMRI7>3.0.CO;2-2 [DOI] [PubMed] [Google Scholar]
- 10. Panigrahy A, Blüml S. Neuroimaging of pediatric brain tumors: from basic to advanced magnetic resonance imaging (MRI). J Child Neurol 2009. 24:1343–65 10.1177/0883073809342129 [DOI] [PubMed] [Google Scholar]
- 11. Muragaki Y, Chernov M, Maruyama T, et al. Low-grade glioma on stereotactic biopsy: how often is the diagnosis accurate? Minim Invasive Neurosurg 2008;51:275–79 10.1055/s-0028-1082322 [DOI] [PubMed] [Google Scholar]
- 12. Ginsberg LE, Fuller GN, Hashmi M, et al. The significance of lack of MR contrast enhancement of supratentorial brain tumors in adults: histopathological evaluation of a series. Surg Neurol 1998;49:436–40 10.1016/S0090-3019(97)00360-1 [DOI] [PubMed] [Google Scholar]
- 13. Gold DR, Cohen BH. Brain tumors in neurofibromatosis. Curr Treat Options Neurol 2003;5:199–206 10.1007/s11940-003-0010-3 [DOI] [PubMed] [Google Scholar]
- 14. Kondziolka D, Lunsford LD, Martinez AJ. Unreliability of contemporary neurodiagnostic imaging in evaluating suspected adult supratentorial (low-grade) astrocytoma. J Neurosurg 1993;79:533–36 [DOI] [PubMed] [Google Scholar]
- 15. Friedman JM. Neurofibromatosis 1. 1998 October 2, Updated 2018 May 17Accessed December 2, 2018 In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A, eds. GeneReviews. Seattle, WA: University of Washington; 1993–2018. https://www.ncbi.nlm.nih.gov/books/NBK1109/. Accessed December 2, 2018. [Google Scholar]
- 16. Pierallini A, Bonamini M, Bozzao A, et al. Supratentorial diffuse astrocytic tumours: proposal of an MRI classification. Eur Radiol 1997;7:395–99 10.1007/s003300050173 [DOI] [PubMed] [Google Scholar]
- 17. Qi C, Yang S, Meng L, et al. Evaluation of cerebral glioma using 3T diffusion kurtosis tensor imaging and the relationship between diffusion kurtosis metrics and tumor cellularity. J Int Med Res 2017;45:1347–58 10.1177/0300060517712654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Miaux Y, Guermazi A, Cornu P, et al. High-intensity lesion on T1-weighted MR images in neurofibromatosis type 1: a case of premalignant lesion. Acta Neurochir (Wien) 1997;139:1085–87 10.1007/BF01411565 [DOI] [PubMed] [Google Scholar]
- 19. Distelmaier F, Fahsold R, Reifenberger G, et al. Fatal glioblastoma multiforme in a patient with neurofibromatosis type I: the dilemma of systematic medical follow-up. Childs Nerv Syst 2007;23:343–37 10.1007/s00381-006-0222-4 [DOI] [PubMed] [Google Scholar]
- 20. Mistry M, Zhukova N, Merico D, et al. BRAF mutation and CDKN2A deletion define a clinically distinct subgroup of childhood secondary high-grade glioma. J Clin Oncol 2015;33:1015–22 10.1200/JCO.2014.58.3922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mukasa A, Aihara K, Gotoh K, et al. Frequent H3F3A K27M mutations in thalamic gliomas from young adult patients. Neuro Oncol 2014;16:140–46 10.1093/neuonc/not144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Neal MT, Ellis TL, Stanton CA. Pleomorphic xanthoastrocytoma in two siblings with neurofibromatosis type 1 (NF-1). Clin Neuropathol 2012;31:54–56 10.5414/NP300424 [DOI] [PubMed] [Google Scholar]
- 23. Prayson RA. Pleomorphic xanthoastrocytoma arising in neurofibromatosis type 1. Clin Neuropathol 2012;31:152–54 10.5414/NP300436 [DOI] [PubMed] [Google Scholar]
- 24. Takei H, Rouah E, Bhattacharjee MB. Cerebellar pleomorphic xanthoastrocytoma in a patient with neurofibromatosis type 1: a case report and literature review. Int J Clin Exp Pathol 2015;8:7570–74 [PMC free article] [PubMed] [Google Scholar]
- 25. Mackay A, Burford A, Molinari V, et al. Molecular, pathological, radiological, and immune profiling of non-brainstem pediatric high-grade glioma from the HERBY Phase II randomized trial. Cancer Cell 2018;33:829–42 10.1016/j.ccell.2018.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Callovini GM. Is it appropriate to redefine the indication for stereotactic brain biopsy in the MRI era? Correlation with final histological diagnosis in supratentorial gliomas. Minim Invasive Neurosurg 2008;51:109–13 10.1055/s-2008-1058096 [DOI] [PubMed] [Google Scholar]
- 27. Huttner AJ, Kieran MW, Yao X, et al. Clinicopathologic study of glioblastoma in children with neurofibromatosis type 1. Pediatr Blood Cancer 2010;54:890–96 10.1002/pbc.22462 [DOI] [PubMed] [Google Scholar]