

Abstract
Background
Aiming to improve treatment options for BRAF wild-type melanoma, we previously conducted the DOC-MEK study of docetaxel with MEK inhibitor (MEKi) selumetinib or placebo, revealing trends to prolongation of progression-free survival (hazard ratio 0.75, P = 0.130), and improved response rates (32% vs 14%, P = 0.059) with docetaxel plus selumetinib. NRAS status did not associate with outcome. Here, the aim was to identify novel biomarkers of response to MEKi.
Methods
A MEK 6 gene signature was quantified using NanoString and correlated with clinical outcomes. Two components of the gene signature were investigated by gene silencing in BRAF/NRAS wild-type melanoma cells.
Results
In melanomas of patients on the selumetinib but not the placebo arm, two gene signature components, dual-specificity protein phosphatase 4 (DUSP4) and ETS translocation variant 4 (ETV4), were expressed more highly in responders than non-responders. In vitro, ETV4 depletion inhibited cell survival but did not influence sensitivity to MEKi selumetinib or trametinib. In contrast, DUSP4-depleted cells showed enhanced cell survival and increased resistance to both selumetinib and trametinib.
Conclusions
ETV4 and DUSP4 associated with clinical response to docetaxel plus selumetinib. DUSP4 depletion induced MEKi resistance, suggesting that DUSP4 is not only a biomarker but also a mediator of MEKi sensitivity.
Clinical Trial Registration
DOC-MEK (EudraCT no: 2009-018153-23).
Subject terms: Melanoma, Cancer therapeutic resistance, Cell signalling
Background
The incidence of melanoma is increasing: it is now the fifth most common cancer in the United Kingdom and the second commonest cancer in adults aged 25–49 (http://www.cancerresearchuk.org/health-professional/cancer-statistics/statistics-by-cancer-type/skin-cancer/). Early detection and treatment with surgical excision is often curative, but ~25% of patients develop local recurrences and/or metastatic disease.1 The prognosis with advanced melanoma is poor, although advances in both immunotherapy and targeted therapy have had a significant impact on overall survival (OS), with median OS more than 24 months in some clinical trials.2 Approximately 40–50% of melanomas harbour BRAF mutations that activate the RAS–RAF–MEK–ERK pathway, also known as the Mitogen-Activated Protein Kinase (MAPK) pathway.3 The most common BRAF mutation is a missense mutation, leading to substitution of valine by glutamic acid at position 600 of the BRAF protein (V600E).4,5 Patients whose melanomas contain these mutations can be treated with BRAF inhibitors, such as vemurafenib or dabrafenib. These drugs achieve ~50% objective response rate (ORR) as monotherapy,6,7 and up to 69% ORR when combined with MEK inhibitors (MEKi), which act downstream of BRAF to inhibit the activity of MEK1/2.8–10 However, treatment resistance and disease progression generally develop within 6–7 months on BRAF inhibitor (BRAFi) monotherapy and 11 months with BRAFi/MEKi combination treatment. For melanoma patients without BRAF mutations, targeted treatments have shown no significant benefit.11,12
We previously reported the outcomes of the multicentre Phase II DOC-MEK trial for patients with advanced BRAF wild-type melanoma.13 Patients were randomised to treatment with docetaxel plus either MEK1/2 inhibitor selumetinib (AZD6244, ARRY-142886) or placebo. The rationales for this combination were, firstly, that selumetinib has demonstrated preclinical efficacy in both BRAF wild-type and mutant melanoma models.14 Secondly, resistance to taxane-induced apoptosis can be mediated by MAPK pathway activation, and thus concurrent MEK1/2 inhibition may potentiate the efficacy of taxane chemotherapy. Thirdly, the combination of selumetinib and docetaxel has been tested in melanoma and colorectal cancer xenografts15,16 and in a Phase I trial,17 with evidence of activity in a Phase II study in patients with non-small-cell lung cancer (NSCLC).18 In the DOC-MEK trial, patients treated with docetaxel plus selumetinib had an ORR of 32% compared with 14% in the docetaxel plus placebo arm (P = 0.059).13 There was a non-significant difference in progression-free survival (PFS) and NRAS mutation was not predictive of the response to MEK inhibition.13
Here, we extend analysis of the DOC-MEK study with reference to a MEK functional activity gene signature, developed as a prognostic and/or predictive biomarker of MEK functional activation across a variety of tumour types.19,20 We confirm no evidence of a predictive role for NRAS mutation status, but demonstrate that there is a correlation between response to treatment with docetaxel plus selumetinib and the MEK 6 gene signature score. Notably, two components of the signature, Dual-specificity protein phosphatase 4 (DUSP4, also known as MKP2) and ETS translocation variant 4 (ETV4), correlate with partial response (PR) or complete response (CR) to docetaxel plus selumetinib but not to docetaxel plus placebo. This suggests that these proteins could act as biomarkers to identify patients likely to respond to the combination treatment. We investigate a possible role for DUSP4 and ETV4 in mediating this response by assessing the effect of their depletion on the sensitivity of BRAF wild-type melanoma cells to MEK inhibition. We show that DUSP4 protein expression is suppressed by MEK inhibition, confirming its status as an ERK-regulated gene.21 Furthermore, we demonstrate that DUSP4 depletion influences response to two MEK inhibitors, selumetinib and trametinib. Thus, DUSP4 expression is not only a potential biomarker for patient response to MEK inhibition but also a mediator of MEK inhibitor sensitivity.
Methods
Tumour mutation analysis by next-generation sequencing
Archival formalin-fixed paraffin-embedded (FFPE) tissue blocks were sectioned and enriched for tumour tissue by macro-dissection. Genomic DNA was extracted by using the QIAamp FFPE Tissue kit and amplified using the Ion AmpliseqTM Library Kit 2.0. DNA sequencing was performed by the IonTorrent Personal Genome Machine (LifeTechnologies, Carlsbad, CA). A targeted cancer hotspot panel (designed using the Ion AmpliseqTM Cancer Primer Pool) was used to detect mutations in 46 known cancer-related genes (Supplementary Table 1).22 The sensitivity of this assay is 5–10% (% of mutant DNA detectable in a background of wild-type DNA). When DNA was of insufficient quality for next-generation sequencing (NGS), pyrosequencing was used to test for mutations in codons 12, 13 and 61 of NRAS.
Gene expression analysis
Tumour FFPE tissue was macro-dissected from 1 to 2 × 5-µm sections, RNA extracted using the RNeasy FFPE kit according to the manufacturer’s instructions and 100 ng of each RNA was analysed using the NanoString nCounter gene expression system.23 The code set was designed by NanoString Inc. (Seattle, WA). Transcript counts were normalised between MEK signature genes and reference genes and transformed using the NanoString Normalisation Tool v2 (AstraZeneca Oncology Bioinformatics http://CRAN.R-project.org/package=NAPPA) in order to generate signature scores.20 Signature scores were calculated blind to clinical outcomes.
Cell lines and reagents
CHL-1 cells were obtained from the American Type Culture Collection (ATCC), and SK-mel-23 cells from Professor V. Cerundolo, Weatherall Institute of Molecular Medicine, University of Oxford. Cultures were maintained in Dulbecco’s Modified Eagle’s Medium with 10% foetal calf serum and 1% penicillin/streptomycin, in a humidified atmosphere of 10% CO2. Both cell lines were negative for mycoplasma (MycoAlert kit, Lonza Rockland Inc., Rockland, USA), and were authenticated by STR genotyping (Eurofins Medigenomix Forensik GmbH). Selumetinib and trametinib (Selleck) were stored as 10 mM solutions in DMSO at −80 °C.
Western blotting and cell survival assays
Cells were incubated with drugs for 60–150 min before harvesting for western blotting as previously described,24 using antibodies to DUSP4 (♯5149, Cell Signaling Technology (CST)), phospho-T202/Y204 ERK 1/2 (♯4377, CST), total ERK 1/2 (♯4695, CST), β-tubulin (T4026, Sigma) and actin (A3854, Sigma). For clonogenic survival assays, drugs or vehicle control were added 24 h after seeding, and cells incubated in the presence of drug for 7–14 days.
Gene silencing by siRNA transfection
Cells were reverse transfected with 50 nM gene-specific or non-silencing Allstars (Qiagen) siRNA on day 1 using Dharmafect 1 (ThermoFisher), forward transfected with 50 nM siRNA on day 2 and re-seeded on day 3 for clonogenic assays. DUSP4 was depleted using siDUSP4_1 (♯4392420-s4372, Ambion) and siDUSP4_2 (J-003963-09, Dharmacon) and ETV4 using siETV4_1 and _2 (♯106636 and ♯106637, Thermofisher).
Quantitative real-time PCR (qRT-PCR)
RNAs were extracted using the ReliaprepTM RNA miniprep kit (Promega) and reverse-transcribed to complementary DNA (cDNA) using Superscript III First-Strand Synthesis Supermix (ThermoFisher). PCRs were performed on a 7500 Fast RT-PCR System (Applied Biosystems) using SYBR Green PCR mastermix (ThermoFisher) with the following primers: DUSP4 forward, 5′-GGGGTCCTGTGGAGATCCTT-3′ and reverse, 5′-GGCAGTCCGAGGAGACATTC-3′; ETV4 forward 5′-GAGCGGAGGATGAAAGCCG-3′ and reverse 5′-CCCATTTCCGGGCGATTTG-3′; TUBA6 (housekeeping gene) forward 5′-CCCCTTCAAGTTCTAGTCATGC-3′ and reverse 5′-ATTGCCAATCTGGACACCA-3′.
Statistical analysis
The Kaplan–Meier method was used to obtain PFS and OS estimates by mutation status. As reported previously, the impact of NRAS mutation status was assessed by adding an interaction term with treatment in the Cox model, adjusting for stratification variables and using a significance level of 0.05.13 Correlations between tumour mutation results, gene signature scores and clinical outcomes were analysed using Microsoft Excel, with significance determined using t tests with a one-tailed distribution. NRAS VAF was compared with gene signature scores using Pearson’s correlation coefficient. Western blot and cell survival data were analysed using GraphPad Prism v5, using t tests to compare two groups and ANOVA for multiple groups in each case with a two-tailed distribution.
Results
Response to MEK inhibition does not correlate with NRAS mutation status
The results from the DOC-MEK Phase II trial in BRAF wild-type advanced melanoma patients13 demonstrated that docetaxel plus selumetinib did not significantly increase PFS or OS compared with docetaxel plus placebo, but there was a trend towards increased ORR in the selumetinib group (P = 0.059). There was no correlation between patient response and NRAS mutation status. Here, we extended this analysis by investigating the mutational status of 45 additional genes in a 46-gene cancer panel. Mutation status was determined for 64 of the 83 patients randomised in the DOC-MEK study, in 59 cases by NGS using targeted sequencing of 46 cancer-associated genes (Fig. 1a, Supplementary Table 1) and in five cases (DM005, DM026, DM029, DM037 and DM083) using pyrosequencing. In total, 84 mutations were found in 49 of the 59 cases analysed using NGS, with two or more concurrent mutations found in 24 cases. The most commonly mutated gene was NRAS (45% of all mutations detected), followed by TP53 (7%; Fig. 1a, Supplementary Table 2). These findings are consistent with an analysis of 699 unselected (i.e. BRAF wild-type and mutated) melanomas using the same 46-gene cancer panel, where, after BRAF mutations (41% of cases), NRAS (22%) and TP53 (17%) were the next commonest mutations.22 In our BRAF wild-type population, four cases tested by NGS had a BRAF mutation that had not been detected during initial screening by hotspot mutation testing (Fig. 1a, Supplementary Table 2). One of these was BRAF V600E, detected at a very low variant allele frequency (VAF) of 5.5%. Three cases harboured non-V600 BRAF mutations: K601E, G466R and N581S, rare mutations reported in the COSMIC database (http://cancer.sanger.ac.uk). Non-V600 BRAF mutations, including K601E, have been associated with sensitivity to MEK inhibition.25,26 The G466R and N581S BRAF mutations were found in melanomas that also harboured mutant NRAS. As activating BRAF and NRAS mutations are considered mutually exclusive,4 these two BRAF mutations are probably non-activating, or may be low-activity BRAF mutants that require upstream RAS activation. The patient with low VAF BRAF V600E (case DM067, Supplementary Table 2) was randomised to docetaxel plus placebo and progressed within 3.3 months of treatment. Those with non-V600 BRAF mutations were all randomised to docetaxel plus selumetinib. The patients with BRAF K601E (case DM072) and BRAF G466R + NRAS Q61H mutant melanoma (case DM071) progressed after 4 and 7 months, respectively, each with stable disease as best response. The patient with BRAF N581S melanoma (case DM077) was found to have two concurrent NRAS mutations (Q61K, Q61R) and initially demonstrated a partial response to treatment, but again progressed quickly after 4 months. The most common concomitant mutations were NRAS and TP53 (Supplementary Table 2). There was no apparent association between the number of concomitant mutations per tumour and median PFS or OS (Supplementary Table 3).
Fig. 1. Correlation of NRAS mutations with MEK 6 gene signature score.

a Distribution of mutations detected in samples from 59 patients using the 46-gene cancer panel. b MEK 6 gene signature scores of patient samples with mutant or wild-type NRAS. c Correlation between MEK 6 gene signature score and variant allele frequency of NRAS.
Relationship between NRAS mutation status and MEK 6 gene expression signature
Forty-eight tumours from the DOC-MEK study were available for transcriptional analysis, using the NanoString platform to quantify the MEK 6 gene score. Supplementary Table 4 summarises these scores, the NRAS mutation data and DOC-MEK clinical outcomes. We tested for associations between the MEK 6 gene score and NRAS status, best overall response and derived benefit. There was a higher mean MEK 6 gene score in NRAS mutant melanomas compared with NRAS wild-type melanomas (P = 0.023, Fig. 1b), but the differences were small and there was considerable overlap between the two groups. Since the levels of MAPK activation may be significantly different between tumours with low vs high mutant NRAS VAF, we assessed the correlation between NRAS VAF where available and MEK 6 gene score and found a modest positive correlation (Pearson’s correlation coefficient, r = 0.51, Fig. 1c).
Response to MEK inhibition correlates with MEK 6 gene expression score
To assess correlations between patient response and MEK 6 gene expression score, patients in the two arms of the trial were analysed separately by comparing those achieving CR/PR with patients having stable disease (SD) or progressive disease (PD) at first assessment (Fig. 2a, b). For patients treated with docetaxel plus placebo, there was no significant difference in MEK 6 gene score between responders and those with SD/PD at first assessment (mean scores 9.28 ± 0.16 and 9.56 ± 0.15, respectively, P = 0.222, Fig. 2a). In contrast, patients achieving CR or PR on docetaxel plus selumetinib had a higher MEK 6 gene score than those with SD/PD at first assessment (mean scores 10.14 ± 0.17 and 9.34 ± 0.31, respectively, P = 0.026, Fig. 2b). However, there was again considerable overlap between the two populations. The absolute difference between mean scores was small, and as there were only four responding patients in the docetaxel plus placebo arm, these results should be interpreted with caution.
Fig. 2. Correlation of response to docetaxel plus selumetinib with MEK 6 gene signature score.

a Best overall response for patients treated with docetaxel + placebo comparing CR and PR with SD and PD. b Best overall response for patients treated with docetaxel + selumetinib as in (a), *p = 0.026 using one-tailed t test. c Derived benefit for patients treated with docetaxel + placebo comparing benefit with no benefit. d Derived benefit for patients treated with docetaxel + selumetinib as in c, *p = 0.038 using one-tailed t test, testing the pre-established hypothesis that a higher MEK 6 gene score correlates with better clinical outcome when treated with a MEK inhibitor.
Since patients with prolonged disease stabilisation can also be considered to have derived benefit from treatment, a second analysis was performed. Gene expression scores were compared between those who “derived benefit”, i.e. had CR, PR or SD ≥ 6 months, and those with “no derived benefit”, i.e. PD at first assessment or SD < 6 months (Fig. 2c, d). As previously, for patients treated with docetaxel plus placebo, there was no difference in the MEK 6 gene score between those who derived benefit and those who did not (mean values of 9.21 ± 0.18 and 9.64 ± 0.17, respectively, P = 0.094, Fig. 2c). With more patients in the “benefit” group, this analysis may be considered more reliable. Patients treated with docetaxel plus selumetinib who were considered to have derived benefit again had a higher MEK 6 gene score than those who did not benefit (mean scores 9.93 ± 0.16 and 9.11 ± 0.58, respectively, P = 0.038, Fig. 2d), with a similar pattern of distribution as in Fig. 2b. Thus, using either approach to categorise clinical benefit, this analysis showed a higher mean MEK 6 gene score in the melanomas of patients responding to docetaxel plus selumetinib, but not to docetaxel plus placebo. However, the absolute difference in mean score using either approach was small.
In a third approach, we analysed expression of individual components of the MEK 6 gene signature, comparing those who responded (CR plus PR) with those with PD at first assessment. Gene expression data were available for 8/13 responders and 2/5 experiencing early progression in the combination treatment arm, and for 3/6 responders and 11/20 experiencing early PD in the docetaxel plus placebo arm. Whilst the numbers are small, the MEK 6 gene score data revealed potentially interesting differences in the expression of DUSP4 and ETV4, which showed significant differences in mean values (p < 0.05) between the responders and nonresponders in the selumetinib group (Table 1), but not in the placebo group (Supplementary Table 5). Thus, patients who responded to treatment had greater expression of both DUSP4 and ETV4 compared with those experiencing early PD, and these differences were found only in the docetaxel plus selumetinib group.
Table 1.
Expression of individual genes in the MEK 6 gene score in melanomas of patients treated with docetaxel plus selumetinib.
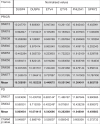 |
Grey shaded rows: patients with PR/CR to docetaxel plus selumetinib combination treatment; unshaded rows: patients with PD at first assessment.
Mean gene expression in both groups was compared using Student’s t test with two-tailed distribution.
aCases where ETV4 expression was below the set limit of detection (mean minus two standard deviations), so accuracy is unclear. Bold values are the mean figures for the gene expression values per gene in each group.
Since higher expression of DUSP4 and ETV4 mRNA was associated with clinical response to MEK inhibition, we hypothesised that depletion of each of these proteins might have the reverse effect and induce resistance to MEK inhibition. We therefore decided to test this in vitro using BRAF wild-type melanoma cell lines CHL-1 and SK-MEL-23, and two MEK inhibitors, namely selumetinib, the MEK inhibitor used in the DOC-MEK clinical trial, and trametinib, the first MEK inhibitor approved by the FDA for use in clinical practice (https://www.cancer.gov/about-cancer/treatment/drugs/fda-trametinib). The aim of using two inhibitors was to check whether any observed changes were likely to be class effects, rather than specific to selumetinib. Western blots for phospho-T202/Y204 ERK 1/2 levels were used as a readout for MEK activity.
MEK inhibition downregulates DUSP4 in BRAF wild-type melanoma cells
We first assessed endogenous expression of DUSP4 and ETV4, which were detectable at the protein and mRNA level in both cell lines (Fig. 3a, b), although CHL-1 showed considerably lower levels of expression of both proteins than SK-MEL-23. We also tested the effect of MEK inhibition on DUSP4 expression, using both selumetinib and trametinib. Selumetinib inhibited ERK phosphorylation at ≥100 nM, whereas trametinib caused inhibition at lower concentrations (≥3 nM) in both cell lines (Fig. 3c, Supplementary Fig. 1A, B), in keeping with the known IC50 values for each drug.14,27 In both cell lines, DUSP4 expression also decreased as inhibitor concentration increased (Fig. 3c, Supplementary Fig. 1A, B), consistent with reported control of its transcription by phosphorylated ERK1.21 Trametinib suppressed ERK phosphorylation for 24 h, with partial return of signal at 48–72 h, while inhibition of DUSP4 expression persisted for at least 72 h (Supplementary Fig. 1C).
Fig. 3. MEK inhibition decreases ERK phosphorylation and DUSP4 expression in BRAF wild-type melanoma cell lines.

a Endogenous expression of DUSP4 and ETV4 protein in duplicate whole-cell extracts of CHL-1 and SK-MEL-23 cells analysed by western blotting. Similar results were obtained in n = 3 independently prepared lysates. A faint ~40-kDa band (arrowhead) just below DUSP4 band was not reduced by DUSP4 depletion (see Fig. 4b, c) so it may be non-specific. b DUSP4 and ETV4 mRNA quantified by qRT-PCR (n = 3 independently prepared cDNAs). c CHL-1 and SK-MEL-23 cells were treated with 100 nM selumetinib or 10 nM trametinib for 1 h before analysis by western blot. Supplementary Fig. 1A–C shows concentration and time dependence of response to MEK inhibition.
ETV4 depletion does not alter sensitivity of BRAF wild-type melanoma cells to MEK inhibition
We assessed the influence of ETV4 on response to MEK inhibition, by depleting ETV4 and measuring cell survival and MEK inhibitor SF50 values (drug concentration suppressing survival to 50% of control values) in clonogenic survival assays. ETV4 knockdown was very effective in CHL-1 cells, with residual ETV4 mRNA values of 1.16 ± 0.53% and 4.14 ± 0.65% for siETV4_1 and siETV4_2, respectively, compared with the Allstars control siRNA transfectants (Supplementary Fig. 2A, left). For SK-MEL-23 cells, the equivalent values were 18.02 ± 13.02% and 4.28 ± 1.84% (Supplementary Fig. 2A, right). ETV4 depletion had a largely detrimental effect on cell survival in SK-MEL-23 but not in CHL-1 cells (Supplementary Fig. 2B). Supplementary Fig. 2C, D shows the results of representative survival assays testing the effects of ETV4 depletion on response to trametinib, and summarises SF50 values from three independent assays in each cell line. We found no evidence that ETV4 depletion influenced the response of either cell line to MEK inhibition by trametinib (Supplementary Fig. 2C, D). However, the presence of relatively few surviving colonies in ETV4-depleted cultures, especially of SK-MEL-23 (Supplementary Fig. 2B, right), could have contributed to the variation in trametinib SF50 data (Supplementary Fig. 2D), so we cannot exclude a small effect on trametinib response.
DUSP4 depletion induces resistance of BRAF wild-type melanoma cells to MEK inhibition
Next, we used siRNAs to deplete DUSP4 and measured cell survival and MEK inhibitor SF50 values. Both DUSP4 siRNAs used induced effective DUSP4 depletion, at both the mRNA and protein level (Fig. 4a, b). We tested the duration of DUSP4 knockdown in CHL-1 cells and demonstrated that depletion lasted at least 7 days (Fig. 4c). Compared with controls, DUSP4-depleted cells showed an increase in cell survival that was significant in CHL-1 cells (**P = 0.0187 and 0.0154 for siDUSP4_1 and _2, respectively, Fig. 4d left), but not in SK-MEL-23 cells (Fig. 4d right).
Fig. 4. Depletion of DUSP4 decreases sensitivity to MEK inhibition in BRAF wild-type melanoma cell lines.

a, b Quantification of DUSP4 depletion following knockdown by siDUSP4_1 and siDUSP4_2 in: left, CHL-1 and right, SK-MEL-23 cells analysed by qRT-PCR (a) and western blot (b). Arrowhead: probable non-specific band not reduced by DUSP4 depletion. c Duration of DUSP4 knockdown in CHL-1 cells, analysed by western blotting at 3, 5 and 7 days after transfection. Representative results are shown for Allstars (AS) control siRNA and siDUSP4_1. ERK is shown as a loading control. d The effect of DUSP4 depletion by siDUSP4_1 and siDUSP4_2 on colony count in: left, CHL-1 and right, SK-MEL-23 cells (mean ± SEM of five independent experiments). e The effect of DUSP4 depletion by siDUSP4_1 and siDUSP4_2 on sensitivity of: left, CHL-1 and right, SK-MEL-23 cells to MEK inhibitors selumetinib and trametinib (mean ± SEM of triplicate values for a single representative experiment). Table 2 shows a summary of fold changes in MEK inhibitor sensitivity (pooled data from six experiments in each case).
Finally, we tested the sensitivity of DUSP4-depleted cells to both MEK inhibitors. The results are shown in Fig. 4e, and the data are summarised in Table 2. In both cell lines with both MEK inhibitors, there was a consistent shift to the right of the MEK inhibitor dose–response curve (Fig. 4e), with 2.5–6.8-fold increase in SF50 values in DUSP4-depleted cells compared with controls (Table 2). These results indicate that DUSP4-depleted cells were less sensitive to both selumetinib and trametinib than the control transfectants. Thus, DUSP4 protein levels are not only affected by MEK inhibition but also alter cellular response to this class of drug.
Table 2.
Effect of DUSP4 depletion on response to MEK inhibition in BRAF wild-type melanoma cells.
| Cell line | CHL-1 | SK-MEL-23 | ||
|---|---|---|---|---|
| MEK inhibitor | Selumetinib | Trametinib | Selumetinib | Trametinib |
| Fold increase in SF50 | 6.71 ± 1.30 | 6.85 ± 2.42 | 2.94 ± 0.40 | 2.48 ± 0.44 |
| p-value | 0.0072 | 0.0605 | 0.0048 | 0.02 |
The table shows mean ± SEM fold increase in SF50 values in DUSP4-depleted CHL-1 and SK-MEL-23 cells (n = 6 in each case; data analysed using two-tailed t test).
Discussion
There are fewer options for treating BRAF wild-type advanced melanoma than for the BRAF-mutated population. DOC-MEK was the first published randomised trial in a selected BRAF wild-type melanoma population, and indicated that a proportion of such patients could benefit from combined treatment with docetaxel and selumetinib.13 During the conduct of the DOC-MEK study, MEK inhibitor binimetinib (MEK162) was reported to have a 20% ORR (6/30) in patients with NRAS-mutated melanoma in a monotherapy Phase II study.28 However, in a retrospective analysis of DOC-MEK data, we observed no correlation between NRAS status and clinical outcome in either treatment arm,13 reflecting previously published data with selumetinib alone29 and selumetinib plus dacarbazine/docetaxel chemotherapy.30
We extended the mutational analysis to a 46-gene cancer panel and found no correlation between the number of concomitant mutations and clinical outcome. We therefore investigated the hypothesis that the expression of genes that correlate with increased MAPK pathway activity may predict for sensitivity to MEK inhibitor therapy. Whilst BRAF and RAS mutations vary across cell lines that are sensitive to MEK inhibition, a MEK 18-gene functional activation signature score was previously found to be consistently elevated in selumetinib-sensitive cell lines, and was higher in BRAF mutant vs wild-type melanomas.19 These 18 genes are DUSP4, DUSP6, ETV4, ETV5, PHLDA1, SPRY2, ELF1, FXYD5, KANK1, LGALS3, LZTS1, MAP2K3, PROS1, S100A6, SERPINB1, SLCO4A1, TRIB2 and ZFP106. After refining the score to six genes (DUSP4, DUSP6, ETV4, ETV5, PHLDA1, and SPRY2), based on reproducibility across tumour types, the MEK 6 gene score has been shown to be higher in KRAS mutant than KRAS wild-type NSCLC.20 This suggests that known activating mutations in the MAPK pathway are associated with higher MEK gene signature scores. Indeed, all the components of the 6-gene score are known transcriptional targets of the MEK–ERK pathway.21,31–36 Furthermore, four of six of these genes (DUSP4, DUSP6, PHLDA1 and SPRY2) are negative regulators of ERK pathway activity, forming part of a regulatory feedback loop.21,31,33,36,37 Consistent with the ability of mutant NRAS to activate MEK–ERK signalling, our results suggest that NRAS mutant melanoma is associated with a higher MEK 6 gene expression score than NRAS wild-type melanoma (Fig. 1b). It is interesting to note that the second highest MEK 6 gene score was in a melanoma found to have two concurrent NRAS mutations, Q61K and Q61R (Supplementary Table 4, patient DM077). However, neither NRAS mutation status nor VAF analysis was sufficiently discriminating to judge dependence on the MAPK pathway and thus potential sensitivity to MEK inhibition. We then assessed the MEK score with respect to patient outcome, and here the data suggested that a higher MEK score did predict for sensitivity to selumetinib plus docetaxel combination therapy, but not docetaxel therapy alone (Fig. 2). To strengthen this conclusion, it would have been preferable to obtain on-treatment biopsies, to confirm that MEK–ERK was indeed inhibited by selumetinib. We also acknowledge that there were only minor differences in MEK signature score by NRAS mutation and clinical response status, limiting the utility of this score as a biomarker for MEK inhibitor response in melanoma. Further data would be required to confirm this trend and fully characterise an optimal threshold.
We report here that two components of the MEK 6 gene score, ETV4 and DUSP4, were expressed at significantly higher levels in melanomas of responders to docetaxel plus selumetinib compared with those who progressed at first assessment. This difference was not found in the docetaxel plus placebo arm. This suggests the possibility that DUSP4 and ETV4 may be potential biomarkers of sensitivity to MEK inhibition. We wished to extend this observation by ascertaining whether DUSP4 or ETV4 might also influence the response of wild-type BRAF melanoma cells to MEK inhibition. ETV4 is a member of the polyomavirus enhancer activator 3 (PEA3) subfamily of the Ets transcription factor family and regulates genes that promote metastasis.38 As well as inducing ETV4 expression, ERK1/2 promotes ETV4 activation by phosphorylation and sumoylation.34,39,40 Previous studies reported inconsistent findings regarding the contribution of ETV4 to cell survival.41,42 Our data indicate that ETV4 depletion inhibited cell survival of BRAF wild-type melanoma cells but did not influence response to MEK inhibition.
DUSP4 was the only other component of the MEK 6 gene score that was expressed at significantly higher levels in the melanomas of patients who responded to selumetinib and docetaxel, compared with those who progressed at first assessment (Table 1). DUSP4 dephosphorylates and thus inactivates ERK1/2 in the nucleus, and may also act on the JNK and p38 pathways.43 There is conflicting evidence regarding the significance of DUSP4 expression in cancer. DUSP4 upregulation has been reported in KRAS mutant rectal cancer,44 and higher DUSP4 levels have been found in melanoma cell lines compared with normal human epidermal melanocytes.45 Conversely, DUSP4 levels are higher in indolent ovarian serous borderline tumours compared with more aggressive serous carcinomas,46 and silencing of DUSP4 plays a key role in the development of glioblastomas,47 suggesting a tumour-suppressor role. In vitro, DUSP4 knockdown increases growth of EGFR mutant lung adenocarcinoma cell lines, whereas in colorectal cancer cell lines DUSP4 overexpression results in increased proliferation.48 There is also conflict in the literature regarding the significance of DUSP4 for predicting response to anticancer therapy. Higher DUSP4 expression has been found to correlate with resistance to anti-EGFR antibody cetuximab in patients with metastatic colorectal cancer, although this may simply reflect the presence of KRAS mutations that activate RAS–MAPK.49 Conversely, lower DUSP4 expression in breast cancer was reported to be associated with reduced response to neoadjuvant chemotherapy, and in breast cancer cell lines, DUSP4 depletion increased resistance to docetaxel and other cytotoxic drugs, while overexpression increased chemotherapy-induced apoptosis.50
Our data are consistent with findings that endogenous DUSP4 levels vary between melanoma cell lines in vitro.51 Relative overexpression of DUSP4 and ETV4 in SK-MEL-23 compared with CHL-1 may reflect the fact that SK-MEL-23 cells harbour amplified wild-type BRAF.52 Both cell lines used in this study harbour wild-type NRAS; given that ~60% of patients in the clinical DOC-MEK study had NRAS mutant melanoma, it may be informative to assess the contribution of DUSP4 and ETV4 to MEK inhibitor response in NRAS mutant cell lines. In the Broad-Novartis Cancer Cell Line Encyclopedia (CCLE) database, melanoma cell lines express the highest level of DUSP4 mRNA of all cancer cell lines tested, likely reflecting the importance of RAS–RAF–MEK activation in melanomas (www.broadinstitute.org/ccle). Of relevance to our study, CCLE data indicate that high DUSP4 expression is predictive of increased sensitivity to selumetinib, with an odds ratio of 2.3.53 Similarly, in a panel of pan-negative (wild-type BRAF, NRAS, KIT, GNAQ and GNA11) melanoma cell lines, cells expressing higher levels of DUSP4 have been reported to show significantly greater sensitivity to MEK inhibition.51 This correlates with our finding that CHL-1 cells that express lower DUSP4 were less sensitive to selumetinib than the higher-expressing SK-MEL-23 cell line (Fig. 4e). We further found that DUSP4 expression was reduced by MEK inhibition, confirming that DUSP4 levels are regulated by the MAPK pathway.21 Consistent with this, and given that DUSP4 is a negative regulator of the MAPK pathway, depleting DUSP4 increased survival of CHL-1 cells. DUSP4 depletion did not influence survival of SK-MEL-23 cells, possibly because residual DUSP4 may have been sufficient to maintain MAPK pathway regulation, consistent with data in Fig. 4b.
Finally, we showed that siRNA-mediated DUSP4 depletion leads to desensitisation to MEK1/2 inhibition. This response parallels the finding in the clinical trial, where higher DUSP4 expression associated significantly with response to MEK inhibition. We observed the MEK sensitisation effect in CHL-1 cells, where DUSP4 depletion had influenced cell survival, and also in SK-MEL-23 cells, where DUSP4-depleted cells showed no significant difference in cell survival compared with controls. These results suggest that the effect of DUSP4 on MEK inhibitor sensitivity is likely independent of the effect on cell survival. Thus, while previous studies showed a correlation between DUSP4 expression and MEK inhibitor sensitivity in pan-tumour cell line panels,53 we report for the first time that this association exists in clinical melanomas, and depleting DUSP4 expression induces resistance to MEK inhibition. These results suggest that DUSP4 is capable of influencing response to drugs that target MEK. Furthermore, given that BRAF amplification reportedly mediates MEK inhibitor resistance54 the ability of DUSP4 depletion to sensitise SK-MEL-23 cells suggests that this approach may have merit in the BRAF-amplified population. However, we recognise the need to be cautious in interpretation of our data, given the small size of the clinical study and in vitro analysis. It may be informative to assess the predictive significance of DUSP4 in a larger clinical dataset.
In summary, our findings suggest that DUSP4 plays a direct role in determining cellular response to MEK inhibition. DUSP4 may therefore be not only a biomarker for, but also a potential determinant of, the response of wild-type BRAF melanomas to MEK inhibition.
Supplementary information
Acknowledgements
We would like to acknowledge all of the patients who participated in the DOC-MEK study and all of the staff involved at the 18 hospitals across the United Kingdom who contributed to the DOC-MEK study.
Author contributions
D.R.H. and M.R.M. designed the clinical study. A.G., M.R.M. and V.M.M. designed the translational research study. A.G., C.T., F.W., R.B., A.C., A.S., R.A., K.M., L.C., A.W., M.R.M. and V.M.M. were responsible for the acquisition of data, including clinical trial management, sample analysis and data interpretation and laboratory experimental work. A.G., F.W., R.B., D.R.H., A.S., P.S, M.R.M. and V.M.M. were responsible for data analysis. A.G., F.W., S.L. and V.M.M. were responsible for statistical analysis. A.G., F.W. and V.M.M. wrote the paper, with input and final approval from all authors.
Ethics approval and consent to participate
All patients who participated in the DOC-MEK study provided written informed consent, including consent to use of archival tissue samples and clinical data for exploratory research analysis. The protocol was approved by the NHS Oxfordshire A Research Ethics Committee. The study was conducted according to UK Clinical Trials Regulations and the ICH guidelines of Good Clinical Practice in accordance with the Declaration of Helsinki.
Consent to publish
Not applicable.
Data availability
The dataset used and analysed for this paper is available from the corresponding author on request.
Competing interests
A.G. has consulted for and received honoraria from BMS and Novartis. R.B., D.R.H., A.S. and P.S. are employees of AstraZeneca. R.B. has shares in AstraZeneca. A.S. has received honoraria from Gilead, Roche, Janssen and Abbvie, and an unrestricted educational grant from Gilead and Janssen. M.R.M. has consulted for and received research funding from AstraZeneca, GlaxoSmithKline and Roche. V.M.M. has consulted for Boehringer Ingelheim.
Funding information
The DOC-MEK trial was supported by the collaboration between AstraZeneca and the UK National Cancer Research Network (Grant number: A12154), for which AstraZeneca provided partial funding. DOC-MEK was sponsored by the University of Oxford and was run by the Oncology Clinical Trials Office (OCTO), Department of Oncology, University of Oxford. This study was supported by funding from the Oxford NIHR Biomedical Research Centre to AS, MRM and VMM, and an Oxfordshire Health Services Research Committee grant to AG. The views expressed are those of the authors and not necessarily those of the NHS, the NIHR or the United Kingdom’s Department of Health. FW and CT were supported by funding to the Preclinical Validation Laboratory from the Cancer Research UK Oxford Cancer Research Centre.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary information is available for this paper at 10.1038/s41416-019-0673-5.
References
- 1.Shah DJ, Dronca RS. Latest advances in chemotherapeutic, targeted, and immune approaches in the treatment of metastatic melanoma. Mayo Clin. Proc. 2014;89:504–519. doi: 10.1016/j.mayocp.2014.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ugurel S, Rohmel J, Ascierto PA, Flaherty KT, GROB JJ, Hauschild A, et al. Survival of patients with advanced metastatic melanoma: the impact of novel therapies. Eur. J. Cancer. 2016;53:125–134. doi: 10.1016/j.ejca.2015.09.013. [DOI] [PubMed] [Google Scholar]
- 3.Amanuel B, Grieu F, Kular J, Millward M, Iacopetta B. Incidence of BRAF p.Val600Glu and p.Val600Lys mutations in a consecutive series of 183 metastatic melanoma patients from a high incidence region. Pathology. 2012;44:357–359. doi: 10.1097/PAT.0b013e3283532565. [DOI] [PubMed] [Google Scholar]
- 4.Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–954. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 5.Wan PT, Garnett MJ, Roe SM, Lee S, Niculescu-Duvaz D, Good VM, et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell. 2004;116:855–867. doi: 10.1016/s0092-8674(04)00215-6. [DOI] [PubMed] [Google Scholar]
- 6.Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med. 2011;364:2507–2516. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hauschild A, Grob JJ, Demidov LV, Jouary T, Gutzmer R, Millward M, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380:358–365. doi: 10.1016/S0140-6736(12)60868-X. [DOI] [PubMed] [Google Scholar]
- 8.Long GV, Stroyakovskiy D, Gogas H, Levchenko E, De Braud F, Larkin J, et al. Dabrafenib and trametinib versus dabrafenib and placebo for Val600 BRAF-mutant melanoma: a multicentre, double-blind, phase 3 randomised controlled trial. Lancet. 2015;386:444–451. doi: 10.1016/S0140-6736(15)60898-4. [DOI] [PubMed] [Google Scholar]
- 9.Robert C, Karaszewska B, Schachter J, Rutkowski P, Mackiewicz A, Stroiakovski D, et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N. Engl. J. Med. 2015;372:30–39. doi: 10.1056/NEJMoa1412690. [DOI] [PubMed] [Google Scholar]
- 10.Larkin J, Ascierto PA, Dreno B, Atkinson V, Liszkay G, Maio M, et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N. Engl. J. Med. 2014;371:1867–1876. doi: 10.1056/NEJMoa1408868. [DOI] [PubMed] [Google Scholar]
- 11.Flaherty KT, Puzanov I, Kim KB, Ribas A, McArthur GA, Sosman JA, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N. Engl. J. Med. 2010;363:809–819. doi: 10.1056/NEJMoa1002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Falchook GS, Long GV, Kurzrock R, Kim KB, Arkenau TH, Brown MP, et al. Dabrafenib in patients with melanoma, untreated brain metastases, and other solid tumours: a phase 1 dose-escalation trial. Lancet. 2012;379:1893–1901. doi: 10.1016/S0140-6736(12)60398-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gupta A, Love S, Schuh A, Shanyinde M, Larkin JM, Plummer R, et al. DOC-MEK: a double-blind randomized phase II trial of docetaxel with or without selumetinib in wild-type BRAF advanced melanoma. Ann. Oncol. 2014;25:968–974. doi: 10.1093/annonc/mdu054. [DOI] [PubMed] [Google Scholar]
- 14.Yeh TC, Marsh V, Bernat BA, Ballard J, Colwell H, Evans RJ, et al. Biological characterization of ARRY-142886 (AZD6244), a potent, highly selective mitogen-activated protein kinase kinase 1/2 inhibitor. Clin. Cancer Res. 2007;13:1576–1583. doi: 10.1158/1078-0432.CCR-06-1150. [DOI] [PubMed] [Google Scholar]
- 15.Haass NK, Sproesser K, Nguyen TK, Contractor R, Medina CA, Nathanson KL, et al. The mitogen-activated protein/extracellular signal-regulated kinase kinase inhibitor AZD6244 (ARRY-142886) induces growth arrest in melanoma cells and tumor regression when combined with docetaxel. Clin. Cancer Res. 2008;14:230–239. doi: 10.1158/1078-0432.CCR-07-1440. [DOI] [PubMed] [Google Scholar]
- 16.Holt SV, Logie A, Odedra R, Heier A, Heaton SP, Alferez D, et al. The MEK1/2 inhibitor, selumetinib (AZD6244; ARRY-142886), enhances anti-tumour efficacy when combined with conventional chemotherapeutic agents in human tumour xenograft models. Br. J. Cancer. 2012;106:858–866. doi: 10.1038/bjc.2012.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.LoRusso PM, Infante JR, Kim KB, Burris HA, 3rd, Curt G, Emeribe U, et al. A phase 1 dose-escalation study of selumetinib in combination with docetaxel or dacarbazine in patients with advanced solid tumours. BMC Cancer. 2017;17:173. doi: 10.1186/s12885-017-3143-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Janne PA, Shaw AT, Pereira JR, Jeannin G, Vansteenkiste J, Barrios C, et al. Selumetinib plus docetaxel for KRAS-mutant advanced non-small-cell lung cancer: a randomised, multicentre, placebo-controlled, phase 2 study. Lancet Oncol. 2013;14:38–47. doi: 10.1016/S1470-2045(12)70489-8. [DOI] [PubMed] [Google Scholar]
- 19.Dry JR, Pavey S, Pratilas CA, Harbron C, Runswick S, Hodgson D, et al. Transcriptional pathway signatures predict MEK addiction and response to selumetinib (AZD6244) Cancer Res. 2010;70:2264–2273. doi: 10.1158/0008-5472.CAN-09-1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brant RG, Sharpe A, Liptrot T, Dry J, Harrington EA, Barrett JC, et al. Clinically viable gene expression assays with potential for predicting benefit from MEK inhibitors. Clin. Cancer Res. 2016;23:1471–1480. doi: 10.1158/1078-0432.CCR-16-0021. [DOI] [PubMed] [Google Scholar]
- 21.Cagnol S, Rivard N. Oncogenic KRAS and BRAF activation of the MEK/ERK signaling pathway promotes expression of dual-specificity phosphatase 4 (DUSP4/MKP2) resulting in nuclear ERK1/2 inhibition. Oncogene. 2013;32:564–576. doi: 10.1038/onc.2012.88. [DOI] [PubMed] [Google Scholar]
- 22.Siroy AE, Boland GM, Milton DR, Roszik J, Frankian S, Malke J, et al. Beyond BRAF(V600): clinical mutation panel testing by next-generation sequencing in advanced melanoma. J. Invest. Dermatol. 2015;135:508–515. doi: 10.1038/jid.2014.366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Geiss GK, Bumgarner RE, Birditt B, Dahl T, Dowidar N, Dunaway DL, et al. Direct multiplexed measurement of gene expression with color-coded probe pairs. Nat Biotechnol. 2008;26:317–325. doi: 10.1038/nbt1385. [DOI] [PubMed] [Google Scholar]
- 24.Chitnis MM, Lodhia KA, Aleksic T, Gao S, Protheroe AS, Macaulay VM. IGF-1R inhibition enhances radiosensitivity and delays double-strand break repair by both non-homologous end-joining and homologous recombination. Oncogene. 2014;33:5262–5273. doi: 10.1038/onc.2013.460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dahlman KB, Xia J, Hutchinson K, Ng C, Hucks D, Jia P, et al. BRAF(L597) mutations in melanoma are associated with sensitivity to MEK inhibitors. Cancer Discov. 2012;2:791–797. doi: 10.1158/2159-8290.CD-12-0097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim KB, Kefford R, Pavlick AC, Infante JR, Ribas A, Sosman JA, et al. Phase II study of the MEK1/MEK2 inhibitor Trametinib in patients with metastatic BRAF-mutant cutaneous melanoma previously treated with or without a BRAF inhibitor. J. Clin. Oncol. 2013;31:482–489. doi: 10.1200/JCO.2012.43.5966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Infante JR, Fecher LA, Falchook GS, Nallapareddy S, Gordon MS, Becerra C, et al. Safety, pharmacokinetic, pharmacodynamic, and efficacy data for the oral MEK inhibitor trametinib: a phase 1 dose-escalation trial. Lancet Oncol. 2012;13:773–781. doi: 10.1016/S1470-2045(12)70270-X. [DOI] [PubMed] [Google Scholar]
- 28.Ascierto PA, Schadendorf D, Berking C, Agarwala SS, Van Herpen CM, Queirolo P, et al. MEK162 for patients with advanced melanoma harbouring NRAS or Val600 BRAF mutations: a non-randomised, open-label phase 2 study. Lancet Oncol. 2013;14:249–256. doi: 10.1016/S1470-2045(13)70024-X. [DOI] [PubMed] [Google Scholar]
- 29.Kirkwood JM, Bastholt L, Robert C, Sosman J, Larkin J, Hersey P, et al. Phase II, open-label, randomized trial of the MEK1/2 inhibitor selumetinib as monotherapy versus temozolomide in patients with advanced melanoma. Clin. Cancer Res. 2012;18:555–567. doi: 10.1158/1078-0432.CCR-11-1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Patel SP, Lazar AJ, Papadopoulos NE, Liu P, Infante JR, Glass MR, et al. Clinical responses to selumetinib (AZD6244; ARRY-142886)-based combination therapy stratified by gene mutations in patients with metastatic melanoma. Cancer. 2013;119:799–805. doi: 10.1002/cncr.27790. [DOI] [PubMed] [Google Scholar]
- 31.Oberst MD, Beberman SJ, Zhao L, Yin JJ, Ward Y, Kelly K. TDAG51 is an ERK signaling target that opposes ERK-mediated HME16C mammary epithelial cell transformation. BMC Cancer. 2008;8:189. doi: 10.1186/1471-2407-8-189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Oh S, Shin S, Janknecht R. ETV1, 4 and 5: an oncogenic subfamily of ETS transcription factors. Biochim. Biophys. Acta. 2012;1826:1–12. doi: 10.1016/j.bbcan.2012.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Caunt CJ, Keyse SM. Dual-specificity MAP kinase phosphatases (MKPs): shaping the outcome of MAP kinase signalling. FEBS J. 2013;280:489–504. doi: 10.1111/j.1742-4658.2012.08716.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fontanet P, Irala D, Alsina FC, Paratcha G, Ledda F. Pea3 transcription factor family members Etv4 and Etv5 mediate retrograde signaling and axonal growth of DRG sensory neurons in response to NGF. J. Neurosci. 2013;33:15940–15951. doi: 10.1523/JNEUROSCI.0928-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Joshi S, Platanias LC. Mnk kinase pathway: Cellular functions and biological outcomes. World J. Biol. Chem. 2014;5:321–333. doi: 10.4331/wjbc.v5.i3.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pratilas C. A., Taylor B. S., Ye Q., Viale A., Sander C., Solit D. B., Rosen N. V600EBRAF is associated with disabled feedback inhibition of RAF-MEK signaling and elevated transcriptional output of the pathway. Proceedings of the National Academy of Sciences. 2009;106(11):4519–4524. doi: 10.1073/pnas.0900780106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ozaki K, Kadomoto R, Asato K, Tanimura S, Itoh N, Kohno M. ERK pathway positively regulates the expression of Sprouty genes. Biochem. Biophys. Res. Commun. 2001;285:1084–1088. doi: 10.1006/bbrc.2001.5295. [DOI] [PubMed] [Google Scholar]
- 38.de Launoit Y, Baert JL, Chotteau-Lelievre A, Monte D, Coutte L, Mauen S, et al. The Ets transcription factors of the PEA3 group: transcriptional regulators in metastasis. Biochim. Biophys. Acta. 2006;1766:79–87. doi: 10.1016/j.bbcan.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 39.Guo B, Sharrocks AD. Extracellular signal-regulated kinase mitogen-activated protein kinase signaling initiates a dynamic interplay between sumoylation and ubiquitination to regulate the activity of the transcriptional activator PEA3. Mol Cell Biol. 2009;29:3204–3218. doi: 10.1128/MCB.01128-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Charlot C, Dubois-Pot H, Serchov T, Tourrette Y, Wasylyk B. A review of post-translational modifications and subcellular localization of Ets transcription factors: possible connection with cancer and involvement in the hypoxic response. Methods Mol. Biol. 2010;647:3–30. doi: 10.1007/978-1-60761-738-9_1. [DOI] [PubMed] [Google Scholar]
- 41.Kherrouche Z, Monte D, Werkmeister E, Stoven L, De Launoit Y, Cortot AB, et al. PEA3 transcription factors are downstream effectors of Met signaling involved in migration and invasiveness of Met-addicted tumor cells. Mol. Oncol. 2015;9:1852–1867. doi: 10.1016/j.molonc.2015.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mesquita D, Barros-Silva JD, Santos J, Skotheim RI, Lothe RA, Paulo P, et al. Specific and redundant activities of ETV1 and ETV4 in prostate cancer aggressiveness revealed by co-overexpression cellular contexts. Oncotarget. 2015;6:5217–5236. doi: 10.18632/oncotarget.2847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen P, Hutter D, Yang X, Gorospe M, Davis RJ, Liu Y. Discordance between the binding affinity of mitogen-activated protein kinase subfamily members for MAP kinase phosphatase-2 and their ability to activate the phosphatase catalytically. J. Biol. Chem. 2001;276:29440–29449. doi: 10.1074/jbc.M103463200. [DOI] [PubMed] [Google Scholar]
- 44.Gaedcke J, Grade M, Jung K, Camps J, Jo P, Emons G, et al. Mutated KRAS results in overexpression of DUSP4, a MAP-kinase phosphatase, and SMYD3, a histone methyltransferase, in rectal carcinomas. Genes Chromosomes Cancer. 2010;49:1024–1034. doi: 10.1002/gcc.20811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Teutschbein J, Haydn JM, Samans B, Krause M, Eilers M, Schartl M, et al. Gene expression analysis after receptor tyrosine kinase activation reveals new potential melanoma proteins. BMC Cancer. 2010;10:386. doi: 10.1186/1471-2407-10-386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sieben NL, Oosting J, Flanagan AM, Prat J, Roemen GM, Kolkman-Uljee SM, et al. Differential gene expression in ovarian tumors reveals Dusp 4 and Serpina 5 as key regulators for benign behavior of serous borderline tumors. J. Clin. Oncol. 2005;23:7257–7264. doi: 10.1200/JCO.2005.02.2541. [DOI] [PubMed] [Google Scholar]
- 47.Waha A, Felsberg J, Hartmann W, Von Dem Knesebeck A, Mikeska T, Joos S, et al. Epigenetic downregulation of mitogen-activated protein kinase phosphatase MKP-2 relieves its growth suppressive activity in glioma cells. Cancer Res. 2010;70:1689–1699. doi: 10.1158/0008-5472.CAN-09-3218. [DOI] [PubMed] [Google Scholar]
- 48.Groschl B, Bettstetter M, Giedl C, Woenckhaus M, Edmonston T, Hofstadter F, et al. Expression of the MAP kinase phosphatase DUSP4 is associated with microsatellite instability in colorectal cancer (CRC) and causes increased cell proliferation. Int. J. Cancer. 2013;132:1537–1546. doi: 10.1002/ijc.27834. [DOI] [PubMed] [Google Scholar]
- 49.Khambata-Ford S, Garrett CR, Meropol NJ, Basik M, Harbison CT, Wu S, et al. Expression of epiregulin and amphiregulin and K-ras mutation status predict disease control in metastatic colorectal cancer patients treated with cetuximab. J. Clin. Oncol. 2007;25:3230–3237. doi: 10.1200/JCO.2006.10.5437. [DOI] [PubMed] [Google Scholar]
- 50.Balko JM, Cook RS, Vaught DB, Kuba MG, Miller TW, Bhola NE, et al. Profiling of residual breast cancers after neoadjuvant chemotherapy identifies DUSP4 deficiency as a mechanism of drug resistance. Nat. Med. 2012;18:1052–1059. doi: 10.1038/nm.2795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hutchinson KE, Johnson DB, Johnson AS, Sanchez V, Kuba M, Lu P, et al. ERBB activation modulates sensitivity to MEK1/2 inhibition in a subset of driver-negative melanoma. Oncotarget. 2015;6:22348–22360. doi: 10.18632/oncotarget.4255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tanami H, Imoto I, Hirasawa A, Yuki Y, Sonoda I, Inoue J, et al. Involvement of overexpressed wild-type BRAF in the growth of malignant melanoma cell lines. Oncogene. 2004;23:8796–8804. doi: 10.1038/sj.onc.1208152. [DOI] [PubMed] [Google Scholar]
- 53.Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603–607. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Villanueva J, Infante JR, Krepler C, Reyes-Uribe P, Samanta M, Chen HY, et al. Concurrent MEK2 mutation and BRAF amplification confer resistance to BRAF and MEK inhibitors in melanoma. Cell Rep. 2013;4:1090–1099. doi: 10.1016/j.celrep.2013.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The dataset used and analysed for this paper is available from the corresponding author on request.