

Identifying human sources of fecal pollution is critical to remediate sanitation concerns. Large financial investments are required to address these concerns; therefore, a high level of confidence in testing results is needed. Human fecal marker genes validated in this study showed high specificity in both sequencing data and qPCR results. Human marker sequences were rarely found in individual animals, and in most cases, the animals had atypical microbial communities. Sequencing also revealed the presence of closely related organisms that could account for nonspecific amplification in certain assays. Both the true cross-reactions and the nonspecific amplification had low signals well below E. coli or Enterococcus levels and likely would not impact the assay’s ability to reliably detect human fecal pollution. No animal source had multiple human/sewage marker genes present; therefore, using a combination of marker genes would increase the confidence of human fecal pollution detection.
KEYWORDS: human fecal indicator, microbial source tracking, animal cross-reaction, next-generation sequencing, qPCR, Lachnospiraceae, Bacteroides, BacV6-21, HF183, Lachno3
ABSTRACT
Quantitative PCR (qPCR) assays for human/sewage marker genes have demonstrated sporadic positive results in animal feces despite their high specificities to sewage and human feces. It is unclear whether these positive reactions are caused by true occurrences of microorganisms containing the marker gene (i.e., indicator organisms) or nonspecific amplification (false positive). The distribution patterns of human/sewage indicator organisms in animals have not been explored in depth, which is crucial for evaluating a marker gene’s true- or false-positive reactions. Here, we analyzed V6 region 16S rRNA gene sequences from 257 animal fecal samples and tested a subset of 184 using qPCR for human/sewage marker genes. Overall, specificities of human/sewage marker genes within sequencing data were 99.6% (BacV6-21), 96.9% (Lachno3), and 96.1% (HF183, indexed by its inferred V6 sequence). Occurrence of some true cross-reactions was associated with atypical compositions of organisms within the genera Blautia or Bacteroides. For human/sewage marker qPCR assays, specificities were 96.7% (HF183/Bac287R), 96.2% (BacV6-21), 95.6% (human Bacteroides [HB]), and 94.0% (Lachno3). Select assays duplexed with either Escherichia coli or Enterococcus spp. were also validated. Most of the positive qPCR results in animals were low level and, on average, 2 orders of magnitude lower than the copy numbers of E. coli and Enterococcus spp. The lower specificity in qPCR assays compared to sequencing data was mainly caused by amplification of sequences highly similar to the marker gene and not the occurrence of the exact marker sequence in animal fecal samples.
IMPORTANCE Identifying human sources of fecal pollution is critical to remediate sanitation concerns. Large financial investments are required to address these concerns; therefore, a high level of confidence in testing results is needed. Human fecal marker genes validated in this study showed high specificity in both sequencing data and qPCR results. Human marker sequences were rarely found in individual animals, and in most cases, the animals had atypical microbial communities. Sequencing also revealed the presence of closely related organisms that could account for nonspecific amplification in certain assays. Both the true cross-reactions and the nonspecific amplification had low signals well below E. coli or Enterococcus levels and likely would not impact the assay’s ability to reliably detect human fecal pollution. No animal source had multiple human/sewage marker genes present; therefore, using a combination of marker genes would increase the confidence of human fecal pollution detection.
INTRODUCTION
Microbial source tracking (MST) methods have been applied to track human fecal pollution in water for about 2 decades (1–4). The basic theory of MST is that detecting the presence of host-specific fecal microorganisms can indicate the source of fecal pollution (5, 6). Bacterial human fecal marker assays were initially developed as PCR assays (4) and subsequently quantitative PCR (qPCR) assays (7–10). Many of these assays target the V2 to V4 hypervariable regions of the 16S rRNA gene of Bacteroides, including one of the most studied markers, HF183, from a human-specific Bacteroides that is detected using a number of similar assays, including human Bacteroides (HB) (4, 9, 11) and HF183/BacR287, that was recently developed into a standard method by the Environmental Protection Agency (EPA) (12, 13). More recently, two assays, designated BacV4V5-1 and BacV6-21, have been designed to target nonfecal Bacteroides in sewage (i.e., Bacteroides that are derived from the sewer pipe environment rather than human or animal feces) (14). These assays provide another measurement for sewage detection independent of human and animal fecal microorganisms. In addition, human fecal marker assays have also been developed from the bacterial family Lachnospiraceae, within which the Lachno3 assay demonstrated high human specificity (15). The BacV4V5-1, BacV6-21, and Lachno3 assays were developed based on next-generation sequencing (NGS) data, which allowed for comparison of animal and sewage microbial communities for marker gene identification.
Perhaps the most important performance characteristic of a human fecal marker assay is host specificity, which refers to the assay’s ability to accurately detect the targeted fecal source (6, 16). Despite the large number of bacterial human fecal marker assays that have been developed, cross-reaction with animal fecal sources have been reported for all. The causes of reduced specificities of previously developed human marker assays (17–19) have not been explored in depth. In the case of assays designed using NGS data, such as Lachno3 (15) and BacV6-21 (14), the host specificities have been validated in silico using an in-depth sequence inventory in addition to qPCR testing of animal samples. A recent study tested the Lachno3, BacV6-21, and HF183/BacR287 (12) assays in a total of 360 animal fecal samples across 14 hosts (20). Although all the marker assays demonstrated specificity over 90%, animal cross-reaction was also observed (20). The reason for positive qPCR results in animal sources is poorly understood, especially for the BacV6-21 assay that is designed to target a sewer pipe-derived Bacteroides rather than human or animal fecal organism. Some hypotheses for the positive results of these human/sewage marker assays in animal sources include (i) the qPCR assay amplifies a sequence that is highly similar but not identical to the marker gene; (ii) the qPCR amplifies a marker gene that is commonly present in an animal host, but this host was not included in initial validation studies; or (iii) the qPCR amplifies the marker gene in an animal individual that has an atypical gut microbial community composition.
To date, most studies have used qPCR to test for marker gene cross-reactions. When sequencing data are available, the presence of a marker gene in an animal fecal sample can also be identified in silico (15, 21). Here, we analyzed V6 NGS data from 257 individual and pooled animal fecal samples collected from the United States and Australia. For 184 samples from hosts that are common sources of fecal pollution in water environments, qPCR experiments were performed using the Lachno3 (15), HF183/BacR287 (12), a duplex Escherichia coli (22)/HB assay (4, 9, 11), and a duplex Enterococcus (23)/BacV6-21 assay (14). By comparing the presence of a marker gene in NGS data and the corresponding qPCR assay, we identified animal fecal samples that were positive for both approaches or only positive for one approach. Mechanisms for human/sewage marker gene cross-reactions in these animals were explored by examining the microbial community compositions of the animal fecal samples for atypical patterns, which suggested shifts in certain genera (i.e., genus Bacteroides and Blautia) toward a human pattern. In rare cases where the marker assays were positive in the absence of the actual marker sequence, sequences closely related to a marker gene could account for qPCR-positive results in animal fecal samples.
RESULTS
Distribution patterns of Lachnospiraceae, Blautia, and Bacteroides and occurrence of human/sewage marker sequences in animals.
We characterized bacterial community composition for human feces (n = 6), untreated sewage (n = 8), and animal fecal samples (n = 257) and identified human marker sequences in the animal fecal samples. Table S1 in the supplemental material summarizes information of animal fecal samples used in this study. In all, human fecal samples contained 124 families, sewage samples contained 249 families, and all animal fecal samples combined comprised 256 families. Fig. 1 shows the relative abundance patterns of families that were over 1% abundance of the whole community across all the different sample types. Both Lachnospiraceae and Bacteroidaceae were abundant in sewage and human fecal samples and present in multiple animal groups. Fig. S1 shows the distribution patterns of these families in individual sewage and human and animal fecal samples.
FIG 1.

Distribution patterns of the 24 most abundant families in all human, sewage, and animal fecal samples (n = 271). Relative abundance values are normalized to the total sequence counts in each host and increase from light blue to red. The x axis shows groups of sewage, human, and animal hosts. The y axis shows bacterial families ranked from the most (top) to the least (bottom) abundant.
We explored the impact of geographical region on the community composition among different host groups (Fig. S2). Three regions that all had cow, deer, dog, horse and pig samples collected (n = 108) were chosen, including Australia, Texas, and Wisconsin. Cow and deer data were combined as the ruminant group. Samples within host groups showed a small geographic effect (R2 = 0.154), but this was negligible compared to separation among host groups (R2 = 0.512), which supports the premise of MST that animals within a host group have a common microbiome and also demonstrates that the set of animals used for this validation appear to be representative for the same hosts across broader geographic regions.
The mammal samples (n = 219) clustered by host type when considering the entire microbial community, as well as select families or genera (i.e., Bacteroides, Lachnospiraceae, or Blautia), with separation between carnivores, herbivores, and omnivores (Fig. 2). We examined the occurrence pattern of human/sewage marker genes in animal fecal communities. The HF183 marker is within the V2 region and was therefore indexed in the NGS data by its associated V6 region sequence (designated as BacV6-4) (14). The Lachno3 marker (Fig. 2B and C) and the BacV6-4 sequence (Fig. 2D) occurred sporadically in animals. These animals demonstrated an atypical pattern compared to other animals within that host group, except for kangaroos, which were clustered together. In the case of the BacV6-4 proxy marker, a number of the positive animals (i.e., rabbits and deer) were shifted toward a human pattern.
FIG 2.

Nonmetric multidimensional scaling (NMDS) analysis of microbial communities of all mammal samples sequenced in this study (n = 219). Analysis is performed for the whole community (A), the family Lachnospiraceae (B), the genus Blautia (C), and the genus Bacteroides (D). The human group is shown in ellipses with a 95% confidence interval. Samples that are positive for Lachno3 or BacV6-4 are marked with an arrow in figures of corresponding genera. Animal host groups are shown in different symbols. Diet groups are indicated in different colors: carnivore is red, omnivore is green, and herbivore is blue.
Overall, the Lachno3 marker had an NGS specificity of 96.9% when compared to animal samples. The Lachno3 marker sequence occurred in one dog, five kangaroos, one cat, and one raccoon. The dog and one kangaroo were positive in qPCR and were considered as true cross-reactions (Fig. 3A). The other animal samples had low levels of the Lachno3 marker sequence (mean sequence count ± standard deviation [SD], 43 ± 74) and were negative by qPCR, indicating that sequencing could be more sensitive than qPCR in detecting organisms of low abundance. The BacV6-4 sequence had an NGS specificity of 96.1%, occurring in two rabbits (PU27 and Pool1), two deer, five chickens, and one raccoon. The two rabbits, one chicken, and one deer were positive in HF183 qPCR assays and were considered as true cross-reactions of the HF183 marker (Fig. 3C). The other four chickens were not tested by qPCR due to inadequate DNA amounts. The sewage-specific Bacteroides marker, BacV6-21, had an overall NGS specificity of 99.6%, occurring only in one chicken sample in low abundance (i.e., 50 sequence counts) that was not tested by qPCR (Fig. 3B). All marker sequence counts in sewage, human, and animal samples are listed in Data Set S1.
FIG 3.
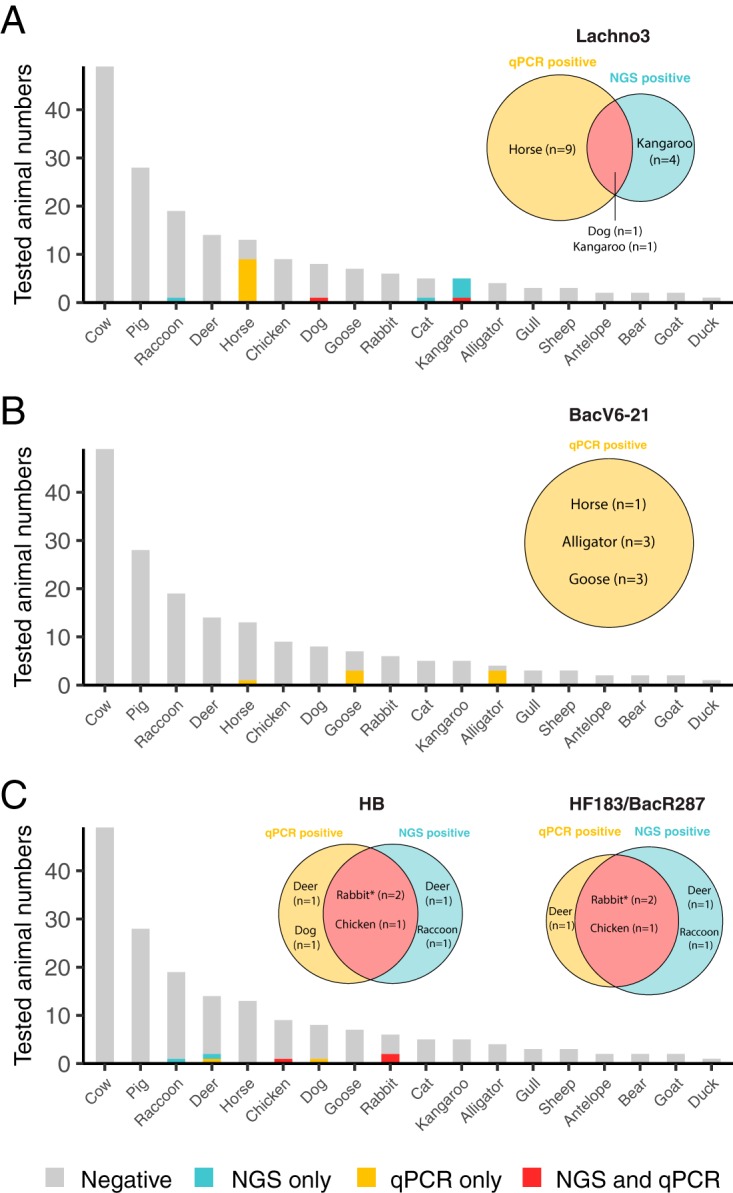
NGS and qPCR validation results in 184 animal samples. Marker genes included are Lachno3 (A), BacV6-21 (B), and BacV6-4 (a proxy of HF183) (C). The x axis shows animal hosts. The y axis shows the total tested animal numbers. Gray bars represent the number of samples that are negative for both NGS and qPCR, blue bars represent the number of samples that are only positive in NGS data, yellow bars represent the number of samples that are only positive in qPCR, and red bars represent the number of samples that are positive in both NGS and qPCR. The Venn diagrams show positive animals of each marker gene in NGS and qPCR data. *, one of the HF183 marker-positive rabbits is sequenced as a pooled sample but tested in qPCR as five individuals.
qPCR human/sewage marker gene specificity and cross-reactions.
For a subset of 184 animal fecal samples, we performed qPCR assays of human/sewage marker genes, including Lachno3, BacV6-21, and two HF183 assays (i.e., HB and HF183/BacR287). Within these animal samples, there were more animal samples positive for one of the human/sewage marker genes when tested by qPCR than when examining the sequence data (Fig. 3). All qPCR assays showed specificity of >90%. Approximately 66% of the qPCR positives had low-level signals (i.e., <102 copy number [CN] per ng of DNA) (Fig. 4). Several rabbit samples had high-level cross-reactions at 103 to 104 CN per ng of DNA when tested with the HF183 assays (Fig. 4). No animal fecal samples were positive for multiple human/sewage marker genes in qPCR assays (i.e., Lachno3, BacV6-21, or HF183).
FIG 4.

Human/sewage marker qPCR assay-positive results. The y axis shows log10-transformed copy number (CN) per nanogram of DNA. The x axis shows tested assays. Each animal host is shown in a different color and shape. The error bar stands for mean ± standard deviation (SD), with the rhombus in the middle representing the mean values.
The Lachno3 qPCR assay had a specificity of 94.0%, showing amplification in nine horses (CN mean ± SD, 101 ± 171 per ng of DNA) that were negative in the NGS data in addition to the kangaroo (CN, 6) and dog (CN, 107) that were positive in the NGS data (Fig. 3A, Fig. 4). The BacV6-21 marker, which was absent in all but one chicken in the NGS data, showed a qPCR assay specificity of 96.2%, with low signals (average CN, 25 ± 21) in three alligators, three geese, and one horse (Fig. 3B, Fig. 4).
For the two HF183 qPCR assays, HB had a specificity of 95.6%, and HF183/BacR287 had a specificity of 96.7%. Six animal samples were positive at similar concentrations for both assays; one dog was positive with the HB assay but negative with the HF183/BacR287 assay (Fig. 3C, Fig. 4). Also, a deer (PU 123) in the NGS data (but not included in the qPCR) in this study was previously tested to have CNs of >103 per ng of DNA for both HB and HF183/BacR287 (15). The qPCR results of all four assays are detailed in Data Set S1.
Mechanisms for human/sewage marker gene occurrence in animals and nonspecific amplification.
We examined the relatedness of sequences within animal microbiomes to Lachno3 or BacV6-21 marker sequences to explore mechanisms for true positives or nonspecific amplification (Fig. 5). The Lachno3 marker was considered to have true cross-reactions in one dog and one kangaroo. These animals also had multiple unique Blautia sequences with over 98% sequence identity to the Lachno3 marker, which were also found in human and sewage samples (Fig. 5A), whereas Lachno3-negative dogs and kangaroos only had Blautia sequences that were < 90% and 92% identity to Lachno3, respectively. These results suggest an overall shift in the Blautia population of positive samples toward organisms found in humans, in addition to the actual marker gene presence.
FIG 5.

Distribution patterns of sequences that are over 90% identical to the human marker genes. (A) Sequences matching the Lachno3 assay’s probe. (B) Sequences matching the BacV6-21 assay’s forward primer and/or probe. For each figure, the y axis shows sequence names with their identity to the marker gene shown from low (green on the top) to high (red at the bottom). The x axis shows hosts of sewage, human, and animals that contain some of the sequences. The heatmap shows the distribution of sequences with original counts increasing from light blue to dark blue. The sequences LC_116 and BC_7, BC_8, and BC_9 are indicated by arrows in panels A and B, respectively.
For animals that lacked the full Lachno3 marker in NGS data but were found to have positive qPCR results (e.g., horse), there were 11 sequences present that matched the Lachno3 probe with varying degrees of similarity to the overall marker sequence (Data Set S2). Ten of these sequences have > 90% similarity to the Lachno3 V6 sequence, including one (LC_116) that was present in 12 out of 13 tested horses, including all qPCR-positive horse samples, in high abundance (Fig. 5A, Data Set S2). Select cows (2 of 49 tested), antelopes (2 of 2 tested), and pigs (5 of 28 tested) also contained sequences >90% similar to Lachno3, but nonspecific amplification did not occur. This suggests that the forward and reverse primer design, which is outside of the Lachno3-sequenced region, is important for the Lachno3 assay specificity.
Similar cases were also observed in the BacV6-21 assay (Fig. 5B, Data Set S2). The qPCR positive-only samples contained several highly similar sequences (i.e., BC_7, BC_8, and BC_9) that only had one base difference with the BacV6-21 marker sequence, either in the forward primer or probe (Fig. 5B, Data Set S2). These sequences were present in alligator, goose, horse, pig, and gull samples. The BacV6-21 qPCR assay was repeated on these animal fecal samples with 1° higher annealing temperature than the original program (i.e., 61°C instead of 60°C). Three out of seven positives were eliminated. Since the HB and the HF183/BacR287 assays target the V2 region, sequence similarity could not be assessed. Although both true and nonspecific amplifications were observed across all of the assays analyzed, specificities of these human and sewage marker assays remained >90%, with the majority of positive signals due to either low sequence occurrence in the community or low CN amplification.
General fecal indicator levels in animals and relationship to human/sewage marker qPCR assays.
Both the E. coli and Enterococcus (ENT) assays had higher CN levels and prevalence in hosts such as alligator, birds, and raccoon and were found in lower levels and had lower prevalence in hosts such as horse, cow, and deer. Fig. S3 shows positive results of the two assays for general fecal indicator bacteria (FIB) in animal host groups. E. coli was positive (CN mean ± SD, 908 ± 2,070 per ng of DNA) in 102 out of 184 animal fecal samples (55%), including all hosts except duck. ENT was positive (CN, 1,850 ± 5,930) in 92 out of 184 samples (50%) in all hosts. Overall, the human/sewage marker assay CNs in animal fecal samples were about 2 orders of magnitude lower than the average CNs of E. coli and ENT qPCR assays (Table S2).
A subset of 83 samples that were negative or positive at low levels for E. coli and/or ENT qPCR assays were retested using the GenBac3 assay to test for the presence/absence of Bacteroidetes organisms to verify that fecal bacteria were present. All samples were positive for the GenBac3 assay with an average CN of 1.51 × 105 ± 3.85 × 105 per ng of DNA, demonstrating that fecal bacteria were present but general indicators were below the limit of detection at the tested DNA concentration (i.e., 1 ng/μl).
DISCUSSION
Microbial source tracking methods have progressed in recent years with extensive qPCR assay validation studies (20, 24–27) and the development of a standardized qPCR method for the HF183 human marker (12, 13, 28). However, studies have shown that no assay is 100% specific for human sources (5, 6, 14), and the underlying mechanism for positive results in animals is poorly understood. Delineating the root causes of positive qPCR results during validation studies will aid in the interpretation of environmental data and improve confidence in results for water resource managers. This latter point is particularly important since large financial resources are required to remediate sewer urban infrastructure or septic system failures (29, 30), and reliable information would enable managers to act.
Human/sewage marker gene occurrence and nonspecific amplification are associated with atypical animal microbiome profiles and shifts toward human patterns.
The composition of gut microbiota can be affected by many factors, including host physiology and environmental factors such as cohabitation and diet (i.e., herbivore, omnivore, and carnivore) (31–33). The influence of host physiology and/or an animal’s diet were consistently observed in our data; for example, the microbial communities of ruminant, horse, pig, and dog were grouped by host species, regardless of the geographic source of the samples, including different continents (Fig. S2 in the supplemental material). General diet and cohabitation also influenced community structure; for example, the microbial community of mammal herbivores was separated from carnivores, and the human fecal microbial community was close to the pet group (Fig. 2). These observations were consistent with other mammal gut microbiome studies, where host species and diet were found to significantly impact patterns when analyzed by network- and UniFrac-based microbial community composition analysis (31, 32).
The animal cross-reactions corresponded to shifts in the entire microbial community as well as shifts within the specific genera of the marker genes. A few animals (i.e., five rabbits and one deer) had Bacteroides that had profiles more closely clustered to the human group, and these samples had high concentrations of the HF183 marker (Fig. 2, Fig. 4). The exact causes of atypical Bacteroides composition are unknown; diet or cohabitation could be a possible driver(s), as all positive rabbit samples were domestic (i.e., from a same farm in Texas with grain and/or alfalfa diet) or pet (i.e., from Wisconsin) samples, while negative samples were all wild rabbits from the same sender and region as the positive Texas samples.
For animals with the Lachno3 marker present in both NGS and qPCR, these samples showed atypical patterns and contained more sequences that were similar to human Lachnospiraceae (i.e., Blautia) than negative animals. This was also true for animals where the actual marker gene was absent in the NGS data but samples were positive by qPCR (e.g., horses). It is well established that there is no phylogenetic core of organisms with a specific 16S rRNA gene sequence within a host, but rather, closely related organisms that share functional attributes (34–36). This is supported by our results that found microorganisms closely related to the indicator organism had a similar host preference (Fig. 5), suggesting they could have similar functions to fulfill the adaptation to the host niches. Similarly, the shift of animal fecal communities toward organisms closely related to the human marker gene may also fulfill certain metabolic functions within these animals. These results demonstrate that cross-reaction is not simply a lack of specificity of the marker gene itself but that some animals have a microbiome that contains a cluster of phylogenetically closely related organisms that resemble human sources. Targeting other human-related Bacteroides or Lachnospiraceae may also result in animal cross-reactions in these particular samples. It is noteworthy that the shifts were observed in either Bacteroides or Lachnospiraceae, but not both in the same animal. These findings support that specificity is greatly improved when using marker genes from two unrelated groups (15, 20, 33, 37).
Interestingly, some animals contained organisms with 16S rRNA gene sequences closely related to the BacV6-21 marker. Previous work has reported this marker matches Bacteroides graminisolvens, an organism commonly found in manure lagoons and other onsite waste disposal systems (14, 38). The presence of closely related organisms in animals suggests there could be host-associated organisms that share similar functional traits with these free-living counterparts.
Implications for cross-reactions of human/sewage marker genes with animals.
Fecal pollution in natural water environments (e.g., rivers and lakes) generally is introduced as a composite of multiple animals from a given source (e.g., agricultural runoff) or humans (i.e., sewage). In practice, the effect of low-level, sporadic occurrence of human markers in individual animals would likely be diluted and not detected. Only in certain cases where a limited number of individuals contribute (e.g., raccoon or dog fecal pollution in stormwater outfalls) might water assessments be affected. For example, when raccoons were positive for the HF183 marker, certain sites within a stormwater system might have positive signals for the HF183 marker. This further supports that using two or three maker genes from different indicator organisms improves confidence for accurately detecting the fecal source, particularly when a small number of animals might be contributing (i.e., stormwater) (15, 39, 40). Further, cross-reaction or nonspecific amplification of human/sewage marker genes in animals was usually at much lower levels compared to their presence in human sources. Also, levels were well below the average concentrations of general FIB in animal fecal samples (Table S2). Therefore, the validated animal cross-reactions could be of smaller effect in urban water testing, where human sources can overwhelm others.
Exploring qPCR-only amplifications of human/sewage markers to improve assay performance.
The qPCR amplification for a certain amplicon actually targets a cluster of organisms and is an inherent problem of 16S rRNA marker gene assays lacking 100% specificity. Non-16S rRNA human marker assays have been developed, such as the HumM2/HumM3 (41), Bacteroides thetaiotaomicron (42), gyrB in Bacteroides fragilis (43), and the esp gene of Enterococcus faecium (44–46). However, these assays were validated in a smaller number of studies, exhibiting animal cross-reactions (41, 42, 45, 47) as well as lower sensitivities in human feces (24, 46, 48). Seeking a marker gene of 100% specificity and sensitivity may be unrealistic; rather, applying multiple validated marker genes of high specificity and sensitivity could be the best approach.
The two HF183 assays showed similar cross-reactions, although the HB assay had slightly lower specificity with low concentration in an extra dog, suggesting that the HB assay cross-reacts with multiple sequences found in dogs. Without NGS data within the V2 region, it is difficult to determine if these qPCR products consisted of the actual HF183 sequence or closely related sequences.
Revising primer or probe design in an assay to exclude a marker gene’s closely related organisms found in certain animals (e.g., horse) may cause a loss of specificity and increase cross-reaction with other animal hosts. Similarly, in the cases of Lachno3 and BacV6-21, increasing the annealing temperature of an assay may reduce nonspecific amplification, but it also decreased assay efficiency (15). The HF183/BacR287 assay has been optimized the most extensively for both primer design and annealing temperature to minimize cross-reactions and is now an EPA standard method (12, 13). Overall, all assays had host specificities above 90%, suggesting all are excellent assays (25, 49).
General fecal indicator qPCR assays may not reflect total fecal pollution.
The general fecal indicator assays only amplified in about 50% of the tested 184 animal fecal samples at 1 ng/μl DNA concentration. The subset of negative samples tested by the GenBac3 assay proved the overall quality of these extracted DNA materials. The patterns of general indicator assays in animal hosts in our work were similar to another study (60), where E. coli and Enterococcus spp. were at about the same order of magnitude as our results for chicken and raccoon and were below the limit of quantification for deer and cow. It might be ambiguous to conclude the level of total fecal pollution by use of these general FIB qPCR assays when animal fecal pollution from these sources was present at low levels. The GenBac3 assay produced a robust signal for fecal bacteria, but this assay may still be affected by nonfecal Bacteroides organisms. The presence of such Bacteroides was reported in environmental samples without apparent fecal pollution (14, 50, 51), including in groundwater and tap water samples (51) detected by the TotBac assay (52) that was used in the GenBac3 assay design. Therefore, more reliable measures of the total amount of fecal pollution in the field would be useful.
Applicability of validation studies of human/sewage maker genes to other geographic regions.
We examined the microbial community of animals from the United States and Australia and found similar patterns within host types, demonstrating representativeness of gut microbiota across a wide geographic range. Animal fecal microbial communities were clustered into host groups with some separation within these groups based on location, which might affect marker development for particular host groups depending on the target chosen but would not be expected to affect validation of human/sewage marker genes given the overall cohesion within animal groups. Therefore, these validation studies for cross-reaction with animals could be extrapolated to other areas. Further, this study, when combined with others (20), provides good coverage of most animal hosts that would be expected to contribute fecal pollution to waterways. Ongoing validation efforts are still needed for human/sewage marker genes, especially for recently developed assays that have been validated in only a small number of studies. Taken together, these results provide a high degree of confidence for specificities of the HF183 (i.e., HB and HF183/BacR287 assays), Lachno3, and BacV6-21 marker assays.
MATERIALS AND METHODS
Sample collection and processing.
A total number of 365 single animal fecal samples across 20 hosts were collected from the United States and Australia and sent to the University of Wisconsin—Milwaukee (Milwaukee, WI, USA) as raw samples or extracted DNA (see Data Set S1 in the supplemental material for detailed information). For raw sample collection, up to 5 ml or 5 g of sample was collected into a 50-ml conical sterile centrifuge tube containing 2 ml buffer ASL (i.e., stool lysis buffer; Qiagen Inc., Valencia, CA). Sample tubes were stored at −80°C and later sent on ice to the University of Wisconsin—Milwaukee. DNA samples were sent freeze-dried. For raw samples, DNA was extracted in individual samples (n = 230) and pooled samples (n = 27 pools). For individual samples, DNA was extracted using the standard protocol of the QIAamp DNA stool mini kit (Qiagen Inc.). For each pooled sample, five individual samples that belonged to the same host type and were from the same sampling event (i.e., from the same location and sender) were combined. DNA extraction of pooled samples combined five lytic stool samples in step 2 of the standard protocol, with each of the lytic samples at one-fifth of the volume of that used in individual sample extraction. One rabbit sample (Rabbit Pool1) was initially extracted and sequenced as a pooled sample but later reextracted as individuals for the HF183 qPCR assays (i.e., HB and HF183/BacR287). A final number of 257 animal fecal samples were prepared for sequencing. A total of 184 samples were tested for human/sewage fecal marker assays. Table S1 summarizes animal hosts and numbers of samples used for sequencing and qPCR.
NGS data analysis.
Fecal DNA of 257 animals was sequenced for the V6 region of the 16S rRNA gene using the Illumina NextSeq sequencing platform at the Marine Biological Laboratory at the University of Chicago. The paired-end, short-reads sequencing method was described in a previous publication (53). NGS data of healthy human fecal samples (n = 6) and untreated U.S. sewage samples (n = 8) were obtained from a public data set (54) and previous studies (15, 55), respectively. Animal hosts were purposely grouped for certain sequence analysis, including cat and dog as the “pet” group; antelope, cow, deer, goat, and sheep as the “ruminant” group; chicken, duck, gull, goose, and parrot as the “bird” group; and bear and raccoon as the “wildlife” group. Other animal hosts, including horse, pig, rabbit, kangaroo, flying fox, and alligator, were not grouped. All sequence data was stored and managed on the Visualization and Analysis of Microbial Population Structures platform (VAMPS) (https://vamps2.mbl.edu) (56). The total number of sequences in each sample was normalized to the median total sequence count of all samples (median, 513,566). Singletons were then removed. In all, 319,418 unique V6 sequences from 137,193,309 reads were analyzed. Analysis of the data set was performed in R (version 3.5.1) (57).
Microbial community and statistical analysis.
Statistical analysis was performed in the vegan package (58) in R. Nonmetric multidimensional scaling (NMDS) analysis was performed based on Bray-Curtis dissimilarities of samples, which were calculated based on counts of unique sequences.
Sample processing control, extraction efficiency, and inhibition testing.
Method blanks (n = 3) were extracted following the standard protocol of QIAamp DNA stool mini kit. Extraction blanks (n = 3) were extracted with 0.24 μg salmon sperm genomic DNA spiked in method blank extractions for a final concentration of 0.2 μg/ml. Standard curves for calculating recovered DNA amounts from the extraction blanks were constructed using cycle threshold (CT) values of serially diluted salmon sperm DNA (i.e., log10[ng of salmon sperm DNA] versus CT values; each concentration level was tested in triplicate). Both method blanks and DNA-grade water were used as negative controls. Extraction efficiency was determined as a recovered DNA amount of extraction blanks in each reaction divided by the theoretical DNA amount in each reaction. The extraction efficiency was calculated as 38.8% ± 7.7% (mean ± SD).
Inhibition testing was performed by spiking about 1,000 copies of salmon sperm genomic DNA into each reaction with animal fecal DNA at 10 ng/μl. A subset of 46 animal fecal samples that included all animal host categories was tested. Four no-template controls that used DNA-grade water were also included. All samples were tested in duplicate. No inhibition was observed.
Single and duplex qPCR experiments.
A subset of animal fecal samples (n = 184) across 18 hosts was tested for human/sewage marker assays using qPCR. Three sets of TaqMan qPCR assays were performed, including the Lachno3 assay (15), a duplex E. coli (22)/human Bacteroides assay (11) (E. coli/HB), a duplex Enterococcus (23)/BacV6-21 assay (14) (ENT/BacV6-21), and the HF183/BacR287 assay (12).
The Lachno3 qPCR experiments were performed as previously described (15). For duplex assays, VIC reporter dye was used for general indicator assay probes, and 6-carboxyfluorescein (FAM) reporter dye was used for human/sewage marker assay probes. For both duplex assays, a 25-μl qPCR system was used, including TaqMan gene expression master mix (Applied Biosystems, Foster City, CA), 1 μM for each primer, 80 nM for each VIC reporter dye probe, and 80 nM for each FAM reporter dye probe; DNA input volume was 5 μl. The amplification program for both duplex assays included one cycle at 50°C for 2 min, followed by one cycle at 95°C for 10 min, and then 40 cycles of 95°C for 15 s followed by 1 min at 60°C. Standard curve validation was carried out using each target’s plasmid or genomic DNA under single assay conditions and under duplex assay conditions (Table S2). Sewage samples (n = 16), as well as animal fecal samples that were known to have no cross-reaction with Lachno3, HB, or BacV6-21 assays (n = 4), were used for comparing single and duplex conditions. Student's t test was performed to test the statistical difference between CT values of single and duplex runs for both sets of duplex assays. The lower limit of quantification (LLOQ) for each assay was defined as the 95% prediction of the upper limit of the 15-copy DNA standard dilution based on the corresponding standard curve. All single and duplex standard curves of the E. coli/HB assay and ENT/BacV6-21 assay showed R2 values of > 0.990. The amplification efficiencies ranged from 92.7% (single BacV6-21 assay) to 100.1% (single HB assay).
Potential competition within duplex assays was also addressed. Six concentrations of two templates were used in concert, with one assay’s template concentration ranging from 1.5 × 106 to 1.5 × 101 copies and the other assay’s template ranging from 1.5 × 101 to 1.5 × 106 copies. Duplex assays and single assays were then run. Effects of competition were observed for the ENT/BacV6-2, where BacV6-21 was not determined at the template concentrations of 1.5 × 102 and 1.5 × 101 copies when enterococci target copies were 1.5 × 105 and 1.5 × 106, respectively. There was no significant difference noted for the remaining template combinations (paired t test; P > 0.05). Similarly, the duplexed E. coli assay at the template concentration of 1.5 × 101 was undetermined when HB target copies were 1.5 × 106 copies, with no significant difference noted for the remaining template combinations (paired t test; P > 0.05). We did not observe competition effects for the inverse experimental regime, i.e., low-ENT and high-BacV6-21 or low-HB and high-E. coli target copies. Competition occurred for the targets, with the larger of the two fragments in the duplex assay, suggesting disparate sizes of the duplexed targets influence efficacy. We concluded that the duplexed assays are reliable when targets are within 3 orders of magnitude for ENT/BacV6-21 and 5 orders of magnitude for E. coli/HB. In practice, the potential inhibition of BacV6-21 in cases of high enterococcus inputs from nonsewage sources would be flagged by HB levels in the E. coli/HB assay, and single reactions could be run for verification.
For Lachno3, E. coli/HB, and ENT/BacV6-21 assays, animal fecal samples were tested at DNA template concentrations of 1 ng/μl, 0.1 ng/μl, and 0.01 ng/μl; for the HF183/BacR287 assay, samples were only tested at a DNA template concentration of 1 ng/μl. Each animal fecal sample was tested in duplicate. Each run included a positive control using DNA from a sewage sample and a blank control of sterile, DNA-grade water. Standard curves were tested in triplicate using plasmids at each concentration, ranging from of 1.5 × 106 to 1.5 copy numbers (CNs) per reaction. Samples that had CNs below the limit of quantification (i.e., 15 copy numbers per reaction) were considered negative for the marker assays. A subset of animal fecal samples that were negative for E. coli and/or ENT (n = 83) were tested using a general Bacteroidetes assay (i.e., GenBac3) (52, 59) at 1 ng/μl template concentration. All qPCR validation results of animal fecal samples are included in Data Set S1. Parameters including the slopes, y intercepts, R2 values, efficiencies, and LLOQ values of the four assays are reported in Table S3.
Data availability.
The V6 region 16S rRNA gene sequencing data were deposited to the NCBI SRA under the accession number SRP225233.
Supplementary Material
ACKNOWLEDGMENTS
We thank the following researchers for assistance with animal fecal sample collection or contribution of animal fecal DNA to this work: Scott E. Henke (Texas A&M University Kingsville), Garret Suen (University of Wisconsin—Madison), Terrance Arthur (U.S. Meat Animal Research Center), Jason Gill (Texas A&M University), Jill Stewart (University of North Carolina), Valerie Harwood (University of South Florida), Karl Miller (University of Georgia), and Melony Wilson (University of Georgia Extension). We thank Keri Lydon for assistance with this study and Melinda J. Bootsma for help with the qPCR inhibition test. We thank Hillary Morrison at the Bay Paul Center, Marine Biological Laboratory (MBL), University of Chicago, for offering expertise in NGS sequencing.
Funding for this study was provided by NIH, grant number R01 AI091829 to S.L.M.
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Wiggins BA. 1996. Discriminant analysis of antibiotic resistance patterns in fecal streptococci, a method to differentiate human and animal sources of fecal pollution in natural waters. Appl Environ Microbiol 62:3997–4002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Parveen S, Portier KM, Robinson K, Edmiston L, Tamplin ML. 1999. Discriminant analysis of ribotype profiles of Escherichia coli for differentiating human and nonhuman sources of fecal pollution. Appl Environ Microbiol 65:3142–3147. doi: 10.1128/AEM.65.7.3142-3147.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bernhard AE, Field KG. 2000. Identification of nonpoint sources of fecal pollution in coastal waters by using host-specific 16S ribosomal DNA genetic markers from fecal anaerobes. Appl Environ Microbiol 66:1587–1594. doi: 10.1128/aem.66.4.1587-1594.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bernhard AE, Field KG. 2000. A PCR assay to discriminate human and ruminant feces on the basis of host differences in Bacteroides-Prevotella genes encoding 16S rRNA. Appl Environ Microbiol 66:4571–4574. doi: 10.1128/aem.66.10.4571-4574.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Harwood VJ, Staley C, Badgley BD, Borges K, Korajkic A. 2014. Microbial source tracking markers for detection of fecal contamination in environmental waters: relationships between pathogens and human health outcomes. FEMS Microbiol Rev 38:1–40. doi: 10.1111/1574-6976.12031. [DOI] [PubMed] [Google Scholar]
- 6.Ahmed W, Hughes B, Harwood VJ. 2016. Current status of marker genes of Bacteroides and related taxa for identifying sewage pollution in environmental waters. Water 8:231. doi: 10.3390/w8060231. [DOI] [Google Scholar]
- 7.Seurinck S, Defoirdt T, Verstraete W, Siciliano SD. 2005. Detection and quantification of the human-specific HF183 Bacteroides 16S rRNA genetic marker with real-time PCR for assessment of human faecal pollution in freshwater. Environ Microbiol 7:249–259. doi: 10.1111/j.1462-2920.2004.00702.x. [DOI] [PubMed] [Google Scholar]
- 8.Reischer GH, Kasper DC, Steinborn R, Mach RL, Farnleitner AH. 2006. Quantitative PCR method for sensitive detection of ruminant fecal pollution in freshwater and evaluation of this method in alpine karstic regions. Appl Environ Microbiol 72:5610–5614. doi: 10.1128/AEM.00364-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kildare BJ, Leutenegger CM, McSwain BS, Bambic DG, Rajal VB, Wuertz S. 2007. 16S rRNA-based assays for quantitative detection of universal, human-, cow-, and dog-specific fecal Bacteroidales: a Bayesian approach. Water Res 41:3701–3715. doi: 10.1016/j.watres.2007.06.037. [DOI] [PubMed] [Google Scholar]
- 10.Haugland RA, Varma M, Sivaganesan M, Kelty C, Peed L, Shanks OC. 2010. Evaluation of genetic markers from the 16S rRNA gene V2 region for use in quantitative detection of selected Bacteroidales species and human fecal waste by qPCR. Syst Appl Microbiol 33:348–357. doi: 10.1016/j.syapm.2010.06.001. [DOI] [PubMed] [Google Scholar]
- 11.Templar HA, Dila DK, Bootsma MJ, Corsi SR, McLellan SL. 2016. Quantification of human-associated fecal indicators reveal sewage from urban watersheds as a source of pollution to Lake Michigan. Water Res 100:556–567. doi: 10.1016/j.watres.2016.05.056. [DOI] [PubMed] [Google Scholar]
- 12.Green HC, Haugland RA, Varma M, Millen HT, Borchardt MA, Field KG, Walters WA, Knight R, Sivaganesan M, Kelty CA, Shanks OC. 2014. Improved HF183 quantitative real-time PCR assay for characterization of human fecal pollution in ambient surface water samples. Appl Environ Microbiol 80:3086–3094. doi: 10.1128/AEM.04137-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.US Environmental Protection Agency. 2019. Method 1696: characterization of human fecal pollution in water by HF183/BacR287 TaqMan quantitative polymerase chain reaction (qPCR) assay. US Environmental Protection Agency, Washington, DC. [Google Scholar]
- 14.Feng S, McLellan SL, Feng S, McLellan SL. 2019. Highly specific sewage-derived Bacteroides qPCR assays target sewage polluted waters. Appl Environ Microbiol 85:e02696-18. doi: 10.1128/AEM.02696-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Feng S, Bootsma M, McLellan SL, Feng S, Bootsma M, McLellan SL. 2018. Human-associated Lachnospiraceae genetic markers improve detection of fecal pollution sources in urban waters. Appl Environ Microbiol 84:e00309-18. doi: 10.1128/AEM.00309-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stoeckel DM, Harwood VJ. 2007. Performance, design, and analysis in microbial source tracking studies. Appl Environ Microbiol 73:2405–2415. doi: 10.1128/AEM.02473-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Okabe S, Okayama N, Savichtcheva O, Ito T. 2007. Quantification of host-specific Bacteroides-Prevotella 16S rRNA genetic markers for assessment of fecal pollution in freshwater. Appl Microbiol Biotechnol 74:890–901. doi: 10.1007/s00253-006-0714-x. [DOI] [PubMed] [Google Scholar]
- 18.Layton A, McKay L, Williams D, Garrett V, Gentry R, Sayler G. 2006. Development of Bacteroides 16S rRNA gene TaqMan-based real-time PCR assays for estimation of total, human, and bovine fecal pollution in water. Appl Environ Microbiol 72:4214–4224. doi: 10.1128/AEM.01036-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee DY, Weir SC, Lee H, Trevors JT. 2010. Quantitative identification of fecal water pollution sources by TaqMan real-time PCR assays using Bacteroidales 16S rRNA genetic markers. Appl Microbiol Biotechnol 88:1373–1383. doi: 10.1007/s00253-010-2880-0. [DOI] [PubMed] [Google Scholar]
- 20.Ahmed W, Gyawali P, Feng S, McLellan SL. 2019. Host specificity and sensitivity of established and novel sewage-associated marker genes in human and nonhuman fecal samples. Appl Environ Microbiol 85:e00641-19. doi: 10.1128/AEM.00641-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tan B, Ng C, Nshimyimana JP, Loh LL, Gin KYH, Thompson JR. 2015. Next-generation sequencing (NGS) for assessment of microbial water quality: current progress, challenges, and future opportunities. Front Microbiol 6:1027. doi: 10.3389/fmicb.2015.01027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li J, McLellan S, Ogawa S. 2006. Accumulation and fate of green fluorescent labeled Escherichia coli in laboratory-scale drinking water biofilters. Water Res 40:3023–3028. doi: 10.1016/j.watres.2006.06.029. [DOI] [PubMed] [Google Scholar]
- 23.Haugland RA, Siefring SC, Wymer LJ, Brenner KP, Dufour AP. 2005. Comparison of Enterococcus measurements in freshwater at two recreational beaches by quantitative polymerase chain reaction and membrane filter culture analysis. Water Res 39:559–568. doi: 10.1016/j.watres.2004.11.011. [DOI] [PubMed] [Google Scholar]
- 24.Shanks OC, White K, Kelty CA, Sivaganesan M, Blannon J, Meckes M, Varma M, Haugland RA. 2010. Performance of PCR-based assays targeting Bacteroidales genetic markers of human fecal pollution in sewage and fecal samples. Environ Sci Technol 44:6281–6288. doi: 10.1021/es100311n. [DOI] [PubMed] [Google Scholar]
- 25.Boehm AB, Van De Werfhorst LC, Griffith JF, Holden PA, Jay JA, Shanks OC, Wang D, Weisberg SB. 2013. Performance of forty-one microbial source tracking methods: a twenty-seven lab evaluation study. Water Res 47:6812–6828. doi: 10.1016/j.watres.2012.12.046. [DOI] [PubMed] [Google Scholar]
- 26.Layton BA, Cao Y, Ebentier DL, Hanley K, Ballesté E, Brandão J, Byappanahalli M, Converse R, Farnleitner AH, Gentry-Shields J, Gidley ML, Gourmelon M, Lee CS, Lee J, Lozach S, Madi T, Meijer WG, Noble R, Peed L, Reischer GH, Rodrigues R, Rose JB, Schriewer A, Sinigalliano C, Srinivasan S, Stewart J, Van De Werfhorst LC, Wang D, Whitman R, Wuertz S, Jay J, Holden PA, Boehm AB, Shanks O, Griffith JF. 2013. Performance of human fecal anaerobe-associated PCR-based assays in a multi-laboratory method evaluation study. Water Res 47:6897–6908. doi: 10.1016/j.watres.2013.05.060. [DOI] [PubMed] [Google Scholar]
- 27.Mayer RE, Reischer GH, Ixenmaier SK, Derx J, Blaschke AP, Ebdon JE, Linke R, Egle L, Ahmed W, Blanch AR, Byamukama D, Savill M, Mushi D, Cristóbal HA, Edge TA, Schade MA, Aslan A, Brooks YM, Sommer R, Masago Y, Sato MI, Taylor HD, Rose JB, Wuertz S, Shanks OC, Piringer H, Mach RL, Savio D, Zessner M, Farnleitner AH. 2018. Global distribution of human-associated fecal genetic markers in reference samples from six continents. Environ Sci Technol 52:5076–5084. doi: 10.1021/acs.est.7b04438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shanks OC, Kelty CA, Oshiro R, Haugland RA, Madi T, Brooks L, Field KG, Sivaganesan M. 2016. Data acceptance criteria for standardized human-associated fecal source identification quantitative real-time PCR methods. Appl Environ Microbiol 82:2773–2782. doi: 10.1128/AEM.03661-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.American Society of Civil Engineers. 2011. Failure to act: the economic impact of current investment trends in electricity infrastructure. American Society of Civil Engineers, Reston, VA. [Google Scholar]
- 30.US Environmental Protection Agency. 2018. Fiscal year 2018–2022, U.S. EPA strategic plan. US Environmental Protection Agency, Washington, DC. [Google Scholar]
- 31.Ley RE, Lozupone CA, Hamady M, Knight R, Gordon JI. 2008. Worlds within worlds: evolution of the vertebrate gut microbiota. Nat Rev Microbiol 6:776–788. doi: 10.1038/nrmicro1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ley RE, Hamady M, Lozupone C, Turnbaugh PJ, Ramey RR, Bircher JS, Schlegel ML, Tucker TA, Schrenzel MD, Knight R, Gordon JI. 2008. Evolution of mammals and their gut microbes. Science 320:1647–1651. doi: 10.1126/science.1155725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fisher JC, Murat Eren A, Green HC, Shanks OC, Morrison HG, Vineis JH, Sogin ML, McLellan SL. 2015. Comparison of sewage and animal fecal microbiomes by using oligotyping reveals potential human fecal indicators in multiple taxonomic groups. Appl Environ Microbiol 81:7023–7033. doi: 10.1128/AEM.01524-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zaneveld JR, Lozupone C, Gordon JI, Knight R. 2010. Ribosomal RNA diversity predicts genome diversity in gut bacteria and their relatives. Nucleic Acids Res 38:3869–3879. doi: 10.1093/nar/gkq066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Segata N, Huttenhower C. 2011. Toward an efficient method of identifying core genes for evolutionary and functional microbial phylogenies. PLoS One 6:e24704. doi: 10.1371/journal.pone.0024704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Langille M, Zaneveld J, Caporaso JG, McDonald D, Knights D, Reyes J, Clemente J, Burkepile D, Vega Thurber R, Knight R, Beiko R, Huttenhower C. 2013. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol 31:814–821. doi: 10.1038/nbt.2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McLellan SL, Eren AM. 2014. Discovering new indicators of fecal pollution. Trends Microbiol 22:697–706. doi: 10.1016/j.tim.2014.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nishiyama T, Ueki A, Kaku N, Watanabe K, Ueki K. 2009. Bacteroides graminisolvens sp. nov., a xylanolytic anaerobe isolated from a methanogenic reactor treating cattle waste. Int J Syst Evol Microbiol 59:1901–1907. doi: 10.1099/ijs.0.008268-0. [DOI] [PubMed] [Google Scholar]
- 39.Eren AM, Sogin ML, Morrison HG, Vineis JH, Fisher JC, Newton RJ, McLellan SL. 2015. A single genus in the gut microbiome reflects host preference and specificity. ISME J 9:90–100. doi: 10.1038/ismej.2014.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ahmed W, Payyappat S, Cassidy M, Besley C. 2019. A duplex PCR assay for the simultaneous quantification of Bacteroides HF183 and crAssphage CPQ_056 marker genes in untreated sewage and stormwater. Environ Int 126:252–259. doi: 10.1016/j.envint.2019.01.035. [DOI] [PubMed] [Google Scholar]
- 41.Shanks OC, Kelty CA, Sivaganesan M, Varma M, Haugland RA. 2009. Quantitative PCR for genetic markers of human fecal pollution. Appl Environ Microbiol 75:5507–5513. doi: 10.1128/AEM.00305-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yampara-Iquise H, Zheng G, Jones JE, Carson CA. 2008. Use of a Bacteroides thetaiotaomicron-specific α-1–6, mannanase quantitative PCR to detect human faecal pollution in water. J Appl Microbiol 105:1686–1693. doi: 10.1111/j.1365-2672.2008.03895.x. [DOI] [PubMed] [Google Scholar]
- 43.Lee CS, Lee J. 2010. Evaluation of new gyrB-based real-time PCR system for the detection of B. fragilis as an indicator of human-specific fecal contamination. J Microbiol Methods 82:311–318. doi: 10.1016/j.mimet.2010.07.012. [DOI] [PubMed] [Google Scholar]
- 44.Scott TM, Jenkins TM, Lukasik J, Rose JB. 2005. Potential use of a host associated molecular marker in Enterococcus faecium as an index of human fecal pollution. Environ Sci Technol 39:283–287. doi: 10.1021/es035267n. [DOI] [PubMed] [Google Scholar]
- 45.Whitman RL, Przybyla-Kelly K, Shively DA, Byappanahalli MN. 2007. Incidence of the enterococcal surface protein (esp) gene in human and animal fecal sources. Environ Sci Technol 41:6090–6095. doi: 10.1021/es070817t. [DOI] [PubMed] [Google Scholar]
- 46.Ahmed W, Stewart J, Powell D, Gardner T. 2008. Evaluation of the host-specificity and prevalence of enterococci surface protein (esp) marker in sewage and its application for sourcing human fecal pollution. J Environ Qual 23:1583–1588. doi: 10.2134/jeq2007.0474. [DOI] [PubMed] [Google Scholar]
- 47.Aslan A, Rose JB. 2013. Evaluation of the host specificity of Bacteroides thetaiotaomicron alpha-1–6, mannanase gene as a sewage marker. Lett Appl Microbiol 56:51–56. doi: 10.1111/lam.12013. [DOI] [PubMed] [Google Scholar]
- 48.Odagiri M, Schriewer A, Hanley K, Wuertz S, Misra PR, Panigrahi P, Jenkins MW. 2015. Validation of Bacteroidales quantitative PCR assays targeting human and animal fecal contamination in the public and domestic domains in India. Sci Total Environ 502:462–470. doi: 10.1016/j.scitotenv.2014.09.040. [DOI] [PubMed] [Google Scholar]
- 49.US Environmental Protection Agency. 2005. Microbial source tracking guide document EPA/600/R-05/064. US Environmental Protection Agency, Washington, DC. [Google Scholar]
- 50.Bower PA, Scopel CO, Jensen ET, Depas MM, McLellan SL. 2005. Detection of genetic markers of fecal indicator bacteria in Lake Michigan and determination of their relationship to Escherichia coli densities using standard microbiological methods. Appl Environ Microbiol 71:8305–8313. doi: 10.1128/AEM.71.12.8305-8313.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Van Der Wielen P, Medema G. 2010. Unsuitability of quantitative Bacteroidales 16S rRNA gene assays for discerning fecal contamination of drinking water. Appl Environ Microbiol 76:4876–4881. doi: 10.1128/AEM.03090-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dick LK, Field KG. 2004. Rapid estimation of numbers of fecal Bacteroidetes by use of a quantitative PCR assay for 16S rRNA genes. Appl Environ Microbiol 71:3179–3183. doi: 10.1128/AEM.70.9.5695-5697.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Eren AM, Vineis JH, Morrison HG, Sogin ML. 2013. A filtering method to generate high quality short reads using Illumina paired-end technology. PLoS One 8:e66643. doi: 10.1371/journal.pone.0066643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vineis JH, Ringus DL, Morrison HG, Delmont TO, Dalal S, Raffals LH, Antonopoulos DA, Rubin DT, Eren AM, Chang EB, Sogin ML. 2016. Patient-specific Bacteroides genome variants in pouchitis. mBio 7:e01713-16. doi: 10.1128/mBio.01713-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Newton RJ, McLellan SL, Dila DK, Vineis JH, Morrison HG, Murat Eren A, Sogin ML. 2015. Sewage reflects the microbiomes of human populations. mBio 6:e02574-14. doi: 10.1128/mBio.02574-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Huse SM, Mark Welch DB, Voorhis A, Shipunova A, Morrison HG, Eren AM, Sogin ML. 2014. VAMPS: a website for visualization and analysis of microbial population structures. BMC Bioinformatics 15:41. doi: 10.1186/1471-2105-15-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.R Core Team. 2018. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. [Google Scholar]
- 58.Oksanen J, Blanchet FG, Friendly M, Kindt R, Legendre P, McGlinn D, Minchin PR, O‘Hara RB, Simpson GL, Solymos P, Henry M, Stevens H, Szoecs E, Wagner H, Oksanen MJ. 2018. vegan: community ecology package 2.4–5. https://CRAN.R-project.org/package=vegan. Accessed 9 July 2018.
- 59.Siefring S, Varma M, Atikovic E, Wymer L, Haugland RA. 2008. Improved real-time PCR assays for the detection of fecal indicator bacteria in surface waters with different instrument and reagent systems. J Water Health 6:225–237. doi: 10.2166/wh.2008.022. [DOI] [PubMed] [Google Scholar]
- 60.Kelty CA, Varma M, Sivaganesan M, Haugland RA, Shanks OC. 2012. Distribution of genetic marker concentrations for fecal indicator bacteria in sewage and animal feces. Appl Environ Microbiol 78:4225–4232. doi: 10.1128/AEM.07819-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The V6 region 16S rRNA gene sequencing data were deposited to the NCBI SRA under the accession number SRP225233.