

Abstract
Colletotrichum species complexes are among the top 10 economically important fungal plant pathogens worldwide because they can infect climacteric and nonclimacteric fruit at the pre and/or postharvest stages. C. truncatum is the major pathogen responsible for anthracnose of green and red bell pepper fruit worldwide. C. brevisporum was recently reported to be a minor pathogen of red bell pepper fruit in Trinidad, but has recently been reported as pathogenic to other host species in other countries. The ability of these phytopathogens to produce and secrete cutinase is required for dismantling the cuticle of the host plant and, therefore, crucial to the necrotrophic phase of their infection strategy. In vitro bioassays using different lipid substrates confirmed the ability of C. truncatum and C. brevisporum isolates from green and red bell peppers to secrete cutinase. The diversity, structure and organization and synteny of the cutinase gene were determined among different Colletotrichum species. Cluster analysis indicated a low level of nucleotide variation among C. truncatum sequences. Nucleotide sequences of C. brevisporum were more related to C. truncatum cutinase nucleotide sequences than to C. gloeosporioides. Cluster patterns coincided with haplotype and there was evidence of significant positive selection with no recombination signatures. The structure of the cutinase gene included two exons with one intervening intron and, therefore, one splice variant. Although amino acid sequences were highly conserved among C. truncatum isolates, diversity “hot spots” were revealed when the 66‐amino acid coding region of 200 fungal species was compared. Twenty cutinase orthologues were detected among different fungal species, whose common ancestor is Pezizomycotina and it is purported that these orthologues arose through a single gene duplication event prior to speciation. The cutinase domain was retained both in structure and arrangement among 34 different Colletotrichum species. The order of aligned genomic blocks between species and the arrangement of flanking protein domains were also conserved and shared for those domains immediately located at the N‐ and C‐terminus of the cutinase domain. Among these were an RNA recognition motif, translation elongation factor, signal peptide, pentatricopeptide repeat, and Hsp70 family of chaperone proteins, all of which support the expression of the cutinase gene. The findings of this study are important to understanding the evolution of the cutinase gene in C. truncatum as a key component of the biotrophic–necrotrophic switch which may be useful in developing gene‐targeting strategies to decrease the pathogenic potential of Colletotrichum species.
Keywords: Colletotrichum, cutinase diversity, evolution, pathogenesis, synteny
Low sequence diversity and high evolutionary conservation of the cutinase gene in Colletotrichum truncatum are key to this phytopathogen's infection strategy.

1. INTRODUCTION
Many plant pathogenic fungi have complex life cycles which enable them to interact differently with their hosts (Horbach, Navarro‐Quesada, Knogge, & Deising, 2011). As such, a two‐stage life cycle is typical of hemibiotrophic fungi: the initial biotrophic phase: the fungus grows and survives in a quiescent state to maintain host viability, therefore, the host remains asymptomatic and without host tissue destruction; the second necrotrophic phase: tissue is decomposed through cellular dismantling and destruction, assimilation of the contents of dead or dying cells which results in symptom manifestation and subsequent death of plant tissue (Brunner, Torriani, Croll, Stukenbrock, & McDonald, 2013; Stone, 2001).
Colletotrichum is one of the most economically important genera of plant pathogenic fungi with a membership of more than 200 species, most of which enjoy a broad host range worldwide, and cause anthracnose of fruit and vegetable crops in tropical and subtropical climates (Udayanga, Manamgoda, Liu, Chukeatirote, & Hyde, 2013). Colletotrichum species are hemibiotrophic plant pathogens that exhibit both forms of nutrient acquisition, that is, utilize sequential biotrophic and necrotrophic infection tactics to invade and cause disease in host plants (Alkan, Friedlander, Ment, Prusky, & Fluhr, 2015; Lee & Rose, 2010; O'Connell et al., 2012). Infection by fungal necrotrophs generally involves stages of conidial attachment, germination, host penetration, primary lesion formation, lesion expansion, and tissue maceration followed by sporulation (Prins et al., 2000). A comparative study involving the genome and transcriptome of C. higginsianum and C. graminicola indicated that (a) both fungal species possessed numerous pathogenicity‐related genes, with genetic footprints reminiscent of biotrophic and necrotrophic pathogens, (b) there are differences in the expression profile of these pathogenicity‐related genes, and (c) induction of gene expression is highly regulated and stage‐specific (Alkan et al., 2015; Damm, O'Connell, Groenewald, & Crous, 2014; Kleemann et al., 2012; Liu et al., 2013; O'Connell et al., 2012).
The interaction between a hemibiotroph and its plant host is highly specialized both structurally and physiologically such that the duration of the biotrophic or necrotrophic phase differs among hemibiotrophic pathogens and is dependent on physiological triggers of the host plant (Laluk & Mengiste, 2010). During fruit ripening, specific physiological changes occur in the host, including those involved in the natural ripening process, in recognition of the fungal pathogen, and in activation of the host's defense response (Alkan et al., 2015). Some of these physiological modifications include remodelling of the cell wall architecture (Brummell et al., 1999; Hückelhoven, 2007), accumulation of soluble sugar, altered bioactivity of phytoalexins, and phytoanticipins (Prusky, 1996); changes in the pH of the cytoplasm of the host plant cell (Prusky, Alkan, Mengiste, & Fluhr, 2013); and cuticle biosynthesis (Bargel & Neinhuis, 2005). Alterations of cuticle composition, structure, and deposition are involved in triggering the switch from a quiescent state to a necrotrophic state which in turn, allow fungal pathogens to penetrate the cuticle, infect and cause destruction of fruit tissue (Agudelo‐Romero et al., 2015; Alkan et al., 2015; Bhadauria et al., 2013; Blanco‐Ulate et al., 2014). These changes are regulated by a complex interplay of hormonal signals which affect both plant defense responses and resistance to pathogen invasion (Giovannoni, 2001; Seymour, Østergaard, Chapman, Knapp, & Martin, 2013; Voisin et al., 2009).
The cuticle is considered to be a “lipidized cell wall region” that is composed of several compounds, with cutin being the most abundant, followed by cuticular waxes which are a combination of organic solvent‐soluble, very long‐chain fatty acid compounds (Lara, Belge, & Goulao, 2015; Martin & Rose, 2014). As a physical barrier, the cuticle surrounds the epidermis of fruits and serves a number of functions including (a) to protect the plant against the physical, environmental and biological stresses and pathogen invasion, (b) to reduce the effects of internal water loss, and (c) to maintain plant organ integrity by providing mechanical support (Belge et al., 2014; Chen et al., 2013; Lara, Belge, & Goulao, 2014). In pathogenicity tests, C. truncatum isolates are pathogenic on Capsicum fruits after wounding the fruit surface, and most produced a low level of infection on nonwounded fruit (De Silva et al., 2019; Ramdial & Rampersad, 2015). The life cycle of C. truncatum in Capsicum sp. included biotrophic and necrotrophic phases of colonization with high susceptibility to lesion development during fruit ripening stages (Ranathunge, Mongkolporn, Ford, & Taylor, 2012; Ranathunge & Sandani, 2016). In climacteric fruits, changes in cuticle thickness and composition change during ripening (Martin & Rose, 2014). However, in nonclimacteric fruits, like bell pepper, once maximum cutin monomer levels are reached during development, it decreases steadily as the fruit ripens (Kosma et al., 2010). This demonstrates the significance of the cuticle to infection by Colletotrichum spp. (Auyong, Ford, & Taylor, 2015).
The cuticle also has active roles in host defense signalling in addition to local and systemic resistance against a variety of pathogenic fungi. Fungal spores adhere to the surface of the host plant which signals production of low levels of cutinase activity which produces small amounts of cutin monomers (Woloshuk & Kolattukudy, 1986). Fungal sensing of cutin monomers leads to (a) production of high levels of cutinase which is required for penetration in Fusarium sp. (Woloshuk & Kolattukudy, 1986), (b) induction of germination and appressorium formation in Magnaporthe grisea and Erysiphe graminis (Francis, Dewey, & Gurr, 1996; Gilbert, Johnson, & Dean, 1996), (c) activation of gene expression of lipid‐induced protein kinase (LIPK) which is essential for infection structure formation in C. trifolii (Dickman, Ha, Yang, Adams, & Huang, 2003). With respect to induction of typical pattern‐triggered immunity (PTI) responses and upregulation of defense‐related genes, recognition of pathogen‐associated molecular patterns (PAMPs) and damage‐associated molecular patterns (DAMPs), enhance resistance to both biotrophic and necrotrophic fungal aggressors (Choi & Klessig, 2016; Kauss, Fauth, Merten, & Jeblick, 1999; Mengiste, Alfen, Leach, & Lindow, 2012; Schweizer, Jeanguenat, Whitacre, Métraux, & Mösinge, 1996). C. gloeosporioides can also induce biosynthesis of ethylene, which is involved in the breaker stage of tomato fruit ripening, that is, a “break” in color from green to tannish‐yellow, pink, or red (Alkan et al., 2015; Blanco‐Ulate, Vincenti, Powell, & Cantu, 2013; Seymour et al., 2013). In this way, the fungus can manipulate the fruit ripening process to engage the switch from biotroph to necrotroph. Thus, the cuticle is a valuable research target for development of early sensing of fungal pathogens and activation of plant defense responses.
Cutinases (EC: 3.1.1.74) are members of the alpha/beta hydrolase family of lipases (Kolattukudy, 1984; Longhi & Cambillau, 1999). The enzyme is able to hydrolyze fatty acids esters and emulsified triacylglycerol as efficiently as lipases, and therefore, it is considered an intermediate between esterases and lipases (https://www.ebi.ac.uk/enzymeportal/ec/3.1.1.74) (Nyyssölä, 2015). The enzyme plays an active role in (a) carbon acquisition for saprophytic growth, (b) adhesion of fungal structures to the host surface, and (c) the early stages of fungal penetration (Auyong et al., 2015). Cutin monomers trigger expression and synthesis of cutinases required for fungal penetration into the plant tissues. Cutinases, therefore, have a critical role in plant surface signalling that elicits differentiation of those fungal structures required for infection (Belbahri, Calmin, Mauch, & Andersson, 2008).
Bell pepper (Capsicum annuum var. grossum (L.) Sendt.) is a member of the Solanaceae family and is one of the most widely cultivated vegetable crops in the world. Over the last decade, the world production and consumption of bell peppers have been steadily increasing. More than 70% of the world's bell peppers are produced in China (FAO, 2017, http://www.fao.org/faostat/en/#data/QC). In Trinidad, bell pepper is grown year‐round and is among the top 10 agricultural commodities in the country. Production, in terms of yield and cost, is directly affected by fruit rot caused primarily by C. truncatum (syn. C. capsici ‐ Damm, Woudenberg, Cannon, & Crous, 2009; Ramdial & Rampersad, 2015) and more recently by C. brevisporum, albeit with lower incidence compared to C. truncatum (Villafana, Ramdass, & Rampersad, 2019). C. truncatum was the predominant species of Colletotrichum associated with anthracnose of chili in Asia and is widely distributed throughout Asia, Australia, and South America (De Silva, Ades, Crous, & Taylor, 2017; De Silva et al., 2019; Diao et al., 2017; Mongkolporn & Taylor, 2018; Sharma, Kumar Pinnaka, & Shenoy, 2014). Multilocus phylogeny revealed that the recently characterized Colletotrichum magnum species complex consists of nine closely‐related species which includes C. brevisporum (Damm et al., 2019). C. brevisporum has also been reported as one of the causal agents of anthracnose disease in chili fruit in China and Brazil and in bell pepper fruit in Trinidad (De Silva, Crous, Ades, Hyde, & Taylor, 2017; Diao et al., 2017; Liu et al., 2016; Villafana et al., 2019).
It is hypothesized that (a) C. truncatum and C. brevisporum are hemibiotrophs that are capable of secreting cutinase as part of their infection strategy, (b) the single cutinase gene structure and arrangement identified in C. truncatum is highly conserved at both the level of the nucleotide and amino acid sequence as opposed to other fungal species with multiple copies of this gene with divergent functions, (c) the cutinase domain has no relationship to synteny of other protein domains flanking this cutinase domain as no information is currently available in the literature concerning flanking amino acid sequences. This study was, therefore, undertaken to (a) assess the ability of C. truncatum and C. brevisporum isolates from green and red bell peppers from Trinidad to produce and secrete cutinase based on in vitro bioassays, and (b) determine the diversity, structure and organization and synteny of cutinase gene sequences and amino acid sequences among different Colletotrichum species and within C. truncatum isolates as a major fungal pathogen of green and red bell pepper fruit worldwide. This work will increase our understanding of the evolution of cutinase genes in fungal–plant host interactions which can be used to develop strategies for disruption of cutinase gene target(s) to reduce diseases caused by these fungal pathogens.
2. MATERIALS AND METHODS
2.1. Collection of isolates
Fifty‐two infected green and red bell pepper fruit with anthracnose lesions were collected from fields across Trinidad. The location of the lesions on the surface of the fruit was recorded according to three regions: the shoulder (region surrounding the peduncle and calyx); the face (largest surface of the fruit); and bottom (apex either acute or truncated). Bell pepper fields located in the main growing areas in Trinidad were visited: Aranguez (north and south), Orange Grove, Maloney, Caura, Caroni, Bon aventure, Penal, Mayo. Thin, transverse slices of the pericarp ~1 cm in length were then cut at the shoulder and face of bell pepper fruit and mounted on slides. Sudan III was used to stain the cuticle an orange to red color for microscopic visualization (Yeung & Chan, 2015).
2.2. Isolation of fungal pathogens
The bell pepper fruits were surface sterilized by rinsing in 70% ethanol for 1 min followed by another rinse in 0.6% sodium hypochlorite solution for 1 min. Samples were then washed three times in sterilized distilled water and dried on sterilized tissue paper. 4‐mm3 blocks of fruit tissue were removed from the margins of the lesions and transferred to potato dextrose agar (PDA) media (Oxoid Ltd., Thermo Fisher Scientific, Inc., USA) supplemented with 50 mg/L streptomycin, tetracycline, and chloramphenicol. Plates were incubated for seven days in the dark at 25°C. Monoconidial cultures of C. truncatum, and C. brevisporum were subsequently obtained and maintained on PDA at 4°C for temporary storage, and as conidial suspensions in 50% glycerol at −70°C for long‐term storage. The identities of C. truncatum and C. brevisporum cultures were confirmed using comparisons of multiple gene sequences (internally transcribed spacer region, ITS1‐5.8S‐ITS2; beta tubulin, TUB2; Actin, ACT‐GenBank Accession Nos. MG822830‐2, KJ780718, KR029613, MG827234‐5, MG839690‐1, MG870320‐1) after PCR amplification of total genomic DNA extracted from each fungal culture according to published protocols (van Poucke et al., 2012; White, Bruns, Lee, & Taylor, 1990).
2.3. Cutinase activity
2.3.1. Tween‐20 and tributyrin opacity test
Gel diffusion bioassays, for example, the Tween and tributyrin opacity test, are based on the ability of lipase‐secreting microbes to break down the lipid substrate incorporated into solid media (see review by Lanka & Latha, 2015). These tests minimize the cost of screening and protect the test microorganisms from the inhibitory effects of various indicator dyes and lipase activity is identified as a clear or turbid zone around the colonies after incubation (Lanka & Latha, 2015). The level of enzyme activity can be evaluated by measuring the diameter of the halo around the colonies. In this study, for the Tween‐20 test, a calcium salt‐free Bacto agar (Oxoid Ltd., Thermo Fisher Scientific Inc.) medium containing Tween‐20 (Sigma Aldrich Inc.) was used (Slifkin, 2000). A 4‐mm3 block taken from the advancing mycelial edge of an actively growing culture of each isolate (eight isolates each of C. truncatum, and C. brevisporum) was plated onto this medium in duplicate. The presence of a white crystalline precipitate around the colony indicated secreted lipase activity. The colony size was measured after three days. The test was repeated.
2.4. Statistical analyses
Data were analyzed using IBM SPSS Statistics version 20. Tables outlining summary statistics for colony diameter (mm) on Tween‐20 and tributyrin, respectively, as well as for halo size (mm) on tributyrin. Analysis of variance tests (One‐way ANOVA) was carried out for “Colony diameter (mm)* Species” for Tween‐20 and tributyrin samples, respectively, and “Halo size (mm)* Species” for tributyrin samples and the appropriate tables were extracted.
2.5. Rhodamine test
Use of a fluorescent indicator dye such as Rhodamine B or G in the presence of olive oil as a lipid substrate has been used to determine lipase‐positive microbes (Lanka & Latha, 2015). Hydrolysis of a lipid substrate (e.g., olive oil) by secreted lipase results in the production of free fatty acids which interacts with Rhodamine fluorescent dye in the medium and causes the formation of yellow to orange fluorescent colonies which are visible upon UV irradiation. A 4‐mm3 block taken from the advancing mycelial edge of an actively growing culture of each isolate (eight isolates each of C. truncatum, and C. brevisporum) was plated onto this medium in duplicate as previously described for the Tween‐20 and tributyrin opacity tests. This test was repeated. Observation of a fluorescent yellow to orange colonies under UV light was recorded after six days.
2.6. DNA extraction, PCR amplification, and sequencing
DNA was extracted from 21 actively growing colonies using the Maxwell®‐16 automated DNA extraction system (Promega) based on magnetic bead capture DNA extraction according to the manufacturer's instructions. PCR amplification was carried out using published protocols for culture identification with modifications (O'Donnell et al., 2004; White et al., 1990). The primers and their use in this study are described in Table 1. For a single 25 µl reaction using GoTaq® Green Master Hot Start Taq DNA Polymerase (Promega Corporation) the PCR components included 1 × master mix, 50 pmoles of each primer (Integrated DNA Technologies), and 5 µl of the 1:4 diluted DNA sample. PCR amplification thermal conditions consisted of an initial denaturation of 5 min at 94°C followed by 35 cycles of 1 min at 94°C, annealing temperature and duration according to primer sequences, 1 min at 72°C with a final extension of 5 min at 72°C. Amplicons were sequenced at MCLAB (Molecular Cloning Laboratories). Sequence identities were verified using the BLAST algorithm in NCBI. Representative cutinase sequences for C. truncatum and C. brevisporum were deposited in GenBank (GenBank Accession Nos. MN473062 and MN473063, respectively). Twenty‐one representative cccut‐F/R‐generated nucleotide sequences were used in the final data set.
Table 1.
Primers used in this study
| Fungus | Gene target | Primer name | Primer orientation | Primer sequence (5′−3′) | Expected band size (bp) | Reference |
|---|---|---|---|---|---|---|
| C. gloeosporioides sensu lato | Species complex‐specific ITS | CgInt | Forward | GGCCTCCCGCCTCCGGGCGG | 450 | Mills, Sreenivasaprasad, and Brown (1992) |
| ITS4 | Reverse | TCCTCCGCTTATTGATATGC | White et al. (1990) | |||
| Fungi | Fungi ITS | ITS5 | Forward | GGAAGTAAAAGTCGTAACAAGG | 550–650 | White et al. (1990) |
| ITS4 | Reverse | TCCTCCGCTTATTGATATGC | White et al. (1990) | |||
| C. truncatum | Species‐specific ITS | Ccap‐F | Forward | GTAGGCGTCCCCTAAAAAGG | 394 | Torres‐Calzada et al. (2011) |
| Ccap‐R | Reverse | CCCAATGCGAGACGAAATG | ||||
| C. gloeosporioides sensu lato | Cutinase | CgCUT‐F | Forward | ATCAGGGTCAGCTAGGTTAGT | 308 | This study |
| CgCUT‐R | Reverse | GGATCGTGAGGCCCTATTTATG | ||||
| C. truncatum | Cutinase | CcCUT‐F | Forward | AGAGTTTCTTCCGACCATTCC | 729 | This study |
| CcCUT‐R | Reverse | GCCCTGTTATAGGAGTCAGTTATC | ||||
| C. gloeosporioides sensu lato | partial Actin | ACT−512 | Forward | ATGTGCAAGGCCGGTTTCGC | 300 | Carbone and Kohn (1999) |
| ACT−738 | Reverse | TACGAGTCCTTCTGGCCCAT | ||||
| Filamentous Ascomycetes | partial β‐Tubulin | Bt2a | Forward | GGTAACCAAATCGGTGCTGCTTTC | 170 | Glass and Donaldson (1995) |
| Bt2b | Reverse | ACCCTCAGTGTAGTGACCCTTGGC |
Cutinase‐specific cccut‐F/R primers were designed against three C. truncatum sequences mined from GenBank (GenBank Accession Nos. M18033, HQ406775 and XM_007602402) using the IDT DNA oligo primer quest tool (https://www.idtdna.com/PrimerQuest/Home/Index). Amplicons were sequenced directly (MCLAB, CA, USA), and sequence identities were verified by BLAST analysis.
2.7. Cutinase nucleotide sequence diversity
Nucleotide sequences were analyzed for sequence diversity and evolutionary maintenance. Sequences were aligned using MAFFT (Multiple Alignment using Fast Fourier Transform; https://www.ebi.ac.uk/Tools/msa/mafft/; Katoh & Standley, 2013), edited in Bioedit (Hall, 1999), and the FASTA alignment files were analyzed in DnaSP (DNA Sequence Polymorphism DnaSP version 6.12.03; Librado & Rozas, 2009; Rozas, 2009) to evaluate DNA polymorphism and evidence of selection.
2.8. Cluster analysis
Cluster analysis of the aligned nucleotide sequences was carried out in MEGA (MEGA7: Molecular Evolutionary Genetics Analysis version 7.0; Kumar, Stecher, & Tamura, 2016) using the maximum likelihood algorithm under the GTR + G + I nucleotide substitution model (based on a test of nucleotide substitution model in MEGA7) with 1,000 replicates. Four reference sequences from GenBank were included in the alignment (GenBank Accession Nos. M18033 and HQ406775–C. truncatum; KP331429 and M21443–C. gloeosporioides; there were no reference cutinase sequences for C. brevisporum).
2.9. Cutinase amino acid sequence diversity
Based on the findings of the cluster analysis, nucleotide sequences of the cccut‐F/R data set were translated to single amino acid sequence and the correct reading frame was identified using the ExPaSY translate tool (https://web.expasy.org/translate/). Two data subsets were analyzed: data subset 1‐ cutinase amino acid sequence (derived from translation of amplified nucleotide sequences by cccut‐F/R primers) curated to 219 amino acids in length and which included exonic and intronic sequences; data subset 2‐cutinase amino acid sequence (derived from translation of amplified nucleotide sequences by cccut‐F/R primers) curated to 66 amino acids in length and only included exonic sequences. The alignment, query coverage, and percent identity were then examined for “within” Colletotrichum species (intraspecific diversity) and “among” Colletotrichum species (interspecific diversity).
Multiple nucleotide sequences were translated to amino acid sequences using the EMBOSS Transeq tool (https://www.ebi.ac.uk/Tools/st/emboss_transeq/). The multiple sequences were then aligned using EMBL‐EBI Clustal Omega (https://www.ebi.ac.uk/Tools/msa/clustalo/). Clustal Omega is a multiple sequence alignment program that uses seeded guide trees and HMM profile–profile techniques to generate alignments among three or more sequences. This alignment was then used to generate an amino acid conservation plot in BioEdit (Hall, 1999), and this conserved sequence was used in subsequent analyses. Protein domain superfamilies in CATH‐Gene3D (http://www.cathdb.info/search/; Dawson et al., 2016; Lewis et al., 2017) have been subclassified into functional families (or FunFams), which are groups of protein sequences and structures with a high probability of sharing the same function(s). Therefore, the functionally important residues in a family are also expected to be highly conserved. Information on conserved positions in CATH‐Gene3D cutinase amino acid sequence alignment was analyzed (http://www.cathdb.info/version/v4_2_0/superfamily/3.40.50.1820/funfam/115401/alignment?task_xml:id=d1db710a5a097f00badbddcb35a91fc5).
2.10. Lineage of cutinase protein based on sequence, structure, and functional diversity
CATH‐Gene3D provides information on the evolutionary relationships of protein domains through sequence, structure, and functional annotation data. The taxonomic lineage of cutinase protein was hypothesized using CATHDB version 4.2 (http://cathdb.info/version/v4_2_0/superfamily/3.40.50.1820/funfam/115401/taxonomy_sunburst) starting with Eukaryota and ending with C. truncatum.
2.11. Structure, organization, and synteny of cutinase gene among 34 Colletotrichum species and among different fungal species
The structure and organization of the cutinase gene and protein domain were analyzed for 34 available Colletotrichum genomes in the MycoCosm fungal genomes database and portal of the Joint Genome Institute (https://mycocosm.jgi.doe.gov/mycocosm/home; Grigoriev et al., 2011, 2014).
3. RESULTS
Lesions caused by C. truncatum were recorded at the shoulder, face, and bottom of the fruit (Figure 1). The frequency of lesion according to location on fruit surface was 44.2% on the shoulder, 50.0% on the face and 53.8% on the bottom. As such, the frequency distribution showed no preference for lesion development at a specific location on the fruit surface (Table A1). Figure 2 shows the difference in cuticle integrity and thickness in green versus red bell pepper fruit.
Figure 1.

Symptoms of (a) Colletotrichum brevisporum infection in red bell pepper fruit; (b) C. truncatum infection in green bell pepper fruit; (c) C. truncatum infection in red bell pepper fruit
Figure 2.

Histological section of bell pepper fruit showing the relative cuticle integrity and thickness in (a) green bell pepper fruit and (b) red bell pepper fruit
3.1. Cutinase bioassays
Tween‐20 Opacity Test: A white crystalline precipitate was observed for all isolates of C. truncatum and C. brevisporum (Figure 3a). Table A2 outlines the summary of statistics for colony diameter (mm) for each Colletotrichum species on Tween‐20. C. truncatum had the larger colony diameter (mm) at 31.84 mm, as well as the higher mean value at 19.53 mm, while C. brevisporum had the smaller colony diameter at 9.21 mm on Tween‐20 media. Based on the analysis of variance (ANOVA) of colony diameter (mm) values for C. truncatum and C. brevisporum on Tween‐20 media, there was a significant difference in colony diameter (mm) at p = .007 (Table A3).
Figure 3.
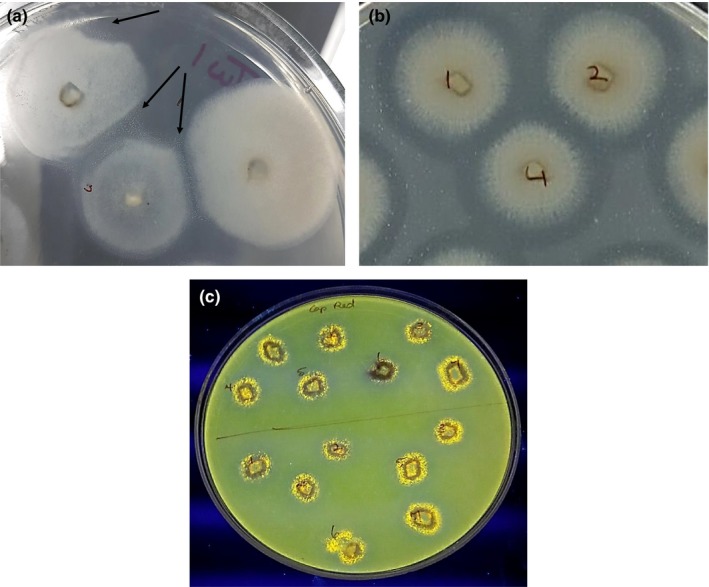
Lipid substrate bioassays; (a) arrows point to white crystalline precipitate on Tween‐20 medium; (b) zone of clearance (halo) around colonies producing secreted lipase (alpha/beta hydrolase/cutinase) on tributyrin medium; (c) yellow–orange fluorescent colonies on Rhodamine medium
Tributyrin Test: The development of a zone of clearance (halo) for all isolates plated on tributyrin agar indicated lipase activity for C. truncatum and C. brevisporum. (Figure 3b). Summary of statistics for colony diameter (mm) for each Colletotrichum species on tributyrin media (Table A4) indicated that C. truncatum had the larger colony diameter (mm) at 15.56 mm, as well as the higher mean diameter value at 6.18 mm. The maximum and minimum halo sizes (mm) measured for C. truncatum were 12.74 mm and 3.42 mm, respectively. C. truncatum halo diameter also had higher mean value at 8.71 mm (Table A5). Analysis of variance of colony diameter for the two Colletotrichum species indicated that there was a significant difference in colony diameter (mm) between C. truncatum and C. brevisporum, (p = .021; Table A6). However, there was no significant difference in halo diameter (mm) for C. truncatum and C. brevisporum (Table A7).
Rhodamine Test: Yellow–orange‐colored fluorescent colonies were observed under UV light, which confirmed secreted cutinase activity for all isolates of C. truncatum and C. brevisporum (Figure 3c).
3.2. Cutinase nucleotide and amino acid sequence analyses
3.2.1. Nucleotide sequence analysis
Nucleotide sequences generated from cccut‐F/R primers (trimmed to a final length of 656 bases) were identified as cutinase gene with 100% sequence coverage, 99.54% and 99.24% sequence identity with an E‐value of 0.0 to C. truncatum (GenBank Accession Nos. HQ406775 and M18011).
The degree of DNA polymorphism and related parameters are presented in Table 2. The nucleotide sequence generated by the cccut‐F/R primer pair consists of exonic and intronic sequences. There were no recombination signatures detected in the aligned nucleotide sequence data set. Tests of neutrality indicated that the nucleotide sequences were under significant (p ≤ .02) positive selection based on Fu and Li's D* statistic (Fu & Li, 1993).
Table 2.
DNA polymorphism analysis of aligned nucleotide sequences generated from cccut‐F/R primers that target the cutinase gene in Colletotrichum truncatum isolates
| cccut‐F/R data set | |
|---|---|
| N | 21 |
| DNA polymorphism | |
| No. of polymorphic sites, S | 17 |
| No. of mutations, Eta | 17 |
| Average no. of nucleotide differences, k | 4.886 |
| Nucleotide diversity, Pi | 0.00748 |
| Haplotype analysis | |
| No. of haplotypes, h | 3 |
| Haplotype diversity, Hd | 0.643 |
| Tests of neutrality | |
| Tajima's D statistic | 0.12559 |
| Fu and Li's D* statistic | 1.55120* |
| Fu and Li's F* statistic | 1.3145 |
| Recombination events, Rm | 0 |
Significant at p ≤ .02
3.2.2. Cluster analysis
For the cccut‐F/R‐generated nucleotide sequence analysis, the clusters were strongly supported (minimum bs ≥ 85%) and were constructed according to haplotype: two haplotypes for the C. truncatum sequences, one haplotype for the C. brevisporum sequences, and two haplotypes that represented the two C. gloeosporioides sequences (Figure 4). The structure of each haplotype cluster was largely polytomic indicating no sequence diversity within a particular cluster. Clustering was irrespective of the maturity of the bell peppers fruit, that is, whether red or green.
Figure 4.

Cluster analysis of cutinase nucleotide sequences designed in this study using the maximum likelihood method based on the GTR + G + I model. The tree with the highest log likelihood is shown. The rate variation model allowed for some sites to be evolutionarily invariable. The tree is drawn to scale, with branch lengths measured in the number of substitutions per site
3.2.3. Cutinase amino acid sequence diversity
Cutinase amino acid sequence was invariable at the intraspecific level with 100% amino acid sequence identity at 100% coverage of the query sequence with C. truncatum (UniProtKB ‐ P10951; GenBank Accession No. ADQ27862). However, variability existed at the interspecific level with 83.3% sequence identity to C. gloeosporioides (GenBank Accession Nos. AAL38030 and AKH80819) at 100% of the query sequence. “Among species” amino acid sequence variability was at minimum for C. gloeosporioides at 83.3% and maximum for C. spinosum at 74.2% (Table 3). For all C. truncatum and C. brevisporum amino acid sequences, there was 100% conservation of the core coding region that corresponded to the cutinase domain despite nucleotide sequence variation in the same region (Figure 5).
Table 3.
Diversity of the cutinase amino acid sequence among 17 Colletotrichum species
| Colletotrichum species | GenBank Accession | Sequence identity % | Aligned aa sequence length | bit score |
|---|---|---|---|---|
| C. truncatum | P10951.1 | 100 | 66 | 2.42E−37 |
| C. capsici | ADQ27862.1 | 100 | 66 | 3.04E−37 |
| C. gloeosporioides | AAL38030.1 | 83.333 | 66 | 1.88E−26 |
| C. gloeosporioides | AKH80819.1 | 81.818 | 66 | 4.49E−26 |
| C. gloeosporioides | P11373.1 | 81.818 | 66 | 5.06E−26 |
| C. simmondsii | KXH53950.1 | 77.612 | 67 | 8.10E−25 |
| C. salicis | KXH52034.1 | 77.612 | 67 | 8.27E−25 |
| C. fiorinae | EXF73863.1 | 77.612 | 67 | 8.82E−25 |
| C. orchidophilium | XP_022472246.1 | 76.119 | 67 | 7.84E−24 |
| C. fructicola | ELA29687.1 | 77.273 | 66 | 3.65E−23 |
| C. orbiculare | TDZ15371.1 | 80.303 | 66 | 2.44E−20 |
| C. trifolii | TDZ54558.1 | 78.788 | 66 | 7.92E−20 |
| C. sidae | TEA20600.1 | 78.788 | 66 | 7.92E−20 |
| C. incanum | KZL82629.1 | 79.412 | 68 | 6.78E−19 |
| C. incanum | OHW94975.1 | 79.412 | 68 | 7.11E−19 |
| C. spinosum | TDZ13928.1 | 74.242 | 66 | 2.22E−18 |
| C. gloeosporioides | 3DCN_A | 93.939 | 33 | 3.07E−12 |
Figure 5.

Conservation plot of cutinase amino acid sequence belonging to Colletotrichum truncatum isolates from Trinidad and C. truncatum reference sequences from GenBank
3.2.4. Conservation of cutinase based on 200 fungal amino acid sequences
An alignment of 200 truncated amino acid sequences of different fungal species with an identified cutinase domain in CATH‐Gene3D protein data bank revealed specific regions in the sequence that may be indicative of “diversity hot spots” (Figure 6). This is in comparison to the conservation plot of aligned amino acid sequences of C. truncatum isolates from Trinidad and two reference C. truncatum sequences mined from GenBank which illustrated 100% conservation. The functionally important residues in this protein superfamily are expected to be highly conserved. CATH indicated that the name of this superfamily (3.40.50.1820), previously called alpha/beta hydrolase, has been modified, and the official domain name is pending.
Figure 6.

Alignment of 66‐amino acid cutinase domain of 200 different fungal species showing sequence diversity “hot spots” in CATHDB. Columns in the alignment are colored where the highly conserved residues are shown in red through to positions that are not conserved at all, shown in blue
3.2.5. Structure of cutinase gene
The structure of the cutinase gene and protein was analyzed across 34 available Colletotrichum genomes in the MycoCosm database. C. truncatum and C. brevisporum were not among the genomes available for reference in the MycoCosm repository. The predicted cutinase gene length for 34 Colletotrichum sequences in the MycoCosm database whose cutinase gene length ranged from 967 bp to 1,296 bp which included two exons interrupted by one intron. Among the 34 genomes, the cutinase gene was composed of two exons interrupted by one intron. Exon 1 varied from 343 to 733 bp in length, exon 2 varied from 556 to 786 bp in length, and the intronic sequence varied in length from 32 to 85 bp. (Figure S1). There was, therefore, one splice variant where the intervening intron between the two coding regions is removed.
3.2.6. Protein signatures
There was a highly conserved domain (Figure S2), characteristic of serine hydrolase, consisting of 194 amino acid residues with a Ser, His, Asp catalytic triad, and 5‐element fingerprint (motifs 1 to 5) among all Colletotrichum species. As such, the enzyme had three identified active sites, ACT_SITE140, ACT_SITE195, and ACT_SITE208 (https://www.ebi.ac.uk/enzymeportal/search/P10951/enzyme), and whose source data are derived from IntEnz (Integrated relational Enzyme database; Fleischmann et al., 2004) and UniProt. There was also evidence of two disulfide bonds (amino acid positions 49 to 198 and 129 to 191) which play a critical role in holding the catalytic residues in juxta‐position; reduction of the disulfide bridges results in the complete inactivation of the enzyme and, therefore, these bonds are involved in posttranslational modification of the enzyme (Figure S2; UniProt P00590, CUT1). Verification of the protein structure and function (molecular function was cutinase activity [GO id 0050525; Interpro id IPR011150] and cellular component was extracellular/secreted [GO id 0005576; Interpro id IPR11150]) was carried out in EnsemblFungi (https://fungi.ensembl.org/index.html; Kersey et al., 2015) by comparing linked cutinase protein sequences in the Gene3D, SPRINTS, SMART, and PROSITE repositories. Each “GO” term was linked to the three major categories “Cellular Component,” “Biological Process,” or “Molecular Function.”
Analysis of the cutinase gene for Colletotrichum sp. (C. gloeosporioides was selected as a model nucleotide sequence, CGGC5_1134; transcript ELA29687, as there were no C. truncatum or C. brevisporum model gene sequences in the EnsemblFungi database). Orthologue analysis identified 20 orthologues of the cutinase gene with other fungal species purportedly as a result of a gene duplication event that occurred ~ 10 Million years ago prior to any speciation event and whose root species belongs to Pezizomycotina (majority of Ascomycetes and lichenized fungi) (Genomicus v. 30.01, http://www.genomicus.biologie.ens.fr; Nguyen, Vincens, Roest Crollius, & Louis, 2017; Figure S3).
3.2.7. Organization and synteny of cutinase gene among Colletotrichum genomes
Synteny analysis in MycoCosm indicated that the cutinase domain was flanked by highly conserved amino acid sequences at both the N‐ and C‐termini in the sequences of 34 Colletotrichum species (Figures 7, 8, 9). At the N‐terminus, there was an uncharacterized but highly conserved hypothetical protein (KOG id 2306) whose amino acid sequence identity ranged from 69.3% (GenBank Accession No. EQB49109) to 97.6% (GenBank Accession No. KXH27358). Model testing revealed that 100% of all external track models returned the same uncharacterized but conserved description for this protein domain. SMART (Simple Modular Architecture Research Tool; https://smart.embl.de/; Letunic & Bork, 2017) using hidden Markov models (HMMs) confidently predicted low complexity regions based on Seg analysis (http://mendel.imp.ac.at/METHODS/seg.server.html), in addition to another domain of unknown function, DUF4210. Proteins containing this DUF4210 domain include the animal FAM214A proteins and fission yeast SPAC3H8.04 proteins. An RNA recognition motif (KOG id 2135) in addition to translation elongation factor 2 (aEF2; KOG id 0469) was also identified at this terminus in immediate proximity to the cutinase domain.
Figure 7.
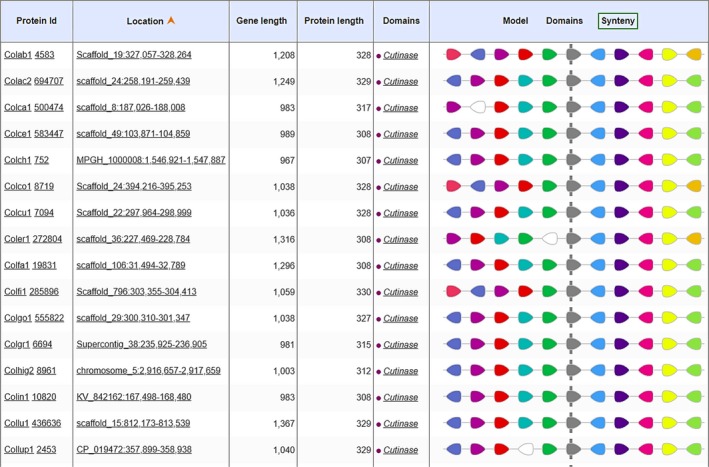
Synteny of cutinase domain and flanking domains in 16 Colletotrichum species as identified in MycoCosm
Figure 8.

Synteny of cutinase domain and flanking domains in 18 Colletotrichum species as identified in MycoCosm
Figure 9.
Key for Colletotrichum species codes analyzed for synteny of the cutinase domain and the KOG key to these domains as identified in MycoCosm
At the C‐terminus, a PPR domain (pentatricopeptide repeat) was identified whose function is hypothetical but predicted to involve RNA stabilization and processing as it retains an amino acid sequence‐specific RNA‐binding site (KOG id 4197). Immediate to this PPR domain, is a signal peptide involved in secretion (KOG id 2460). Identification of this signal peptide [VAA‐AP] at position 16 and 17 of a 66‐amino acid sequence, translated from a core coding region of the test isolates’ amino acid sequence, was verified using SignalP 5.0 (http://www.cbs.dtu.dk/services/SignalP/; Armenteros et al., 2019) and Phobius (http://phobius.sbc.su.se/; Madeira et al., 2019) software. The signal peptide prediction is consistent with the database annotation (Figure S4–S6). There was no identified transmembrane domain. Molecular chaperones belonging to the HSP70 family of heat shock proteins and which are involved in posttranslational modification and protein folding (KOG id 0102) were located closer to the C‐terminus. WoLFPSORT (https://www.genscript.com/wolf-psort.html; Horton et al., 2007) was used in protein subcellular localization prediction which confirmed that the enzyme product, deduced from in‐frame amino acid sequence of test isolates, is extracellular or secreted. The identified domains at the both termini were conserved both in their structure and strict arrangement immediate to and relative to the cutinase domain.
4. DISCUSSION
This study investigated the diversity, structure, organization, and synteny of a single cutinase gene within C. truncatum and C. brevisporum isolates from bell pepper fruit in Trinidad and among different Colletotrichum species. Evidence of secreted cutinase capability was first tested among the C. truncatum and C. brevisporum isolates from Trinidad based on bioassays using different lipid substrates.
4.1. Lipid substrate bioassays
Bioassays confirmed the ability of C. truncatum and C. brevisporum to produce and secrete cutinase enzyme in lipid substrate media. Growth characteristics for the identification of secreted lipase activity using Tween‐20, Rhodamine, and tributyrin agar plates were congruent with the results obtained from other studies (Lanka & Latha, 2015; Ramnath, Sithole, & Govinden, 2017; Wadia & Jain, 2017). As both Tween‐20 and Tributyrin can test positive with esterases, the positive fluorescence with Rhodamine B was used as the determining factor for the presence of lipase activity (Lanka & Latha, 2015; Tomulescu et al., 2015). Bornscheuer (2002) explained that lipases can be differentiated from carboxyl esterases based on their substrate spectra: p‐nitrophenyl palmitate (pNPP) is hydrolyzed by lipases; p‐nitrophenyl butyrate (pNPB) is hydrolyzed by esterases. However, Chen, Liang, Zhang, and Rodrigues (2006) reported that C. kahawae and C. gloeosporioides isolates demonstrated cutinase activity on tributyrin media but this activity was significantly reduced with p‐nitrophenyl butyrate assays. Conversely, Bonnen and Hammerschmidt (1989) stated that a purified cutinase enzyme, extracted from C. lagenarium isolates, was active on both cutin and p‐nitrophenylbutyrate as substrates. For this reason, three bioassays using different lipid substrates were used to assess cutinase activity in this study.
4.2. Number of cutinase genes in C. truncatum compared with other fungal genomes
Only one cutinase gene has been identified in C. truncatum (Auyong et al., 2015), and this gene was the target under study. Primers were designed to amplify a portion of this cutinase gene for genetic analysis of C. truncatum and C. brevisporum isolates infecting bell pepper in Trinidad. Most plant pathogenic fungi possess multiple copies of genes that encode a family of cutinase enzymes: 14 to 17 cutinase genes in Magnaporthe oryzae, 13 in Curvularia lunata, 13 in V. dahliae, 12 in F. graminearum, 11 in Botrytis cinerea, seven in Rhizoctonia solani, and six in R. cerealis (Skamnioti, Furlong, & Gurr, 2008; Liu et al., 2016; Gui et al., 2018; Lu, Rong, Massart, & Zhang, 2018). The adaptive advantage of multiple gene copies may include (a) an increase in the amount of gene product generated, (b) copies of the same gene may retain their original functions, but are expressed at different levels and at different times in response to different triggers, (c) one or more copies of the gene can evolve to carry out a new important functional role (neofunctionalization), so that conservative selection acts to preserve multiple versions of the gene (Altenhoff et al., 2019).
4.3. Cutinase nucleotide and amino acid sequence variation among only C. truncatum
Cluster analysis indicated a low level of nucleotide variation among C. truncatum sequences. Wang et al., (2017) also found high homology among cutinase (CUT1) gene sequences of C. gloeosporioides. The cutinase nucleotide sequences of C. brevisporum in this study, were more related to C. truncatum cutinase nucleotide sequences than to C. gloeosporioides. Cluster patterns coincided with haplotype with moderate to high bootstrap support. Zhu and Freeland (2006) purported that the degenerate nature of the genetic code enables amino acid changes that conserve certain amino acid properties. As such, the 66‐amino acid sequence translated from the partial coding region of the cutinase gene was highly conserved within C. truncatum and C. brevisporum species. Pathogenesis is actualized through cutinase production and secretion among C. truncatum isolates, therefore, evolutionary maintenance of those amino acids in the primary protein structure that are important for folding, structural stability, and are required to form a substrate binding site(s) which guarantees the catalytic ability of the enzyme (Altenhoff et al., 2019; Rudnicki, Mroczek, & Cudek, 2014).
4.4. Cutinase sequence diversity
The cutinase gene family in various genera of fungal genomes demonstrates a high degree of coding‐sequence variation reflective of the diverse of roles attributed to cutinases and which perhaps, allowed adaptation to diverse ecological niches over time (Deng, Carbone, & Dean, 2007; Skamnioti et al., 2008). However, the relative degree of nucleotide and amino acid sequence diversity in addition to the role of these genes and their gene products in pathogenesis can vary (Liu et al., 2016; Skamnioti & Gurr, 2007; Sweigard, Chumley, & Valent, 1992). For example, in M. oryzae, CUT1 gene is essential for pathogenicity and CUT2 gene is required for cuticle sensing and formation of infection structures (Skamnioti et al., 2008). It is also reasonable to associate comparatively high cutinase gene family diversity in the genomes of M. oryzae, B. cinerea, and F. graminearum because of their alternate saprophytic/pathogenic lifestyle; wide host range and concomitant exposure to cutin from a number of host plant species and their subsequent acquired ability to sense and degrade cuticle in different plant species and organ types. In this study, diversity hot spots were identified in the cutinase amino acid sequence when 200 fungal species were compared and this could be explained by divergent evolution allowing different functions of cutinase enzyme in plant pathogenic fungi. Gui et al. (2017) conducted functional analysis of 13 cutinase genes (VdCUTs) and found significant sequence divergence in cutinase family members in the genome of Verticillium dahliae Vd991.
4.5. Cutinase gene structure within C. truncatum and among Colletotrichum sp
Within C. truncatum sequences, the cutinase gene structure as cutinase 1 (CUT1) was maintained as two exons with a single intervening intron which is spliced out during posttranscriptional modification resulting in a single splice variant based on comparative clustering. The relative positions of the two exons and the intervening intron varied within base pairs among Colletotrichum species and this is in keeping with the findings of Liu et al. (2016).
4.6. Orthologue detection in 200 fungal genomes and implications of gene duplications
Twenty cutinase orthologues detected among different fungal species and whose common ancestor is Pezizomycotina were identified in this study. Using the CATH‐Gene3D hierarchical classification approach which grouped cutinase protein domains according to sequence, structure, and functional diversity, confident predictions of the likely evolutionary relatives indicated that a hypothesized gene duplication event is one mechanism by which the cutinase gene in C. truncatum evolved. Gene duplication is an evolutionary tool by which new genes and genetic novelty can be generated in eukaryotes which in turn, would serve to increase the plasticity of a genome or species as an adaptive advantage to changing environments. Several factors impact upon whether a duplicated copy of this gene is fixed in the lineage of given pathogen population through positive selection forces. Primarily, the copy must serve some selective advantage to the pathogen, for example, two copies of one gene may increase the rate of expression and production of gene products or the duplicated copy may have evolved through neofunctionalization and is now adjunct to the function of the original gene (Magadum, Banerjee, Murugan, Gangapur, & Ravikesavan, 2013; Savory, Leonard, & Richards, 2015). Among test isolates in this study, the aligned cutinase nucleotide sequences, generated from the cccut‐F/R primer pair, demonstrated signatures of significant positive selection according to Fu and Li's D* statistic which indicates the importance of this gene's functions during host pathogen interaction (Tiffin & Moeller, 2006).
It is suggested that the hypothesized lineage of the cutinase gene emerged after its transference between distantly related plant‐degrading microbes through lateral gene transfer events (LGTs) (Belbahri et al., 2008). Genes that evolved from what is considered to be an “ancient” duplication event (i.e., duplication before speciation as in the case of Pzezizomycotina and subsequent speciation events that occurred after a single duplication event) may have diverged to an extent that created new functions (Altenhoff et al., 2019). Gene function annotation of 13 Colletotrichum species with 11 other fungal species indicated that the evolutionary features involved in pathogenesis of Colletotrichum fungi existed at multiple taxonomic levels, some of which were facilitated by fungus‐to‐fungus lateral transfer which suggests that the success of Colletotrichum fungi as plant pathogens are due in part to shared, as well as lineage‐specific virulence factors (Liang et al., 2018). These findings provide insight into the acquisition of virulence factors in these important plant pathogens (Belbahari et al., 2008).
4.7. Synteny among 34 Colletotrichum genomes
It is useful to investigate synteny, in the context of genome structure evolution, because it demonstrates the comparative framework by which conservation of protein‐coding genes and gene order are enabled between genomes of different species. This type of analysis is carried out under the presumption that genome assembly of good contiguation is used in the analysis (Liu, Hunt, & Tsai, 2018). The findings revealed that the cutinase domain was retained both in structure and arrangement among Colletotrichum species. The order of aligned genomic blocks between species and the arrangement of flanking protein domains were also conserved and shared for those domains immediately located at the N‐ and C‐terminus of the cutinase domain. No synteny is suggestive of genomic rearrangements during speciation in order to colonize different niches (Bhadauria, Vijayan, Wei, & Banniza, 2017). Among the conserved domains that flanked the cutinase block were signal peptide and chaperone proteins. One explanation for this finding is the production of cutinase is enhanced by signal peptide optimization and chaperone expression (Yao, Su, Li, & Wu, 2019). It is important that the function of the highly conserved region located at the immediate N‐terminus of the cutinase domain found in this study, be characterized because its high interspecific conservation in arrangement strongly suggests a contributory role in cutinase expression and function among Colletotrichum species. Further, these conserved genomic blocks can be alternative targets for cutinase function disruption and possibly disease control.
In this study, it was found that positive selection of low diversity cutinase gene sequence, preservation of amino acid sequence with conservation of protein function and synteny have been identified as evolutionary mechanisms that maintain the cutinase gene in the genomes of Colletotrichum species. This information is important to developing strategies to enhance plant defense and decrease the pathogenic potential of fungal phytopathogens by targeting and disrupting the cutinase genomic block identified in this study for Colletotrichum species (Brunner et al., 2013). This may be particularly important for the management of C. truncatum diseases as there is only one cutinase gene in C. truncatum available for targeted gene disruption.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
AUTHOR CONTRIBUTIONS
RTV: Conducted the experiments, analyzed the data, and wrote the manuscript; SNR: Conceptualized the experiments, analyzed the data, and wrote the manuscript.
Supporting information
ACKNOWLEDGMENTS
Funding for this work was provided by The University of the West Indies, St. Augustine, Campus Research and Publication Fund, Grant #CRP.3.MAR16.12. The authors wish to thank Mr. Stephen Narine and Mr. Sumair Mahabir for field assistance.
APPENDIX 1.
Table A1.
Frequency count with respect to the location of lesions caused by Colletotrichum truncatum on green bell pepper fruit
| Location on the Fruit | Count (yes) | Count (no) |
|---|---|---|
| Shoulder | 29 | 23 |
| Face | 26 | 26 |
| Bottom | 24 | 28 |
Table A2.
Summary of statistics for colony diameter (mm) for each Colletotrichum species on Tween‐20
| Colletotrichum truncatum | Colletotrichum brevisporum | |
|---|---|---|
| Mean | 19.5379 | 11.5925 |
| SD | 6.58016 | 1.85959 |
| SE Mean | 1.34316 | 0.75917 |
| Minimum | 13.22 | 9.21 |
| Maximum | 31.84 | 13.62 |
Table A3.
Analysis of variance (ANOVA) of colony diameter (mm) values for Colletotrichum truncatum and C. brevisporum on Tween‐20
| SS | DF | MS | F | P | |
|---|---|---|---|---|---|
| Between | 303.022 | 1 | 303.022 | 8.375 | 0.007 |
| Within | 1,013.147 | 28 | 36.184 | ||
| Total | 1,316.169 | 29 |
DF, Degrees of freedom; F, F statistic; MS, Mean square; P‐Significance level at p ≤ .05; SS, Sum of squares.
Table A4.
Summary of statistics for colony diameter (mm) for each Colletotrichum species on tributyrin
| C. truncatum | C. brevisporum | |
|---|---|---|
| Mean | 6.1815 | 0.567 |
| SD | 6.05387 | 0.12695 |
| SE Mean | 1.23574 | 0.05183 |
| Minimum | 0 | 0 |
| Maximum | 15.56 | 0.32 |
Table A5.
Summary of statistics for halo size (mm) for each Colletotrichum species on tributyrin
| C. truncatum | C. brevisporum | |
|---|---|---|
| Mean | 8.7121 | 8.8267 |
| SD | 3.0399 | 2.3646 |
| SE mean | 0.6205 | 0.94977 |
| Minimum | 3.42 | 6.32 |
| Maximum | 12.74 | 12.28 |
Table A6.
Analysis of variance (ANOVA) of colony diameter (mm) values for C. truncatum and C. brevisporum on tributyrin
| SS | DF | MS | F | P | |
|---|---|---|---|---|---|
| BETWEEN | 180.063 | 1 | 180.063 | 5.981 | 0.021 |
| WITHIN | 843.017 | 28 | 30.108 | ||
| TOTAL | 1,023.080 | 29 |
Abbreviations: DF, Degrees of freedom; F, F statistic; MS, Mean square; P‐Significance level at p ≤ .05; SS, Sum of squares.
Table A7.
Analysis of variance (ANOVA) of halo size (mm) values for C. truncatum and C. brevisporum
| SS | DF | MS | F | P | |
|---|---|---|---|---|---|
| BETWEEN | 0.063 | 1 | 0.063 | 0.007 | 0.932 |
| WITHIN | 239.430 | 28 | 8.551 | ||
| TOTAL | 239.493 | 29 |
Abbreviations: DF, Degrees of freedom; F, F statistic; MS, Mean square; P‐Significance level at p ≤ .05; SS, Sum of squares.
Villafana RT, Rampersad SN. Diversity, structure, and synteny of the cutinase gene of Colletotrichum species. Ecol Evol. 2020;10:1425–1443. 10.1002/ece3.5998
DATA AVAILABILITY STATEMENT
DNA sequences: Genbank accessions numbers: MG822830‐2, KJ780718, KR029613, MG827234‐5, MG839690‐1, MG870320‐1, MN473062, and MN473063.
REFERENCES
- Agudelo‐Romero, P. , Erban, A. , Rego, C. , Carbonell‐Bejerano, P. , Nascimento, T. , Sousa, L. , … Fortes, A. M. (2015). Transcriptome and metabolome reprogramming in Vitis vinifera cv. Trincadeira berries upon infection with Botrytis cinerea . Journal of Experimental Botany, 66, 1769–1785. 10.1093/jxb/eru517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkan, N. , Friedlander, G. , Ment, D. , Prusky, D. , & Fluhr, R. (2015). Simultaneous transcriptome analysis of Colletotrichum gloeosporioides and tomato fruit pathosystem reveals novel fungal pathogenicity and fruit defense strategies. New Phytologist, 205, 801–815. 10.1111/nph.13087 [DOI] [PubMed] [Google Scholar]
- Altenhoff, A. M. , Levy, J. , Zarowiecki, M. , Tomiczek, B. , Vesztrocy, A. W. , Dalquen, D. A. , … Dessimoz, C. (2019). OMA standalone: O rthologuey inference among public and custom genomes and transcriptomes. Genome Research, 29, 1152–1163. 10.1101/gr.243212.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armenteros, J. J. A. , Tsirigos, K. D. , Sønderby, C. K. , Petersen, T. N. , Winther, O. , Brunak, S. , … Nielsen, H. (2019). SignalP 5.0 improves signal peptide predictions using deep neural networks. Nature Biotechnology, 37, 420. [DOI] [PubMed] [Google Scholar]
- Auyong, A. S. , Ford, R. , & Taylor, P. W. (2015). The role of cutinase and its impact on pathogenicity of Colletotrichum truncatum . Journal of Plant Pathology and Microbiology, 6, 259–270. 10.4172/2157-7471.1000259 [DOI] [Google Scholar]
- Bargel, H. , & Neinhuis, C. (2005). Tomato (Lycopersicon esculentum Mill.) fruit growth and ripening as related to the biomechanical properties of fruit skin and isolated cuticle. Journal of Experimental Botany, 56, 1049–1060. 10.1093/jxb/eri098 [DOI] [PubMed] [Google Scholar]
- Belbahri, L. , Calmin, G. , Mauch, F. , & Andersson, J. O. (2008). Evolution of the cutinase gene family: Evidence for lateral gene transfer of a candidate phytophthora virulence factor. Gene, 408, 1–8. 10.1016/j.gene.2007.10.019 [DOI] [PubMed] [Google Scholar]
- Belge, B. , Llovera, M. , Comabella, E. , Gatius, F. , Guillén, P. , Graell, J. , & Lara, I. (2014). Characterization of cuticle composition after cold storage of “celeste” and “somerset” sweet cherry fruit. Journal of Agricultural and Food Chemistry, 62, 8722–8729. 10.1021/jf502650t [DOI] [PubMed] [Google Scholar]
- Bhadauria, V. , Bett, K. E. , Zhou, T. , Vandenberg, A. , Wei, Y. , & Banniza, S. (2013). Identification of Lens culinaris defense genes responsive to the anthracnose pathogen Colletotrichum truncatum . BMC Genetics, 14, 31 10.1186/1471-2156-14-31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhadauria, V. , Vijayan, P. , Wei, Y. , & Banniza, S. (2017). Transcriptome analysis reveals a complex interplay between resistance and effector genes during the compatible lentil‐Colletotrichum lentis interaction. Scientific Reports, 7, 42338 10.1038/srep42338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco‐Ulate, B. , Morales‐Cruz, A. , Amrine, K. C. , Labavitch, J. M. , Powell, A. L. , & Cantu, D. (2014). Genome‐wide transcriptional profiling of Botrytis cinerea genes targeting plant cell walls during infections of different hosts. Frontiers in Plant Science, 5, 435 10.3389/fpls.2014.00435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco‐Ulate, B. , Vincenti, E. , Powell, A. L. , & Cantu, D. (2013). Tomato transcriptome and mutant analyses suggest a role for plant stress hormones in the interaction between fruit and Botrytis cinerea . Frontiers in Plant Science, 4, 142 10.3389/fpls.2013.00142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnen, A. M. , & Hammerschmidt, R. (1989). Cutinolytic enzymes from Colletotrichum lagenarium . Physiological and Molecular Plant Pathology, 35, 463–474. 10.1016/0885-5765(89)90088-X [DOI] [Google Scholar]
- Bornscheuer, U. T. (2002). Microbial carboxyl esterases: Classification, properties and application in biocatalysis. FEMS Microbiology Reviews, 26, 73–81. 10.1111/j.1574-6976.2002.tb00599.x [DOI] [PubMed] [Google Scholar]
- Brummell, D. A. , Harpster, M. H. , Civello, P. M. , Palys, J. M. , Bennett, A. B. , & Dunsmuir, P. (1999). Modification of expansin protein abundance in tomato fruit alters softening and cell wall polymer metabolism during ripening. The Plant Cell, 11, 2203–2216. 10.1105/tpc.11.11.2203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunner, P. C. , Torriani, S. F. , Croll, D. , Stukenbrock, E. H. , & McDonald, B. A. (2013). Coevolution and life cycle specialization of plant cell wall degrading enzymes in a hemibiotrophic pathogen. Molecular Biology and Evolution, 30, 1337–1347. 10.1093/molbev/mst041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbone, I. , & Kohn, L. M. (1999). A method for designing primer sets for speciation studies in filamentous ascomycetes. Mycologia, 91(3), 553–556. [Google Scholar]
- Chen, L. , Niu, K. , Wu, Y. , Geng, Y. , Mi, Z. , Flynn, D. F. , & He, J. S. (2013). UV radiation is the primary factor driving the variation in leaf phenolics across Chinese grasslands. Ecology and Evolution, 3, 4696–4710. 10.1002/ece3.862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, Z. , Liang, J. , Zhang, C. , & Rodrigues, C. J. (2006). Epicatechin and catechin may prevent coffee berry disease by inhibition of appressorial melanization of Colletotrichum kahawae . Biotechnology Letters, 28, 1637–1640. 10.1007/s10529-006-9135-2 [DOI] [PubMed] [Google Scholar]
- Choi, H. W. , & Klessig, D. F. (2016). DAMPs, MAMPs, and NAMPs in plant innate immunity. BMC Plant Biology, 16, 232 10.1186/s12870-016-0921-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damm, U. , O'Connell, R. J. , Groenewald, J. Z. , & Crous, P. W. (2014). The Colletotrichum destructivum species complex–hemibiotrophic pathogens of forage and field crops. Studies in Mycology, 79, 49–84. 10.1016/j.simyco.2014.09.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damm, U. , Woudenberg, J. H. C. , Cannon, P. F. , & Crous, P. W. (2009). Colletotrichum species with curved conidia from herbaceous hosts. Fungal Diversity, 39, 45. [Google Scholar]
- Dawson, N. L. , Lewis, T. E. , Das, S. , Lees, J. G. , Lee, D. , Ashford, P. , … Sillitoe, I. (2016). CATH: An expanded resource to predict protein function through structure and sequence. Nucleic Acids Research, 45, D289–D295. 10.1093/nar/gkw1098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Silva, D. D. , Ades, P. K. , Crous, P. W. , & Taylor, P. W. J. (2017). Colletotrichum species associated with chili anthracnose in Australia. Plant Pathology, 66, 254–267. 10.1111/ppa.12572 [DOI] [Google Scholar]
- De Silva, D. D. , Crous, P. W. , Ades, P. K. , Hyde, K. D. , & Taylor, P. W. (2017). Life styles of Colletotrichum species and implications for plant biosecurity. Fungal Biology Reviews, 31, 155–168. 10.1016/j.fbr.2017.05.001 [DOI] [Google Scholar]
- De Silva, D. D. , Groenewald, J. Z. , Crous, P. W. , Ades, P. K. , Nasruddin, A. , Mongkolporn, O. , & Taylor, P. W. (2019). Identification, prevalence and pathogenicity of Colletotrichum species causing anthracnose of Capsicum annuum in Asia. IMA Fungus, 10, 1 10.1186/s43008-019-0001-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng, J. , Carbone, I. , & Dean, R. A. (2007). The evolutionary history of cytochrome P450 genes in four filamentous ascomycetes. BMC Evolutionary Biology, 7, 30 10.1186/1471-2148-7-30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diao, Y. Z. , Zhang, C. , Liu, F. , Wang, W. Z. , Liu, L. , Cai, L. , & Liu, X. L. (2017). Colletotrichum species causing anthracnose disease of chili in China. Persoonia: Molecular Phylogeny and Evolution of Fungi, 38, 20 10.3767/003158517X692788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickman, M. B. , Ha, Y. S. , Yang, Z. , Adams, B. , & Huang, C. (2003). A protein kinase from Colletotrichum trifolii is induced by plant cutin and is required for appressorium formation. Molecular Plant‐Microbe Interactions, 16, 411–421. 10.1094/MPMI.2003.16.5.411 [DOI] [PubMed] [Google Scholar]
- Fleischmann, A. , Darsow, M. , Degtyarenko, K. , Fleischmann, W. , Boyce, S. , Axelsen, K. B. , … Apweiler, R. (2004). IntEnz, the integrated relational enzyme database. Nucleic Acids Research, 32, D434–D437. 10.1093/nar/gkh119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis, S. A. , Dewey, F. M. , & Gurr, S. J. (1996). The role of cutinase in germling development and infection by Erysiphe graminisf. sp. hordei . Physiological and Molecular Plant Pathology, 49, 201–211. 10.1006/pmpp.1996.0049 [DOI] [Google Scholar]
- Fu, Y. X. , & Li, W. H. (1993). Statistical tests of neutrality of mutations. Genetics, 133, 693–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert, R. D. , Johnson, A. M. , & Dean, R. A. (1996). Chemical signals responsible for appressorium formation in the rice blast fungus Magnaporthe grisea . Physiological and Molecular Plant Pathology, 48, 335–346. 10.1006/pmpp.1996.0027 [DOI] [Google Scholar]
- Giovannoni, J. (2001). Molecular biology of fruit maturation and ripening. Annual Review of Plant Biology, 52, 725–749. 10.1146/annurev.arplant.52.1.725 [DOI] [PubMed] [Google Scholar]
- Glass, N. L. , & Donaldson, G. C. (1995). Development of primer sets designed for use with the PCR to amplify conserved genes from filamentous ascomycetes. Appl. Environ. Microbiol., 61(4), 1323–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigoriev, I. V. , Cullen, D. , Goodwin, S. B. , Hibbett, D. , Jeffries, T. W. , Kubicek, C. P. , … Baker, S. (2011). Fueling the future with fungal genomics. Mycology, 2, 192–209. 10.1080/21501203.2011.584577 [DOI] [Google Scholar]
- Grigoriev, I. V. , Nikitin, R. , Haridas, S. , Kuo, A. , Ohm, R. , Otillar, R. , … Shabalov, I. (2014). MycoCosm portal: Gearing up for 1000 fungal genomes. Nucleic Acids Research, 42, D699–D704. 10.1093/nar/gkt1183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gui, Y. J. , Zhang, W. Q. , Zhang, D. D. , Zhou, L. , Short, D. P. , Wang, J. et al (2017). A Verticillium dahliae extracellular cutinase modulates plant immune responses. Molecular Plant‐Microbe Interactions, 31, 260–273. 10.1094/MPMI-06-17-0136-R [DOI] [PubMed] [Google Scholar]
- Hall, T. A. (1999). BioEdit: A user‐friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symposium Series, 41, 95–98. [Google Scholar]
- Horbach, R. , Navarro‐Quesada, A. R. , Knogge, W. , & Deising, H. B. (2011). When and how to kill a plant cell: Infection strategies of plant pathogenic fungi. Journal of Plant Physiology, 168, 51–62. 10.1016/j.jplph.2010.06.014 [DOI] [PubMed] [Google Scholar]
- Horton, P. , Park, K. J. , Obayashi, T. , Fujita, N. , Harada, H. , Adams‐Collier, C. J. , & Nakai, K. (2007). WoLF PSORT: Protein localization predictor. Nucleic Acids Research, 35, W585–W587. 10.1093/nar/gkm259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hückelhoven, R. (2007). Cell wall–associated mechanisms of disease resistance and susceptibility. Annual Review of Phytopathology, 45, 101–127. 10.1146/annurev.phyto.45.062806.094325 [DOI] [PubMed] [Google Scholar]
- Katoh, K. , & Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Molecular Biology and Evolution, 30, 772–780. 10.1093/molbev/mst010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauss, H. , Fauth, M. , Merten, A. , & Jeblick, W. (1999). Cucumber hypocotyls respond to cutin monomers via both an inducible and a constitutive H2O2‐generating system. Plant Physiology, 120, 1175–1182. 10.1104/pp.120.4.1175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kersey, P. J. , Allen, J. E. , Armean, I. , Boddu, S. , Bolt, B. J. , Carvalho‐Silva, D. , … Staines, D. M. (2015). Ensembl Genomes 2016: More genomes, more complexity. Nucleic Acids Research, 44, D574–D580. 10.1093/nar/gkv1209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleemann, J. , Rincon‐Rivera, L. J. , Takahara, H. , Neumann, U. , van Themaat, E. V. L. , van der Does, H. C. , … O'Connell, R. J. (2012). Sequential delivery of host‐induced virulence effectors by appressoria and intracellular hyphae of the phytopathogen Colletotrichum higginsianum . PLoS Path, 8, e1002643 10.1371/journal.ppat.1002643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolattukudy, P. E. (1984). Cutinases from fungi and pollen In Borgstrbm B., & Brockman H. L. (Eds.) Lipases (pp. 471–504). Amsterdam, The Netherlands: Elsevier; 10.1016/0076-6879(81)71078-4 [DOI] [Google Scholar]
- Kosma, D. K. , Parsons, E. P. , Isaacson, T. , Lü, S. , Rose, J. K. C. , & Jenks, M. A. (2010). Fruit cuticle lipid composition during development in tomato ripening mutants. Physiologia Plantarum, 107–117. 10.1111/j.1399-3054.2009.01342.x [DOI] [PubMed] [Google Scholar]
- Kumar, S. , Stecher, G. , & Tamura, K. (2016). MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Molecular Biology and Evolution, 33, 1870–1874. 10.1093/molbev/msw054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laluk, K. , & Mengiste, T. (2010). Necrotroph attacks on plants: Wanton destruction or covert extortion? The Arabidopsis Book/American Society of Plant Biologists, 8, 1–24. 10.1199/tab.0136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanka, S. , & Latha, J. N. L. (2015). A short review on various screening methods to isolate potential lipase producers: Lipases‐the present and future enzymes of biotech industry. International Journal of Biology and Chemistry, 9, 207–219. 10.3923/ijbc.2015 [DOI] [Google Scholar]
- Lara, I. , Belge, B. , & Goulao, L. F. (2014). The fruit cuticle as a modulator of postharvest quality. Postharvest Biology and Technology, 87, 103–112. 10.1016/j.postharvbio.2013.08.012 [DOI] [Google Scholar]
- Lara, I. , Belge, B. , & Goulao, L. F. (2015). A focus on the biosynthesis and composition of cuticle in fruits. Journal of Agricultural and Food Chemistry, 63, 4005–4019. 10.1021/acs.jafc.5b00013 [DOI] [PubMed] [Google Scholar]
- Lee, S. J. , & Rose, J. K. (2010). Mediation of the transition from biotrophy to necrotrophy in hemibiotrophic plant pathogens by secreted effector proteins. Plant Signaling and Behavior, 5, 769–772. 10.1021/acs.jafc.5b00013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letunic, I. , & Bork, P. (2017). 20 years of the SMART protein domain annotation resource. Nucleic Acids Research, 46, D493–D496. 10.1093/nar/gkx922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis, T. E. , Sillitoe, I. , Dawson, N. , Lam, S. D. , Clarke, T. , Lee, D. , … Lees, J. (2017). Gene3D: Extensive prediction of globular domains in proteins. Nucleic Acids Research, 46, D435–D439. 10.1093/nar/gkx1069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang, X. , Wang, B. , Dong, Q. , Li, L. , Rollins, J. A. , Zhang, R. , & Sun, G. (2018). Pathogenic adaptations of Colletotrichum fungi revealed by genome wide gene family evolutionary analyses. PLoS ONE, 13, e0196303 10.1371/journal.pone.0196303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Librado, P. , & Rozas, J. (2009). DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics, 25, 1451–1452. 10.1093/bioinformatics/btp187 [DOI] [PubMed] [Google Scholar]
- Liu, D. , Hunt, M. , & Tsai, I. J. (2018). Inferring synteny between genome assemblies: A systematic evaluation. BMC Bioinformatics, 19, 26 10.1186/s12859-018-2026-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, F. , Tang, G. , Zheng, X. , Li, Y. , Sun, X. , Qi, X. , … Gong, G. (2016). Molecular and phenotypic characterization of Colletotrichum species associated with anthracnose disease in peppers from Sichuan Province, China. Scientific Reports, 6, 32761 10.1038/srep32761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, L. , Zhao, D. , Zheng, L. , Hsiang, T. , Wei, Y. , Fu, Y. , & Huang, J. (2013). Identification of virulence genes in the crucifer anthracnose fungus Colletotrichum higginsianum by insertional mutagenesis. Microbial Pathogenesis, 64, 6–17. 10.1016/j.micpath.2013.06.001 [DOI] [PubMed] [Google Scholar]
- Longhi, S. , & Cambillau, C. (1999). Structure‐activity of cutinase, a small lipolytic enzyme. Biochimica Et Biophysica Acta ‐Molecular and Cell Biology of Lipids, 1441, 185–196. 10.1016/S1388-1981(99)00159-6 [DOI] [PubMed] [Google Scholar]
- Lu, L. , Rong, W. , Massart, S. , & Zhang, Z. (2018). Genome‐wide identification and expression analysis of cutinase gene family in Rhizoctonia cerealis and functional study of an active cutinase RcCUT1 in the fungal‐wheat interaction. Frontiers in Microbiology, 9, 1813 10.3389/fmicb.2018.01813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madeira, F. , Park, Y. M. , Lee, J. , Buso, N. , Gur, T. , Madhusoodanan, N. , … Lopez, R. (2019). The EMBL‐EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Research, 47, W636–W641. 10.1093/nar/gkz268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magadum, S. , Banerjee, U. , Murugan, P. , Gangapur, D. , & Ravikesavan, R. (2013). Gene duplication as a major force in evolution. Journal of Genetics, 92, 155–161. 10.1007/s12041-013-0212-8 [DOI] [PubMed] [Google Scholar]
- Martin, L. B. , & Rose, J. K. (2014). There's more than one way to skin a fruit: Formation and functions of fruit cuticles. Journal of Experimental Botany, 65, 4639–4651. 10.1093/jxb/eru301 [DOI] [PubMed] [Google Scholar]
- Mengiste, T. , Van Alfen, N. , Leach, J. , & Lindow, S. (2012). Plant immunity to necrotrophs. Annual Review Phytopathology, 50, 267–294. 10.1146/annurev-phyto-081211-172955 [DOI] [PubMed] [Google Scholar]
- Mills, P. R. , Sreenivasaprasad, S. , & Brown, A. E. (1992). Detection and differentiation of Colletotrichum gloeosporioides isolates using PCR. FEMS Microbiology Letters, 98(1–3), 137–143. [DOI] [PubMed] [Google Scholar]
- Mongkolporn, O. , & Taylor, P. W. J. (2018). Chili anthracnose: Colletotrichum taxonomy and pathogenicity. Plant Pathology, 67, 1255–1263. 10.1111/ppa.12850 [DOI] [Google Scholar]
- Nguyen, N. T. T. , Vincens, P. , Roest Crollius, H. , & Louis, A. (2017). Genomicus 2018: Karyotype evolutionary trees and on‐the‐fly synteny computing. Nucleic Acids Research, 46, D816–D822. 10.1093/nar/gkx1003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nyyssölä, A. (2015). Which properties of cutinases are important for applications? Applied Microbiology and Biotechnology, 99, 4931–4942. 10.1007/s00253-015-6596-z [DOI] [PubMed] [Google Scholar]
- O'Connell, R. J. , Thon, M. R. , Hacquard, S. , Amyotte, S. G. , Kleemann, J. , Torres, M. F. , … Vaillancourt, L. J. (2012). Lifestyle transitions in plant pathogenic Colletotrichum fungi deciphered by genome and transcriptome analyses. Nature Genetics, 44, 1060 10.1038/ng.2372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Donnell, K. , Sutton, D. A. , Rinaldi, M. G. , Magnon, K. C. , Cox, P. A. , Revankar, S. G. , … Robinson, J. S. (2004). Genetic diversity of human pathogenic members of the Fusarium oxysporum complex inferred from multilocus DNA sequence data and amplified fragment length polymorphism analyses: Evidence for the recent dispersion of a geographically widespread clonal lineage and nosocomial origin. Journal of Clinical Microbiology, 42, 5109–5120. 10.1128/JCM.42.11.5109-5120.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prins, T. W. , Tudzynski, P. , von Tiedemann, A. , Tudzynski, B. , Ten Have, A. , Hansen, M. E. , … van Kan, J. A. (2000). Infection strategies of Botrytis cinerea and related necrotrophic pathogens In Kronstad J. W. (Eds.), Fungal pathology (pp. 33–64). Dordrecht, The Netherlands: Springer; 10.1007/978-94-015-9546-9 [DOI] [Google Scholar]
- Prusky, D. (1996). Pathogen quiescence in postharvest diseases. Annual Review of Phytopathology, 34, 413–434. 10.1146/annurev.phyto.34.1.413 [DOI] [PubMed] [Google Scholar]
- Prusky, D. , Alkan, N. , Mengiste, T. , & Fluhr, R. (2013). Quiescent and necrotrophic lifestyle choice during postharvest disease development. Annual Review of Phytopathology, 51, 155–176. 10.1146/annurev-phyto-082712-102349 [DOI] [PubMed] [Google Scholar]
- Ramdial, H. , & Rampersad, S. N. (2015). Characterization of Colletotrichum spp. causing anthracnose of bell pepper (Capsicum annuum L.) in Trinidad. Phytoparasitica, 43, 37–49. 10.1007/s12600-014-0428-z [DOI] [Google Scholar]
- Ramnath, L. , Sithole, B. , & Govinden, R. (2017). Identification of lipolytic enzymes isolated from bacteria indigenous to eucalyptus wood species for application in the pulping industry. Biotechnology Reports, 15, 114–124. 10.1016/j.btre.2017.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranathunge, N. P. , Mongkolporn, O. , Ford, R. , & Taylor, P. W. J. (2012). Colletotrichum truncatum pathosystem on Capsicum spp: Infection, colonization and defence mechanisms. Australasian Plant Pathology, 41, 463–473. 10.1007/s13313-012-0156-0 [DOI] [Google Scholar]
- Ranathunge, N. P. , & Sandani, H. B. P. (2016). Deceptive behaviour of Colletotrichum truncatum: Strategic survival as an asymptomatic endophyte on non‐host species. Journal of Plant Protection Research, 56, 157–162. 10.1515/jppr-2016-0026 [DOI] [Google Scholar]
- Rozas, J. (2009). DNA sequence polymorphism analysis using DnaSP In Posada D. (Ed.), Bioinformatics for DNA sequence analysis (pp. 337–350). Totowa, NJ: Humana Press; 10.1007/978-1-59745-251-9_17 [DOI] [PubMed] [Google Scholar]
- Rudnicki, W. R. , Mroczek, T. , & Cudek, P. (2014). Amino acid properties conserved in molecular evolution. PLoS ONE, 9, e98983 10.1371/journal.pone.0098983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savory, F. , Leonard, G. , & Richards, T. A. (2015). The role of horizontal gene transfer in the evolution of the oomycetes. PLoS Path, 11, e1004805 10.1371/journal.ppat.1004805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweizer, P. , Jeanguenat, A. , Whitacre, D. , Métraux, J. P. , & Mösinge, E. (1996). Induction of resistance in barley against Erysiphe graminisf. sp. hordeiby free cutin monomers. Physiological and Molecular Plant Pathology, 49, 103–120. 10.1006/pmpp.1996.0043 [DOI] [Google Scholar]
- Seymour, G. B. , Østergaard, L. , Chapman, N. H. , Knapp, S. , & Martin, C. (2013). Fruit development and ripening. Annual Review of Plant Biology, 64, 219–241. 10.1146/annurev-arplant-050312-120057 [DOI] [PubMed] [Google Scholar]
- Sharma, G. , Kumar Pinnaka, A. , & Shenoy, B. D. (2014). Infra‐specific diversity of Colletotrichum truncatum associated with chilli anthracnose in India based on microsatellite marker analysis. Archives of Phytopathology and Plant Protection, 47, 2509–2523. 10.1080/03235408.2014.880577 [DOI] [Google Scholar]
- Skamnioti, P. , Furlong, R. F. , & Gurr, S. J. (2008). Evolutionary history of the ancient cutinase family in five filamentous ascomycetes reveals differential gene duplications and losses and in Magnaporthe grisea shows evidence of sub‐and neo‐functionalization. New Phytologist, 180, 711–721. 10.1111/j.1469-8137.2008.02598.x [DOI] [PubMed] [Google Scholar]
- Skamnioti, P. , & Gurr, S. J. (2007). Magnaporthe grisea cutinase 2 mediates appressorium differentiation and host penetration and is required for full virulence. The Plant Cell, 19, 2674–2689. 10.1105/tpc.107.051219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slifkin, M. (2000). Tween 80 opacity test responses of various Candida species. Journal of Clinical Microbiology, 38, 4626–4628. 10.1128/JCM.38.12.4626-4628.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone, J. K. (2001). Necrotroph In: Maloy O. C., & Murray T. D. (Eds.), Encyclopaedia of plant pathology (pp. 676–677). New York, NY: Wiley. [Google Scholar]
- Sweigard, J. A. , Chumley, F. G. , & Valent, B. (1992). Cloning and analysis of CUT1, a cutinase gene from Magnaporthe grisea . Molecular and General Genetics, 232, 174–182. 10.1007/BF00279994 [DOI] [PubMed] [Google Scholar]
- Tiffin, P. , & Moeller, D. A. (2006). Molecular evolution of plant immune system genes. Trends in Genetics, 22, 662–670. 10.1016/j.tig.2006.09.011 [DOI] [PubMed] [Google Scholar]
- Tomulescu, C. , Moscovici, M. , Ghiorghita, A. , Petrescu, M. , Vladu, M. , Tamaian, R. , & Vamanu, A. (2015). Microbial screening for lipase and amylase production using newly isolated strains from various biotopes. Scientific Bulletin. Series F. Biotechnologies, 19, 271–278. [Google Scholar]
- Torres-Calzada, C. , Tapia-Tussell, R. , Quijano-Ramayo, A. , Martin-Mex, R. , Rojas-Herrera, R. , Higuera- Ciapara, I. , & Perez-Brito, D. (2011). A Species-Specific Polymerase Chain Reaction Assay for Rapid and Sensitive Detection of Colletotrichumcapsici. Molecular biotechnology, 49(1), 48–55. [DOI] [PubMed] [Google Scholar]
- Udayanga, D. , Manamgoda, D. S. , Liu, X. , Chukeatirote, E. , & Hyde, K. D. (2013). What are the common anthracnose pathogens of tropical fruits? Fungal Diversity, 61, 165–179. 10.1007/s13225-013-0257-2 [DOI] [Google Scholar]
- van Poucke, K. , Monbaliu, S. , Munaut, F. , Heungens, K. , De Saeger, S. , & van Hove, F. (2012). Genetic diversity and mycotoxin production of Fusarium lactis species complex isolates from sweet pepper. International Journal of Food Microbiology, 153, 28–37. 10.1016/j.ijfoodmicro.2011.10.011 [DOI] [PubMed] [Google Scholar]
- Villafana, R. T. , Ramdass, A. C. , & Rampersad, S. N. (2019). Selection of Fusarium trichothecene toxin genes for molecular detection depends on TRI gene cluster organization and gene function. Toxins, 11, 36 10.3390/toxins11010036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voisin, D. , Nawrath, C. , Kurdyukov, S. , Franke, R. B. , Reina‐Pinto, J. J. , Efremova, N. , … Yephremov, A. (2009). Dissection of the complex phenotype in cuticular mutants of Arabidopsis reveals a role of SERRATE as a mediator. PLoS Genetics, 5, e1000703 10.1371/journal.pgen.1000703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wadia, T. , & Jain, S. K. (2017). Isolation, screening and identification of lipase producing fungi from oil contaminated soil of Shani Mandir Ujjain. International Journal of Current Microbiology and Applied Sciences, 6, 1872–1878. 10.20546/ijcmas.2017.607.223 [DOI] [Google Scholar]
- Wang, X. , Guan, Y. , Zhang, D. , Dong, X. , Tian, L. , & Qu, L. Q. (2017). A β‐ketoacyl‐CoA synthase is involved in rice leaf cuticular wax synthesis and requires a CER2‐LIKE protein as a cofactor. Plant Physiology, 173, 944–955. 10.1104/pp.16.01527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White, T. J. , Bruns, T. , Lee, S. J. W. T. , & Taylor, J. (1990). Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. PCR Protocols: A Guide to Methods and Applications, 18, 315–322. 10.1016/b978-0-12-372180-8.50042-1 [DOI] [Google Scholar]
- Woloshuk, C. P. , & Kolattukudy, P. E. (1986). Mechanism by which contact with plant cuticle triggers cutinase gene expression in the spores of Fusarium solani f. sp. pisi . Proceedings of the National Academy of Sciences, 83(6), 1704–1708. 10.1073/pnas.83.6.1704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao, D. , Su, L. , Li, N. , & Wu, J. (2019). Enhanced extracellular expression of Bacillus stearothermophilus α‐amylase in Bacillus subtilis through signal peptide optimization, chaperone overexpression and α‐amylase mutant selection. Microbial Cell Factories, 18, 69 10.1186/s12934-019-1119-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung, E. C. , & Chan, C. K. (2015). The glycol methacrylate embedding resins—technovit 7100 and 8100 In Yeung E. C. T., Stasolla C., Sumner M. J., & Huang B. Q. (Eds.), Plant microtechniques and protocols (pp. 67–82). New York, NY: Springer; 10.1007/978-3-319-19944-3_4 [DOI] [Google Scholar]
- Zhu, W. , & Freeland, S. (2006). The standard genetic code enhances adaptive evolution of proteins. Journal of Theoretical Biology, 239, 63–70. 10.1016/j.jtbi.2005.07.012 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
DNA sequences: Genbank accessions numbers: MG822830‐2, KJ780718, KR029613, MG827234‐5, MG839690‐1, MG870320‐1, MN473062, and MN473063.