

Abstract
PlsX is the first enzyme in the pathway that produces phosphatidic acid in Gram-positive bacteria. It makes acylphosphate from acyl-acyl carrier protein (acyl-ACP) and is also involved in coordinating phospholipid and fatty acid biosyntheses. PlsX is a peripheral membrane enzyme in Bacillus subtilis, but how it associates with the membrane remains largely unknown. In the present study, using fluorescence microscopy, liposome sedimentation, differential scanning calorimetry, and acyltransferase assays, we determined that PlsX binds directly to lipid bilayers and identified its membrane anchoring moiety, consisting of a hydrophobic loop located at the tip of two amphipathic dimerization helices. To establish the role of the membrane association of PlsX in acylphosphate synthesis and in the flux through the phosphatidic acid pathway, we then created mutations and gene fusions that prevent PlsX's interaction with the membrane. Interestingly, phospholipid synthesis was severely hampered in cells in which PlsX was detached from the membrane, and results from metabolic labeling indicated that these cells accumulated free fatty acids. Because the same mutations did not affect PlsX transacylase activity, we conclude that membrane association is required for the proper delivery of PlsX's product to PlsY, the next enzyme in the phosphatidic acid pathway. We conclude that PlsX plays a dual role in phospholipid synthesis, acting both as a catalyst and as a chaperone protein that mediates substrate channeling into the pathway.
Keywords: glycerophospholipid, Gram-positive bacteria, acyltransferase, membrane biogenesis, lipid binding protein, acylphosphate, PlsX, substrate channeling
Introduction
Most bacteria synthesize phosphatidic acid (PA),5 the central precursor of membrane phospholipids, using an unusual acylphosphate intermediate in a three-step pathway mediated by the PlsX, PlsY, and PlsC enzymes (1, 2) (Fig. 1A). PlsX is an acyl-ACP:phosphate transacylase that converts acyl-ACP made by FASII into acylphosphate (acyl-PO4). Next, PlsY, a glycerol 3-phosphate (G3P) acyltransferase (GPAT), which uses exclusively acyl-PO4, transfers the acyl group of acyl-PO4 to position 1 of G3P to produce lysophosphatidic acid (LPA). The last step of the pathway is catalyzed by PlsC, an LPA acyltransferase (AGPAT), which uses acyl-ACP to convert LPA into PA (1, 3). PlsX and PlsY are found exclusively in bacteria and are essential in important human pathogens (1), therefore, these enzymes are considered promising targets for new antibacterial molecules.
Figure 1.

Phospholipid synthesis in bacteria and the structure of PlsX. A, phospholipid synthesis pathway in Gram-positive bacteria. B, diagram of the B. subtilis PlsX protein showing the position of the putative AH (232–249) and the hydrophobic exposed region (254–263) and lysine-rich region (264–276). C, right, structural model of B. subtilis PlsX dimer performed by Swiss Model homology-modeling server (31) (using PDB 1VI1 as a template), indicating the localization of the hydrophobic loop and the lysine-rich helix α10. Left, zoomed-in view of the α9 and α10 helices in a PlsX monomer.
Even though the enzymology of PA synthesis is well-understood, much less is known about other aspects of this pathway. One important but often unappreciated challenge of PA synthesis is the topology of the process. The amphipathic and segregated nature of the membrane bilayer makes the chemistry of its synthesis less straightforward than the metabolic reactions involving soluble molecules. Membranes grow by the in situ synthesis of new phospholipid molecules from soluble precursors (G3P, acyl-ACP) that are made in the cytoplasm (4). How these precursors get channeled into the membrane and how acyltransferases manage to access simultaneously substrates that are embedded within the bilayer and substrates that come from the cytoplasm was poorly understood until the recent determination of the crystal structures of PlsY (5) and PlsC (6). PlsY is a typical integral membrane protein with seven transmembrane segments delimiting a V-shaped active site that extends deeply into the bilayer. Access of its substrate acyl-PO4 to the active site seems to occur from within the membrane, through a putative lateral gate between transmembrane helices 2 and 5, whereas G3P enters the active site via its cytoplasmic opening (5). The crystal structure of PlsC, on the other hand, showed that the protein lacks transmembrane segments and is instead anchored to the cytoplasmic leaflet of the bilayer via a novel motif comprised of two α helices that lay parallel to the membrane (6). This mode of anchoring places the active site of PlsC above the bilayer in a way that it can simultaneously access LPA residing within the membrane and acyl-ACP from the cytoplasm (6). Despite these advances, topological information is still not available for the first reaction in the pathway, acyl-PO4 formation by PlsX.
Based on sequence analysis and fractionation, PlsX was originally thought to be a soluble enzyme but subsequent work showed that it is a peripheral membrane protein (3, 7, 8). One of these publications reported that PlsX localization on membranes followed that of the bacterial cell division apparatus but our group has challenged this conclusion by showing that PlsX is evenly distributed on the membrane of proliferating cells, and that its localization was independent of cell division proteins (8). We also showed that PlsX association with the membrane was independent of PlsY, and that both inhibition of phospholipid synthesis and changes in membrane potential perturbed PlsX localization (8). These data suggested that PlsX associates directly with the phospholipid bilayer, but this possibility has not been tested experimentally.
Currently, two crystal structures of PlsX are available, those from Bacillus subtilis (9) and from the human pathogen Enterococcus faecalis (10). In both structures PlsX forms an S-shaped dimer. Dimerization is mainly mediated by hydrophobic contacts between a long amphipathic helix (α9) and α10 in each monomer, which together form a tip that protrudes away from the core of the enzyme. This dimeric structure is further stabilized by additional hydrophobic interactions with the enzyme core and several intrasubunit salt bridges on the protein surface. The core domain of the enzyme comprises a long β-strand surrounded by α-helices, generating a deep groove where the active site is located (10). Both structures suggest that this tip might participate into the association of PlsX with the membrane, however, the membrane-binding mechanism and how membrane association allows PlsX to perform its transacylase reaction are largely unknown.
In this study, we report that PlsX binds directly to lipid bilayers and identified a loop rich in hydrophobic residues localized at the tip of PlsX, which plays a critical role in mediating protein-membrane interaction. We then exploited mutations and gene fusions in PlsX to assess the role of membrane association in acyl-PO4 production and flux through the PA pathway. Mutations that severely affect the association of PlsX to the membrane do not eliminate its transacylase enzymatic activity. Strikingly, PA synthesis is severely hampered in these cells, probably because membrane association is required for the proper delivery of PlsX's product, acyl-PO4, to PlsY, its partner protein. Thus, PlsX plays a dual role in PA synthesis, acting both as a catalyst and as a chaperone protein that mediates substrate channeling into the pathway.
Results
Identification of the membrane-interacting domain of PlsX
To determine how PlsX associates with membranes, we analyzed its structure (9, 10) and amino acid sequence searching for typical membrane-interacting domains. PlsX lacks terminal amphipathic helices like those mediating the association of peripheral proteins such as MinD and FtsA with the membrane (11, 12), but we found a hydrophobic loop (containing 70% hydrophobic residues, H = 0.648 and GRAVY value of 1.56, over 10 amino acids), which is solvent-exposed and next to a stretch of cationic amino acids (lysine residues) (Figs. 1B and 2A). This loop, which connects helices α9 and α10 and is disordered in the crystal structure, could play a role in targeting PlsX to the membrane (Fig. 1C). To test this possibility, we replaced some of the hydrophobic residues in the loop to glutamates and some of the basic amino acids in the adjacent α10 helix to alanine residues and investigated how these mutations affected PlsX's ability to interact with phospholipids in vitro. We expressed and purified each mutant with a C-terminal His6 tag and assayed their association with liposomes made with B. subtilis lipids. As shown in Fig. 2B, PlsX-His6 sedimented with liposomes but when the same experiment was repeated with mutants in the hydrophobic loop (PlsXL254E, PlsXL258E-A259E, and PlsXV262E), little or no protein was detected in the pellet, indicating the importance of these residues for lipid binding. In contrast, PlsX mutants with substitutions in positive residues (PlsXK264A and PlsXK271A) and in tyrosine 276 (PlsXY276E) of the α10 helix sedimented with liposomes (Fig. 2B).
Figure 2.

Mutations in the hydrophobic loop perturb PlsX membrane interaction. A, mutated amino acid residues of the hydrophobic loop and helix α10 of PlsX highlighted on the structure. B, cosedimentation assay showing the interaction of WT PlsX and different mutants with liposomes prepared with B. subtilis total lipid extracts. Data represent the mean ± S.D. from two independent experiments. C, effects of PlsX and selected mutants on the thermotropic behavior of lipid bilayer membranes probed by DSC. Left, representative DSC traces of DMPG LUVs in the absence and presence of different concentrations of PlsX. Right, excess heat capacity of DMPG in the absence (black) and presence of PlsXL254E, PlsXL258E/A259E, PlsXK264A, and PlsXK271A mutants at lipid-to-protein (L/P) molar ratio of 136:1. Lipid concentration was 800 μm in all cases. D, fluorescence microscopy images showing the subcellular localization of GFP-PlsX wt (strain DS69; Pxyl-plsX amyE::Physpank-gfp-plsX) and PlsXL254E (strain DS160; Pxyl-plsX amyE::Physpank-gfp-plsXL254E) and PlsXK271A (DS161; (Pxyl-plsX amyE::Physpank-gfp-plsXK271A); site-directed mutant. Images are representative of at least 100 cells analyzed for each condition in at least two different days. All images are at the same magnification. Scale bar = 5 μm.
We also investigated the interaction between PlsX and membranes by differential scanning calorimetry (DSC). Fig. 2C shows the heat capacity profiles of 1,2-dimiristoyl-sn-glycero-3-phospho-(1′-rac glycerol) (DMPG) large unilamellar vesicles (LUVs) mixed with PlsX at different lipid-to-protein (L/P) molar ratios. Binding of PlsX to DMPG LUVs decreased the enthalpy change (ΔH) associated with the lipid phase transition (Table 1), and made this transition broader and less cooperative without changing the transition melting temperature (Tm). DSC was also employed to examine the role of head group charge in the PlsX-membrane interaction. As illustrated in Fig. S1, WT PlsX binds to LUVs made with another anionic phospholipid (phosphatidylserine, 1,2-dimyristoyl-sn-glycero-3-phospho-l-serine (DMPS)), but does not bind to LUVs made with a zwitterionic lipid (phosphatidylcholine, 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC)). This result highlights the importance of electrostatic interactions for PlsX-membrane association.
Table 1.
Thermodynamic parameters associated to the DMPG phase transition
The phase transition temperature (Tm), the calorimetric enthalpy change (ΔH), and the linewidth at half height (ΔT½) were obtained from analysis of the thermograms with MicroCal Origin software. Lipid-to-protein molar ratios (L/P) are indicated. Data shown are representative of two independent experiments.
| Sample | L/P | Tm °C | ΔH kcal/mol | ΔT½ °C |
|---|---|---|---|---|
| DMPG | ||||
| Blank | 23.3 (2)a | 5.0 (2) | 0.7 (2) | |
| + PlsX wt | 500 | 22.9 (1) | 5.0 (1) | 0.5 (1) |
| + PlsX wt | 400 | 22.8 (1) | 4.5 (1) | 0.6 (1) |
| + PlsX wt | 320 | 23.7 (2) | 4.6 (2) | 2.9 (1) |
| + PlsX wt | 250 | 23.6 (3) | 4.2 (2) | 4.4 (2) |
| + PlsX wt | 136 | 23.6 (3) | 2.9 (3) | 5.1 (2) |
| + PlsX L254E | 136 | 22.8 (1) | 4.5 (1) | 0.6 (1) |
| + PlsX L258E-A259E | 136 | 23.7 (2) | 4.6 (2) | 2.9 (1) |
| + PlsX K264A | 136 | 23.6 (3) | 4.2 (2) | 4.4 (2) |
| + PlsX K271A | 136 | 23.6 (3) | 2.9 (3) | 5.1 (2) |
a The numbers in parentheses correspond to the uncertainties in the last significant digit.
We next probed our mutants with DSC and observed behaviors that were in complete agreement with the sedimentation experiments: both lysine residue mutants (PlsXK264A and PlsXK271A) perturbed phase transition like the WT protein (Fig. 2C and Table 1), whereas the double mutant PlsXL258E-A259E showed a partial effect compatible with its residual sedimentation with liposomes. Interestingly, the PlsXL254E mutant had no effect on phase transition, confirming that this mutant has a severely compromised interaction with membranes (Fig. 2C).
To confirm that the hydrophobic loop identified in vitro was indeed necessary for membrane localization of PlsX in vivo we tagged the PlsXL254E and PlsXK271A mutant proteins with GFP and determined their subcellular localization by fluorescence microscopy. We used a strategy in which the GFP-tagged mutants were integrated at the nonessential amyE locus and expressed from an IPTG-responsive promoter (Physpank) in a strain where endogenous PlsX could be depleted by growth in the absence of xylose (DS28 background). Thus, the localization of mutant GFP-PlsX proteins was evaluated without the interference of endogenous PlsX, which could potentially hetero-oligomerize with the mutant proteins. As shown in Fig. 2D, the mutant proteins localized as expected from their ability to bind to lipids in vitro: GFP-PlsXK271A was found in the membrane, like WT GFP-PlsX (8), whereas GFP-PlsXL254E exhibited a cytosolic localization. Altogether, our data indicate that the hydrophobic loop between helices α9 and α10 plays a prominent role in the association and localization of PlsX to the membrane, both in vitro and in vivo.
Dimerization of PlsX is important for membrane interaction
The crystal structures of B. subtilis and E. faecalis PlsX showed that the protein is a dimer maintained mostly by interactions involving helices α9 and α10, which flank the hydrophobic loop responsible for membrane insertion (9, 10) (Fig. 1C). Helix α9, in particular, is a long amphipathic helix (AH) whose hydrophobic side makes many of the contacts that stabilize the PlsX dimer. To test the role of dimerization for membrane targeting we constructed three mutants: (i) PlsXL235E in which a conserved Leu in the middle of α9 was mutated to a negatively charged Glu residue; (ii) PlsXAH(+2) in which two residues (Ala-Glu) were inserted between Ile242 and Phe243 to maintain the helicity but destroy the polar-apolar residue phasing of α9; and (iii) PlsXAH(+3) in which the insertion of three residues (Glu-Glu-Leu) between Ile242 and Phe243 should maintain both the helicity and the amphipathic phasing of α9 (Fig. 3A). The three mutants were expressed and purified and their oligomerization state was analyzed by gel filtration alongside the WT protein (Fig. 3B). As expected from the crystal structures and previously shown for Streptococcus pneumoniae PlsX (1), WT PlsX and PlsXL254E eluted from the S-200 column with the expected size of a dimer (molecular mass of ∼80 kDa, Fig. 3B, Fig. S2). Significant changes in the elution pattern were observed for mutants PlsXAH(+2) and PlsXL235E, which displayed retention times corresponding to the monomer (∼40 kDa), in addition to large aggregates (Fig. 3B), which probably arise because the mutant proteins can no longer dimerize. In contrast, mutant PlsXAH(+3), which bears a larger insertion than PlsXAH(+2) but whose amphipathicity is maintained, behaved as a mixture of both monomeric and dimeric species (Fig. 3B).
Figure 3.

The amphipathic helix α9 is essential for the dimerization and proper localization of PlsX. A, helical wheel representation of the H9 amphipathic helix located at amino acids 232–249 of PlsX. Hydrophobic residues are marked in yellow. Mutation in one of the leucines (L235E), and insertion of two (AH(+2)) and three residues (AH(+3)) mutants are represented. Diagrams generated using the web server HELIQUEST (32) B, gel filtration chromatography assays. Purified PlsX wt (red) was a dimeric protein, whereas PlsXAH(+2) (blue) and PlsXL235E (black) presented monomeric and aggregated states. PlsXAH(+3) (green) present both dimeric and monomeric states. Data shown are representative of two independent experiments. C, fluorescence microscopy showing subcellular localization of GFP-tagged PlsX wt (strain DS69; Pxyl-plsX amyE::Physpank-gfp-plsX), PlsXAH(+2) (strain DS34; Pxyl-plsX amyE::Physpank-gfp-plsXAH(+2)), PlsXAH(+3) (strain DS48; Pxyl-plsX amyE::Physpank-gfp-plsXAH(+2)), and PlsXL235E (strain DS58; Pxyl-plsX amyE::Physpank-gfp-plsXL235E) mutants. Images are representative of at least 100 cells analyzed for each condition in at least two different days. All images are at the same magnification. Scale bar = 5 μm.
To examine whether dimerization of PlsX is relevant for membrane association, the above mutants were fused to GFP and their cellular distribution was examined, employing the same strategy used to study the localization of the hydrophobic loop mutants (expression of mutant GFP-PlsX fusions from an IPTG-inducible promoter in the absence of endogenous PlsX). As shown in Fig. 3C, the two mutants that no longer dimerized, PlsXL235E and PlsXAH(+2), had severely compromised membrane localization, as inferred by their cytoplasmic staining. In contrast, mutant PlsXAH(+3), which is partially a dimer in vitro (Fig. 3B), exhibited a mixed localization pattern, with both membrane and cytoplasmic staining (Fig. 3C). The importance of dimerization for membrane association was corroborated by in vitro experiments in which we observed that monomeric PlsXL235E no longer co-sedimented with liposomes, whereas the partially dimeric PlsXAH(+3) still sedimented, albeit to a lesser extent than the WT protein (Fig. S3).
Membrane association is necessary for PlsX function in vivo
The availability of mutants with severe defects in membrane association provided an opportunity to investigate if PlsX's membrane localization was necessary for its functioning in vivo. The function of PlsX mutants was monitored by their ability to support growth in the absence of WT PlsX. We created merodiploid strains with mutant PlsX alleles controlled by IPTG in our PlsX depletion strain background and assayed growth by spotting dilutions on plates containing IPTG but no xylose. As shown in Fig. 4A, there was a clear correlation between membrane association and the ability to support growth: mutants that were affected in membrane association because of alterations in the hydrophobic loop (PlsXL254E) or because of impaired dimerization (PlsXL235E, PlsXAH(+2)) supported little or no growth in the absence of endogenous PlsX, mutant PlsXAH(+3), which exhibited less affected membrane localization, promoted intermediate levels of growth, and mutant PlsXK271A, which associates with the membrane, was able to fully support growth (Fig. 4A).
Figure 4.

Proper membrane association is required for PlsX function in vivo but not for catalysis in vitro. A, growth complementation assays. Cells of B. subtilis DS28 (Pxyl-plsX), DS69 (Pxyl-plsX amyE::Physpank-gfp-plsX), DS160 (Pxyl-plsX amyE::Physpank-gfp-plsXL254E), DS34 (Pxyl-plsX amyE::Physpank-gfp-plsXAH(+2)), DS48 (Pxyl-plsX amyE::Physpank-gfp-plsXAH(+3)), and DS58 (Pxyl-plsX amyE::Physpank-gfp-plsXL235E) were grown in liquid LB to logarithmic phase, serially diluted and spotted (4 μl) onto LB + 0.5 mm IPTG plates, with or without 0.4% xylose, and incubated at 37 °C for 24 h. B, in vitro acyltransferase activity assay of PlsX wt and mutants. The assay detects the synthesis of acyl-ACP from 16:0-PO4 and holo-ACP by the appearance of a slow migrating band in conformationally sensitive gel electrophoresis (15% PAGE, 2 m urea). Data shown are representative of three independent experiments. Note that the PlsX AH (2) sample was run in a different gel and thus is depicted separate from the other samples.
An important control to determine the role of PlsX localization in vivo was to rule out that the mutations affecting membrane association may have also affected catalysis by the enzyme. To study catalysis, we developed an in vitro acyltransferase assay measuring the reverse of the reaction normally executed by PlsX. We employed palmitoyl-PO4 and holo-ACP as substrates and monitored the appearance of palmitoyl-ACP by conformation-sensitive gel electrophoresis. As shown in Fig. 4B, mutations that impaired dimerization (PlsXL235E, PlsXAH(+2)) abolished the production of palmitoyl-ACP by PlsX in vitro. This suggests that catalysis requires dimerization, a conclusion supported also by the fact that a mutation that rescues dimerization (PlsXAH(+3)) is catalytically active, even though it introduces a larger insertion in helix α9 than one that is inactive (PlsXAH(+2)). Importantly, however, a mutation that compromises membrane association without affecting dimerization (PlsXL254E) did not affect catalysis in vitro (Fig. 4B). Therefore, the PlsXL254E mutation demonstrates that membrane binding and catalysis can be uncoupled in PlsX and we further characterized this mutation to understand why membrane localization is necessary for PlsX function in vivo.
Removal of PlsX from the membrane blocks phospholipid synthesis and leads to free fatty acid accumulation
The growth impairment of cells expressing PlsXL254E was an indication that the phospholipid synthesis pathway becomes blocked when PlsX cannot appropriately associate with the membrane. To test this possibility, we investigated if PlsXL254E growth could be rescued by exogenous fatty acids, as was shown for a plsX null mutant (13). In Gram-positive bacteria such as Bacillus and Staphylococcus, exogenous fatty acids can bypass the need for PlsX because they are phosphorylated directly by fatty acid kinase (Fig. 1A). Indeed, we found that supplementation of a strain expressing PlsXL254E with a mixture of branched-chain fatty acids rescued its growth as well as it rescued growth of a plsX null mutant (Fig. 5A), a strong indication that the growth defect of the PlsXL254E mutant was due to inhibition of phospholipid synthesis.
Figure 5.

Removal of PlsX from the membrane blocks phospholipid synthesis. A, rescue of cells expressing PlsXL254E by supplementation with B. subtilis fatty acids. Strains DS28 (Pxyl-plsX), DS69 (Pxyl-plsX amyE::Physpank-gfp-plsX), and DS160 (Pxyl-plsX amyE::Physpank-gfp-plsXL254), were grown in LB medium containing 0.5 mm IPTG and 1 mg/ml of BSA, and in the presence or absence of 0.4% xylose and/or fatty acid methyl esters (FAMEs) (2 mg/ml). −FAMEs, black lines/squares; + FAMEs, red lines/circles. Data are representative of at least two independent experiments. B, lipid synthesis in cells expressing PlsX mutants. Strains were grown in LB medium in the presence of 1 mm IPTG to induce the expression of the different PlsX variants. The cultures were supplemented with xylose (0.4%) to induce the expression of WT PlsX (PlsXwt) or PlsY (in strain VD211 Pxyl-plsY). Strains DS28, DS160, DS171, and VD211 were labeled with 2 μCi/ml of [14C]acetate for 30 min during the transition from log to stationary phase. Strains DS69 and DS170 grown to midexponential phase were labeled with 2 μCi/ml of [14C]acetate for 30 min. Cells were harvested and total lipids were quantified in a scintillation counter. Bars represent the mean ± S.D. from three independent experiments. C, fatty acids accumulate in strains expressing PlsX that does not associate with the membrane. Total lipids of cells labeled with[14C]acetate as described in panel B were analyzed by TLC on preadsorbent Silica Gel G layers (Merck) developed with hexane-ethyl ether-acetic acid (60/40/1, v/v/v). Each lane was loaded with about 1,000 cpm of labeled lipids. The radioactive lipid species were identified by their co-migration with standards. FFA, free fatty acid; DAGs, diacylglycerols; PL, phospholipid; Unk, unknown. The example shown is typical of three independent experiments.
We envisaged two possible scenarios to explain why phospholipid synthesis was compromised in cells expressing PlsXL254E: (i) the mutant protein is not catalytically active in vivo, or (ii) it is catalytically active but membrane association of PlsX is necessary for efficient delivery of acyl-PO4 to PlsY to proceed with phospholipid synthesis. We reasoned that the two possibilities could be distinguished by comparing the de novo lipid synthesis profile of a PlsXL254E mutant with those of plsX− and plsY− mutants. If PlsXL254E is inactive in vivo it should exhibit a plsX− phenotype, but if the block in the pathway is at the level of product delivery, PlsXL254E should exhibit a plsY− phenotype. As reported previously by our group, a plsX− strain has a strong block in de novo lipid synthesis, presumably because the accumulation of acyl-ACP shuts down FASII (3). In contrast, de novo lipid synthesis is only partially inhibited in a plsY− strain, but this mutant accumulates free fatty acids, likely derived from the acyl-PO4 produced by PlsX and which cannot be used further in the absence of PlsY (3). Therefore, we used [14C]acetate labeling to analyze lipid synthesis in a strain expressing GFP-PlsXL254E, and compared it with a strain expressing a WT GFP-PlsX fusion, a conditional plsX mutant (plsX−) and a conditional plsY mutant (plsY−). As show in Fig. 5B, [14C]acetate incorporation in a strain expressing GFP-PlsXL254E was partially inhibited, reaching ∼30% of the WT levels, a value much higher than the incorporation observed in the conditional plsX mutant (7.0%) but similar to that observed in a conditional plsY mutant (Fig. 5B) (3). The inhibition of the GFP-PlsXL254E mutant was not due to a negative effect of the GFP fusion because lipid synthesis was normal in a strain expressing a WT GFP-PlsX fusion (Fig. 5B). In addition, TLC analysis of the newly synthesized lipids showed that the GFP-PlsXL254E mutant accumulated free fatty acids to a level comparable with the conditional plsY mutant (Fig. 5C). Thus, collectively, these data are most consistent with the hypothesis that GFP-PlsXL254E is catalytically active in vivo and that the main defect of this mutant protein is in channeling its product into the next step of the pathway.
Mistargeting as a way to test the role of membrane localization in PlsX function
Another way to test if membrane localization is necessary for PlsX function is to artificially target the protein to a different subcellular compartment without mutating the protein's amino acid sequence. We attempted to do that by fusing PlsX to HBsu, a histone-like protein that binds to the bacterial nucleoid with great affinity, such that normally all HBsu in the cell is nucleoid-associated (14). The high affinity of HBsu for the nucleoid and the fact that this structure is spatially segregated from the membrane suggested that fusing PlsX to HBsu might suffice to “peel” it from the membrane (Fig. 6A). However, when we imaged a strain expressing a triple GFP-PlsX-HBsu fusion we observed that the protein localized as foci or patches that overlapped with the nucleoid but that also reached the membrane, indicating that the fused protein manages to bind simultaneously to both structures (Fig. 6, B and C). In retrospect, this observation is not unexpected because it is known that DNA loops are frequently found in close proximity to the bacterial membrane (15, 16).
Figure 6.

Subcellular localization of PlsX and PlsXL254E fused to GFP and HBsu. A, schematic representation of the triple fusion GFP-PlsX-HBsu, designed as an attempt to remove PlsX from the cytoplasmic membrane, due to the interaction of HBsu histone-like protein with the nucleoid. B, fluorescence microscopy showing localization of GFP-PlsX wt and GFP-PlsXL254E fused and unfused to Hbsu in B. subtilis strains DS69 (Pxyl-plsX amyE::Physpank-gfp-plsX), DS160 (Pxyl-plsX amyE::Physpank-gfp-plsXL254), DS170 (Pxyl-plsX amyE::Physpank-gfp-plsX-hbsu), and DS171 (Pxyl-plsX amyE::Physpank-gfp-plsX L254E-hbsu). C, fluorescence microscopy showing subcellular localization of GFP-PlsX-HBsu fusions in B. subtilis DS170 (Pxyl-plsX amyE::Physpank-gfp-plsX-hbsu) and DS171 (Pxyl-plsX amyE::Physpank-gfp-plsX L254E-hbsu) using FM4-64 and DAPI to stain membrane and nucleoid, respectively. Images are representative of at least 100 cells analyzed for each condition in at least two different days. All images are at the same magnification. Scale bar = 5 μm. D, growth complementation assays of strains expressing PlsX-HBsu fusions. Cells of DS28 (Pxyl-plsX), DS69 (Pxyl-plsX amyE::Physpank-gfp-plsX), DS160 (Pxyl-plsX amyE::Physpank-gfp-plsXL254), DS170 (Pxyl-plsX amyE::Physpank-gfp-plsX-hbsu), and DS171 (Pxyl-plsX amyE::Physpank-gfp-plsXL254E-hbsu) were grown in liquid LB to logarithmic phase, serially diluted and spotted (4 μl) onto LB + 0.5 mm IPTG plates, with or without 0.4% xylose, and incubated at 37 °C for 24 h. Data shown are representative of at least three independent experiments.
Although the strategy of fusing PlsX to HBsu did not work to remove the WT protein from the membrane, we reasoned it could still be employed to further evaluate the importance of membrane association for PlsX function. Even though the PlsXL254E mutant displays a strongly reduced interaction with the membrane, it still supports residual growth when expressed in place of endogenous PlsX (Fig. 4A, compare spots of PlsXL254E and the parental depletion strain). Furthermore, a detailed biophysical analysis of the PlsXL254E mutant showed that it still retains the ability to bind superficially to membranes, probably via electrostatic interactions (33). Therefore, it is certainly possible that the cytoplasmic molecules of PlsXL254E will exhibit transient or superficial contact with the membrane sufficient to allow some transfer of acyl-PO4 to the bilayer and maintain a low level of phospholipid synthesis. Alternatively, the residual growth may result from acyl-PO4 produced in the cytoplasm diffusing and spontaneously inserting itself in the membrane. We reasoned we could distinguish between these possibilities by fusing PlsXL254E to HBsu to completely restrict access of the protein to the membrane. In fact, as shown in Fig. 6, B and C, fusing PlsXL254E to HBsu led to a dramatic change in its localization, which went from cytoplasmic to being exclusively associated with the nucleoid. We then asked if this protein, which now had no access to the membrane, was capable of supporting growth in the absence of endogenous PlsX. As shown in Fig. 6D, GFP-PlsXL254E-HBsu was slightly worse than GFP-PlsXL254E at complementing the lack of PlsX. The similar phenotypes of GFP-PlsXL254E and GFP-PlsXL254E-HBsu were also evident in the [14C]acetate labeling experiments: both mutants exhibited comparable inhibition of lipid synthesis (Fig. 5B). However, the strain expressing GFP-PlsXL254E-HBsu accumulated 2-fold more free fatty acids (Fig. 5C), a sign that phospholipid synthesis is indeed more impaired when PlsX is completely prevented from accessing the membrane. The worse performance of GFP-PlsXL254E-HBsu cannot be explained as a general negative effect of HBsu, because fusion of WT PlsX to HBsu (GFP-PlsX-HBsu) had little if any impact on growth (Fig. 6D) or lipid synthesis (Fig. 5, B and C). We also carried out a control, which showed that all protein fusions were successfully expressed (Fig. S4). Therefore, these results reinforce our conclusion that PlsX membrane association is crucial for the efficient channeling of acyl-PO4 into the phospholipid synthesis pathway.
Discussion
Peripheral proteins associate with the membrane in a wide variety of ways but two of the most common motifs are amphipathic helices and protruding hydrophobic loops (17). Here we showed that B. subtilis PlsX associates with the membrane via a hydrophobic loop comprising residues 254–263 that projects from the end of its amphipathic dimerization helices. Typical loops have exposed hydrophobic residues at their tips, flanked by cationic residues that interact favorably with the negative head groups of lipids (18, 19). In agreement with our prediction that the loop is the motif that targets PlsX to the membrane, mutations in hydrophobic residues (L254E, L258E/A259E, and V262E) perturb to varying degrees membrane interaction. In contrast, we failed to disrupt membrane binding when single lysine residues (i.e. Lys-264 and Lys-271) of the adjacent α10 helix were mutated. This suggests that the energetic contribution of each individual positive residue for membrane binding is modest, but we cannot rule out the possibility that the α10 helix is not directly involved in membrane binding.
The existence of a hydrophobic loop flanked by positive residues in helix α10 is a feature conserved in all PlsX homologues (Fig. S5). In proteobacteria, this region is slightly longer (15 residues, instead of 10) and contains more aromatic and positive residues, but still has the features expected of a membrane-binding hydrophobic loop. Hence, we expect that all PlsX proteins use similar motifs to associate with the membrane.
While this work was in preparation, Jiang and colleagues (20) described a new PlsX crystal structure in which the hydrophobic loop of one protomer is folded into a short amphipathic α helix, suggesting that the region we have so far described as a hydrophobic loop may in fact change structure to become helical upon membrane interaction. This plausible model was supported by mutations that, similarly to ours, perturbed membrane interaction, but more detailed structural analysis in the presence of membranes will be necessary to ascertain the exact structure of PlsX. Irrespective of this structural detail, both works reinforce each other's conclusion that PlsX employs a stretch of hydrophobic and positively charged amino acids at the tip of its stalk as its membrane binding motif.
Another important observation of our work was that removal of PlsX from the membrane via mutation or the fusion to HBsu resulted in inhibition of phospholipid synthesis. This observation may be explained if membrane association was necessary for PlsX catalysis, but we find this very unlikely for two reasons: (i) PlsX catalysis can be detected in vitro in the absence of lipids or detergents (see Refs. 1 and 20, and this work), and (ii) we detect free fatty acid accumulation in cells expressing PlsX variants that do not localize to the membrane, confirming that the enzyme is catalytically active even when removed from the membrane. Why, then, would PlsX need to associate with the membrane to be fully functional? Being at the membrane interface makes sense for enzymes that act on substrates that are embedded in the bilayer, such as PlsC, which uses LPA, but this is not the case for PlsX whose substrates, acyl-ACP and Pi, are accessible in the cytoplasm. Instead, we propose that PlsX association with the membrane is needed to channel acyl-PO4 efficiently into the next step of the pathway, which is carried out by PlsY, an integral membrane protein that likely access acyl-PO4 only after it has partitioned into the membrane (5) (Fig. 7).
Figure 7.
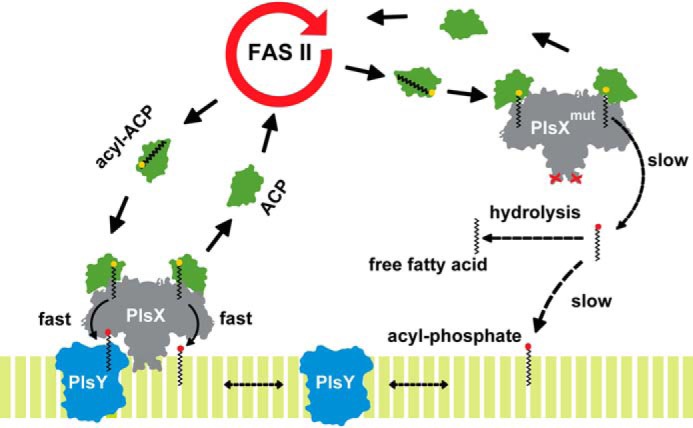
PlsX membrane association is crucial for acyl-PO4 channeling to PlsY. Membrane association creates a path shielded from solvent for the transfer of acyl-PO4 to the bilayer. Channeling may also involve transfer directly to PlsY, via protein-protein interaction. PlsX mutants that do not associate with the membrane still produce acyl-PO4 but this cannot reach PlsY and is hydrolyzed to free fatty acids in the cytosol.
Acylphosphates are less water soluble than acyl-ACP (2) and this may represent a barrier to its diffusion from the cytosolic compartment and into the membrane. Although amphipathic molecules like acyl-PO4, or even fatty acids, will spontaneously partition into the membrane from a water phase, the kinetics of the process may not be fast enough to support highly active pathways such as phospholipid synthesis (Fig. 7). In fact, the residual growth of strains containing PlsX variants that do not bind to the membrane (Figs. 4A and 6B) probably reflects the slow membrane partitioning of acyl-PO4 that is being produced by PlsX molecules located in the cytoplasm. There are several examples of proteins that participate in cytoplasmic transport of fatty acids, whose diffusion is limited by their poor solubility (21). ACP itself can be thought of as an analogous acyl transporter, in the sense of avoiding solubility problems, because the acyl chain in an acyl-ACP molecule is tucked into a hydrophobic groove in the protein (22–24). Thus, it seems that acyl intermediates are not usually found freely surrounded by water inside cells, and PlsX would be the protein in charge of keeping acyl-PO4 away from water as well. Finally, acylphosphates are very labile and the channeling (or chaperoning) role of PlsX may also be relevant to protect these molecules from hydrolysis, as pointed out by Li et al. (5). Consistent with this idea, we observe the accumulation of free fatty acids in strains in which PlsX is cytoplasmic and no longer capable of efficiently channeling acyl-PO4 into the membrane (Fig. 5B).
The detailed mechanism of acyl-PO4 channeling by PlsX remains to be defined, but our data allows us to speculate that it depends on the proper and stable insertion of PlsX in the membrane. This can be concluded from the biophysical analysis of the PlsXL254E mutant presented in the accompanying paper (33), which showed that this protein can interact with membranes only superficially. A similar conclusion is also supported by in vivo experiments, which showed that the phenotype of the PlsXL254E mutant is not much worse than the phenotype of a strain in which PlsX can never access the membrane (PlsXL254E-HBsu) (Fig. 6C). Together, these results mean that superficially accessing the membrane is not sufficient to ensure efficient transfer of acyl-PO4. Stable insertion in the membrane may be necessary to create a path shielded from solvent for acyl-PO4 to leave the active site and insert into the bilayer. This path may also involve the physical association between PlsX and PlsY, as these proteins were shown to interact in two-hybrid experiments (25). Proper interaction between PlsX and PlsY may only happen if PlsX is stably inserted in the membrane.
In conclusion, we have identified a loop rich in hydrophobic residues localized at the tip of the stem of PlsX that plays a critical role in mediating protein-membrane interaction and used this information to advance our understanding of bacterial phospholipid biosynthesis. On the basis of our combined results, we propose that the proper membrane interaction and localization of PlsX plays a key topological role in the channeling of acyl substrates from the cytoplasm and into the phospholipid synthesis pathway. These results highlight the importance of spatial organization for the proper functioning of certain biochemical pathways within cells. In addition, they contribute to the field of antibacterial drug discovery, considering the importance of bacterial lipid synthesis as a target for new antimicrobial compounds.
Experimental procedures
Bacterial strains and growth conditions
All bacterial strains used in this study were listed in Table S1. Drug concentrations for selection were: erythromycin, 1 μg ml−1; lincomycin 25 μg ml−1; kanamycin, 50 μg ml−1; chloramphenicol, 5 μg ml−1; spectinomycin, 100 μg ml−1; ampiciline 100 μg ml−1. In general, B. subtilis and Escherichia coli were grown in Luria-Bertani (LB) broth medium or on plates at 37 °C with aeration.
General molecular techniques
Molecular cloning techniques were performed as described (26). DNA fragments were obtained by PCR using Phusion DNA polymerase (New England Biolabs). Oligonucleotide primers were purchased from Exxtend SRL (Campinas, SP, Brazil) and Thermo Fisher Scientific (São Paulo, SP, Brazil) and are listed in Table S2. PCR products of the expected sizes were purified from gels using an GeneJETTM Gel Extraction Kit (Fermentas), ligated into the pTZ57R/T cloning vector (Fermentas) and transformed into E. coli DHα (Table S1). Plasmid DNA was prepared using a GeneJET Plasmid Miniprep kit (Thermo Scientific). Sequencing was carried out using the Big Dye terminator cycle sequencing kit (Applied Biosystems) by the sequencing service of the Departamento de Bioquímica, IQ-USP (SP, Brazil). Transformation of B. subtilis was carried out by the method of Dubnau and Davidoff-Abelson (27). The amy phenotype was assayed for colonies grown for 24 h in LB-starch plates by flooding the plates with 1% I2, KI solution.
Plasmid and strain construction
All plasmids used in this study are listed in Table S1. The plsX gene from B. subtilis was amplified and inserted into pET24b (T7 promotor) within NdeI and XhoI sites to generate pET24-plsX, which expressed BsPlsX with a C-terminal His6 tag. Site-directed mutantagenesis was performed using Phusion Site-directed Mutagenesis Kit (Thermo Fisher Scientific). GFP fusion constructs were designed for fluorescence microscopy. Construction of GFP-PlsX and GFP-PlsX-HBsu into pDR111 vector containing the double or triple fusion within NheI and SphI sites to generate pDR111-gfp-plsX and pDR111-gfp-plsX-hbsu vectors to integrate in amyE locus of B. subtilis DS28 strains.
Expression and purification of BsPlsX
Vectors pET24b-plsX were used to transform E. coli BL21 (DE3) pRIL-competent cells for protein expression. BsPlsX WT and variant mutants were induced with 0.8 mm IPTG for 4 h at 37 °C. Cells were collected by centrifugation (6000 rpm, 4 °C, 15 min), and cell pellets were lysed with a sonicator. Soluble proteins were applied to a Ni2+-agarose column and washed with buffer: 50 mm Tris-HCl (pH 7.5), 300 mm NaCl, and 50 mm imidazole). His-tagged proteins were eluted with the same buffer containing 300 mm imidazole. Purified proteins were applied to a P10 desalting column and eluted with buffer A without imidazole. The proteins were stored at −80 °C.
Gel filtration assay
The PlsX WT protein and variant mutants were purified by Ni2+ column and then were subjected to gel filtration chromatography using Superdex 200 Increase 10/300 GL resin in a HPLC column (10.0 × 300 mm) and employing the AKTA pure system (GE Healthcare). Proteins were eluted with buffer 50 mm Tris-HCl (pH 7.5), 300 mm NaCl at a flow rate of 0.5 ml/min and elution was monitored at 280 nm.
Fluorescence microscopy
For fluorescence microscopy, overnight precultures of B. subtilis were grown in LB medium containing antibiotics diluted in fresh medium without antibiotics to an optical density at 600 nm of 0.05 and grown at 37 °C. At appropriate time intervals, 5 μl of living cells was applied directly on glass slides on a thin film of 1.5% LB-agarose and examined using a ×100 oil immersion objective on a Nikon Eclipse Ti microscope, equipped with GFP BrightLineR Set (Semrock). Images were acquired on a system with the Nikon digital camera (Nikon, Tokyo, Japan). All images were captured using NIS software version 3.07 (Nikon) and fluorescence signals were analyzed using ImageJ software.
Synthesis of palmitoyl-PO4
The 16:0-PO4 was synthesized by a new mechanochemical method for the synthesis of acylphosphates.6 Palmitoyl chloride (370 μl, 1,2 mmol) was added to a mixture of tripotassium phosphate (K3PO4, 297 mg, 1.4 mmol) and 10 mol % tetra-n-butylammonium bromide (Bu4NBr, 40 mg), followed by trituration. Trituration was continued until the reaction was completed as evident from the formation of a solid (about 5 min). The resulting mixture was filtered in a celite pad, and the retained solid was washed with chloroform (3 × 10 ml). The chloroform-soluble material was dried under vacuum to afford 400 mg of 16:0-PO4 as a white powder (50% yield). 1H NMR (CDCl3, 300 MHz), d 0.92 (t, 3H, J = 6.0 Hz), 1.31 (bs, 26 H), 1.65 (m, 2H), 2.46 (t, 2H, J = 7.0 Hz). 13C NMR (CDCl3, 300MHz), d 12.5, 21.8, 23.6, 28.6, 28.7, 28.8, 31.1, 34.0, 168.3. 31P NMR (CDCl3) −7.2 ppm MS(ESI), m/z = 359 [M + Na]+; MS(ESI), m/z = 335 [M 2 H]2.
PlsX activity assay
The acyltransferase activity was determined according to the procedures described by Lu et al. (1). The reaction for acyl chain transfer from 16:0-PO4 to ACP contained 0.1 m HEPES (pH 7.4), 0.05% Tween 20, 2 mm MgCl2, 40 μm 16:0-PO4, and 40 μm ACP. Reaction was started by adding BsPlsX (5–30 μm) and was incubated at 37 °C for 1 h with agitation. Samples were analyzed by conformationally sensitive gel electrophoresis in 15% polyacrylamide gels containing 2 m urea (34, 35) and staining with Coomassie Brillant Blue.
Western blotting
Samples were prepared as described Weart et al. (28). Briefly, harvested cells were resuspended in 50 mm Tris-HCl (pH 7.5), 300 mm NaCl, 1 mm phenylmethylsulfonyl fluoride, 2 mg ml−1 of lysozyme. Cells were incubated at 37 °C for 30 min and lysates were normalized by OD600 at cell harvest, resuspended in sample buffer, and subjected to SDS-PAGE. GFP-PlsX was detected using the polyclonal rabbit anti-GFP antibody (29). FtsZ was detected using the polyclonal anti-FtsZ antibody (laboratory stock).
Liposome sedimentation assay
Initially, total phospholipids from B. subtilis PY79 were extracted with the Bligh and Dyer method (30). Liposomes were made by resuspension of the lipid film in 50 mm Tris-HCl (pH 7.5), 300 mm NaCl. For sedimentation assays, protein and liposomes were mixed at different concentrations in the assay buffer to a final volume of 150 μl. After incubation at room temperature for 30 min, samples were centrifuged at 65,000 rpm for 15 min in a Beckman TLA100 rotor. Supernatants were removed immediately and pellets were resuspended in an equal volume of buffer. Samples from supernatant and pellet were subjected to 12% SDS-PAGE and proteins were detected by Coomassie Blue staining phospholipid vesicles.
DSC experiments
The effects of PlsX on lipid bilayer membranes composed of DMPG, DMPC, and DMPS were analyzed by monitoring the thermotropic phase behavior of lipid and lipid-protein samples using DSC. The thermograms were recorded in a VP-DSC MicroCal MicroCalorimeter (Microcal, Northampton, MA) using a heating rate of 23.4 °C/h. The analyses of the heat capacity curves were performed using Microcal Origin software (OriginLab Corporation, Northampton, MA). Lipid vesicles were prepared as follows: aliquots of lipid stock solutions in chloroform/methanol, 1:1 (v/v), were added into a glass tube, dried under a N2 flow, and ultracentrifuged under vacuum for 5 h to remove traces of organic solvent. The resulting lipid film was hydrated in 50 mm Tris-HCl buffer (pH 7.5), 300 mm NaCl at a temperature above the main phase transition of the lipid for 2 h and submitted to six freeze-thaw cycles. LUV were obtained by extruding the preformed multilamellar dispersions using polycarbonate filters with a pore size of 100 nm (Nuclepore Corp., Cambridge, CA). An aliquot of the protein stock solution was added into the LUVs, gently vortexed for few seconds, and kept at either 37 °C (DMPC and DMPG) or 40 °C (DMPS) for 1 h prior to the DSC experiment. The phospholipids were purchased from Avanti Polar Lipids, Inc. (Alabaster, AL) and used without further purification.
Author contributions
D. E. S., D. d. M., and F. G.-F. conceptualization; D. E. S., A. A. P., L. G. B., J. S. B. P., F. M., D. A., M. V.A. S.N., D. d. M., and F. G.-F. formal analysis; D. E. S., A. A. P., L. G. B., J. S. B. P., C. G. C. M. N., F. M., and D. A. investigation; D. E. S., L. G. B., C. G. C. M. N., F. M., and D. A. methodology; D. E. S., D. d. M., and F. G.-F. writing-original draft; D. E. S., D. A., D. d. M., and F. G.-F. writing-review and editing; M. V. A. S. N. resources; M. V. A. S. N. and F. G.-F. funding acquisition; F. G.-F. supervision.
Supplementary Material
This work was supported by Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP) Grant 2016/05203-5 (to F. J. G.-F.), Agencia Nacional de Promocion Cientifica y Tecnologica Grant PICT 2016-1594 (to D. d. M.), FAPESP Postdoctoral Fellowship Grant 2014/13411-1 (to D. E. S), FAPESP Grant 2015/21583-0 (to M. V. A. S. N), and FAPESP scholarship Grant 2014/00206-0 (to L. G. M. B.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S5 and Tables S1 and S2.
Marques Netto, unpublished data.
- PA
- phosphatidic acid
- LPA
- lysophosphatidic acid
- G6P
- glycerol 3-phosphate
- DSC
- differential scanning calorimetry
- LUV
- large unilamellar vesicle
- DMPG
- 1,2-dimiristoyl-sn-glycero-3-phospho-(1′-rac glycerol)
- DMPS
- 1,2-dimyristoyl-sn-glycero-3-phospho-l-serine
- DMPC
- 1,2-dimyristoyl-sn-glycero-3-phosphocholine
- IPTG
- isopropyl 1-thio-β-d-galactopyranoside
- AH
- amphipathic helix.
References
- 1. Lu Y. J., Zhang Y. M., Grimes K. D., Qi J., Lee R. E., and Rock C. O. (2006) Acyl-phosphates initiate membrane phospholipid synthesis in Gram-positive pathogens. Mol. Cell 23, 765–772 10.1016/j.molcel.2006.06.030 [DOI] [PubMed] [Google Scholar]
- 2. Yao J., and Rock C. O. (2013) Phosphatidic acid synthesis in bacteria. Biochim. Biophys. Acta 1831, 495–502 10.1016/j.bbalip.2012.08.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Paoletti L., Lu Y. J., Schujman G. E., de Mendoza D., and Rock C. O. (2007) Coupling of fatty acid and phospholipid synthesis in Bacillus subtilis. J. Bacteriol. 189, 5816–5824 10.1128/JB.00602-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Parsons J. B., and Rock C. O. (2013) Bacterial lipids: metabolism and membrane homeostasis. Prog. Lipid Res. 52, 249–276 10.1016/j.plipres.2013.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Li Z., Tang Y., Wu Y., Zhao S., Bao J., Luo Y., and Li D. (2017) Structural insights into the committed step of bacterial phospholipid biosynthesis. Nat. Commun. 8, 1691 10.1038/s41467-017-01821-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Robertson R. M., Yao J., Gajewski S., Kumar G., Martin E. W., Rock C. O., and White S. W. (2017) A two-helix motif positions the lysophosphatidic acid acyltransferase active site for catalysis within the membrane bilayer. Nat. Struct. Mol. Biol. 24, 666–671 10.1038/nsmb.3436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Takada H., Fukushima-Tanaka S., Morita M., Kasahara Y., Watanabe S., Chibazakura T., Hara H., Matsumoto K., and Yoshikawa H. (2014) An essential enzyme for phospholipid synthesis associates with the Bacillus subtilis divisome. Mol. Microbiol. 91, 242–255 10.1111/mmi.12457 [DOI] [PubMed] [Google Scholar]
- 8. Sastre D. E., Bisson-Filho A., de Mendoza D., and Gueiros-Filho F. J. (2016) Revisiting the cell biology of the acyl-ACP:phosphate transacylase PlsX suggests that the phospholipid synthesis and cell division machineries are not coupled in. Bacillus subtilis. Mol. Microbiol. 100, 621–634 10.1111/mmi.13337 [DOI] [PubMed] [Google Scholar]
- 9. Badger J., Sauder J. M., Adams J. M., Antonysamy S., Bain K., Bergseid M. G., Buchanan S. G., Buchanan M. D., Batiyenko Y., Christopher J. A., Emtage S., Eroshkina A., Feil I., Furlong E. B., Gajiwala K. S., et al. (2005) Structural analysis of a set of proteins resulting from a bacterial genomics project. Proteins 60, 787–796 10.1002/prot.20541 [DOI] [PubMed] [Google Scholar]
- 10. Kim Y., Li H., Binkowski T. A., Holzle D., and Joachimiak A. (2009) Crystal structure of fatty acid/phospholipid synthesis protein PlsX from. Enterococcus faecalis. J. Struct. Funct. Genomics 10, 157–163 10.1007/s10969-008-9052-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Marston A. L., Thomaides H. B., Edwards D. H., Sharpe M. E., and Errington J. (1998) Polar localization of the MinD protein of Bacillus subtilis and its role in selection of the mid-cell division site. Genes Dev. 12, 3419–3430 10.1101/gad.12.21.3419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pichoff S., and Lutkenhaus J. (2005) Tethering the Z ring to the membrane through a conserved membrane targeting sequence in FtsA. Mol. Microbiol. 55, 1722–1734 10.1111/j.1365-2958.2005.04522.x [DOI] [PubMed] [Google Scholar]
- 13. Parsons J. B., Frank M. W., Jackson P., Subramanian C., and Rock C. O. (2014) Incorporation of extracellular fatty acids by a fatty acid kinase-dependent pathway in Staphylococcus aureus. Mol. Microbiol. 92, 234–245 10.1111/mmi.12556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Köhler P., and Marahiel M. A. (1997) Association of the histone-like protein HBsu with the nucleoid of Bacillus subtilis. J. Bacteriol. 179, 2060–2064 10.1128/JB.179.6.2060-2064.1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Roggiani M., and Goulian M. (2015) Chromosome-membrane interactions in bacteria. Annu. Rev. Genet. 49, 115–129 10.1146/annurev-genet-112414-054958 [DOI] [PubMed] [Google Scholar]
- 16. Adams D. W., Wu L. J., and Errington J. (2015) Nucleoid occlusion protein Noc recruits DNA to the bacterial cell membrane. EMBO J. 34, 491–501 10.15252/embj.201490177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Whited A. M., and Johs A. (2015) The interactions of peripheral membrane proteins with biological membranes. Chem. Phys. Lipids 192, 51–59 10.1016/j.chemphyslip.2015.07.015 [DOI] [PubMed] [Google Scholar]
- 18. Malmberg N. J., Van Buskirk D. R., and Falke J. J. (2003) Membrane-docking loops of the cPLA2 C2 domain: detailed structural analysis of the protein-membrane interface via site-directed spin-labeling. Biochemistry 42, 13227–13240 10.1021/bi035119+ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gamsjaeger R., Johs A., Gries A., Gruber H. J., Romanin C., Prassl R., and Hinterdorfer P. (2005) Membrane binding of beta2-glycoprotein I can be described by a two-state reaction model: an atomic force microscopy and surface plasmon resonance study. Biochem. J. 389, 665–673 10.1042/BJ20050156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jiang Y., Dai X., Qin M., and Guo Z. (2019) Identification of an amphipathic peptide sensor of the Bacillus subtilis fluid membrane microdomains. Commun. Biol. 2, 316 10.1038/s42003-019-0562-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cuypers M. G., Subramanian C., Gullett J. M., Frank M. W., White S. W., and Rock C. O. (2019) Acyl-chain selectivity and physiological roles of Staphylococcus aureus fatty acid-binding proteins. J. Biol. Chem. 294, 38–49 10.1074/jbc.RA118.006160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Weisiger R. A. (2007) Mechanisms of intracellular fatty acid transport: role of cytoplasmic-binding proteins. J. Mol. Neurosci. 33, 42–44 10.1007/s12031-007-0047-y [DOI] [PubMed] [Google Scholar]
- 23. Roujeinikova A., Simon W. J., Gilroy J., Rice D. W., Rafferty J. B., and Slabas A. R. (2007) Structural studies of fatty acyl-(acyl carrier protein) thioesters reveal a hydrophobic binding cavity that can expand to fit longer substrates. J. Mol. Biol. 365, 135–145 10.1016/j.jmb.2006.09.049 [DOI] [PubMed] [Google Scholar]
- 24. Cronan J. E. (2014) The chain-flipping mechanism of ACP (acyl carrier protein)-dependent enzymes appears universal. Biochem. J. 460, 157–163 10.1042/BJ20140239 [DOI] [PubMed] [Google Scholar]
- 25. Hara Y., Seki M., Matsuoka S., Hara H., Yamashita A., and Matsumoto K. (2008) Involvement of PlsX and the acyl-phosphate dependent sn-glycerol-3-phosphate acyltransferase PlsY in the initial stage of glycerolipid synthesis in Bacillus subtilis. Genes Genet. Syst. 83, 433–442 10.1266/ggs.83.433 [DOI] [PubMed] [Google Scholar]
- 26. Sambrook J., Fritsch E. F., and Maniatis T. (1989) Molecular cloning: a laboratory manual, 2nd Ed., Cold Spring Harbor Laboratory, Cold Spring Harbor, NY [Google Scholar]
- 27. Dubnau D., Davidoff-Abelson R., and Smith I. (1969) Transformation and transduction in Bacillus subtilis: evidence for separate modes of recombinant formation. J. Mol. Biol. 45, 155–179 10.1016/0022-2836(69)90097-7 [DOI] [PubMed] [Google Scholar]
- 28. Weart R. B., Lee A. H., Chien A. C., Haeusser D. P., Hill N. S., and Levin P. A. (2007) A metabolic sensor governing cell size in bacteria. Cell 130, 335–347 10.1016/j.cell.2007.05.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rudner D. Z., and Losick R. (2002) A sporulation membrane protein tethers the pro-σK processing enzyme to its inhibitor and dictates its subcellular localization. Genes Dev. 16, 1007–1018 10.1101/gad.977702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bligh E. G., and Dyer W. J. (1959) A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 37, 911–917 10.1139/o59-099 [DOI] [PubMed] [Google Scholar]
- 31. Waterhouse A., Bertoni M., Bienert S., Studer G., Tauriello G., Gumienny R., Heer F. T., de Beer T. A. P., Rempfer C., Bordoli L., Lepore R., and Schwede T. (2018) SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 46, W296–W303 10.1093/nar/gky427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gautier R., Douguet D., Antonny B., and Drin G. (2008) HELIQUEST: a web server to screen sequences with specific α-helical properties. Bioinformatics. 24, 2101–2102 10.1093/bioinformatics/btn392 [DOI] [PubMed] [Google Scholar]
- 33. Sastre D. E., Basso L. G. M., Trastoy B., Cifuente J. O., Contreras X., Gueiros-Filho F., de Mendoza D., Navarro M. V. A. S., and Guerin M. E. (2019) Membrane fluidity adjusts the insertion of the transacylase PlsX to regulate phospholipid biosynthesis in Gram-positive bacteria. J. Biol. Chem. 294, 2136–2147 10.1074/jbc.RA119.011122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cronan J. E., Jr. (1982) Molecular properties of short chain acyl thioesters of acyl carrier protein. J. Biol. Chem. 257, 5013–5017 [PubMed] [Google Scholar]
- 35. Rock C. O., Cronan J. E. Jr., and Armitage I. M. (1981) Molecular properties of acyl carrier protein derivatives. J. Biol. Chem. 256, 2669–2674 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.