

Abstract
Cell scaffolding and signaling are governed by protein–protein interactions. Although a particular interaction is often defined by two specific domains binding to each other, this interaction often occurs in the context of other domains in multidomain proteins. How such adjacent domains form supertertiary structures and modulate protein–protein interactions has only recently been addressed and is incompletely understood. The postsynaptic density protein PSD-95 contains a three-domain supramodule, denoted PSG, which consists of PDZ, Src homology 3 (SH3), and guanylate kinase-like domains. The PDZ domain binds to the C terminus of its proposed natural ligand, CXXC repeat–containing interactor of PDZ3 domain (CRIPT), and results from previous experiments using only the isolated PDZ domain are consistent with the simplest scenario for a protein–protein interaction; namely, a two-state mechanism. Here we analyzed the binding kinetics of the PSG supramodule with CRIPT. We show that PSG binds CRIPT via a more complex mechanism involving two conformational states interconverting on the second timescale. Both conformational states bound a CRIPT peptide with similar affinities but with different rates, and the distribution of the two conformational states was slightly shifted upon CRIPT binding. Our results are consistent with recent structural findings of conformational changes in PSD-95 and demonstrate how conformational transitions in supertertiary structures can shape the ligand-binding energy landscape and modulate protein–protein interactions.
Keywords: PDZ domain, protein conformation, protein–protein interaction, conformational change, kinetics, CXXC repeat–containing interactor of PDZ3 domain (CRIPT), postsynaptic density protein, PSD-95, supertertiary structure, supramodule
Introduction
Detailed characterization of protein-mediated interactions is a critical task for understanding cellular processes as well as for identification of novel drug targets. From an experimental perspective, a quantitative description of protein-mediated networks demands detection of specific interactions, followed by an attempt to understand larger systems. It should be noted, however, that describing protein interaction networks is a daunting task complicated by several factors that could be hard to estimate within the cellular environment, such as concentrations of individual proteins and compartmentalization. Additionally, proteins may experience conformational changes that may be difficult to capture but nevertheless modulate their function. Thus, a combination of structural, functional, and computational approaches is generally necessary to understand the role of conformational changes.
Postsynaptic density protein-95 (PSD-95) is a multidomain protein abundant in the postsynaptic density of neurons, where it acts as a scaffolding protein (1). Starting from the N terminus, this protein contains three PDZ domains followed by an Src homology 3 (SH3)3 domain and a guanylate kinase-like (GK) domain (2). The PDZ domain is one of the most common protein interaction domains in our proteome (3). It presents a binding groove on its surface in which protein ligands can bind as a β-strand to form an extended β-sheet with the PDZ domain. PSD-95 contains a supertertiary structure (4); the first two PDZ domains form one supramodule (5), whereas the third PDZ domain (PDZ3) forms another supramodule together with the SH3 and GK domains (denoted PSG) (6–9) (Fig. 1). This feature is shared by other membrane-associated GK family proteins (10), to which PSD-95 belongs. In general, PDZ domains display similar affinity for a range of protein ligands (11) so that specificity must rely on spatial and temporal control in the cell. Because of this, it is often difficult to pinpoint natural ligands of PDZ domains, but a likely native ligand of PDZ3 from PSD-95 and of PDZ3 from its closely related paralog SAP97 is CXXC repeat–containing interactor of PDZ3 domain (CRIPT) (12, 13). PDZ3 binds the C terminus of CRIPT with a Kd of around 0.3 μm, and this affinity is achieved mainly via interactions between the six C-terminal residues of CRIPT (YKQTSV) and the binding groove of PDZ3 (14–16). However, several recent studies have highlighted the role of context dependence and more or less well-defined interactions outside of the binding motif for modular protein–protein interactions (17–20), mechanisms for which conformational transitions are likely important.
Figure 1.
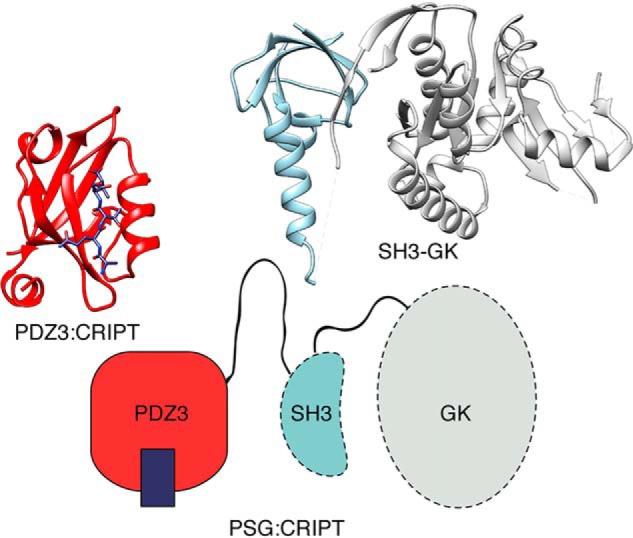
Structure of the PSG supramodule from PSD-95. Schematic supertertiary structure of the PSG supramodule from PSD-95 together with crystal structures of PDZ3 with a bound CRIPT peptide (PDB code 1BE9) and of the SH3-GK tandem (PDB code 1JXO).
In this study, we investigated the binding kinetics of the PSG supramodule from human PSD-95 with a CRIPT peptide and found that the simplest mechanistic model consistent with the data involves two distinct conformational states of PSG. The two interconverting states can both bind a short CRIPT peptide with a similar affinity, but only one of the states can bind a longer CRIPT peptide. Moreover, the rate constants differ so that one conformer binds CRIPT faster than the other one, reflecting changes around the binding groove associated with the conformational transition. Previous work demonstrated conformational transitions within the PSG using structural approaches (8, 9), and more recently, functional oligomerization has been shown to be dependent on the PSG supramodule (17, 21–23). The binding kinetics observed in this study could be related to these phenomena.
Results
The binding kinetics of PSG and a six-amino-acid CRIPT peptide displayed biphasic behavior
We monitored binding of CRIPT to PSG using an N-terminal dansyl group attached to the six C-terminal residues of CRIPT (D-CRIPT6). The dansyl quenches the fluorescence of an engineered Trp in PSG. The Trp was situated in the same position in PDZ3 (residue 337) as in previous studies of the isolated PDZ3 domain, where it did not affect affinity or kinetics of binding (16). Quenching of the Trp upon binding resulted in kinetic traces with high signal to noise (Fig. 2A). We previously performed extensive experiments with isolated PDZ3 and D-CRIPT6 that yielded single exponential traces as pseudo-first-order conditions were approached, consistent with an apparent two-state binding mechanism (16, 24). However, fitting of the kinetic binding data of the PSG supramodule and D-CRIPT6 to a single exponential function consistently yielded residuals displaying a clear nonrandom distribution around zero, even at D-CRIPT6 concentrations 10-fold higher than those of PSG (Fig. 2), suggesting a more complex mechanism than two-state. A double-exponential equation fitted well to the experimental traces recorded over 1 s, and the observed rate constants, kobs1 and kobs2, obtained from the fit were determined over a range of D-CRIPT6 concentrations (Fig. 2C). The apparently linear dependence of both rate constants on D-CRIPT6 concentration suggests that the PSG sample contained two distinct species, both of which could bind D-CRIPT6, one with an association rate constant, kon1, of 5.8 × 106 m−1 s−1 (from kobs1) and the other with a kon2 of ∼0.56 × 106 m−1 s−1 (from kobs2). In terms of amplitudes of the kinetic traces, kobs1 had a relatively large and kobs2 a relatively small amplitude (Fig. 2D). The koff values could not be determined directly from these binding data because they were too low to allow accurate extrapolation of kobs to zero D-CRIPT6 concentration.
Figure 2.

Binding kinetics of the PSG supramodule and CRIPT. A, example of kinetic transients for the association of PSG with D-CRIPT6. PSG (1 μm final concentration) was mixed rapidly in the stopped flow with D-CRIPT6 at different concentrations. B, the residuals are from fits to single- and double-exponential functions, respectively, at 4 μm final concentration of D-CRIPT6. C, the experiment was repeated over a range of D-CRIPT6 concentrations, and the kobs values obtained from the fit to a double-exponential function were plotted versus [D-CRIPT6]. kobs values of more than 150 s−1 were omitted because they very slightly but progressively deviated from a straight line because of instrumental limitations in the mixing. D, kinetic amplitudes associated with the respective kobs value.
Displacement experiments corroborated the presence of two distinct molecular species
To investigate the dissociation kinetics of the PSG:CRIPT complex, we performed displacement experiments in which a preformed complex of PSG:D-CRIPT6 (1 and 5 μm final concentrations) was rapidly mixed with an excess (100–200 μm) of unlabeled CRIPT peptide. In contrast to what we observed previously for the isolated PDZ3 domain (16, 24), the dissociation kinetics of PSG:D-CRIPT6 were also biphasic (Fig. 3A), which strongly supports the presence of two distinct binding-competent PSG conformers. In fact, in a displacement experiment, any sequential mechanism, such as induced fit, usually yield kinetic dissociation transients that are well-described by single-exponential kinetics. The observed rate constants, which approximate the overall apparent dissociation rate constants (koffapp), were approximately koff1app = 1.35 s−1 and koff2app = 0.17 s−1, respectively (Fig. 3B). The errors in the amplitudes were large because of covariation of the parameters, a common phenomenon in curve fitting of complex models (25). Nevertheless, based on six independent experiments, we found that the fast phase (described by koff1app) was associated with a relatively smaller amplitude and the slow phase (described by koff2app) with a relatively higher amplitude (Fig. 3C).
Figure 3.
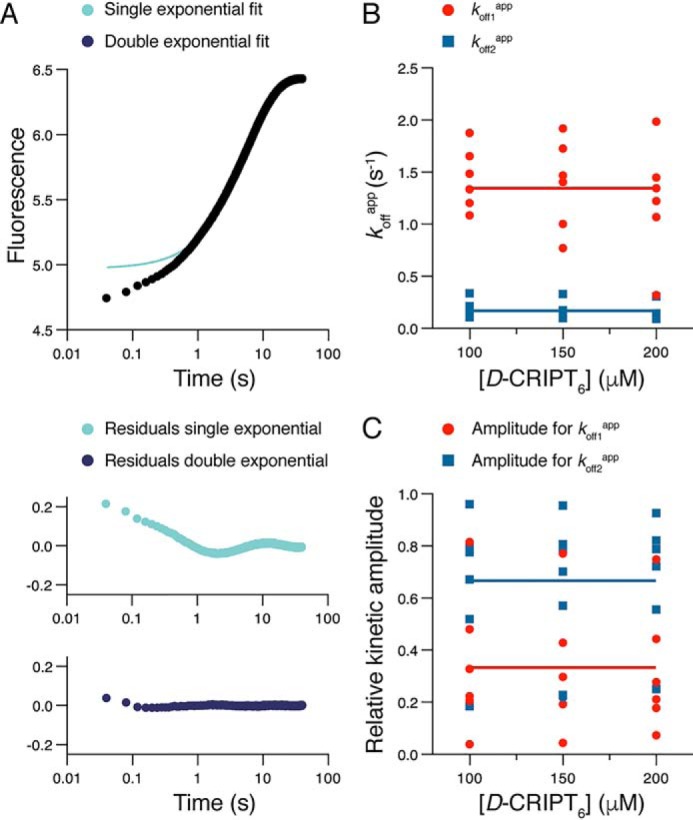
Dissociation kinetics of the PSG supramodule and D-CRIPT6. A, example of kinetic trace from an experiment where a complex of PSG and D-CRIPT6 (2 and 10 μm) was rapidly mixed with an excess of unlabeled CRIPT (150 μm). The resulting trace was fitted to a single or double exponential, and the residuals from the respective fit are presented below the trace. The double-exponential fit yielded the following parameters: koff1app = 1.35 s−1 (amplitude = 0.32) and koff2app = 0.16 s−1 (amplitude = 1.4). B and C, this experiment was repeated six times, each time at three different concentrations of unlabeled CRIPT (100, 150, and 200 μm). The double-exponential fit yielded koffapp values (B) and their associated amplitudes (C, normalized), which were plotted versus CRIPT concentration. The large scatter in the points, in particular for the amplitudes, reflects the covariation of parameters in the curve fitting. The average of all 18 koffapp values for each kinetic phase yielded the following parameters: koff1app = 1.35 ± 0.42 s−1 and koff2app = 0.17 ± 0.08 s−1 (indicated by horizontal lines in B). The average of all 18 amplitudes yielded the following values: Amp1 = 0.33 ± 0.24 and Amp2 = 0.67 ± 0.24 (indicated by horizontal lines in C).
Affinity between PSG and CRIPT, determined by isothermal titration calorimetry, confirmed an affinity in the low micromolar range
From the kinetic data, it is not clear which koffapp belongs to which kon. We therefore performed ITC experiments to obtain an independent estimate of Kd. ITC experiments at the temperature of the stopped flow (10 °C) resulted in poor thermograms, but experiments at 20 °C, 25 °C, 30 °C, and 35 °C yielded good data (Fig. 4). The Kd values at 20 °C–30 °C were all around 0.2 μm, and we deem it likely that the affinity is similar at 10 °C. The two Kd values obtained from the ratios of rate constants, koff1app/kon1 (0.23 μm) and koff2app/kon2 (0.30 μm), respectively, are in very good agreement with the ITC data. This Kd is also close to that obtained for the isolated PDZ3 domain (15, 16). On the other hand koff1app/kon2 and koff2app/kon1 would give Kd values of 29 nm and 2.4 μm, respectively. Thus, the equilibrium binding data were most consistent with a scenario where koff1app and kon1 are associated with the binding to one conformer (denoted PSGA) and koff2app and kon2 to the other conformer (PSGB).
Figure 4.
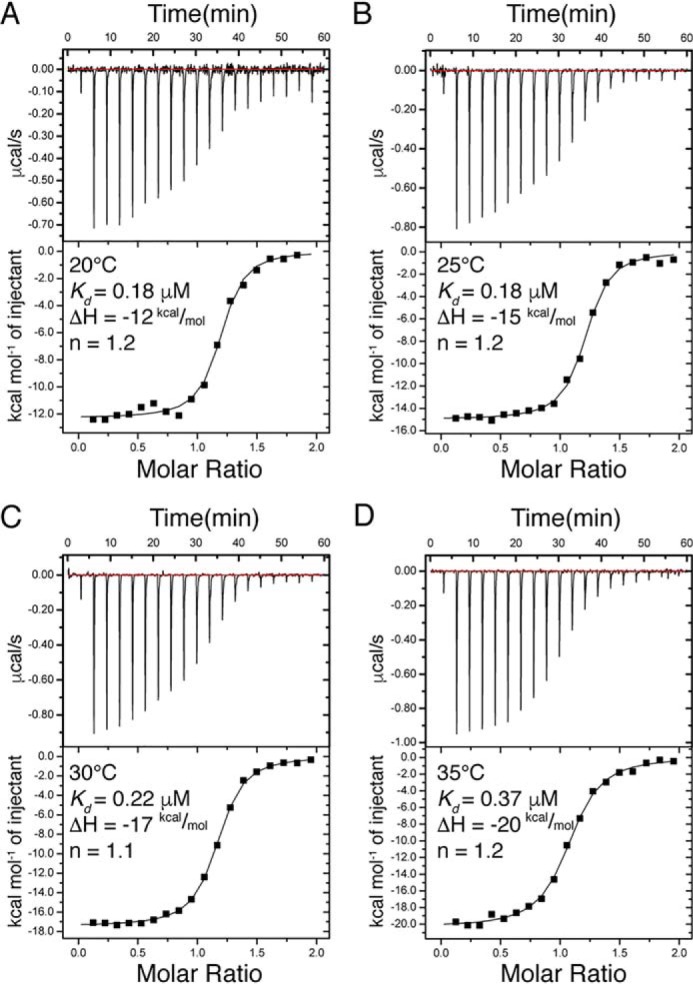
Isothermal titration calorimetry experiments with PSG and D-CRIPT6. A–D, ITC experiments were performed at four different temperatures (20 °C–35 °C). The parameters shown were obtained by fitting a 1:1 binding model using the software provided with the instrument. The Kd values from these experiments agreed well with those calculated from the kinetic experiments at 10 °C (koff1app/kon1 or koff2app/kon2).
However, a model with two noninterconverting conformers predicts that the larger association amplitude should be connected to the larger dissociation amplitude. This is inconsistent with the relative amplitudes of the observed kinetic phases. Although the large variation in dissociation amplitudes precludes a detailed quantitative analysis, it is qualitatively clear that the kinetic amplitude of koff2app (Fig. 3C) is more prominent than that associated with kobs2 in the binding experiment (Fig. 2D). The most likely explanation for this discrepancy is that there is an equilibrium between PSGA and PSGB and that this equilibrium is shifted so that relatively more PSGB is present upon binding of D-CRIPT6 (Fig. 5).
Figure 5.

A kinetic scheme consistent with the binding kinetics of PSG and D-CRIPT6. A scenario with two conformations, PSGA and PSGB, which are in equilibrium and can both bind CRIPT, is consistent with the kinetic and equilibrium data. Access to the binding groove is more restricted in the PSGB conformation.
Direct detection of the conformational transition in a double-jump interrupted binding experiment
In an attempt to directly monitor a transition between PSGA and PSGB in their bound conformation according to the scheme in Fig. 5, we performed interrupted binding experiments in a double-jump setup in the stopped flow instrument (Fig. 6A). 2 μm PSG was mixed with 4 μm D-CRIPT6 in the “first jump” (concentrations after 1:1 mixing). After a defined delay (or aging) ranging between 10 ms and 200 s, this complex was dissociated by mixing with a large excess of unlabeled CRIPT in the “second jump.” Following this second jump, the apparently irreversible dissociation kinetics of PSG:D-CRIPT6 were monitored as described for a regular dissociation experiment, which, in practice, corresponds to an interrupted binding experiment with a very long delay time. We collected 14 kinetic traces from these double-jump experiments, with delay times between 10 ms and 200 s. The trace at 10 ms delay time was excluded because the mismatch between expected and actual delay time was deemed too large. Next, all remaining dissociation traces (delay times between 24 ms and 200 s) were fitted simultaneously to a double-exponential function with shared values for koff1app and koff2app, either locked to the average values reported in Fig. 3B or free-fitted (Fig. 6B). This is possible because the final conditions under which kinetics were measured were identical for each double-jump experiment. However, it is also necessary to perform a simultaneous fit because the kinetic amplitudes at short delay times are too small for accurate free-fitting of koff1app and koff2app. Next, the kinetic amplitudes associated with koff1app and koff2app were plotted as a function of delay time (Fig. 6, C and D). The change in the kinetic amplitudes over time followed biphasic behavior, as clearly seen for the amplitude related to koff1app. This biphasic behavior reflects the initial build-up of the two complexes, PSGA:D-CRIPT6 and PSGB:D-CRIPT6, respectively, followed by their equilibration. The change in the amplitudes over time thus follows exponential kinetics. We could fit a double-exponential function to the amplitude data and obtain two observed rate constants. However, the errors were very large, and this experiment is best interpreted qualitatively or semiquantitatively; kobs1DJ (DJ, double jump) was around 20–40 s−1 and kobs2DJ in the range of 0.1–0.8 s−1, depending on which set of amplitudes was used. Thus, at short delay times, both amplitudes increased with increasing delay time with an apparent rate constant, which corresponds to the binding of D-CRIPT6 to PSG and which, at least for kobs1DJ, is in excellent agreement with the parameters obtained from single-jump binding experiments (kobs1DJ ≈ kon1 × 4 μm + koff1app ≈ 25 s−1). At longer delay times, the kinetic phase associated with koff1app decreases, whereas the phase associated with koff2app should increase (the latter is not as clear as the former because of scatter in the data). This reflects the redistribution of conformations in the bound complex occurring over 1–10 s with a net flow from PSGA: D-CRIPT6 to PSGB:D-CRIPT6 (Fig. 5).
Figure 6.

Interrupted binding experiments. A, schematic of the double-jump setup. In the first jump, 4 μm PSG was mixed with 8 μm D-CRIPT6 (i.e. the final concentrations after mixing were 2 μm and 4 μm, respectively) and incubated for a certain delay time in an aging loop. Following the set delay time, any formed complex was dissociated by a large excess of unlabeled CRIPT in a second jump (final concentrations were 1 μm PSG, 2 μm D-CRIPT6, and 50 μm unlabeled CRIPT). B, the dissociation kinetics were monitored in the flow cell. The kinetic traces were fitted simultaneously to a double-exponential function with either locked kobs values (the two average koffapp values from single-jump experiments, 1.35 s−1 and 0.17 s−1, respectively) or shared and free-fitted kobs values (koff1app = 2.1 s−1 and koff2app = 0.35 s−1). The best fit curves in the figure correspond to the latter fit. C and D, the kinetic amplitudes from the respective fits in B were plotted against delay time. This reflects the build up of PSGA:D-CRIPT6 and PSGB:D-CRIPT6, respectively, with time. These amplitude data, in turn, were fitted to a double-exponential function to obtain two “observed double-jump rate constants” for the build up of PSGA and PSGB, respectively. From the fit, kobsDJ values of around 20–40 s−1 and 0.1–0.8 s−1, respectively, were obtained. Although the parameters are underdetermined, it is clear that there is an initial increase in the respective population, followed by a decrease in PSGA:D-CRIPT6. The expected concomitant increase in PSGB:D-CRIPT6 is lost in the experimental noise.
Global fitting of a minimal model for the interaction between PSG and D-CRIPT6
To test the robustness of the kinetic scheme in Fig. 5, we attempted a global fit of kinetic transients from binding and dissociation experiments using the Kintek software (26, 27). Global fitting with numerical integration takes information from both rate constants and kinetic amplitudes into consideration. The information from kinetic amplitudes is often overlooked but can, in favorable cases, be used to distinguish between kinetic models. However, in the case of fluorescence-monitored measurements, this requires knowledge about the fluorescence yields of all molecular species, and those of intermediates are usually unknown. Here, the information from the kinetic amplitudes was used to restrain the parameters in the model. Thus, a global fit of a dataset, including seven binding curves from Fig. 2C and all dissociation curves from the double-jump experiment (Fig. 6), was performed, and it resulted in reasonable kinetic parameters (Fig. 7).
Figure 7.
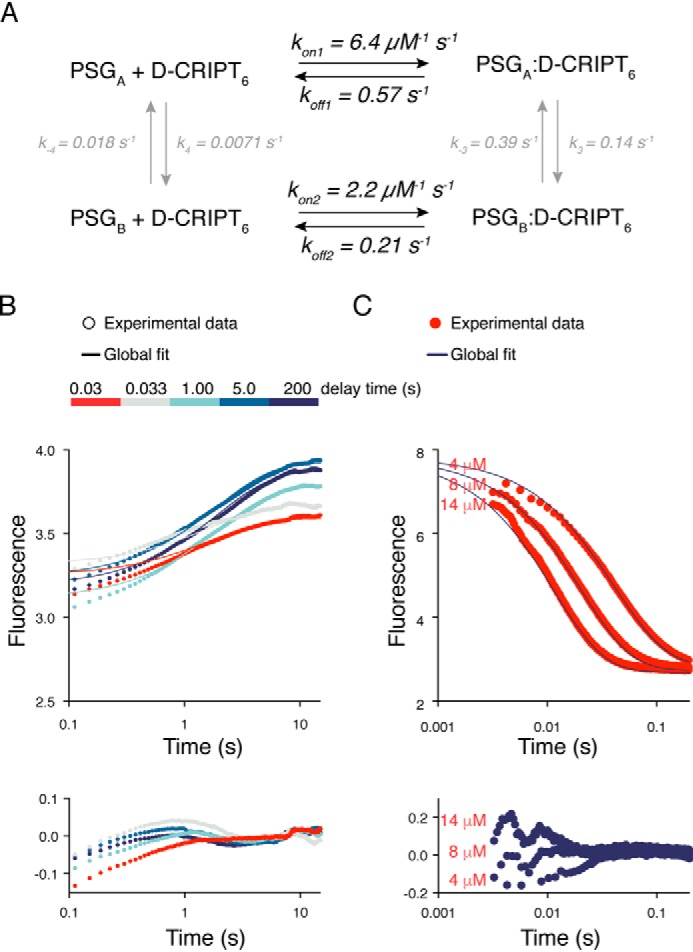
Global fitting to a minimal scheme describing PSG:CRIPT binding. Data from binding and dissociation kinetic experiments were fitted globally with KinTek Explorer. A, the scheme shows the fitted square model with the best fit parameters and their standard errors. The amplitude factors for PSGA:D-CRIPT6 and PSGB:D-CRIPT6 were fitted to 0.9 ± 0.1 and 0.13 ± 0.03, respectively. It should be noted that the reported standard errors for all parameters are likely underestimated. We used the global fit to qualitatively test our suggested minimal model. Thus, less emphasis should be put on the estimates of the microscopic rate constants, which are, in some cases, poorly constrained. kon2 is likely overestimated because we could only use lower D-CRIPT6 concentrations in the global fit to capture the fast phase (Fig. 2). B, fit of interrupted binding stopped flow data to the minimal model in A. Five kinetic traces are shown for clarity. The residuals of the fits are shown below the curve. C, fit of the single-jump stopped-flow data to the model, with residuals shown below the curves. The fits to kinetic traces of three different concentrations of D-CRIPT6 are shown for clarity.
A comparison of the globally fitted rate constants with those obtained from fitting binding and dissociation transients individually shows that, although kon1 is similar, kon2 from the global fit is 4-fold larger than in Fig. 2C. The main reason is that we could not include the data points at high concentrations of D-CRIPT6 in the global curve fitting because the binding step described by kon1 and koff1app gets too fast. It is clear that kon2 is closer to 0.6 μm−1 s−1 (Fig. 2C) than to 2.2 μm−1 s−1 obtained from the global fit. koff1 and koff2 are also more similar to each other in the global fit than koff1app and koff2app in the individual fits presented in Fig. 3. However, the values of koff1 and koff2 in the global fit are dependent on the relative values of k3 and k−3, which are poorly defined. The rate constants k−3 and k3 agree well with the fit of the double-jump data in Fig. 6 insofar as their sum is 0.53 s−1. Finally, k−4 and k4 are fitted as 0.007–0.018 s−1, but they are obviously very poorly defined, except that this step must be relatively slow relative to the other ones. In summary, despite low accuracy for several of the fitted rate constants, global fitting (Fig. 7) shows that the experimental data are overall consistent with the proposed minimal model depicted in Fig. 5 for the interaction between PSG and D-CRIPT6.
Binding kinetics of a 15-mer CRIPT peptide are monophasic
To further investigate the conformational transition in the PSG supramodule, we performed binding experiments with an N-dansylated peptide corresponding to the 15 C-terminal residues of CRIPT, D-CRIPT15. In contrast to D-CRIPT6, we only observed single exponential kinetics with D-CRIPT15 in both binding and displacement experiments (Fig. 8). The apparent rate constants (kon and koff) for D-CRIPT15 were more similar to kon1 and koff1app, associated with the fast phase for D-CRIPT6, and giving a similar Kd (0.42 μm) as D-CRIPT6. The Kd determined by ITC for D-CRIPT15 was 1.6 ± 0.2 μm at a higher temperature (25 °C), in fair agreement with Kd from kinetics. One explanation, considering the close match of the kon values, would be that D-CRIPT15 can only bind to PSGA but not to PSGB. However, even with D-CRIPT6, the kinetic amplitude of the slow phase is low, and for the longer D-CRIPT15 it may not be visible. Regardless of whether D-CRIPT15 binds to PSGB, this conformation is present in equilibrium with PSGA, as monitored by the smaller D-CRIPT6, which can likely access a more narrow binding pocket present in PSGB.
Figure 8.
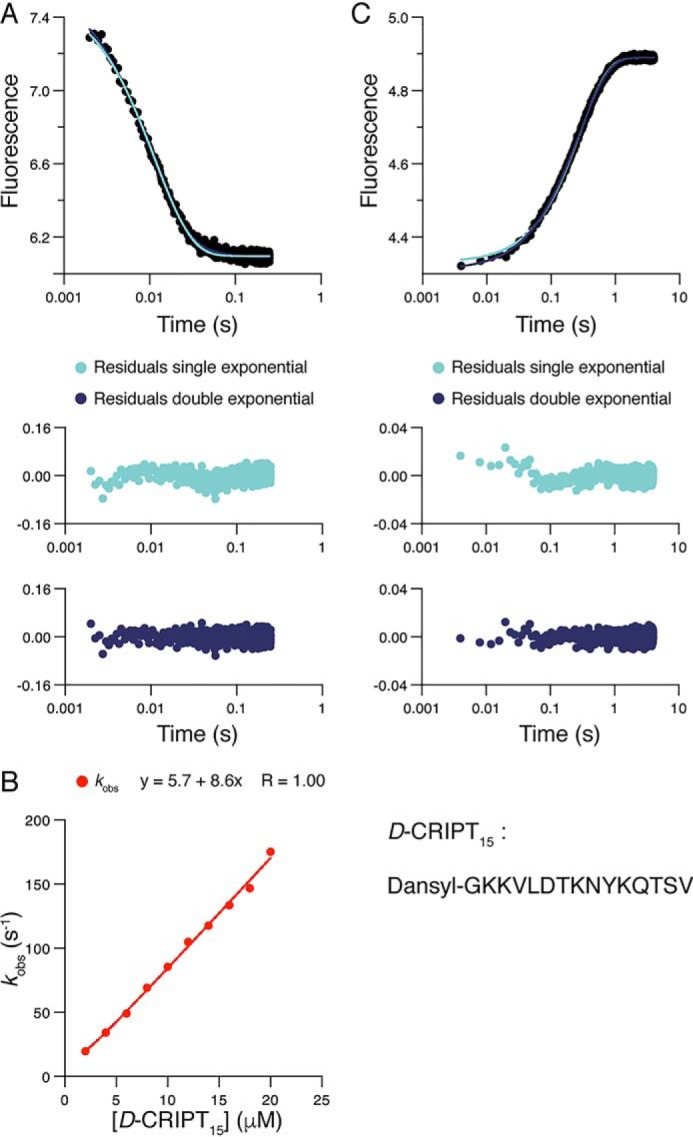
Binding kinetics of the PSG supramodule and D-CRIPT15. A, PSG (1 μm final concentration) was mixed rapidly in the stopped flow with D-CRIPT15 (10 μm final concentration). A single-exponential function well describes the experimental transient, as shown by comparison of residuals from fit to single- and double-exponential functions. B, the experiment was repeated over a range of D-CRIPT15 concentrations, and kobs obtained from the fit to a single-exponential function was plotted versus [D-CRIPT15] to obtain konapp = (8.4 ± 0.2) × 106 m−1 s−1 (fitting error). C, dissociation of the complex. PSG:D-CRIPT15 (2 and 10 μm) was rapidly mixed with an excess of unlabeled CRIPT (150 μm). The resulting trace was fitted to both single- and double-exponential functions. The residuals from the fits show that the dissociation kinetics follow single exponential kinetics. The average of three experiments using 100, 150, and 200 μm CRIPT, respectively, for displacement yielded koffapp = 3.5 ± 0.011 s−1 (± S.D.).
Discussion
Much of our current knowledge regarding protein-mediated interactions is based on study of isolated domains in solution. Nevertheless, it is clear that, because proteins are dynamic entities, the interaction between contiguous domains may play a role in their structure and function. A hallmark of these interactions is represented by hemoglobin, a system where it was established how the intricate interplay between the four subunits results in very complex binding schemes that ultimately mediate allosteric regulation and fine-tune oxygen (un)binding to optimize its transport from the lungs to tissues. Although the dynamics of allosteric multisubunit proteins have a long history (28), recent work has been focused on multidomain proteins in which several distinct protein domains occur in one single polypeptide as beads on a string. It is becoming clear that such proteins often display supertertiary structures (4) where supramodules could form from two or several domains, which may then cooperate positively or negatively in ligand binding or other functions. Similarly to hemoglobin, simplified models must be used to account for the behavior of such multidomain proteins.
The MAGUK family of proteins, to which PSD-95 belongs, is one class of multidomain proteins that has been well-studied, in particular from the point of view of single domains but also from the perspective of its two supramodules. We previously determined binding rate constants for the isolated PDZ3 domain and established that its interaction with CRIPT is a one-step process (16, 24). Here we extended these studies to the PSG supramodule, which consists of PDZ3 and two more domains, SH3 and GK, and found that a model with two conformations, denoted PSGA and PSGB, can satisfactorily describe the data. Although PSGA binds CRIPT with similar rate constants as PDZ3, PSGB appears to display a more restricted binding pocket.
Previous work on PSG from the PSD-95 family or other homologs has not assessed the mechanism with direct kinetic methods. However, multiple complementary methods, including crystallography, NMR, small-angle X-ray scattering, single-molecule FRET, binding experiments, and modeling, have demonstrated that the three domains form a well-defined and dynamic supertertiary structure (8–10, 29, 30). For example, single-molecule FRET in PSD-95 showed that PDZ3 displays motional averaging relative to the SH3 domain (8). Furthermore, NMR, small-angle X-ray scattering, and simulations were consistent with distinct structural ensembles in free PSG and CRIPT-bound PSG, where the ensemble of bound conformations was shifted toward extended rather than compact species (9). In more detail, the peptide-binding groove of PDZ3 has been shown to interact weakly with the SH3 domain (or the linker between them) so that, in the ligand-free state, the SH3 domain restricts ligand binding to PDZ3. Binding of a ligand to PDZ3 would thus promote a more open conformation of PSG where SH3 is released from PDZ3. These conformational transitions could be the basis of the biphasic kinetics observed in this study. From a functional perspective, PSD-95 forms oligomers in the postsynaptic density, and one way is via the SH3 and GK domains (6). This oligomerization is mediated by ligand binding to PDZ3 (21). Furthermore, ligand binding to PDZ3 promotes binding of GK to other proteins, such as the G protein subunit Gnb5 (23) but restricts binding of another protein, GukHolder (29). These previous studies are consistent with our kinetic data insofar as the binding kinetics suggest that the PSGB conformation is less accessible than PSGA, as mirrored by the differences in kon1 (PSGA) and kon2 (PSGB) upon binding to D-CRIPT6, and that D-CRIPT15 apparently only binds to PSGA. A more restricted binding site provides fewer possibilities of productive initial encounters, reducing the apparent association rate constant. It is possible that the more open PSGA is the conformation allowing SH3-GK-mediated oligomerization and binding between GK and the G protein subunit Gnb5 (17, 21–23).
In conclusion, there is a firm structural basis for a dynamic supertertiary structure of the PSG from PSD-95 (8, 9). This dynamic behavior is characterized by some distinctive kinetic signatures, as highlighted by our kinetic analysis. Furthermore, our experiments estimate a timescale of these transitions of about 0.5 s−1 at 10 °C. We speculate that such conformational transitions in the PSG are related to the previously reported conformational changes and are most likely connected to its function, as demonstrated previously in several studies (17, 21–23, 29). Future work based on site-directed variants will further explore the functional role of these transitions.
Experimental procedures
Protein purification
The PSD-95 PDZ3-SH3-GK (PSG) supramodule (residues 308–724) contained an engineered Trp in the same position (F337W) as in previous studies of the isolated PDZ3 domain (16, 24). PSG was expressed from a modified pRSET plasmid (Invitrogen), which was transformed into Escherichia coli BL21(DE3) pLys cells. The cells were grown in lysogeny broth (LB) medium at 37 °C until A600 of the culture reached 0.6. Then overexpression of protein was induced with 1 mm isopropyl β-d-thiogalactopyranoside, and the bottles were incubated overnight in a rotary shaker at 18 °C. Cells were harvested by centrifugation at 4 °C, and the pellet was resuspended in 50 mm Tris (pH 7.8), 100 mm NaCl, and 10% glycerol and stored at −20 °C until purification. The pellet was thawed and sonicated twice for 4 min each time, followed by centrifugation. The supernatant was filtered and loaded onto a nickel-Sepharose Fast Flow column (GE Healthcare) pre-equilibrated with 50 mm Tris, 100 mm NaCl, 10% glycerol, and 0.5 mm DTT. The column was washed with equilibration buffer, and bound protein was eluted with 250 mm imidazole. The protein sample was dialyzed overnight into 50 mm Tris, 2 mm DTT, 100 mm NaCl, and 10% glycerol. The PSG sample was concentrated and further purified using size exclusion chromatography (S-100, GE Healthcare). Protein purity was quantified by SDS-PAGE and identity by MALDI-TOF MS. Concentrations of protein samples were measured by absorbance at 280 nm and theoretical extinction coefficients based on the amino acid composition. Far-UV CD spectra (200–260 nm) of the protein (10 μm) were recorded to ensure that it was folded. CD experiments were performed in 50 mm sodium phosphate (pH 7.45) and 21 mm KCl (I = 150) at 10 °C on a JASCO J-1500 spectropolarimeter using the average of five scans.
Kinetic methods
All kinetic experiments were performed in 50 mm sodium phosphate (pH 7.45), 21 mm KCl (I = 150 mm), 0.5 mm tris(2-carboxyethyl)phosphine at 10 °C. Kinetic binding and dissociation experiments were performed in an upgraded SX-17 MV stopped-flow spectrophotometer (Applied Photophysics, Leatherhead, UK) and carried out as described previously (16). Briefly, kon was obtained from binding experiments approaching pseudo-first-order conditions ([PSG] < [D-CRIPT]). PSG (1 μm) was rapidly mixed with different concentrations of N-terminally dansyl-labeled CRIPT. The shorter peptide, dansyl-YKQTSV, was denoted D-CRIPT6, and the longer one, dansyl-GKKVLDTKNYKQTSV, was denoted D-CRIPT15. The binding reaction was monitored by fluorescence, where the Trp at position 337 in PDZ3 was quenched by the dansyl group of the CRIPT peptide. Trp 337 was excited at 280 nm, and emission was followed at 330 nm (using a 330 ± 25 nm interference filter). Kinetic traces were recorded and averaged (a minimum of five individual traces were used at each concentration) and fitted to single- or double-exponential functions to obtain the kobs for each CRIPT concentration. To estimate kon values, kobs1 and kobs2 (for PSG with D-CRIPT6) or kobs (for PSG with D-CRIPT15) were plotted versus CRIPT concentration and fitted to a linear function, where the slope corresponds to kon. Because of slow dissociation rates, koff could not be accurately determined by extrapolation of kobs to zero CRIPT in the binding experiments. Thus, displacement experiments were performed to obtain more accurate estimates of koff. In these experiments, PSG (2 μm) was preincubated with D-CRIPT6 (10 μm). D-CRIPT6 was then displaced from PSG by rapidly mixing the complex with an excess of unlabeled CRIPT6 (100, 150, and 200 μm). The high excess of unlabeled peptide ensured an irreversible dissociation reaction, allowing koff to be estimated from the average of three experiments in which kobs (≈ koff) did not change with CRIPT6 concentration. Similar to binding, all displacement experiments performed with PSG and D-CRIPT6 were fitted to a double-exponential function, yielding two distinct observed dissociation rate constants.
Double-jump mixing experiments were performed on an Applied Photophysics SX18-MV stopped-flow instrument according to the manufacturer's instructions with regard to mixing volumes. In particular, a solution containing 4 μm PSG in the presence of 50 mm phosphate buffer (pH 7.5) was first mixed 1:1 with D-CRIPT6 peptide at an initial concentration of 8 μm. Then, after a controlled delay time (between 10 ms and 200 s), the solution was challenged with a high concentration of unlabeled CRIPT6 in a second mixing event (the initial concentration of CRIPT6 was 100 μm. Thus, the final concentrations in the displacement reaction were 1 μm PSG, 2 μm D-CRIPT6, and 50 μm unlabeled CRIPT. In analogy to single-mix stopped-flow experiments, the double-jump experiment was carried out at 10 °C and analyzed as described under “Results.”
Global fitting of kinetic data
Kinetic transients from single- and double-jump stopped-flow experiments were fitted globally to a minimal model (Fig. 5) using KinTek Explorer (26, 27). This software utilizes numerical integration and allows fitting of raw kinetic data directly to a mechanistic model. The interrupted binding experiments, with delay times varying from 24 ms to 200 s, were included in the global fit together with binding experiments in which [D-CRIPT6] varied between 2–14 μm. For practical reasons, the lower boundary of k4 and k−4 was set to 10−5 s−1 in the fitting procedure. kon for the CRIPT6 peptide used in dissociation experiments (with no dansyl group) was locked to be the same as for D-CRIPT6, and koff was locked to 0 (irreversible binding is practically achieved at concentrations where CRIPT6 ≫ D-CRIPT6). The remaining parameters in the model were fitted without constrains. To account for baseline shifts in the double-jump kinetic experiments, constants were added to the output expressions. A scaling factor was used to account for the difference in total fluorescence output between the single-jump and double-jump stopped-flow experiments.
Isothermal titration calorimetry
Isothermal titration calorimetry (ITC) experiments were performed in 50 mm sodium phosphate (pH 7.45), 21 mm KCl (I = 150), and 0.5 mm tris(2-carboxyethyl)phosphine at 20 °C, 25 °C, 30 °C, and 35 °C in an iTC200 instrument (Malvern). D-CRIPT6 and PSG were dialyzed overnight in the same buffer to minimize artifacts from buffer mismatch. D-CRIPT6 (220 μm) was titrated (16–19 injections) into a cell containing 22 μm PSG, and saturation was obtained by having approximately a two times excess of D-CRIPT6. Baseline correction was performed manually to minimize the χ value in the curve fitting using the software provided with the iTC200 instrument.
Author contributions
L. L. data curation; L. L., E. K., S. G., and P. J. formal analysis; L. L. and E. K. investigation; L. L. and E. K. visualization; L. L., E. K., and S. G. methodology; L. L., E. K., S. G., and P. J. writing-review and editing; S. G. and P. J. conceptualization; P. J. resources; P. J. supervision; S. G. and P. J. funding acquisition; P. J. writing-original draft; P. J. project administration.
This work was funded in part by the European Union's Horizon 2020 Research and Innovation Program under Marie Sklodowska-Curie Grant Agreement 675341 (to S. G. and P. J.) and by Swedish Research Council Grant 2016-04965 (to P. J.). The authors declare that they have no conflicts of interest with the contents of this article.
- SH
- Src homology
- GK
- guanylate kinase-like
- CRIPT
- CXXC repeat–containing interactor of PDZ3 domain
- ITC
- isothermal titration calorimetry
- DJ
- double jump.
References
- 1. Sheng M., and Kim E. (2011) The postsynaptic organization of synapses. Cold Spring Harb. Perspect. Biol. 3, pii: a005678 10.1101/cshperspect.a005678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Feng W., and Zhang M. (2009) Organization and dynamics of PDZ-domain-related supramodules in the postsynaptic density. Nat. Rev. Neurosci. 10, 87–99 10.1038/nrn2540 [DOI] [PubMed] [Google Scholar]
- 3. Ivarsson Y. (2012) Plasticity of PDZ domains in ligand recognition and signaling. FEBS Lett. 586, 2638–2647 10.1016/j.febslet.2012.04.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tompa P. (2012) On the supertertiary structure of proteins. Nat. Chem. Biol. 8, 597–600 10.1038/nchembio.1009 [DOI] [PubMed] [Google Scholar]
- 5. Long J.-F., Tochio H., Wang P., Fan J.-S., Sala C., Niethammer M., Sheng M., and Zhang M. (2003) Supramodular structure and synergistic target binding of the N-terminal tandem PDZ domains of PSD-95. J. Mol. Biol. 327, 203–214 10.1016/S0022-2836(03)00113-X [DOI] [PubMed] [Google Scholar]
- 6. McGee A. W., Dakoji S. R., Olsen O., Bredt D. S., Lim W. A., and Prehoda K. E. (2001) Structure of the SH3-guanylate kinase module from PSD-95 suggests a mechanism for regulated assembly of MAGUK scaffolding proteins. Mol. Cell. 8, 1291–1301 10.1016/S1097-2765(01)00411-7 [DOI] [PubMed] [Google Scholar]
- 7. Tavares G. A., Panepucci E. H., and Brunger A. T. (2001) Structural characterization of the intramolecular interaction between the SH3 and guanylate kinase domains of PSD-95. Mol. Cell. 8, 1313–1325 10.1016/S1097-2765(01)00416-6 [DOI] [PubMed] [Google Scholar]
- 8. McCann J. J., Zheng L., Rohrbeck D., Felekyan S., Kühnemuth R., Sutton R. B., Seidel C. A., and Bowen M. E. (2012) Supertertiary structure of the synaptic MAGuK scaffold proteins is conserved. Proc. Natl. Acad. Sci. 109, 15775–15780 10.1073/pnas.1200254109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhang J., Lewis S. M., Kuhlman B., and Lee A. L. (2013) Supertertiary structure of the MAGUK core from PSD-95. Structure 21, 402–413 10.1016/j.str.2012.12.014 [DOI] [PubMed] [Google Scholar]
- 10. Pan L., Chen J., Yu J., Yu H., and Zhang M. (2011) The structure of the PDZ3-SH3-GuK tandem of ZO-1 protein suggests a supramodular organization of the membrane-associated guanylate kinase (MAGUK) family scaffold protein core. J. Biol. Chem. 286, 40069–40074 10.1074/jbc.C111.293084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tonikian R., Zhang Y., Sazinsky S. L., Currell B., Yeh J.-H., Reva B., Held H. A., Appleton B. A., Evangelista M., Wu Y., Xin X., Chan A. C., Seshagiri S., Lasky L. A., Sander C., et al. (2008) A specificity map for the PDZ domain family. PLoS Biol. 6, e239 10.1371/journal.pbio.0060239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Niethammer M., Valtschanoff J. G., Kapoor T. M., Allison D. W., Weinberg R. J., Craig A. M., and Sheng M. (1998) CRIPT, a novel postsynaptic protein that binds to the third PDZ domain of PSD-95/SAP90. Neuron 20, 693–707 10.1016/S0896-6273(00)81009-0 [DOI] [PubMed] [Google Scholar]
- 13. Zhang L., Jablonski A. M., Mojsilovic-Petrovic J., Ding H., Seeholzer S., Newton I. P., Nathke I., Neve R., Zhai J., Shang Y., Zhang M., and Kalb R. G. (2017) SAP97 binding partner CRIPT promotes dendrite growth in vitro and in vivo. eNeuro 4, pii: ENEURO.0175–17.2017 10.1523/ENEURO.0175-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Doyle D. A., Lee A., Lewis J., Kim E., Sheng M., and MacKinnon R. (1996) Crystal structures of a complexed and peptide-free membrane protein-binding domain: molecular basis of peptide recognition by PDZ. Cell 85, 1067–1076 10.1016/S0092-8674(00)81307-0 [DOI] [PubMed] [Google Scholar]
- 15. Saro D., Li T., Rupasinghe C., Paredes A., Caspers N., and Spaller M. R. (2007) A thermodynamic ligand binding study of the third PDZ domain (PDZ3) from the mammalian neuronal protein PSD-95. Biochemistry 46, 6340–6352 10.1021/bi062088k [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gianni S., Haq S. R., Montemiglio L. C., Jürgens M. C., Engström Å., Chi C. N., Brunori M., and Jemth P. (2011) Sequence-specific long range networks in PSD-95/discs large/ZO-1 (PDZ) domains tune their binding selectivity. J. Biol. Chem. 286, 27167–27175 10.1074/jbc.M111.239541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zeng M., Ye F., Xu J., and Zhang M. (2018) PDZ ligand binding-induced conformational coupling of the PDZ-SH3-GK tandems in PSD-95 family MAGUKs. J. Mol. Biol. 430, 69–86 10.1016/j.jmb.2017.11.003 [DOI] [PubMed] [Google Scholar]
- 18. Pascoe H. G., Gutowski S., Chen H., Brautigam C. A., Chen Z., Sternweis P. C., and Zhang X. (2015) Secondary PDZ domain-binding site on class B plexins enhances the affinity for PDZ-RhoGEF. Proc. Natl. Acad. Sci. U.S.A. 112, 14852–14857 10.1073/pnas.1508931112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ivarsson Y., and Jemth P. (2019) Affinity and specificity of motif-based protein-protein interactions. Curr. Opin. Struct. Biol. 54, 26–33 [DOI] [PubMed] [Google Scholar]
- 20. Erlendsson S., Thorsen T. S., Vauquelin G., Ammendrup-Johnsen I., Wirth V., Martinez K. L., Teilum K., Gether U., and Madsen K. L. (2019) Mechanisms of PDZ domain scaffold assembly illuminated by use of supported cell membrane sheets. eLife 8, e39180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rademacher N., Kunde S.-A., Kalscheuer V. M., and Shoichet S. A. (2013) Synaptic MAGUK multimer formation is mediated by PDZ domains and promoted by ligand binding. Chem. Biol. 20, 1044–1054 10.1016/j.chembiol.2013.06.016 [DOI] [PubMed] [Google Scholar]
- 22. Zeng M., Shang Y., Araki Y., Guo T., Huganir R. L., and Zhang M. (2016) Phase transition in postsynaptic densities underlies formation of synaptic complexes and synaptic plasticity. Cell 166, 1163–1175.e12 10.1016/j.cell.2016.07.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rademacher N., Kuropka B., Kunde S.-A., Wahl M. C., Freund C., and Shoichet S. A. (2019) Intramolecular domain dynamics regulate synaptic MAGUK protein interactions. eLife 8, e41299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gianni S., Engström A., Larsson M., Calosci N., Malatesta F., Eklund L., Ngang C. C., Travaglini-Allocatelli C., and Jemth P. (2005) The kinetics of PDZ domain-ligand interactions and implications for the binding mechanism. J. Biol. Chem. 280, 34805–34812 10.1074/jbc.M506017200 [DOI] [PubMed] [Google Scholar]
- 25. Bagshaw C. R. (2017) Biomolecular kinetics: A Step-by-Step Guide, CRC Press, Boca Raton, FL [Google Scholar]
- 26. Johnson K. A., Simpson Z. B., and Blom T. (2009) Global kinetic explorer: a new computer program for dynamic simulation and fitting of kinetic data. Anal. Biochem. 387, 20–29 10.1016/j.ab.2008.12.024 [DOI] [PubMed] [Google Scholar]
- 27. Johnson K. A., Simpson Z. B., and Blom T. (2009) FitSpace explorer: an algorithm to evaluate multidimensional parameter space in fitting kinetic data. Anal. Biochem. 387, 30–41 10.1016/j.ab.2008.12.025 [DOI] [PubMed] [Google Scholar]
- 28. Monod J., Wyman J., and Changeux J. P. (1965) On the nature of allosteric transitions: a plausible model. J. Mol. Biol. 12, 88–118 10.1016/S0022-2836(65)80285-6 [DOI] [PubMed] [Google Scholar]
- 29. Qian Y., and Prehoda K. E. (2006) Interdomain interactions in the tumor suppressor discs large regulate binding to the synaptic protein GukHolder. J. Biol. Chem. 281, 35757–35763 10.1074/jbc.M607057200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nomme J., Fanning A. S., Caffrey M., Lye M. F., Anderson J. M., and Lavie A. (2011) The Src homology 3 domain is required for junctional adhesion molecule binding to the third PDZ domain of the scaffolding protein ZO-1. J. Biol. Chem. 286, 43352–43360 10.1074/jbc.M111.304089 [DOI] [PMC free article] [PubMed] [Google Scholar]