

Abstract
Dental enamel comprises interwoven arrays of extremely long and narrow crystals of carbonated hydroxyapatite called enamel rods. Amelogenin (AMELX) is the predominant extracellular enamel matrix protein and plays an essential role in enamel formation (amelogenesis). Previously, we have demonstrated that full-length AMELX forms higher-order supramolecular assemblies that regulate ordered mineralization in vitro, as observed in enamel rods. Phosphorylation of the sole AMELX phosphorylation site (Ser-16) in vitro greatly enhances its capacity to stabilize amorphous calcium phosphate (ACP), the first mineral phase formed in developing enamel, and prevents apatitic crystal formation. To test our hypothesis that AMELX phosphorylation is critical for amelogenesis, we generated and characterized a hemizygous knockin (KI) mouse model with a phosphorylation-defective Ser-16 to Ala-16 substitution in AMELX. Using EM analysis, we demonstrate that in the absence of phosphorylated AMELX, KI enamel lacks enamel rods, the hallmark component of mammalian enamel, and, unlike WT enamel, appears to be composed of less organized arrays of shorter crystals oriented normal to the dentinoenamel junction. KI enamel also exhibited hypoplasia and numerous surface defects, whereas heterozygous enamel displayed highly variable mosaic structures with both KI and WT features. Importantly, ACP-to-apatitic crystal transformation occurred significantly faster in KI enamel. Secretory KI ameloblasts also lacked Tomes' processes, consistent with the absence of enamel rods, and underwent progressive cell pathology throughout enamel development. In conclusion, AMELX phosphorylation plays critical mechanistic roles in regulating ACP-phase transformation and enamel crystal growth, and in maintaining ameloblast integrity and function during amelogenesis.
Keywords: biomineralization, tooth, tooth development, extracellular matrix protein, phosphorylation, amelogenesis, amelogenin, dental enamel, knock-in mouse model
Introduction
Dental enamel, the hardest and most highly mineralized tissue in the human body, is comprised of parallel arrays of extremely long and narrow crystals of carbonated hydroxyapatite (HA),4 called enamel rods, which form an intricate interwoven (decussating) structure. This key structural feature of mammalian enamel provides enamel with a unique combination of high hardness and fracture toughness (1–3) that support its vital function. The enamel structure is the product of amelogenesis, a process tightly regulated by specialized epithelial cells, called ameloblasts, involving the secretion, self-assembly, and proteolytic processing of extracellular enamel matrix proteins (EMPs) that control the size, shape, and organization of enamel mineral crystals within each rod (4). Importantly, during enamel secretion each ameloblast forms a highly specialized secretory apparatus called a Tomes' process, which is responsible for the formation of one enamel rod (5, 6). Highly coordinated movements of ameloblasts during the appositional growth of enamel lead to the formation of the decussating pattern (7, 8). Key EMPs that include amelogenin (AMELX) (9, 10), the major (90%) component of the extracellular enamel matrix, and lesser amounts of ameloblastin (AMBN) (11) and enamelin (ENAM) (12, 13), have each been shown to be essential for dental enamel formation, through the use of knockout and genetically modified mouse models. Mutations in these and other extracellular matrix molecules have also been associated with genetic defects in human enamel (14). These essential EMPs undergo proteolysis by matrix metalloproteinase 20 (MMP20) during the secretory stage of amelogenesis (13, 15–18), when the initial enamel rod structure forms. This organized structure, which serves as a template for mature enamel, is comprised of long thin ribbons of mineral particles within an enamel matrix that is 80–90% (by volume) protein. Once the full thickness of the enamel layer is established, resident proteinases, including kallikrein 4 (KLK4) (19), bring about the almost complete removal of the extracellular enamel matrix during the maturation stage of amelogenesis, resulting in the volumetric growth of the initially formed enamel ribbons leading to a tissue that is >95% mineral (by weight) and only 1–2% water and proteins. MMP20 (20) and KLK4 (19) that regulate proteolysis during amelogenesis have been also shown to be essential for proper enamel formation.
Studies from our laboratories have focused on elucidating the role AMELX plays in amelogenesis. The primary sequence of AMELX is comprised of three amino acid domains: a 45-amino acid N-terminal domain that is rich in tyrosine, a large central domain that is primarily hydrophobic, and an 11-amino acid hydrophilic C terminus (reviewed in Ref. 4). The primary structure of the N- and C-terminal domains are almost completely conserved across mammalian species, whereas variations occur in the central portion of the protein. Of note, the highly conserved N terminus contains the only post-translational modification in AMELX, i.e. the phosphorylation of Ser-16. The conservation of the primary structures of the N and C termini lead to the suggestion that these AMELX domains play important roles in amelogenesis (21–23). Consistent with this idea, we have shown that full-length nonphosphorylated (24–26) and phosphorylated (albeit at lower concentrations) (27, 28) AMELXs have the capacity to guide the formation of bundles of aligned apatitic crystals in vitro, similar to those found in developing enamel, unlike their proteolytic cleavage products that lack the hydrophilic C-terminal domain. Similar behavior was observed with the leucine-rich amelogenin peptide (LRAP) (29), an alternative splice product of AMELX comprised of the N- and C-terminal domains of AMELX. We have also found that native (porcine) phosphorylated full-length AMELX (30) and its primary phosphorylated proteolytic cleavage product (26) have enhanced capacities to stabilize amorphous calcium phosphate (ACP) mineral phase precursors and to inhibit apatite crystal growth in vitro (25–28, 30, 31), compared with their recombinant nonphosphorylated counterparts. These findings lead us to propose that the stabilization of ACP may serve as a means to control enamel mineral formation (32). Studies by us (31, 33) and others (34) suggest that the single phosphorylation at Ser-16 in AMELX may alter protein conformation and protein-mineral interactions that could enhance its capacity to stabilize ACP and inhibit apatitic crystal formation (29, 35). These in vitro findings are of significant biological importance, as ACP is the first mineral phase that forms in newly deposited enamel mineral, which subsequently undergoes transformation to HA-like enamel crystals during the secretory stage of amelogenesis (36, 37). Importantly, this enamel formation strategy, involving metastable transient mineral phases, appears to be universally utilized in the development of mineralized tissues (38, 39). These prior findings provide us with a strong scientific basis on which to evaluate the impact of a key mutation in native AMELX (i.e. the lack of amelogenin phosphorylation) on the mechanism of enamel formation.
Based on these collective findings we hypothesize that phosphorylation of Ser-16 in AMELX plays an essential role in regulating enamel mineral formation. To test this hypothesis in vivo, we have developed a novel knockin (KI) mouse that lacks AMELX phosphorylation. Here, we demonstrate for the first time that AMELX phosphorylation is essential for proper tooth enamel formation, based on the analyses of this new KI mouse model. Our present findings show that AMELX phosphorylation plays a critical role in regulating the rate of enamel mineral phase transformation, appositional enamel crystal growth, and in maintaining the integrity and functional capacity of the ameloblast cell layer to form the enamel rod structure.
Results
Generation and validation of KI mice carrying a phosphorylation-incompetent S16A mutation in the Amelx gene
KI mice, carrying a single point mutation of AGC to GCC (S16A) in the exon 3 of the Amelx gene, were successfully generated as described under “Experimental procedures” and in Fig. 1A. The S16A mutation and the targeting vector integration were confirmed by PCR (Fig. 1B) and also by Southern blotting (Fig. S1). Both heterozygous (HET) female and hemizygous KI male mice had normal fertility and growth without apparent developmental defects, once weaned from a gel-diet at 5 weeks.
Figure 1.

Generation of AMELX-S16A knockin mice. A, schematic diagram of WT AMELX locus, targeted and neomycin resistance cassette-deleted alleles. Exon 3 was replaced with the knockin construct containing the S16A point mutation (black bar) with FRT-flanked neomycin selection cassette. FRT sites are represented by gray triangles. Not drawn to scale. B, PCR using F1/R1 primers amplify the 2.60-kb fragment. ES clones 1–4 were identified as positive and selected for expansion. DNA from an individual clone and no DNA were used as positive (+) and negative (−) controls, respectively. Confirmation of the point mutation was performed by PCR using F2/R2 primers. This reaction produces a product 0.99 kb in size. Primer set F3/R3 was used to screen F1 female mice for the deletion of the Neo cassette. The PCR product for the WT is 254 bp and for the Neo-deleted, 289 bp. Heterozygous female mice were set up to mate with WT C57BL/6 males to generate offspring.
To assess AMELX expression in the secretory stage of developing enamel and confirm its phosphorylation status in KI and HET mice, protein extracts of 5-day-old mouse molars were similarly subjected to SDS-PAGE and Western blotting analyses. As shown with SDS-PAGE analyses (Fig. 2A), relative protein levels appear equivalent in wildtype (WT), HET, and KI molar enamel extracts. The absence of AMELX phosphorylation in KI enamel was verified by Western blot and immunofluorescence analyses, using a polyclonal antibody specific for the amino acid sequence surrounding the phosphorylated Ser-16 of native mouse AMELX (anti-pS16AMELX), as described under “Experimental procedures.” The specificity of the anti-pS16AMELX antibody was first established by Western blot analyses using nonphosphorylated full-length recombinant AMELX, rM179, and 5-day-old mouse molar extracts (M5d) of WT enamel. As shown in Fig. 2B, anti-rM179 (40) detected both recombinant nonphosphorylated rM179 and a 5-day-old WT mouse molar extracts that is comprised of native phosphorylated AMELXs. In contrast, anti-pS16AMELX only detected native phosphorylated AMELXs in the WT enamel extracts. In Western blot analyses using anti-rM179 and anti-pS16AMELX, phosphorylated AMELX levels are shown to be significantly reduced in HET enamel, relative to that seen in the WT, and undetectable in KI enamel extracts, as expected (Fig. 2C), validating the efficacy of the knockin S16A mutation. These findings were further confirmed in paraffin sections of developing enamel using immunofluorescence. The fluorescent signal of AMELX using anti-AMELX (FL-191) in enamel was detected in both WT and KI incisor sections (Fig. 2, D and E), whereas there is a clear absence of phosphorylated AMELX in the KI enamel layer (Fig. 2G), unlike in the WT (Fig. 2F), as assessed using anti-pS16AMELX.
Figure 2.

Verification of the absence of AMELX phosphorylation in the AMELX-S16A mouse model. A, SDS-PAGE analyses of 5-day molar (M5d) enamel extracts of WT, HET, and KI enamel. B, Western blot analyses to validate of the specificity of anti-pS16AMELX (anti-pS16) that selectively reacts with native phosphorylated mouse amelogenins from M5d extracts of WT enamel, but does not react with recombinant nonphosphorylated full-length mouse AMELX, rM179 (molecular mass = 20.2 kDa (40)). C, using anti-rM179 and anti-pS16AMELX (anti-pS16), Western blot analyses show that phosphorylated AMELX levels are significantly reduced in HET enamel, relative to the WT, and undetected in KI enamel extracts. Using anti-AMELX (FL-191), immunofluorescence (red) shows the presence of AMELX in both WT (D, mid-to-late secretory stage) and KI enamel (E, mid-to-late secretory stage), whereas the use of anti-pS16AMELX confirms the absence of phosphorylated AMELX in KI enamel (G, early-to-mid secretory stage), compared with that seen in WT enamel (F, mid-to-late secretory stage). Observed differences in enamel in the mid-to-late secretory stage in WT (D) and KI (E) are described under “Results.”
The AMELX-S16A mutation has a dramatic impact on mature and developing enamel structures
Light microscope and scanning electron microscopy (SEM) analyses of erupted enamel
Marked differences in the appearance of erupted mandibular incisors from adult WT, HET, and KI mice were observed at low magnification using a stereomicroscope. In sharp contrast to WT mouse incisors that exhibit a smooth, shiny and naturally pigmented enamel surface, KI incisor surfaces appear to be hypoplastic, rough, and covered with numerous protrusions, with a notable loss of the characteristic yellow iron pigmentation seen in WT enamel (41), as shown in Fig. 3 (upper panels). The tips of KI incisors also appear to be more rounded, compared with that seen for WT incisors, reflecting differences in mechanical wear properties (Fig. 3, upper and lower panels). Of note, HET incisor surfaces show a range of phenotypes, exhibiting varying proportions of the characteristic features associated with WT and KI enamel surfaces (Fig. 3, upper and lower panels). Differences in enamel surfaces attributed to the AMELX-S16A mutation were also observed using scanning electron microscopy (SEM). WT incisors again exhibit smooth enamel surfaces (Fig. 4A), whereas numerous large mineral nodules were present on KI enamel surfaces (Fig. 4B). SEM again shows that HET enamel exhibits a wide range of phenotypes reflecting varying contributions of both WT, with few to no surface defects (Fig. 4C), and KI enamel surface characteristics (Fig. 4D). Similar surface defects were also seen on KI molars (Fig. S2). SEM images of KI molars showed the presence of large mineralized nodules, in contrast to the smooth enamel surfaces observed for WT molars. The cusps of KI molars also appear rounded compared with the sharp, chisel-like appearance of WT molar cusps, suggesting that the enamel of KI cusps is much thinner than that of the WT.
Figure 3.

Comparison of erupted mandibular incisors from adult WT, HET, and KI mice. Appearance of 8-week-old incisor surfaces (upper panel) and profiles (lower panels) at low magnification using a stereomicroscope reveal differences in the shape and surface characteristics of enamel layers of each genotype, as described in the text.
Figure 4.
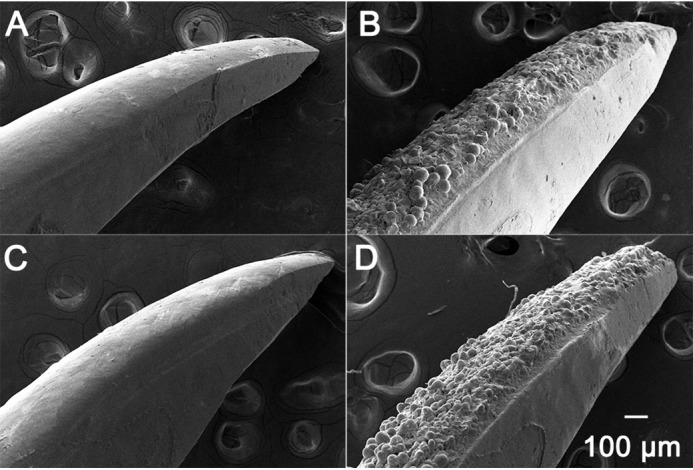
SEM images of enamel surfaces from WT, KI, and HET mandibular incisors. WT incisors exhibit a smooth enamel surface (A), whereas KI incisor surfaces are characterized by a rough enamel surface comprised of numerous spherical nodules (B). Examples of enamel surfaces of HET incisors show that HET enamel exhibits a wide range of phenotypes with few to no surface defects (C), as in WT enamel, to surfaces that are highly defective and remarkably similar to those of KI enamel (D).
Most notably, SEM examination of underlying enamel structures (Fig. 5, A–C) reveal that KI and some HET enamel layers lack the characteristic decussating enamel rod structure, the key structural component of mammalian enamel, as found in WT enamel (Fig. 5A). As shown in Fig. 5C, KI mineral particles grow abnormally in a direction that is perpendicular to the dentinoenamel junction (DEJ). Again, the HET enamel ultrastructure has a highly variable appearance, exhibiting defects that range from severe (Fig. 5B), resembling highly defective KI enamel, to mild (Fig. 5D), with a WT-like enamel structure, or with a mosaic pattern of WT-like and KI-like features within a single tooth (Fig. 5E). Based on SEM analyses of both fractured and cut/polished incisors (Fig. 5F), KI (41.7 ± 6.7 μm, not including surface deposits; n = 8) enamel layers are significantly (p < 0.001) thinner than those of the WT (112.4 ± 13.7 μm; n = 8). Interestingly, in HET incisors, enamel thicknesses within areas in which rod-like structures (116.4 ± 18.4 μm; n = 5) are predominant and within areas that mostly lack enamel rods (46.9 ± 4.9 μm; n = 6) are similar to those found in WT and KI incisors, respectively, and significantly (p < 0.01) different from each other.
Figure 5.

Examples of SEM images of polished and etched cross-sections of WT, HET, and KI enamel from mandibular incisors. WT enamel shows (A) a characteristic interwoven decussating enamel rod pattern, whereas KI enamel (C) lacks the critically important rod structure, as does this particular example of HET enamel (B). Additional SEM examples of cross-sections of HET enamel again illustrate the variability of the enamel phenotype found in HET mice. D, an example of HET enamel with a decussating rod pattern, similar to that seen in WT (A) enamel that is clearly present throughout the HET enamel layer. E, a cross-section image of HET enamel that exhibits a mosaic structure, comprised of both a decussating enamel rod structure and a middle area that lacks a rod structure, as seen in KI (C) enamel. F, comparison of enamel thicknesses measured in WT (n = 8), KI (n = 8), and HET (rod (n = 5) and rodless (n = 6) enamel areas); *, p < 0.001; **, p < 0.01.
Microcomputed tomography (μCT) analyses of mature enamel
The relative mineral density of enamel of WT, HET, and KI mice at maturation was compared using μCT. Analyses show that for both KI and some HET incisors enamel layers at maturation are under-mineralized compared with the WT that exhibits a thick, homogeneous and dense enamel layer (e.g. Fig. 6). Notably, μCT of mutant HET and KI incisors show thin layers of inhomogeneous enamel with uneven surfaces, with a relatively more dense layer of mineral directly adjacent to the DEJ. In addition, KI enamel and some HET enamel specimens (as shown) exhibit ectopic mineral deposits (Fig. 6, arrows) that appear to correspond to the protrusions on the enamel surface that are observed by light stereoscopy (Fig. 3) and SEM (Fig. 4, B and D).
Figure 6.

μCT analyses of mature enamel from WT, HET, and KI enamel. Analyses show that KI enamel layers at maturation are under-mineralized compared with the thick WT enamel layer. The example of the HET enamel shown is similar to KI enamel. As discussed, KI enamel and some HET enamel specimens (as shown) exhibit ectopic mineral deposits (arrows). Note the presence of molar M1 roots, indicative of the enamel maturation stage. These images were obtained using an Xradia MicroXCT-200 instrument (see “Experimental procedures”).
Transmission electron microscopy (TEM) and selected area electron diffraction (SAED) analyses of enamel development in WT, HET, and KI incisors
WT enamel mineral particles initially grow perpendicular to the DEJ at the beginning of the secretory stage of amelogenesis (grid 1, see “Experimental procedures” for method details) to form a thin rodless (aprismatic) enamel layer on top of dentin (Fig. 7A, Fig. S3). This process is regulated by presecretory ameloblasts (42, 43). SAED results indicate that this thin mineral layer is amorphous (ACP) in nature (Fig. S4A). As enamel development progresses, the characteristic decussating enamel rod structure that is reflected in mature erupted WT enamel (Fig. 5A), begins to form at grid 2 (early secretory stage: data not shown), with each interwoven enamel rod comprised of bundles of long thin ribbons of forming enamel mineral, as more clearly seen by the midsecretory stage (grid 5) where the enamel layer has grown in thickness (Fig. 7C). As shown in Fig. 7E, SAED analyses reveal a diffuse ring diffraction pattern, characteristic of ACP, in the outer three selected areas (e.g. at location 1) indicating that newly deposited enamel close to the enamel surface/ameloblast interface is amorphous in WT enamel. Distinct reflections, which begin to appear at the next selected area from the enamel surface (Fig. S4, B and G) indicate that the amorphous enamel particles have begun to transform into crystals. From this point on, diffraction patterns characteristic of apatite become more pronounced as measurements get closer to the DEJ where the oldest enamel is found. Near the DEJ (e.g. Fig. 7C at location 2), electron diffraction shows well-defined narrow arcs of the 002 and 004 reflections of HA (Fig. 7E, location 2), consistent with the presence of well-aligned crystals with a narrow angular spread along their c-axes, as we have previously reported (36, 37). A change in directional growth of enamel (relative to the DEJ) shown in Fig. 7C from that seen in Fig. 7A occurs as the enamel rod structure (Fig. 7C) forms on top of the aprismatic enamel layer (Fig. 7A). This change that takes place as secretory ameloblasts develop Tomes' processes (see below), a specialized secretory apparatus responsible for the formation of the enamel rod structure (42, 44), and change their movement trajectories, leading to the formation of the decussating rod pattern (Fig. 7C).
Figure 7.

Selected TEM images and SAED results of the developing enamel matrix of WT and KI maxillary incisors. A, early secretory stage (grid 1) in WT enamel exhibiting a rodless enamel layer on more densely mineralized dentin. B, early secretory stage of enamel formation (grid 1) in KI enamel, where a rodless enamel layer is again observed, as seen in WT enamel (A). C, mid-secretory stage (grid 5) in WT enamel. A thicker enamel layer with an enamel rod structure is clearly observed. White circle and diamond-shaped markers represent locations of SAED measurements. SAED results at diamond-shaped marker locations are presented in E. Additional results are found in Fig. S4. D, mid-secretory stage (grid 4) of KI enamel, as throughout KI enamel development, grows perpendicular to the DEJ and lacks an enamel rod structure. E, selected SAED findings near the enamel surface (C, location 1; D, location 3) and close to the DEJ (C, location 2; D, location 4). F, comparison of rates of ACP transformation to apatitic crystals in developing WT (n = 3), HET (n = 2), and KI (n = 3) enamel. Mean ± S.D. with different letter notations are significantly different (p < 0.05). The biological and mechanistic importance of noted differences in ACP stabilization and rates of mineral phase transformation are discussed in the text.
In KI enamel, initial mineral deposits were again found to grow perpendicular to the DEJ at the beginning of the secretory stage (Fig. 7B, grid 1), as in the WT. In addition, mineral deposits closer to the enamel surface were found to be primarily amorphous, whereas SAED results showed some diffraction near the DEJ, indicating the presence of crystals (Fig. S4, C and G). These results are more clearly seen as KI development continues, as shown in the beginning (Fig. S4D) and at the end (Fig. S4, E and G) of grid 2 (early secretory stage). In grid 2 (Fig. S4E), we began to see some breakage and loss of mineral above the DEJ. A greater loss of mineral was seen as KI enamel development continues (Fig. S4, D–F), as shown in the early/middle secretory stage (Fig. 7D, grid 4). Nevertheless, consistent findings on mineral phase transformation in KI enamel were obtained, as the outer enamel layer was found to remain intact. Importantly, in contrast to WT enamel, TEM reveals (e.g. Fig. 7D and Fig. S4F) that the characteristic enamel rod structure does not form at any point during KI enamel development as enamel continues to grow in a direction perpendicular to the DEJ, consistent with SEM findings (Fig. 5C). Loss of KI enamel mineral above the DEJ is possibly due to a fragile nature of KI enamel and lack of the characteristic enamel rod structure. Newly deposited mineral near the KI enamel surface (Figs. 7, D and E, location 3) is again found to be amorphous, based on the diffuse ring appearance of the diffraction patterns, whereas older enamel nearer to the DEJ is comprised of aligned apatitic crystals as observed in WT secretory enamel (Fig. 7, D and E, location 4), based on the presence of narrow arcs of the 002 and 004 reflections of HA. Upon closer examination at higher magnification, however, KI enamel was found to lack exceptionally long and aligned enamel crystal ribbons that are characteristic of WT enamel rods (Fig. 8A) and, rather, appears to be comprised of less organized arrays of substantially shorter crystals (Fig. 8B).
Figure 8.

Comparison of TEM images of WT and KI enamel at higher magnification. A, WT enamel rods, with an individual rod running from the lower left to the upper right of the image, in the mid-secretory stage (grid 8) and B, KI enamel also in the mid-secretory stage (grid 7). Results show that KI enamel (B) lacks the very long enamel crystal ribbons that are characteristic of WT enamel rods (A), and is less organized than WT enamel and comprised of shorter crystals (black arrows).
As found in mature enamel by SEM (Fig. 5, B, D, and E), TEM analyses show that developing HET enamel can exhibit either a WT-like (i.e. a decussating enamel rod structure, as seen in Fig. S5A) or a KI-like (i.e. a lack of rod structure and growth perpendicular to the DEJ, as seen in Fig. S5B) enamel structure, or sometimes a mosaic combination of the two phenotypes, as seen in Fig. S5C. In this latter example of a mosaic HET structure, it is interesting to note that enamel growth in the KI-like rodless area and, to some extent, in the noted “prismatic area,” appears to take place in a direction that is perpendicular to the DEJ and not at an angle, as seen in the WT decussating enamel rod pattern (Fig. 7C).
The AMELX-S16A mutation affects the rate of ACP transformation to apatitic crystals in developing enamel
In support of our working hypothesis, we have found that the observed ACP to apatitic crystal transformation described in the preceding section occurs significantly (p < 0.05) faster (i.e. closer to the enamel surface/ameloblast cell interface where EMP secretion takes place) in KI (2.3 ± 0.6 μm; n = 3) enamel that lacks phosphorylated AMELX compared with that found in WT (8.1 ± 1.7 μm; n = 3) and HET (4.8 ± 0.14 μm; n = 2) enamel. This key finding is presented graphically in Fig. 7F.
Light and electron microscopy observations of ameloblast cell layers in WT and KI enamel
Histological analyses show that early ameloblasts from 8-week-old KI incisors appear to be normal at the presecretory and early secretory stage of enamel deposition. Specifically, KI ameloblasts appear as highly polarized cells forming a well-organized epithelial layer at these early stages of development, similar to WT ameloblasts (Fig. 9, A, B, F, and G). Shortly after the onset of enamel deposition, Tomes' processes start to develop at the secretory end of WT ameloblasts, signifying the transition from presecretory to secretory ameloblasts (Fig. 9, B, C (inset), and D), However, in contrast, Tomes' processes fail to develop in KI ameloblasts (Fig. 9, G and H). By the time enamel reached a thickness of ∼30 μm at the midsecretory stage, the KI ameloblast layer became less organized and abnormal secretory aggregates (SA) started to form inside the ameloblast layer (Fig. 9H). This disruption of the KI ameloblast layer became more prominent in the late secretory stage, at which point ameloblasts exhibited a loss of polarity and an increase in the number of SA (Fig. 9I). At the maturation stage the KI ameloblast layer was completely disintegrated, however, proteolysis and removal of enamel matrix processes remained active, based on the decrease of histological staining in the extracellular space (Fig. 9J).
Figure 9.

Histological analyses of ameloblasts from 8-week-old WT (A–E) and KI incisors (F–J), as a function of stages of enamel development. A and F, presecretory; B and G, early secretory; C and H, mid-secretory; D and I, late secretory; E and J, maturation. C (inset, scale bar: 10 μm) shows the presence of Tomes' processes in WT ameloblasts at higher magnification. AB, ameloblasts; DE, dentin; EN, enamel; OD, odontoblasts; P, pulp; PAB, pre-ameloblast; PL, papillary layer; SI, stratum intermedium; arrowheads, Tomes' processes; asterisks (*), SA.
TEM studies of secretory stage ameloblasts of WT incisors revealed highly organized rough endoplasmic reticulum (ER) in the distal portion of ameloblasts and well-established distal junctional complexes, which demarcate the boundaries of the Tomes' processes (Fig. 10A). The ameloblasts also featured well-developed distal Tomes' processes (DTPs) secreting enamel rods (Fig. 10, A and B). The proximal Tomes' processes (PTP) and distal portions of ameloblast cell bodies contained large secretory compartments (LSC), responsible for secretion of the interrod (Fig. 10, A and B). The ultrastructural organization of secretory ameloblasts in WT incisors observed here is in consensus with results of earlier studies (45–47). In contrast to the WT ameloblasts, early secretory ameloblasts in KI incisors lacked DTPs (Fig. 10, C and D). Furthermore, although the region of cytoplasm corresponding to PTP of WT ameloblasts, distal to the junctional complexes, was present, it is structurally different from the WT PTP. Specifically, LSCs in the KI ameloblast layers appear to locate intercellularly (Fig. 10, C and D) and not intracellularly, as in the WT ameloblast layer (Fig. 10, A and B). The ER in the ameloblast cell bodies is less organized than in the WT (Fig. 10, A, C, and D). Consistent with the lack of DTPs in KI early secretory ameloblasts, a rod pattern is not observed in KI enamel. We also investigated mid-to-late secretory enamel of KI incisors. Similarly to the early secretory KI ameloblasts, mid-to-late secretory ameloblasts lacked DTPs, while maintaining the area of cytoplasm, distal to junctional complexes corresponding to the PTP (Fig. 11, A, C, and D). The ameloblasts secreted a homogeneous matrix into the extracellular space, consistent with the rodless KI enamel layer described earlier. Furthermore, the ameloblast layer started to lose its integrity and large SA started to form by ameloblasts that lost their axial polarity and were found to bend their distal end ∼90° from the cell axis (Fig. 11, A and B). These ultrastructural features are indicative of progressive cell pathology developing in ameloblasts during the secretory stage of amelogenesis.
Figure 10.

TEM micrographs of demineralized and resin-embedded sections of WT and KI incisors during the secretory stage of enamel formation. A, WT secretory stage ameloblasts. B, close up of the boxed area in A. C, KI early secretory stage ameloblasts. D, close up of boxed area in C. EN, enamel; ER, rough endoplasmic reticulum; DTP, distal Tomes' process; IR, interrod; LSC, large secretory compartments; MD, mantle dentin; PTP, proximal Tomes' process; arrows, distal junctional complexes; double-headed arrow, area of PTP in WT secretory ameloblasts; SV, secretory vesicles.
Figure 11.

TEM micrographs of demineralized and resin-embedded sections of KI incisors during the secretory stage of enamel formation. A, KI mid-to-late secretory stage ameloblasts. B, close up of upper boxed area in A. C, close up of lower boxed area in A. D, close up of the boxed area in C. EN, enamel; ER, rough endoplasmic reticulum; MT, mitochondria; PTP, proximal Tomes' process; arrows, distal junctional complexes.
Discussion
Dental enamel development is an exquisite example of biomineralization, the formation of organized mineralized structures through highly-regulated cellular and molecular processes, given its intricate decussating enamel rod structure formed under the control of ameloblasts that undergo concerted morphological and functional changes as development progresses (e.g. Ref. 43). Ameloblasts regulate the secretion, self-assembly, and proteolytic processing of EMPs that guide initial mineral formation during the secretory stage of amelogenesis. During the maturation stage, ameloblasts are responsible for the near complete removal of extracellular matrix components (43) to allow for the volumetric growth of initial enamel crystals to produce the hardest mineralized tissue in the human body that has evolved to last for a lifetime of use. Although the organic matrix of forming enamel is transient, which is uncommon in biomineralization (48), setting amelogenesis apart from other biomineralization processes, the interplay between the extracellular enamel matrix and ameloblast function is evident during the protein-rich secretory stage of amelogenesis, when the enamel rod structure is established in WT enamel, but fails to form within the KI enamel matrix that lacks phosphorylated AMELXs. As has been shown in X-linked enamel disorders caused by other AMELX mutations (10, 49), HET enamel exhibits a wide range of highly variable defective structures, due to X-chromosome inactivation (lyonization) (50, 51) and the random distribution of ameloblasts that secrete either phosphorylated or nonphosphorylated AMELXs, again reflecting the impact of the interplay between the nature of the extracellular matrix and ameloblast function in controlling enamel structural organization.
Our initial in vitro findings (26, 30) lead us to the present study using KI mice to test the hypothesis that Ser-16 AMELX phosphorylation plays an essential role in amelogenesis. Although other yet to be determined factors may be involved, we have shown for the first time that the highly-conserved single phosphorylated serine site in AMELX plays an essential role in regulating enamel crystal formation, enamel thickness, and in maintaining the integrity and functional capacity of the ameloblast cell layer to form the characteristic enamel rod pattern.
The key discovery of our study is that AMELX phosphorylation plays an important mechanistic role in the stabilization of an ACP mineral phase precursor in vivo, during the secretory stage of enamel development. This finding is in excellent agreement with the results of our in vitro experiments (26, 30), which show that phosphorylation at Ser-16 enhances the capacity of AMELX to stabilize ACP and inhibit its transformation into apatitic crystals. Based on studies using the AMELX alternative splice product LRAP, we (31, 33) and others (34) have obtained results that suggest single-site AMELX phosphorylation at Ser-16 may alter protein conformation and protein-mineral interactions in a manner that enhances the ACP stabilization properties of phosphorylated AMELX (29, 35). In the present study, we provide strong ultrastructural and crystallographic evidence (Fig. 7) that support our conclusion that amelogenin phosphorylation fulfills an important mechanistic function in the regulation of mineral phase transformation during the secretory stage of amelogenesis through its enhanced capacity to stabilize ACP. Importantly, we have found that the rate of transformation of ACP to apatitic crystals (Fig. 7F) is significantly greater (by a factor of 3.5) in KI enamel than in WT enamel. In addition, we have found that KI enamel crystals (Fig. 8B) fail to elongate to the remarkable extent observed for WT enamel crystals (Fig. 8A). Therefore, the absence of Ser-16 phosphorylated AMELX in KI enamel leads to a decrease in the capacity of the extracellular matrix to slow the rate of the ACP to apatitic crystal transformation process (Fig. 7F) and to guide the appositional growth (elongation) of forming enamel crystals. These findings are most significant to our understanding of the mechanism of enamel development, as discussed below, given the fact that we have previously shown that the shape and organization of developing enamel crystals that comprise WT enamel rods are established prior to ACP transformation to crystalline material (36, 37).
The formation and transformation of amorphous mineral phases in both vertebrate (calcium phosphate based) and invertebrate (calcium carbonate based) systems represents a universal strategy used in regulating biological mineralization (38), including dental enamel, bone, mollusk shells, and echinoderm skeletal elements (36, 39, 52–56). Our current in vivo findings that demonstrate the role of AMELX phosphorylation in stabilizing ACP in secretory enamel are consistent with earlier TEM studies that suggested ACP as the first mineral phase in forming enamel. More recently (36), using multiple physical characterization approaches, we have unequivocally demonstrated that transient ACP is the initial mineral phase in secretory enamel and provided new insight into the mechanism of enamel formation (see below).
As noted earlier, full-length AMELXs can guide the formation of aligned bundles of apatitic crystals in vitro, similar to those found in developing enamel (24–27, 32, 57). Full-length AMELXs have also been found to organize initially formed mineral nanoparticles into linear chain-like structures prior to the formation of parallel arrays of mineral particles in vitro (25, 26, 57). Moreover, full-length AMELXs, in the absence of mineral, can assemble into higher-order structures such as nanospheres and their chain-like assemblies, in contrast to amelogenins that lack the hydrophilic C terminus (24, 25, 30, 39, 58–60). Accordingly, based on these findings, we previously proposed that super-assemblies of full-length amelogenin play a key role in guiding the formation of linear arrays of ACP nanoparticles, the first mineral phase found to form in developing enamel, which subsequently align, fuse, and transform into bundles of ribbon-like apatitic crystals during the secretory stage of amelogenesis (39). Together with our current findings, a modified version of the mechanism of protein-mediated enamel development is now presented (Fig. 12). This model emphasizes the importance of the enhanced capacity of AMELX phosphorylation in stabilizing ACP, while promoting enamel mineral ribbon elongation prior to ACP transformation to crystalline material. The model is consistent with the one proposed by Robinson and co-workers (61–64) based on the examination of developing rat incisor enamel prepared using a freeze fracturing technique (64). Results obtained using that approach lead these investigators to propose that the formation of enamel crystals proceeded through the linear assembly of observed globular structures (50 nm in diameter) that contained enamel matrix proteins and amorphous mineral particles, although the amorphous nature of the mineral phase was not established. It was further proposed that these nanomineral deposits subsequently fuse and transform into characteristically long apatitic enamel crystals, following proteolytic processing of matrix components. Although much remains to be learned, there is a growing body of evidence to support particle attachment mechanisms associated with synthetic, geologic, and biologic mineral formation (65). Accordingly, our proposed model involving protein-mediated mineral particle attachment (Fig. 12) is further supported by our prior cryo-EM studies of the growth of calcium phosphate crystals in the presence of amelogenin in vitro in which this stepwise process was observed (25, 57).
Figure 12.

Proposed mechanisms for the growth of mineral ribbons in WT (A) and KI (B) enamel. A, native phosphorylated AMELXs stabilize ACP nanoparticles (1) during the secretory stage of amelogenesis. These nanoparticles undergo assembly to form ACP clusters that align to generate linear mineral arrays that grow from the DEJ by protein-mediated particle attachment (2 and 3), as ameloblasts recede. These arrays then fuse to form stable, long and well-organized ACP ribbons (4) within developing enamel rods. Subsequently, transformation of ACP ribbons to parallel arrays of apatitic crystals begins (5) in older enamel near the DEJ. This transformation may be promoted by the processing of enamel matrix components, as previously proposed (36, 39, 62) and noted in the text. During this process, phosphorylated AMELXs (symbolized by curved and straight, black lines) stabilize ACP particles and prevent their transformation to apatitic crystals. B, in the absence of AMELX phosphorylation in KI enamel, S16A mutated AMELXs (symbolized by curved and straight gray lines) transiently stabilize initially formed ACP nanoparticles, during the secretory stage of amelogenesis. As in the WT, these nanoparticles undergo assembly to form ACP clusters that align to form linear arrays, as ameloblasts recede. In contrast to the WT, however, ACP clusters in KI enamel undergo more rapid fusion and phase transformation to form relatively short arrays of well-aligned crystal ribbons (6), which are less organized (7) than the longer enamel crystal ribbons seen in WT enamel (5). As discussed in the text, a reduction in the capacity of nonphosphorylated AMELXs to stabilize ACP leads to a decrease in the ability of the mutated extracellular matrix in KI enamel to sustain enamel crystal elongation, as illustrated here.
Given the fact that the enamel structural organization is established prior to its transformation from ACP to apatitic crystals in WT enamel and our current observations, it is reasonable to assume that a reduction in the capacity of the extracellular enamel matrix to stabilize ACP would affect the kinetics of mineral cluster fusion and lead to a decrease in the ability of the mutated extracellular matrix to foster and sustain enamel crystal elongation. This suggestion is consistent with the observed enhanced rate of ACP transformation to apatitic crystals (Fig. 7F) and the formation of shorter enamel crystals in KI enamel (Fig. 8), as we have illustrated (Fig. 12). This proposal is further supported by in vitro evidence (66) that has demonstrated (in the absence and presence of biological additives) that the formation of higher-order assemblies of nano-HA particles was only possible when nano-HA particles have a surface shell of ACP. The ACP shell serves to link the nanoparticles prior to ACP transformation to HA, leading to the generation of larger single crystals. Alternatively, the lack of AMELX phosphorylation and the associated reduced capacity of the enamel matrix to suppress crystallization may allow for the formation of more critical mineral nuclei during the early secretory stage that subsequently lead to the generation of numerous and shorter enamel crystals in KI enamel. Interestingly, despite the lack of AMELX phosphorylation, the shorter arrays of enamel crystals in KI enamel are well-aligned, based on SAED analyses, as are the much longer crystals in WT enamel (Fig. 7). This finding is consistent with our prior in vitro demonstration that nonphosphorylated full-length AMELXs (24–26), like native phosphorylated full-length AMELXs (27, 28), can guide the formation of aligned bundles of apatitic crystals, despite the fact that nonphosphorylated full-length AMELXs have a much lower capacity to stabilize ACP (26). Notably, in perfect agreement with these in vitro observations and in support of our main hypothesis, our current in vivo studies clearly demonstrate that the presence of phosphorylated amelogenin is critical for reducing the rate of ACP to HA transformation. Moreover, the capacity of the enamel matrix to stabilize ACP enamel mineral precursors is essential for the appositional growth of the very long and well-aligned enamel mineral crystals in WT enamel rods (Fig. 12).
As we have shown, the failure to form an enamel rod structure during the secretory stage of amelogenesis in KI enamel coincides with the lack of DTP that are required for the development of the enamel rod structure (47, 67), where each enamel rod is generated by a Tomes' process of a single ameloblast (5). Our ultrastructural findings also indicate progressive cell pathology of ameloblasts during the secretory stage of amelogenesis in KI enamel, further illustrating the critical importance of AMELX phosphorylation in enamel formation. Although intracellular factors involved in AMELX S16A mutant enamel development may affect ameloblast cell biology and function, it has been suggested that extracellular “stimuli” generated by proper enamel formation are essential for ameloblasts to sustain their phenotypic Tomes' processes (68). As in the present study, the lack of a rod structure in mature Mmp20-KO (69) and Amelx-KO (10) enamel is associated with the loss or absence of ameloblast Tomes' processes, respectively. Amelx-KO mice similarly exhibit mineralized surface nodules and a reduced enamel thickness (20.3 ± 3.3 μm) that is approximately one-half of that observed in AMELX S16A KI mice. The Amelx-KO mouse enamel layer was also characterized as being most highly mineralized near the DEJ, as we have found for both HET and KI enamel (Fig. 6). The dramatic KI enamel phenotype, in these respects similar to enamel produced in the complete absence of AMELX (Amelx-KO), further supports the notion that phosphorylation of the single Ser-16 residue is absolutely essential for the function of this predominant enamel matrix protein. However, in further comparison to AMELX S16A KI enamel, in addition to being two times thinner, the Amelx-KO enamel layer was found to contain fan-shaped structures comprised of plate-like enamel crystals (10). Prompted by the unusual appearance of these plate-like crystals, these investigators used X-ray diffraction to find that the predominant mineral phase in Amelx-KO enamel is octacalcium phosphate (OCP), unlike WT enamel mineral that corresponded to HA. In contrast, such abnormal mineral deposits were not observed by us in either KI or WT enamel. Although KI and WT enamel crystals appear similarly apatitic in nature, additional testing is needed to check for the presence of octacalcium phosphate in KI enamel. Nevertheless, apparent differences between KI and Amelx-KO enamel structures likely reflect the fact that native AMELX has other key functional domains in addition to the phosphorylated N terminus, as has been demonstrated (e.g. Refs. 24–26 and 70).
Although factors that influence the interplay between the extracellular enamel matrix and ameloblasts are not well-understood, we have recently suggested that local changes in mineral ion chemistry in the extracellular enamel environment brought about by uncharacteristic mineral formation in Mmp20-KO enamel (e.g. abnormal changes in ion activities such as calcium and hydrogen (pH)), as also seen in the present study and in the Amelx-KO, may affect molecular signaling, leading to disrupted ameloblast cell biology, including loss of Tomes' processes, and function (37). More specifically, factors that alter mineralization in developing enamel, as found in the present study, may induce a loss of function effect on the capacity of the ameloblast layer to form the enamel rod structure. The fact that presecretory and early secretory stage ameloblasts develop normally (Fig. 9, A, B, F, and G) and start to deposit rodless enamel in KI enamel, but fail to form DTP and later develop cell pathology (Fig. 9, H–J), supports the notion that the lack of AMELX phosphorylation alters the extracellular environment and that these changes affect ameloblast cell biology. The proposed impact of the extracellular environment on ameloblast function, however, may not simply be an indirect response to altered mineralization but, rather, a further reflection of the highly concerted cellular and extracellular nature of the amelogenesis process.
It is also worth noting that Ser-16 is a part of the highly conserved N-terminal amino acid sequence that is involved in protein-protein interactions (71). Our earlier studies indicate that there are small but potentially important changes in AMELX structure and assembly, associated with its phosphorylation (30, 57). It is likely, therefore, that differences in assembly kinetics and the structural organization of phosphorylated versus nonphosphorylated AMELX in the developing enamel matrix may also contribute to differences in the structural organization of enamel mineral at the nano- and mesoscale. Protein-protein interactions of native AMELX with other essential EMPs (i.e. AMBN, ENAM), which are believed to play important roles in enamel formation (72–74), may also be affected by AMELX phosphorylation.
The phosphorylation of AMELX, along with other key EMPs present in lesser amounts, i.e. AMBN and ENAM, occurs intracellularly by casein kinase FAM20C (also called Golgi casein kinase) (75). Prior studies indicate that phosphorylation of both AMBN and ENAM plays an important role in enamel formation. Altered serine phosphorylation sites (S216L) in ENAM have been shown to cause severe amelogenesis imperfecta (76). Using a transgenic mouse model, it has also been suggested that serine phosphorylation (with three putative phosphorylation sites) is an essential component of ABMN function in enamel formation (77). The present study is the first to show that phosphorylation of AMELX, the predominant enamel matrix protein, is critically important for proper enamel formation. Consistent with the importance of EMP phosphorylation in amelogenesis, FAM20C has been found to be essential for enamel formation (78). Recently, mutations in FAM20A, a binding partner of FAM20C (79, 80) and a member of a family of genes including, FAM20B and FAM20C, which encode kinases, have been associated with amelogenesis imperfecta and gingival fibrosis syndrome (AIGFS) (75). Subsequent studies with families with AIGFS (later diagnosed to have enamel-renal syndrome (ERS), due to the detection of calcifications in the kidney) demonstrate that FAM20A−/− molars lack true enamel with a complete absence of long thin crystals and evidence of an enamel rod structure (75). Enamel surfaces of molars of these ERS subjects also exhibit surface regions that show resorption pits or missing enamel, along with “knob-like” calcifications, similar to the nodular surface defects we observe in AMELX-S16A mutant enamel (Fig. 4, Fig. S2). Additional studies (81) have shown that erupted enamel from an ERS patient was mostly comprised of smaller and less organized enamel crystals, compared with normal enamel, which was reported to cover a thin “inner-most” layer of prismatic enamel adjacent to the DEJ. The enamel phenotypes observed in our S16A mutant mice that lack AMELX phosphorylation are quite similar to the reported enamel phenotypes in humans affected by mutations in a key kinase that is believed to play a critical role in amelogenesis. This association further emphasizes the biological and potential clinical importance of our present findings regarding amelogenin phosphorylation.
In summary, we have shown that Ser-16 phosphorylation of AMELX plays an essential role in enamel formation, including the stabilization of ACP, the regulation of ACP transformation and enamel crystal growth, enamel thickness, and in maintaining the integrity and functional capacity of the ameloblast cell layer to form the decussating enamel rod pattern, the hallmark of mammalian enamel structure. Of particular importance, our present findings show that Ser-16 AMELX phosphorylation plays a critical role in the regulation of the appositional growth of the extremely long and well-aligned enamel mineral crystals in WT enamel. The enhanced stabilization of ACP enamel mineral precursors by phosphorylated AMELXs appears to be essential for the regulation of this latter process, as described in our model (Fig. 12). On the basis of our present findings, we are lead to the conclusion that the sole post-translational phosphorylation of Ser-16 imparts to AMELX critical aspects of its essential functional capacity in amelogenesis (9, 10). As demonstrated in this study, given the significant impact of AMELX phosphorylation in amelogenesis, the AMELX-S16A KI mouse serves as a means to provide new insight into the overall mechanism of enamel formation and the importance of the kinetic control of biological mineralization through the stabilization of ACP and the inhibition of mineral formation by elements of the extracellular organic matrix (32).
Experimental procedures
Animal protocol approval
Animal care and use were carried out in accordance with protocols approved by The Forsyth Institute's Institutional Animal Care and Use Committee. The mice were housed in facilities approved by the Association for Assessment and Accreditation of Laboratory Animal Care.
Generation of KI mice lacking AMELX phosphorylation
Working with ingenious Targeting Laboratory (iTL) (Ronkonkoma, NY), we generated gene-targeted KI mice carrying a point mutation (Ser-16 to Ala-16: AGC ≫ GCC; S16A) on exon 3 in the X-chromosomal AMELX gene from WT C57BL/6 genomic DNA (Fig. 1A). The gene-targeting vector consisted of exons 1–5, where exon 3 contained the desired S16A point mutation with FRT-flanked neomycin selection cassette (Fig. 1A). The linearized targeting vector was transfected by electroporation into C57Bl/6 (B6) stem cells. After selection with G418 antibiotics, surviving clones were expanded for PCR analysis (Fig. 1B) to identify recombinant ES clones. The point mutation was confirmed by DNA sequencing and the targeted constructs were further verified by Southern blot analysis and the confirmed clones were injected into blastocysts to generate mouse chimeras. The male chimeras were then bred with female Flp recombinase expressing mice to excise the FRT-flanked neomycin selection cassette. These procedures were carried out at ingenious Targeting Laboratory. Neomycin-deleted AMELX-S16A heterozygous mice were then transferred to the animal facilities at The Forsyth Institute and bred with C57Bl/6 mice to obtain WT male and female, HET female, and KI male littermates. As reported for other enamel-defective mouse models (19), some KI mice had difficulty surviving after weaning at 3 weeks, presumably from difficulty eating solid food due to discomfort resulting from malformed enamel. Therefore, all mouse littermates were weaned at 4 weeks and were provided with a gel diet and powdered food for 1 week. Mice were then given solid and powdered food, and found to grow normally. As described in the following sections, developing and mature enamel tissues from WT, HET, and KI littermates were characterized using multiple approaches.
Antibody development for native phosphorylated AMELXs
To validate that the AMELX-S16A KI mouse lacks AMELX phosphorylation using Western blot analyses of extracts of developing enamel (see below), we first developed a rabbit polyclonal antibody specific for the amino acid sequence surrounding phosphorylated Ser-16 of native mouse AMELX (anti-pS16AMELX), using a 15-amino acid synthetic peptide (NH2-SPGYINLSPYEVLTPL). Anti-pS16AMELX antibodies were generated in rabbits (Pocono Rabbit Farm and Laboratory, Inc., Canadensis, PA), following 4 immunizations using the peptide, which was synthesized and HPLC-purified at the Tufts University Core Facility for Peptide Synthesis (Boston, MA). The serum obtained was purified by affinity chromatography using columns containing the nonphosphorylated AMELX peptide. Anti-pS16AMELX was also used for immunohistochemistry studies. As shown under “Results,” the novel anti-pS16AMELX antibody was found to react specifically with phosphorylated mouse AMELXs.
Western Blot analyses of enamel extracts from WT, HET, and KI mouse molars
First molars were extracted from WT, HET, and KI mice at 5 days postnatal using a dissecting microscope. Mouse molar enamel development at this postnatal age is in the secretory stage of amelogenesis (17, 82). After removing pulp tissues, molars were dissolved in 0.17 m HCl, 0.98% formic acid for 2 h at 4 °C with rocking. Following the removal of undissolved materials by centrifugation at 3,500 × g for 5 min at 4 °C, the molar extracts were concentrated using Amicon 3K Ultracentrifugal Filters (Millipore, Germany). The concentrated extracts were then subjected to SDS-PAGE and Western blot analyses, according to published procedures (40). SDS-PAGE (4–10 μg of protein/well) was run using 12% BisTris gels (Life Technologies) and stained with GelCode Blue Safe Protein Stain (Thermo Scientific). Replica gels were transblotted to polyvinylidene difluoride membrane (LC2002, Life Technologies) and immunostained using two different anti-AMELX primary antibodies, anti-rM179 (40) and anti-pS16AMELX, as described above. Anti-rM179 (40) that reacts with both phosphorylated and nonphosphorylated AMELXs (e.g. Fig. 2), a gift from Dr. James Simmer, was also used to aid in the characterization of developing enamel tissues, using Western blot analyses. Anti-rabbit IgG conjugated with horseradish peroxidase was used as a secondary antibody (Thermo Scientific). The membranes were then analyzed using an enhanced chemiluminescence method with a Luminatra Forte Western HRP Substrate (Millipore) and FluoroChem Q system (Alpha Innotech). Recombinant full-length mouse AMELX, rM179, supplied by Dr. James Simmer, was used as a control and to aid in the identification of protein bands.
Assessment of tooth morphology and characterization of WT, HET, and KI enamel structures
Light microscope and SEM analyses
Dissected hemimandibles from ∼8-week-old adult WT, HET, and KI mice were prepared and the remaining soft tissues were removed in a beetle chamber. The specimens were then rinsed in 70% ethanol and air dried for subsequent visual and SEM assessments. Erupted portions of incisor enamel were examined and photographed under a Zeiss stereomicroscope equipped with a Zeiss Axiocam HRc camera. For SEM analyses, erupted portions of incisors were fractured by hand along the transverse plane. The specimens (at least 6 specimens of each genotype) were mounted on aluminum stubs, sputter-coated with gold (Desk V; Denton Vacuum), and visualized using a Zeiss Evo LS10 SEM. In addition, hemimandibles were embedded in LR White resin (Electron Microscopy Sciences). Embedded incisors were then cut along the transverse plane, polished to 0.25 μm using diamond suspensions (Electron Microscopy Sciences, Hatfield, PA), etched with 30% phosphoric acid for 12 s, gold-coated and imaged, based on published procedures (83, 84). SEM images of fractured and acid-etched incisors (n = 5–8, for each genotype) were also used to determine enamel thickness. Where possible, multiple enamel thickness measurements were made and averaged for each incisor.
TEM and SAED analyses
The effect of the AMELX-S16A mutation on the development of enamel mineral structure during the secretory stage of amelogenesis was systematically assessed by TEM and SAED at defined intervals (i.e. within numbered TEM grid spacings (“grids”), ∼130 μm across, that outline developing incisor enamel structures), as we have previously described (37) and as shown in Fig. S3. These analyses take advantage of the continuously erupting mouse incisor in which all stages of enamel development can be seen in a single section, e.g. (43). Analyses were conducted using ultrathin sagittally cut sections of WT and mutant maxillary incisors starting from the first sign of mineral formation (grid 1) to early enamel maturation (grids 12–13). Maxillary incisors from 6- to 8-week-old adult mice of each genotype were isolated by dissection and immediately fixed with 70% ethanol followed by dehydration through graded ethyl alcohol treatment. The incisors were then infused with a 1:1 solution of 100% ethyl alcohol and LR White resin (Hard Grade, Electron Microscopy Sciences) overnight, followed by two repeated 2-h infusions with the LR White solution. Treated incisors were embedded in the LR White resin using a cold cure method. Ultra-thin incisor sections (70–100 nm thick) were cut in the sagittal plane with an ultramicrotome (PowerTome XL; RMC Products) using a diamond knife (DiATOME) and floated into a pool of distilled water pre-saturated with HA with a few added drops of ethanol. The sections were immediately mounted on 200-mesh carbon-coated copper TEM grids (Electron Microscopy Sciences). Developing enamel structures from each genotype were examined using TEM (1200EX, JEOL) in bright-field mode operated at 100 kV. SAED analyses of selected areas were also carried out to assess the nature of the enamel mineral phase and crystal organization. All images were captured by an AMT CCD camera. At least 5 maxillary incisors from each genotype group were examined.
Assessment of the rate of ACP transformation to apatitic enamel crystals
In selected sections prepared for TEM from different mice (3 for WT, 2 for HET, and 3 for KI), SAED measurements were carried out systematically from the enamel surface to the DEJ, with a 1–5– μm spacing between each measurement. SAED measurements traced the appositional growth of the enamel layer, as illustrated in Fig. 7, C and D. By carefully analyzing each SAED image, we determined whether the enamel particles within each SAED measurement area were comprised of ACP only (SAED only showing diffuse ring patterns) or a mixture of ACP and apatitic crystals (SAED patterns also showing diffraction dots and/or arc patterns), indicating that crystallization has occurred. The distance from the enamel surface at which ACP transformation to apatitic crystals took place was defined as the mean value of the furthest distance from the enamel surface at which only ACP was detected and the first measurement point that showed evidence that crystallization had begun. The distance of ACP transformation to apatite crystals were measured in several trajectories in each section, and the results were averaged and compared among each genotype. As reported under “Results,” the midportion of developing KI enamel is brittle and breaks away during the preparation of TEM sections using an ultramicrotome. However, as shown, the outer portion of the KI enamel layer generally remains intact and can be used for SAED measurements to determine the distance from the enamel surface where ACP transformation to apatitic crystals occurs in KI enamel.
μCT analyses of hemimandibles from WT, HET, and KI mice
μCT analyses were carried out using 8-week-old adult mouse incisors to compare enamel mineral of WT (n = 6), HET (n = 4), and KI (n = 4) mice at maturation. Hemimandibles free of soft tissues (described under “Experimental procedures”) were scanned using a Scanco Medical (Wayne, PA) μCT 40 at 70 kV, 114 mA, and 6-μm resolution. Images were processed with μCT 40 evaluation software and ImageJ was used to orient the images of incisors and molars for observation at the maturation stage, just before eruption and enclosed by bone (adapted from Ref. 83). Representative samples were also scanned using an Xradia MicroXCT-200 (Harvard School of Dental Medicine, Boston, MA) at 80 kV, 8 watts, and <1 μm resolution. The latter images were obtained and similarly assessed at the maturation stage prior to eruption, as signified by the presence of the roots of M1 molars, as shown.
Histology and immunohistochemistry of developing enamel in mandibular incisors of WT, HET, and KI mice
Dissected hemimandibles of adult mice were immediately fixed in buffered Zn-formalin (Thermo Fisher Scientific, Waltham, MA) for 18–24 h, rinsed with running water for 3–4 h, and decalcified in 10% EDTA, pH 8.0, with 0.2% paraformaldehyde and 0.05% glutaraldehyde for 3 weeks at 4 °C with rocking and changing to fresh solution every other day. The tissues were then dehydrated in a series of graded ethanol solutions and embedded in paraffin for sagittal sectioning. Sections (6 μm thick) were mounted onto glass slides and stained with hematoxylin and eosin for detailed morphological analyses. For immunofluorescence labeling, tissue sections were subjected to antigen retrieval by incubation in 10 mm sodium citrate, pH 6.0, using microwave heating. After washing with distilled deionized water and TBS, sections were blocked with 1% BSA with pre-immune serum and then incubated overnight with a primary antibody (either anti-AMELX (FL-191) (sc-32892, from Santa Cruz Biotechnology (Santa Cruz, CA)) or anti-pS16AMELX) at 4 °C, followed by secondary antibody incubation. For the secondary antibody, a goat anti-rabbit antibody with Alexa Fluor 647 dye (Life Technologies, Carlsbad, CA) was used. Prepared sections were mounted with ProLong Gold Antifade Mountant with 4′,6-diamidino-2-phenylindole (Thermo Fisher Scientific) and visualized using a Zeiss Axio Observer inverted wide-field fluorescence microscope.
TEM analyses of demineralized incisor sections
Hemimandibles were obtained from 6- to 8-week-old adult mice of each genotype, and immediately stored in Karnovsky fixative (2% glutaraldehyde, 2% formaldehyde in 10 mm PBS) and kept at 4 °C. After 24 h of fixation at 4 °C, samples were placed into a demineralization solution, containing 0.1 m EDTA (pH 7.2–7.4) for 1 to 2 weeks. The demineralization solution was changed every other day. For TEM studies, after demineralization, mandibular bone around the incisors was trimmed and the molars were removed. The distal fragments of the jaws containing apical portions of the incisors were further cross-sectioned into 1–1.5–mm thick pieces, processed, and embedded in LR White or Embed 812 (catalog numbers 14381 and 14120, Electron Microscopy Sciences, Hatfield, PA) according to published protocols (85). In brief, for Embed 812 processing, incisor pieces were post-fixed in 1% ferrocyanide-reduced osmium tetroxide for 1 h, washed in PBS, dehydrated in graded ethanol, and infiltrated with propylene oxide. The samples were embedded in Embed-812 and cured at 65 °C for 2 days. For LR White processing, some of the samples were post-fixed in osmium tetroxide, whereas others were not. Incisor pieces were washed in PBS, dehydrated in graded ethanol, embedded in LR White, and cured at 60 °C for 1–2 days. Resin blocks were sectioned into 70-nm thick sections using a Leica EM UC7 ultramicrotome (Leica Biosystems, Buffalo, IL) equipped with diamond knife (Electron Microscopy Sciences, Hatfield, PA). The sections were mounted on carbon-coated nickel grids (Electron Microscopy Sciences), allowed to dry, and then stained using 1% uranyl acetate staining and 1% lead citrate staining for 10 and 5 min, respectively.
Statistical analyses
Comparisons of measurements of enamel thickness and rates of ACP transformation to enamel crystals made between genotypes were evaluated using analysis of variance and/or t tests, with a level of significance set at a minimum of p = 0.05.
Author contributions
N.-Y. S. and H. Y. data curation; N.-Y. S., H. Y., E. B., X. Y., and H. C. M. formal analysis; N.-Y. S., H. Y., E. B., X. Y., S. S. M., and H. C. M. investigation; N.-Y. S., H. Y., E. B., X. Y., S. S. M., M. K. P., J. P. S., and H. C. M. methodology; N.-Y. S., H. Y., E. B., and H. C. M. writing-original draft; N.-Y. S., H. Y., E. B., X. Y., S. S. M., M. K. P., J. P. S., and H. C. M. writing-review and editing; E. B. and H. C. M. supervision; E. B. and H. C. M. project administration; S. S. M., M. K. P., J. P. S., and H. C. M. conceptualization; J. P. S. resources; H. C. M. funding acquisition.
Supplementary Material
Acknowledgments
We are grateful to Justine Dobeck (The Forsyth Institute) for help with mouse dissection, and the Center for Biological Imaging and the Nanofabrication and Nanocharacterization Core at the University of Pittsburgh (Pittsburgh, PA) for providing access to TEM. We also acknowledge John Martin for data collection and micro-CT analyses using the Xradia MicroXCT-200 instrument and the Harvard School of Dental Medicine for access to its micro-CT Core Facility.
This work was supported by National Institutes of Health NIDCR Grant R21-DE023425 (to H. C. M.), The Forsyth Institute, and the University of Pittsburgh School of Dental Medicine. The authors declare that they have no conflicts of interest with the contents of this article.The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S5.
- HA
- hydroxyapatite
- ACP
- amorphous calcium phosphate
- AIGFS
- amelogenesis imperfecta and gingival fibrosis syndrome
- AMBN
- ameloblastin
- AMELX
- amelogenin
- DEJ
- dentinoenamel junction
- DTP
- distal Tomes' process
- ENAM
- enamelin
- EMP
- enamel matrix protein
- ER
- endoplasmic reticulum
- ERS
- enamel-renal syndrome
- HET
- heterozygous mouse
- KI
- knockin mouse
- KLK4
- kallikrein 4
- KO
- knockout mouse
- LRAP
- leucine-rich amelogenin peptide
- LSC
- large secretory compartments
- MMP20
- matrix metalloproteinase-20
- PTP
- proximal Tomes' processes
- rM179
- recombinant non-phosphorylated full-length mouse amelogenin
- SA
- secretory aggregates
- SAED
- selected area electron diffraction
- SEM
- scanning electron microscopy
- TEM
- transmission electron microscopy
- WT
- wildtype
- μCT
- microcomputed tomography
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol.
References
- 1. Chai H., Lee J. J., Constantino P. J., Lucas P. W., and Lawn B. R. (2009) Remarkable resilience of teeth. Proc. Natl. Acad. Sci. U.S.A. 106, 7289–7293 10.1073/pnas.0902466106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bajaj D., and Arola D. D. (2009) On the R-curve behavior of human tooth enamel. Biomaterials 30, 4037–4046 10.1016/j.biomaterials.2009.04.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Baldassarri M., Margolis H. C., and Beniash E. (2008) Compositional determinants of mechanical properties of enamel. J. Dent. Res. 87, 645–649 10.1177/154405910808700711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Margolis H. C., Beniash E., and Fowler C. E. (2006) Role of macromolecular assembly of enamel matrix proteins in enamel formation. J. Dent. Res. 85, 775–793 10.1177/154405910608500902 [DOI] [PubMed] [Google Scholar]
- 5. Skobe Z. (2006) SEM evidence that one ameloblast secretes one keyhole-shaped enamel rod in monkey teeth. Eur. J. Oral Sci. 114, 338–350, 382 10.1111/j.1600-0722.2006.00305.x [DOI] [PubMed] [Google Scholar]
- 6. Leblond C. P., and Warshawsky H. (1979) Dynamics of enamel formation in the rat incisor tooth. J. Dent. Res. 58, 950–975 10.1177/00220345790580024901 [DOI] [PubMed] [Google Scholar]
- 7. Nishikawa S. (2017) Cytoskeleton, intercellular junctions, planar cell polarity, and cell movement in amelogenesis. J. Oral Biosci. 59, 197–204 10.1016/j.job.2017.07.002 [DOI] [Google Scholar]
- 8. Smith C. E., Hu Y., Hu J. C., and Simmer J. P. (2019) Characteristics of the transverse 2D uniserial arrangement of rows of decussating enamel rods in the inner enamel layer of mouse mandibular incisors. J. Anat. 235, 912–930 10.1111/joa.13053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gibson C. W., Yuan Z.-A., Hall B., Longenecker G., Chen E., Thyagarajan T., Sreenath T., Wright J. T., Decker S., Piddington R., Harrison G., and Kulkarni A. B. (2001) Amelogenin-deficient mice display an amelogenesis imperfecta phenotype. J. Biol. Chem. 276, 31871–31875 10.1074/jbc.M104624200 [DOI] [PubMed] [Google Scholar]
- 10. Hu Y., Smith C. E., Cai Z., Donnelly L. A., Yang J., Hu J. C., and Simmer J. P. (2016) Enamel ribbons, surface nodules, and octacalcium phosphate in C57BL/6 Amelx−/− mice and Amelx+/− lyonization. Mol. Genet. Genomic Med. 4, 641–661 10.1002/mgg3.252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Fukumoto S., Kiba T., Hall B., Iehara N., Nakamura T., Longenecker G., Krebsbach P. H., Nanci A., Kulkarni A. B., and Yamada Y. (2004) Ameloblastin is a cell adhesion molecule required for maintaining the differentiation state of ameloblasts. J. Cell Biol. 167, 973–983 10.1083/jcb.200409077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hu J. C., Hu Y., Smith C. E., McKee M. D., Wright J. T., Yamakoshi Y., Papagerakis P., Hunter G. K., Feng J. Q., Yamakoshi F., and Simmer J. P. (2008) Enamel defects and ameloblast-specific expression in Enam knock-out/lacZ knock-in mice. J. Biol. Chem. 283, 10858–10871 10.1074/jbc.M710565200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hu J. C., Hu Y., Lu Y., Smith C. E., Lertlam R., Wright J. T., Suggs C., McKee M. D., Beniash E., Kabir M. E., and Simmer J. P. (2014) Enamelin is critical for ameloblast integrity and enamel ultrastructure formation. PLoS ONE 9, e89303 10.1371/journal.pone.0089303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Smith C. E. L., Poulter J. A., Antanaviciute A., Kirkham J., Brookes S. J., Inglehearn C. F., and Mighell A. J. (2017) Amelogenesis imperfecta: genes, proteins, and pathways. Front. Physiol. 8, 435–435 10.3389/fphys.2017.00435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bartlett J., and Simmer J. (1999) Proteinases in developing dental enamel. Crit. Rev. Oral Biol. Medicine 10, 425–441 10.1177/10454411990100040101 [DOI] [PubMed] [Google Scholar]
- 16. Nagano T., Kakegawa A., Yamakoshi Y., Tsuchiya S., Hu J.-C., Gomi K., Arai T., Bartlett J. D., and Simmer J. P. (2009) Mmp-20 and Klk4 cleavage site preferences for amelogenin sequences. J. Dent. Res. 88, 823–828 10.1177/0022034509342694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yamakoshi Y., Richardson A. S., Nunez S. M., Yamakoshi F., Milkovich R. N., Hu J. C., Bartlett J. D., and Simmer J. P. (2011) Enamel proteins and proteases in Mmp20 and Klk4 null and double-null mice. Eur. J. Oral Sci. 119, 206–216 10.1111/j.1600-0722.2011.00866.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fukae M., and Tanabe T. (1998) Degradation of enamel matrix proteins in porcine secretory enamel. Connect. Tissue Res. 39, 123–129, 141–149 10.3109/03008209809023918 [DOI] [PubMed] [Google Scholar]
- 19. Simmer J. P., Hu Y., Lertlam R., Yamakoshi Y., and Hu J. C. (2009) Hypomaturation enamel defects in Klk4 knockout/LacZ knockin mice. J. Biol. Chem. 284, 19110–19121 10.1074/jbc.M109.013623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Caterina J. J., Skobe Z., Shi J., Ding Y., Simmer J. P., Birkedal-Hansen H., and Bartlett J. D. (2002) Enamelysin (matrix metalloproteinase 20)-deficient mice display an amelogenesis imperfecta phenotype. J. Biol. Chem. 277, 49598–49604 10.1074/jbc.M209100200 [DOI] [PubMed] [Google Scholar]
- 21. Fincham A. G., Moradian-Oldak J., and Simmer J. P. (1999) The structural biology of the developing dental enamel matrix. J. Struct. Biol. 126, 270–299 10.1006/jsbi.1999.4130 [DOI] [PubMed] [Google Scholar]
- 22. Paine M. L., Zhu D.-H., Luo W., Bringas P. Jr., Goldberg M., White S. N., Lei Y.-P., Sarikaya M., Fong H. K., and Snead M. L. (2000) Enamel biomineralization defects result from alterations to amelogenin self-assembly. J. Struct. Biol. 132, 191–200 10.1006/jsbi.2000.4324 [DOI] [PubMed] [Google Scholar]
- 23. Paine C. T., Paine M. L., and Snead M. L. (1998) Identification of tuftelin- and amelogenin-interacting proteins using the yeast two-hybrid system. Connect Tissue Res. 38, 257–267; discussion 295–303 10.3109/03008209809017046 [DOI] [PubMed] [Google Scholar]
- 24. Beniash E., Simmer J. P., and Margolis H. C. (2005) The effect of recombinant mouse amelogenins on the formation and organization of hydroxyapatite crystals in vitro. J. Struct. Biol. 149, 182–190 10.1016/j.jsb.2004.11.001 [DOI] [PubMed] [Google Scholar]
- 25. Fang P. A., Conway J. F., Margolis H. C., Simmer J. P., and Beniash E. (2011) Hierarchical self-assembly of amelogenin and the regulation of biomineralization at the nanoscale. Proc. Natl. Acad. Sci. U.S.A. 108, 14097–14102 10.1073/pnas.1106228108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kwak S.-Y., Wiedemann-Bidlack F. B., Beniash E., Yamakoshi Y., Simmer J. P., Litman A., and Margolis H. C. (2009) Role of 20-kDa amelogenin (P148) phosphorylation in calcium phosphate formation in vitro. J. Biol. Chem. 284, 18972–18979 10.1074/jbc.M109.020370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kwak S. Y., Yamakoshi Y., Simmer J. P., and Margolis H. C. (2016) MMP20 proteolysis of native amelogenin regulates mineralization in vitro. J. Dent. Res. 95, 1511–1517 10.1177/0022034516662814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kwak S.-Y., Kim S., Yamakoshi Y., Simmer J. P., Beniash E., and Margolis H. C. (2014) Regulation of calcium phosphate formation by native amelogenins in vitro. Connect. Tissue Res. 55, 21–24 10.3109/03008207.2014.923853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Le Norcy E., Kwak S. Y., Wiedemann-Bidlack F. B., Beniash E., Yamakoshi Y., Simmer J. P., and Margolis H. C. (2011) Leucine-rich amelogenin peptides regulate mineralization in vitro. J. Dent. Res. 90, 1091–1097 10.1177/0022034511411301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wiedemann-Bidlack F. B., Kwak S.-Y., Beniash E., Yamakoshi Y., Simmer J. P., and Margolis H. C. (2011) Effects of phosphorylation on the self-assembly of native full-length porcine amelogenin and its regulation of calcium phosphate formation in vitro. J. Struct. Biol. 173, 250–260 10.1016/j.jsb.2010.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Le Norcy E., Kwak S.-Y., Allaire M., Fratzl P., Yamakoshi Y., Simmer J. P., and Margolis H. C. (2011) Effect of phosphorylation on the interaction of calcium with leucine-rich amelogenin peptide. Eur. J. Oral Sci. 119, 97–102 10.1111/j.1600-0722.2011.00900.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Margolis H. C., Kwak S.-Y., and Yamazaki H. (2014) Role of mineralization inhibitors in the regulation of hard tissue biomineralization: relevance to initial enamel formation and maturation. Front. Physiol. 5, 339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yamazaki H., Beniash E., Yamakoshi Y., Simmer J. P., and Margolis H. C. (2017) Protein phosphorylation and mineral binding affect the secondary structure of the leucine-rich amelogenin peptide. Front. Physiol. 8, 450 10.3389/fphys.2017.00450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Masica D. L., Gray J. J., and Shaw W. J. (2011) Partial high-resolution structure of phosphorylated and non-phosphorylated leucine-rich amelogenin protein adsorbed to hydroxyapatite. J. Phys. Chem. C Nanomater. Interfaces 115, 13775–13785 10.1021/jp202965h [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Connelly C., Cicuto T., Leavitt J., Petty A., Litman A., Margolis H. C., and Gerdon A. E. (2016) Dynamic interactions of amelogenin with hydroxyapatite surfaces are dependent on protein phosphorylation and solution pH. Colloids Surf. B Biointerfaces 148, 377–384 10.1016/j.colsurfb.2016.09.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Beniash E., Metzler R. A., Lam R. S., and Gilbert P. U. (2009) Transient amorphous calcium phosphate in forming enamel. J. Struct. Biol. 166, 133–143 10.1016/j.jsb.2009.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yamazaki H., Tran B., Beniash E., Kwak S. Y., and Margolis H. C. (2019) Proteolysis by MMP20 prevents aberrant mineralization in secretory enamel. J. Dent. Res. 98, 468–475 10.1177/0022034518823537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Weiner S., and Addadi L. (2011) Crystallization Pathways in Biomineralization. Annu. Rev. Materials Res. 41, 21–40 10.1146/annurev-matsci-062910-095803 [DOI] [Google Scholar]
- 39. Margolis H. C., and Beniash E. (2010) The role of amelogenin in dental enamel formation: a universal strategy for protein-mediated biomineralization. in Amelogenins: Multifaceted Proteins for Dental and Bone Formation and Repair, pp. 133–142, Bentham ebooks 10.2174/978160805171711001010133 [DOI] [Google Scholar]
- 40. Simmer J. P., Lau E. C., Hu C. C., Aoba T., Lacey M., Nelson D., Zeichner-David M., Snead M. L., Slavkin H. C., and Fincham A. G. (1994) Isolation and characterization of a mouse amelogenin expressed in Escherichia coli. Calcif. Tissue Int. 54, 312–319 10.1007/BF00295956 [DOI] [PubMed] [Google Scholar]
- 41. Smith C. E. (1998) Cellular and chemical events during enamel maturation. Crit. Rev. Oral Biol. Med. 9, 128–161 10.1177/10454411980090020101 [DOI] [PubMed] [Google Scholar]
- 42. Nanci A. (2007) Ten Cate's Oral Histology: Development, Structure, and Function, Elsevier Health Sciences, New York [Google Scholar]
- 43. Hu J. C., Chun Y. H., Al Hazzazzi T., and Simmer J. P. (2007) Enamel formation and amelogenesis imperfecta. Cells Tissues Organs 186, 78–85 10.1159/000102683 [DOI] [PubMed] [Google Scholar]
- 44. Warshawsky H., and Smith C. E. (1971) A three-dimensional reconstruction of the rods in rat maxillary incisor enamel. Anat. Rec. 169, 585–591 10.1002/ar.1091690308 [DOI] [PubMed] [Google Scholar]
- 45. Franklin D. L., Severs N. J., and Katchburian E. (1991) Development of the distal end and Tomes' processes of ameloblasts observed by freeze-fracture and ultrathin section electron microscopy. J. Anat. 174, 103–114 [PMC free article] [PubMed] [Google Scholar]
- 46. Kallenbach E. (1973) The fine structure of Tomes' process of rat incisor ameloblasts and its relationship to the elaboration of enamel. Tissue Cell 5, 501–524 10.1016/S0040-8166(73)80041-2 [DOI] [PubMed] [Google Scholar]
- 47. Kallenbach E. (1977) Fine structure of secretory ameloblasts in the kitten. Am. J. Anat. 148, 479–511 10.1002/aja.1001480406 [DOI] [PubMed] [Google Scholar]
- 48. Weiner S., and Lowenstam H. (1986) Organization of extracellularly mineralized tissues: a comparative study of biological crystal growth. Crit. Rev. Biochem. 20, 365–408 10.3109/10409238609081998 [DOI] [PubMed] [Google Scholar]
- 49. Hart P. S., Aldred M. J., Crawford P. J., Wright N. J., Hart T. C., and Wright J. T. (2002) Amelogenesis imperfecta phenotype–genotype correlations with two amelogenin gene mutations. Arch. Oral. Biol. 47, 261–265 10.1016/S0003-9969(02)00003-1 [DOI] [PubMed] [Google Scholar]
- 50. Witkop C. J. (1967) Partial expression of sex-linked recessive amelogenesis imperfecta in females compatible with the Lyon hypothesis. Oral Surg. Oral Med. Oral Pathol. 23, 174–182 10.1016/0030-4220(67)90092-8 [DOI] [PubMed] [Google Scholar]
- 51. Wright J. T. (2006) The molecular etiologies and associated phenotypes of amelogenesis imperfecta. Am. J. Med. Genet. A 140, 2547–2555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Beniash E., Aizenberg J., Addadi L., and Weiner S. (1997) Amorphous calcium carbonate transforms into calcite during sea urchin larval spicule growth. Proc. R. Soc. B Biol. Sci. 264, 461–465 10.1098/rspb.1997.0066 [DOI] [Google Scholar]
- 53. Weiss I. M., Tuross N., Addadi L., and Weiner S. (2002) Mollusc larval shell formation: amorphous calcium carbonate is a precursor phase for aragonite. J. Exp. Zool. 293, 478–491 10.1002/jez.90004 [DOI] [PubMed] [Google Scholar]
- 54. Mahamid J., Sharir A., Addadi L., and Weiner S. (2008) Amorphous calcium phosphate is a major component of the forming fin bones of zebrafish: indications for an amorphous precursor phase. Proc. Natl. Acad. Sci. U.S.A. 105, 12748–12753 10.1073/pnas.0803354105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Crane N. J., Popescu V., Morris M. D., Steenhuis P., and Ignelzi M. A. Jr. (2006) Raman spectroscopic evidence for octacalcium phosphate and other transient mineral species deposited during intramembranous mineralization. Bone 39, 434–442 10.1016/j.bone.2006.02.059 [DOI] [PubMed] [Google Scholar]
- 56. Mahamid J., Aichmayer B., Shimoni E., Ziblat R., Li C., Siegel S., Paris O., Fratzl P., Weiner S., and Addadi L. (2010) Mapping amorphous calcium phosphate transformation into crystalline mineral from the cell to the bone in zebrafish fin rays. Proc. Natl. Acad. Sci. U.S.A. 107, 6316–6321 10.1073/pnas.0914218107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Fang P.-A., Margolis H. C., Conway J. F., Simmer J. P., and Beniash E. (2013) CryoTEM study of effects of phosphorylation on the hierarchical assembly of porcine amelogenin and its regulation of mineralization in vitro. J. Struct. Biol. 183, 250–257 10.1016/j.jsb.2013.05.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Fincham A. G., Moradian-Oldak J., Simmer J. P., Sarte P., Lau E. C., Diekwisch T., and Slavkin H. C. (1994) Self-assembly of a recombinant amelogenin protein generates supramolecular structures. J. Struct. Biol. 112, 103–109 10.1006/jsbi.1994.1011 [DOI] [PubMed] [Google Scholar]
- 59. Moradian-Oldak J., Du C., and Falini G. (2006) On the formation of amelogenin microribbons. Eur. J. Oral Sci. 114, 327–329, 382 10.1111/j.1600-0722.2006.00285.x [DOI] [PubMed] [Google Scholar]
- 60. Aichmayer B., Margolis H. C., Sigel R., Yamakoshi Y., Simmer J. P., and Fratzl P. (2005) The onset of amelogenin nanosphere aggregation studied by small-angle X-ray scattering and dynamic light scattering. J. Struct. Biol. 151, 239–249 10.1016/j.jsb.2005.06.007 [DOI] [PubMed] [Google Scholar]
- 61. Robinson C., and Connell S. D. (2017) Crystal initiation structures in developing enamel: possible implications for caries dissolution of enamel crystals. Front. Physiol. 8, 405 10.3389/fphys.2017.00405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Robinson C., Shore R. C., Wood S. R., Brookes S. J., Smith D. A., Wright J. T., Connell S., and Kirkham J. (2003) Subunit structures in hydroxyapatite crystal development in enamel: implications for amelogenesis imperfecta. Connect. Tissue Res. 44, 65–71 [PubMed] [Google Scholar]
- 63. Robinson C. (2007) Self-oriented assembly of nano-apatite particles: a subunit mechanism for building biological mineral crystals. J. Dent. Res. 86, 677–679 10.1177/154405910708600801 [DOI] [PubMed] [Google Scholar]
- 64. Robinson C., Fuchs P., and Weatherell J. A. (1981) The appearance of developing rat incisor enamel using a freeze fracturing technique. J. Cryst. Growth 53, 160–165 10.1016/0022-0248(81)90062-2 [DOI] [Google Scholar]
- 65. De Yoreo J. J., Gilbert P. U., Sommerdijk N. A., Penn R. L., Whitelam S., Joester D., Zhang H., Rimer J. D., Navrotsky A., Banfield J. F., Wallace A. F., Michel F. M., Meldrum F. C., Cölfen H., and Dove P. M. (2015) Crystallization by particle attachment in synthetic, biogenic, and geologic environments. Science 349, aaa6760 10.1126/science.aaa6760 [DOI] [PubMed] [Google Scholar]
- 66. Tao J., Pan H., Zeng Y., Xu X., and Tang R. (2007) Roles of amorphous calcium phosphate and biological additives in the assembly of hydroxyapatite nanoparticles. J. Phys. Chem. B 111, 13410–13418 10.1021/jp0732918 [DOI] [PubMed] [Google Scholar]
- 67. Lacruz R. S., Nakayama Y., Holcroft J., Nguyen V., Somogyi-Ganss E., Snead M. L., White S. N., Paine M. L., and Ganss B. (2012) Targeted overexpression of amelotin disrupts the microstructure of dental enamel. PLoS ONE 7, e35200 10.1371/journal.pone.0035200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Hu Y., Hu J. C., Smith C. E., Bartlett J. D., and Simmer J. P. (2011) Kallikrein-related peptidase 4, matrix metalloproteinase 20, and the maturation of murine and porcine enamel. Eur. J. Oral Sci. 119, 217–225 10.1111/j.1600-0722.2011.00859.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Bartlett J. D., Yamakoshi Y., Simmer J. P., Nanci A., and Smith C. E. (2011) MMP20 cleaves E-cadherin and influences ameloblast development. Cells Tissues Organs 194, 222–226 10.1159/000324205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Pugach M. K., Li Y., Suggs C., Wright J. T., Aragon M. A., Yuan Z. A., Simmons D., Kulkarni A. B., and Gibson C. W. (2010) The amelogenin C-terminus is required for enamel development. J. Dent. Res. 89, 165–169 10.1177/0022034509358392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Wald T., Spoutil F., Osickova A., Prochazkova M., Benada O., Kasparek P., Bumba L., Klein O. D., Sedlacek R., Sebo P., Prochazka J., and Osicka R. (2017) Intrinsically disordered proteins drive enamel formation via an evolutionarily conserved self-assembly motif. Proc. Natl. Acad. Sci. U.S.A. 114, E1641–E1650 10.1073/pnas.1615334114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Gallon V., Chen L., Yang X., and Moradian-Oldak J. (2013) Localization and quantitative co-localization of enamelin with amelogenin. J. Struct. Biol. 183, 239–249 10.1016/j.jsb.2013.03.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Mazumder P., Prajapati S., Lokappa S. B., Gallon V., and Moradian-Oldak J. (2014) Analysis of co-assembly and co-localization of ameloblastin and amelogenin. Front. Physiol. 5, 274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Mazumder P., Prajapati S., Bapat R., and Moradian-Oldak J. (2016) Amelogenin-ameloblastin spatial interaction around maturing enamel rods. J. Dent. Res. 95, 1042–1048 10.1177/0022034516645389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Wang S.-K., Samann A. C., Hu J. C. C., and Simmer J. P. (2013) FAM20C functions intracellularly within both ameloblasts and odontoblasts in vivo. J. Bone Miner. Res. 28, 2508–2511 10.1002/jbmr.1990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Chan H.-C., Mai L., Oikonomopoulou A., Chan H. L., Richardson A. S., Wang S.-K., Simmer J. P., and Hu J. C. (2010) Altered enamelin phosphorylation site causes amelogenesis imperfecta. J. Dent. Res. 89, 695–699 10.1177/0022034510365662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Ma P., Yan W., Tian Y., He J., Brookes S. J., and Wang X. (2016) The importance of serine phosphorylation of ameloblastin on enamel formation. J. Dent. Res. 95, 1408–1414 10.1177/0022034516661513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Wang X., Wang S., Lu Y., Gibson M. P., Liu Y., Yuan B., Feng J. Q., and Qin C. (2012) FAM20C plays an essential role in the formation of murine teeth. J. Biol. Chem. 287, 35934–35942 10.1074/jbc.M112.386862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Ohyama Y., Lin J.-H., Govitvattana N., Lin I. P., Venkitapathi S., Alamoudi A., Husein D., An C., Hotta H., Kaku M., and Mochida Y. (2016) FAM20A binds to and regulates FAM20C localization. Sci. Rep. 6, 27784–27784 10.1038/srep27784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Cui J., Zhu Q., Zhang H., Cianfrocco M. A., Leschziner A. E., Dixon J. E., and Xiao J. (2017) Structure of Fam20A reveals a pseudokinase featuring a unique disulfide pattern and inverted ATP-binding. eLife 6, e23990 10.7554/eLife.23990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Lignon G., Beres F., Quentric M., Rouzière S., Weil R., De La Dure-Molla M., Naveau A., Kozyraki R., Dessombz A., and Berdal A. (2017) FAM20A gene mutation: amelogenesis or ectopic mineralization? Front. Physiol. 8, 267 10.3389/fphys.2017.00267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Shin M., Hu Y., Tye C. E., Guan X., Deagle C. C., Antone J. V., Smith C. E., Simmer J. P., and Bartlett J. D. (2014) Matrix metalloproteinase-20 over-expression is detrimental to enamel development: a Mus musculus model. PLoS ONE 9, e86774 10.1371/journal.pone.0086774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Xia Y., Ren A., and Pugach M. K. (2016) Truncated amelogenin and LRAP transgenes improve Amelx null mouse enamel. Matrix Biol. 52–54, 198–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Verdelis K., Szabo-Rogers H. L., Xu Y., Chong R., Kang R., Cusack B. J., Jani P., Boskey A. L., Qin C. L., and Beniash E. (2016) Accelerated enamel mineralization in Dspp mutant mice. Matrix Biol. 52–54, 246–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Zalzal S. F., Smith C. E., and Nanci A. (2008) Ameloblastin and amelogenin share a common secretory pathway and are co-secreted during enamel formation. Matrix Biol. 27, 352–359 10.1016/j.matbio.2008.01.003 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.