

Abstract
The small-vessel disorder cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) arises from mutations in the human gene encoding NOTCH3 and results in vascular smooth muscle cell degeneration, stroke, and dementia. However, the structural changes in NOTCH3 involved in CADASIL etiology are unclear. Here, we discovered site-specific fragmentation of NOTCH3 protein in pathologically affected vessels of human CADASIL-affected brains. EM-based experiments to pinpoint NOTCH3 localization in these brains indicated accumulation of NOTCH3 fragmentation products in the basement membrane, collagen fibers, and granular osmiophilic material within the cerebrovasculature. Using antibodies generated against a disease-linked neo-epitope found in degenerating vascular medium of CADASIL brains, we mapped the site of fragmentation to the NOTCH3 N terminus at the peptide bond joining Asp80 and Pro81. Cleavage at this site was predicted to separate the first epidermal growth factor (EGF)-like domain from the remainder of the protein. We found that the cleavage product from this fragmentation event is released into the conditioned medium of cells expressing recombinant NOTCH3 fragments. Mutagenesis of Pro81 abolished the fragmentation, and low pH and reducing conditions enhanced NOTCH3 proteolysis. Furthermore, substitution of multiple cysteine residues of the NOTCH3 N terminus activated proteolytic release of the first EGF-like repeat, suggesting that the elimination of multiple disulfide bonds in NOTCH3 accelerates its fragmentation. These characteristics link the signature molecular genetic alterations present in individuals with CADASIL to a post-translational protein alteration in degenerating brain arteries. The cellular consequences of these pathological NOTCH3 fragments are an important area for future investigation.
Keywords: Notch protein, vascular smooth muscle cells, proteolysis, protein processing, stroke, arteries, cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL), cysteine residues, NOTCH3, small-vessel disease
Introduction
Cerebral small-vessel disease has emerged as a key neuropathological factor in age-related brain disorders. As a leading cause of stroke (1–4) and dementia (5–8), vascular disease of the brain is caused by numerous complex factors, including genetic factors. The most common genetic causes of cerebral small-vessel disease are mutations in NOTCH3, which result in cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL)3 (9, 10). In a vast majority of CADASIL patients, NOTCH3 mutations alter the number of cysteines in the ectodomain of the gene product (11).
NOTCH3 is a receptor protein that is expressed in vascular smooth muscle cells of brain arteries (12). Its structure, which is highly similar to related Notch receptors (13), includes a large N-terminal extracellular domain that is enzymatically proteolyzed within its membrane-spanning domain (14). The extracellular domain remains attached to the C-terminal domain via protein-protein interactions. On exposure to ligands at the cell surface, NOTCH3 is again enzymatically cleaved, releasing the intracellular C-terminal domain that functions within the nucleus to alter transcription (13, 15).
All CADASIL-causing mutations in NOTCH3 thus far have been mapped to the N-terminal extracellular domain, which is composed of a tandem series of 34 epidermal growth factor (EGF)-like domains (9, 11, 16). The NOTCH3 ectodomain that harbors these mutations accumulates in the cerebral blood vessels of CADASIL patients (12) and is a component of granular osmiophilic material (GOM) (17), a characteristic ultrastructural pathological feature. A leading hypothesis is that structural changes caused by CADASIL mutations in the EGF-like domains of the ectodomain lead to accumulation of NOTCH3 protein that, by unknown mechanisms, results in smooth muscle cell failure (18).
The potential structural changes in NOTCH3 that are induced by point mutations have not been extensively characterized. With uncommon exceptions (19, 20), point mutations that cause CADASIL do not cause changes in signaling properties (21). But point mutations in recombinant NOTCH3 ectodomain have been shown to alter NOTCH3 homophilic interactions (22). It is likely therefore that undiscovered molecular changes in the NOTCH3 protein are found in CADASIL blood vessels and that these structural alterations trigger new pathological processes.
In other disorders, post-translational processing of disease proteins has been the subject of enormous degrees of attention. For example, Aβ from amyloid plaques in Alzheimer's disease (AD) is derived from enzyme-mediated cleavage of the amyloid precursor protein (APP) (23). The importance of this event is underscored by the ability of Aβ to aggregate and oligomerize, which deleteriously affects neuronal and vascular cell homeostasis (24). The enzymatic machinery required to generate Aβ from APP includes γ-secretase, an enzyme complex that is also required for physiological processing of NOTCH proteins and driving Notch cell signaling by regulated proteolysis (25). Impaired physiological enzymatic cleavage of NOTCH3 in CADASIL is not suspected because the ectodomain (but not the C-terminal intracellular domain) of NOTCH3 accumulates in CADASIL vessels (12). Pathological cleavage or modification of the NOTCH3 protein has not yet been described to our knowledge. Here, we describe the discovery of non-enzymatic fragmentation of NOTCH3 in CADASIL vessels and characterize the molecular underpinnings of this disease-related process.
Results
Novel monoclonal antibodies against NOTCH3 protein react with CADASIL blood vessels
NOTCH3 has been shown to accumulate in the vasculature of CADASIL brains. Our laboratory has previously generated polyclonal antibodies that preferentially recognize pathological forms of NOTCH3 in diseased cerebral blood vessels (26). To define specific disease-relevant NOTCH3 epitopes, we generated monoclonal antibodies in rabbits against an N-terminal peptide of the NOTCH3 ectodomain containing the first EGF-like domain. Two of the monoclonal antibodies that reacted with the immunogen and demonstrated disease-relevant recognition are described herein: UMI-D and UMI-F. In immunohistochemical studies, both UMI-D and UMI-F intensely and broadly stained the degenerating medial layer of CADASIL leptomeningeal vessels and the penetrating white matter blood vessels (19 of 19 CADASIL samples; Fig. 1, A–D and G–J). Moreover, we saw little to no antibody recognition of normal-appearing leptomeningeal and penetrating white matter blood vessels of 10 control patients (Fig. 1, E, F, K, and L). In controls, only sporadic thickened vessels demonstrated reactivity with these antibodies. We compared the staining patterns of UMI-D and UMI-F with that of a NOTCH3 antibody generated against EGF-like repeats 17–21 (1E4; EMD Millipore). We observed that UMI-D and UMI-F resulted in stronger staining than 1E4 in all CADASIL tissues stained, but not in control tissues (Fig. 2).
Figure 1.

Localization of novel NOTCH3 antigens by monoclonal antibodies UMI-D and UMI-F in CADASIL arteries. UMI-D (A, B, E–G, H, K, and L) and UMI-F (C, D, I, and J) were used for immunohistochemical localization of NOTCH3 antigen in frontal lobe of 19 genetically verified CADASIL and 10 control brains. Photographs were captured of representative leptomeningeal vessels (LM; A–F) of the gray matter and of penetrating arteries of the white matter (WM; G–L) at the magnifications shown. Both antibodies demonstrated staining in the medial layers of cerebral arteries in CADASIL patients (A–D and G–J) but not in pathologically normal vessels from controls (E, F, K, and L).
Figure 2.
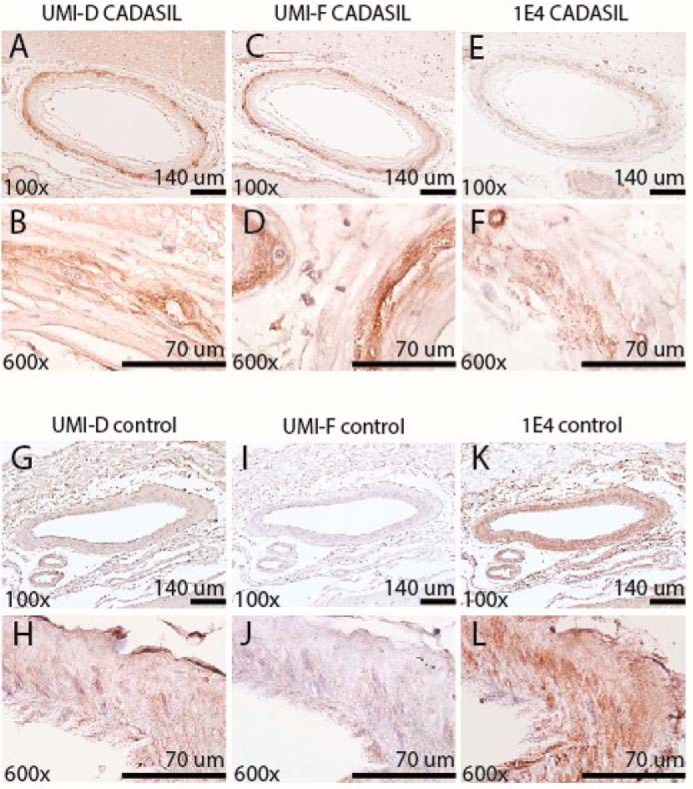
Comparison of NOTCH3 staining by monoclonal antibodies UMI-D and UMI-F and a standard NOTCH3 antibody (1E4, EMD Millipore). UMI-D (A, B, G, and H), UMI-F (C, D, I, and J), and 1E4 (E, F, K, and L) were used for immunohistochemical localization of NOTCH3 antigen in the frontal lobe of four genetically verified CADASIL (A–F) and four control brains (G–L). Representative photographs were captured from serial sections of representative leptomeningeal vessels at the magnifications shown. UMI-D and UMI-F (A–D) reacted more strongly with NOTCH3 in CADASIL leptomeningeal vessels, compared with 1E4 (E and F). UMI-D and UMI-F demonstrated little to no staining in the medial layers of cerebral arteries in control patients (G–J), whereas 1E4 demonstrated increased staining of control vessels (K and L).
To ultrastructurally localize the antigen, we performed transmission EM (TEM) and immunogold labeling with both antibodies, UMI-D and UMI-F, on two post-mortem human brain samples from CADASIL patients and one control patient. Numerous GOMs were identified in the extracellular space around smooth muscle cells in both CADASIL samples (Fig. 3A), whereas none were visualized in the control sample by TEM.
Figure 3.

Subcellular localization of UMI-F–reactive antigens. Two CADASIL brain samples were processed for transmission EM. Cross-sections of CADASIL cerebral blood vessels (A) demonstrated the presence of multiple GOMs (black arrows) found in between the basement membrane (BM) and vascular smooth muscle cell (SMC) plasma membrane (black arrowheads) in CADASIL. We also identified red blood cells (RBC) and endothelial cells (EC) by TEM. The same two CADASIL samples were processed for immunogold labeling in parallel. Samples were labeled with UMI-F (B–D), followed by UltraSmall secondary (Electron Microscopy Sciences) and silver enhancement (Aurion). UMI-F demonstrated binding to antigen in the extracellular space: basement membrane (B; BM), extracellular collagen fibers (C), and GOMs (D). Clusters of gold labeling are marked with white arrows. Approximately 30% of GOMs from randomly selected vessels showed labeling (compare B and D). Labeling experiments were done at least two times with similar results.
In both CADASIL samples, UMI-F immunoreactive antigen was found in the extracellular space, including the basement membrane (Fig. 3B), collagen fibers (Fig. 3C), and GOMs (Fig. 3D). We randomly selected three to four blood vessels from each patient and quantified the proportion of GOMs that contained immunogold labeling. We found that 19 of 62 (30.7%) and 10 of 36 (27.8%) GOMs had immunolabeling with UMI-F in each CADASIL patient sample, respectively. We obtained similar findings for UMI-D, where localization of antigen was concentrated in structures of the extracellular matrix (Fig. S1, A–C). Quantification of GOMs from 3–4 vessels/patient revealed that 62 of 154 (40.3%) and 6 of 28 (21.4%) GOMs demonstrated immunogold reactivity with UMI-D in each patient sample. Interestingly, GOMs of smaller cross-sectional area occasionally demonstrated increased amounts of gold labeling (Fig. S1D). The control patient sample consistently demonstrated decreased sporadic reactivity with UMI-D and UMI-F that was localized to the basement membrane. Each immunogold labeling experiment was repeated at least two times for each antibody with similar findings.
A series of experiments were pursued to determine the target of these antibodies. UMI-D and UMI-F failed to react with reduced, purified recombinant NOTCH3 ectodomain fragments on dot blots and Western blots. UMI-D and UMI-F also did not react in immunohistochemical experiments with recombinant NOTCH3 ectodomain proteins overexpressed in 293 cell lines. In contrast, we discovered that UMI-D reacted with recombinant NOTCH3 ectodomain protein that was treated with a combination of heat and acid, but not either alone (Fig. 4A). This led us to investigate whether UMI-D targets a proteolytic fragment of NOTCH3.
Figure 4.

Characterization of novel NOTCH3 antigens recognized by UMI-D and UMI-F. A, UMI-D reactivity against NOTCH3 under chemical stress. Purified, recombinant WT NOTCH3 ectodomain fused to Fc was exposed to either heat or acid (50 mm HCl) for up to 18 h, as indicated. Proteins were then dotted onto nitrocellulose that was probed using UMI-D. B, UMI-D reactivity against NOTCH3 fragments fused to the C terminus of GFP. cDNA clones were prepared to generate GFP ending with a series of NOTCH3 sequences. On the left, the base sequence is shown at the top. Letters indicate the C-terminal extent of the clone. For example, the Q clone contains GWVGERCQ; the L clone contains GWVGERCQL, and so forth. The P clone encodes GWVGERCQLEDP, which blocks the Asp80 residue with a proline. On the right, GFP extension clones were prepared to encode the base sequence at the top, terminating in Asp80 (clone D), Pro81 (clone P), Cys82 (clone C), and so forth. 293 cells were transfected with each clone, and lysates were probed with UMI-D (top panels) or GFP. C, map of NOTCH3 showing location of sequences used in B. The top panel shows the large scale map of human NOTCH3, which is divided into a series of EGF-like domains. The area of focus is the EGF-like domain 1, which terminates in the Asp80 residue. A summary of binding is shown to the right. The bottom line depicts an N-terminal fusion of the target sequence, which does not bind to UMI-D (data not shown). D, dot blot analysis of UMI-D and UMI-F targets. Peptides were synthesized that correspond to sequences around the end of EGF-like domain 1 of NOTCH3. The sequences are shown on the right. Each peptide was spotted onto replicate membranes that were then probed with the antibodies indicated. 1E7 was generated against the first peptide but binds to all of the tested molecules and serves as a protein-spotting control. All experiments were conducted at least four times with similar results.
To test whether UMI-D targets a fragmentation site, we generated recombinant constructs encoding pieces of the NOTCH3 sequence used for immunization to the C terminus of GFP; we then analyzed the ability of UMI-D to bind to recombinant GFP fusion proteins on Western blots. UMI-D was found to readily bind to proteins ending in Asp80 but failed to interact with proteins ending in Gln77, Leu78, and Glu79 (Fig. 4, B (left) and C). GFP fusion proteins terminating with Pro81, Cys82, His83, or Ser84 were not strongly bound to UMI-D (Fig. 4, B (right) and C). NOTCH3 sequences ending in Asp80 that were fused at the N terminus of GFP did not react with UMI-D.
Peptides corresponding to the same region of NOTCH3 were used to confirm the target specificity of UMI-D. Fig. 4D shows that UMI-D binds to peptides ending in Asp80. UMI-D again failed to recognize synthetic sequences missing the Asp80 residue and did not bind to peptides with extended sequences beyond Asp80. Furthermore, we observed that N-terminal deletions did not affect UMI-D. An independent NOTCH3 mAb 1E7 recognized all of the peptides tested. In total, these immunoblotting studies indicate that UMI-D recognizes a neo-epitope resulting from cleavage of NOTCH3, C-terminal to Asp80.
UMI-F did not bind proteins in Western blotting experiments but exhibited the same peptide-binding specificity as UMI-D (Fig. 4D), suggesting that both UMI-D and UMI-F recognize the same linear sequence that contains C-terminal Asp80. Together, these studies demonstrate a novel NOTCH3 fragmentation site C-terminal to Asp80 in human CADASIL tissue samples.
The NOTCH3 cleavage product can be found in both the intracellular and extracellular space
To determine whether the cleavage process occurs inside or outside the cell, we transfected recombinant constructs for Fc-NOTCH3 proteins containing the first two EGF-like repeats into 293 cells. Cell lysates and conditioned medium of transfected cells were collected and analyzed by Western blotting. We identified the presence of NOTCH3 cleavage product both in the cell lysate and in the medium (Fig. 5A). The intensity of UMI-D–reactive cleavage product normalized to the Fc tag was 20-fold higher in the lysate compared with the medium (Fig. 5B), supporting the possibility that cleavage occurs inside and outside cells. All experiments were performed at least three times with similar results.
Figure 5.
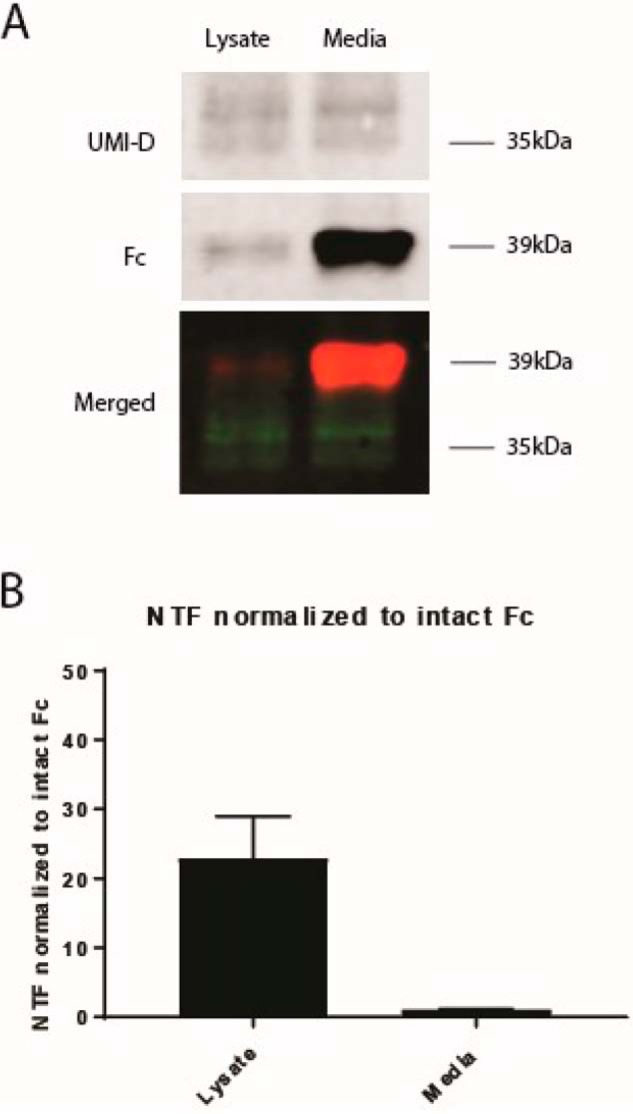
Identification of NOTCH3 fragmentation product in both intracellular and extracellular spaces. 293 cells were transiently transfected with recombinant plasmids encoding Fc fused at the C terminus to NOTCH3 fragments containing 2 EGF-like domains. After transfection, proteins from both the conditioned medium of the 293 cells and the 293 cell lysates were purified by protein A–agarose pulldown and analyzed for UMI-D reactivity. In A, the NOTCH3 cleavage product as detected by UMI-D was identified in both the cell lysate and the medium. In the merged image, UMI-D reactivity (top) is shown in green, and Fc reactivity (middle) is shown in red. B, demonstrates that the intensity of UMI-D–reactive cleavage product normalized to the Fc tag was roughly 20-fold higher in the lysate compared with the medium. All experiments were conducted at least three times with similar results. Error bars, S.D.
The role of proline 81 in NOTCH3 fragmentation
To characterize the cleavage process responsible for NOTCH3 fragmentation in living cells, we transfected recombinant constructs into 293 cells to generate Fc-NOTCH3 proteins containing the first EGF-like domain (Fc-E1), the first two EGF-like domains (Fc-E2), the first three EGF-like domains (Fc-E3), or the first eight EGF-like domains (Fc-E8) at the C terminus. Conditioned medium of transfected cells was collected and analyzed by immunoblotting. In addition to the expected Fc fusion protein (Fig. 6A, bottom, lane marked WT), each group generated a common Fc fragment of a size corresponding to protein resulting from fragmentation at Asp80, which was reactive with UMI-D (Fig. 6A, top, lanes marked WT). This demonstrates that overexpressed NOTCH3 protein is cleaved in living cells at the same position as in CADASIL tissues and that cleavage at Asp80 does not require the entire NOTCH3 protein.
Figure 6.

Mapping and sequence specificity of NOTCH3 fragmentation of protein produced in cells. Proteins were produced by transfection of 293 cells with recombinant plasmids encoding Fc fused at the C terminus to NOTCH3 EGF-like domains. The number of EGF-like domains is x for each Fc-Ex construct. After transfection, protein was concentrated by incubation with protein A–agarose. The captured proteins were analyzed by Western blotting using UMI-D to detect a band at the size expected after cleavage between EGF-like domains 1 and 2 at Asp80. Fc-E1 was used as a size control. In A, secreted proteins from cells transfected with constructs listed were concentrated using protein A beads, eluted, and analyzed for generation of UMI-D reactivity at the size corresponding to Fc-E1. The blots were also probed for Fc to demonstrate capture of protein. In B, Fc-E3 proteins were mutated at NOTCH3 residue 81 (normally Pro81) using constructs containing multiple cysteine mutations in EGF-like repeats 1 and 2. As in A, cells were transfected and then tested for UMI-D–reactive protein. Fc blots verified the successful production of secreted protein. All experiments were conducted at least four times with similar results.
The ability to detect cutting of NOTCH3 sequences in cell culture allowed us to investigate residues required for cleavage at the Asp80–Pro81 bond. The aspartate-proline sequence has been shown to be susceptible to cleavage in other proteins (27–32). To determine the importance of Pro81 for NOTCH3 cleavage, we mutated this residue to alanine in the same NOTCH3 fusion constructs (Fc-E1, -E2, -E3, and -E8). Cells transfected with P81A mutant proteins synthesized similar amounts of full-length protein but generated markedly reduced levels of truncated protein that reacted with UMI-D (Fig. 6A, lanes marked DA).
We also mutated Pro81 to the each of the remaining 19 amino acids and studied the formation of the NOTCH3 fragment recognized by UMI-D in a mutant Fc-E3 that normally exhibits high levels of fragmentation; we found that cleavage and neo-epitope formation was strongest with proline at the cleavage site but also occurred with cysteine and modestly with alanine, leucine, and glycine substitutions (Fig. 6B). Other amino acid substitutions did not generate the neo-epitope (Fig. 6B). These studies demonstrate that proline at the cleavage junction is critical for efficient NOTCH3 cleavage and resultant UMI-D neo-epitope generation.
NOTCH3 protein can be non-enzymatically cleaved by acidic and reducing conditions
Low pH can trigger the autocatalytic cleavage of proteins (27–30, 32). To test the effect of acidic conditions on NOTCH3 protein, purified Fc-E2 was incubated in buffers at different pH. There is increased NOTCH3 fragmentation detected by the UMI-D antibody (Fig. 7A) in progressively more acidic conditions. Similarly, when the reducing agent (disulfide bond breaker) TCEP at neutral pH was added to Fc-E2, there was a dose-dependent increase in fragmentation of NOTCH3 as detected by UMI-D (Fig. 7B). Longer fragments of NOTCH3 proteins with either three EGF-like repeats (Fc-E3) or eight EGF-like repeats (Fc-E8) also demonstrate increased N-terminal fragmentation under reducing conditions (Fig. 7C). Because all of the reactions occurred with purified proteins, we conclude that exogenous enzymes are not required for NOTCH3 fragmentation in acidic and reducing conditions.
Figure 7.

The effect of pH and redox state on fragmentation of NOTCH3 at Asp80. A, purified protein (Fc-E2) was exposed to buffers at specific pH for 1 h at 37 ºC, boiled in reducing sample buffer, and electrophoresed on gels. Western blot analysis was performed using UMI-D to detect fragmentation (top) and Fc to detect total protein input (bottom). There was a clear increase in fragmentation at modestly acidic conditions. B, the same purified protein was exposed to a series of TCEP concentrations for 1 h at 37 ºC and analyzed as in A. There was a consistent increase in fragmentation with increasing concentration of TCEP. C, purified proteins with different numbers of EGF-like repeats were exposed to TCEP or vehicle prior to Western blot analysis as in A. All proteins exhibited increased fragmentation with TCEP incubation. All experiments were performed four or more times and showed the same findings.
Effect of NOTCH3 mutations on protein fragmentation
Mutations that cause CADASIL create an odd number of cysteines that may affect disulfide bonding of NOTCH3 protein. Because chemical reduction increases NOTCH3 fragmentation, we investigated the fragmentation of NOTCH3 at Asp80 in cysteine mutants in Fc-E3. Constructs containing zero, one, two, four, five, or six cysteine mutations in EGF1 were transfected into cells, and proteins from the conditioned medium were evaluated for fragmentation at Asp80. Only mutants containing five or six cysteine-to-serine mutations exhibited increased N-terminal fragment generation compared with WT NOTCH3 (Fig. 8A, compare summed fragmented products (top) with total protein (bottom)). The promotion of fragmentation by multiple cysteine mutations was also observed for GST-tagged NOTCH3 protein. In this case, the addition of a reducing agent, TCEP, cooperated with multiple cysteine mutations to increase N-terminal fragmentation of the bacterially produced protein (Fig. 8B).
Figure 8.

Assessment of fragmentation of NOTCH3 at Asp80 in cysteine mutants of NOTCH3. A, cysteines were mutated to serines within the first EGF-like repeats of NOTCH3 in Fc-E3 fusions (Fc fused to the first three EGF-like repeats of NOTCH3). The number of cysteines mutated in each protein is indicated above each lane; the cysteine-to-serine mutations were located as follows. Lane 1, cysteine 1; lane 2, cysteines 2 and 3; lane 4, cysteines 1–3 and 6; lane 5, cysteines 1–3 and 5 and 6; lane 6, all six cysteines. After transient transfection, the proteins from conditioned medium of 293 cells were purified by protein A–agarose pulldown and analyzed for UMI-D reactivity (top) and total Fc reactivity (bottom) to assess the effects of progressively larger numbers of cysteine mutations. Higher numbers of mutations resulted in both a shift of molecular weight up and in the total reactivity to UMI-D normalized to Fc. B, several GST-E2 (GST fused to the first two EGF-like repeats) fusion proteins were prepared in E. coli with progressively increased numbers of cysteine to serine mutations. The E1 sample contains GST fused to the first EGF-like repeat, as a size marker for the cleavage product. The E2 sample is GST-E2. The E2* sample is GST-E2 with a single mutation, P81A, in human NOTCH3. The 2× mutant (2x mut) contains serine substitutions for the fifth and sixth cysteines of EGF-like repeat 1. The 4× mutant (4x mut) contains serine substitutions for four cysteine residues: the fifth and sixth cysteines of EGF-like repeat 1 and the first and second cysteines of EGF-like repeat 2. After purification using GSH magnetic beads, proteins were analyzed as in A, except that some protein was treated without detergent with TCEP first before electrophoresis. Protein yields were assessed using GST Western blotting. There was an increase in fragmented protein with more mutations that was enhanced by the addition of TCEP prior to adding sample buffer. All experiments were performed four or more times with similar findings.
Discussion
Novel molecules found to accumulate in disease tissues provide possible mechanistic insights and potential treatment targets. Here, we describe a new process by which a vascular disease molecule is produced: non-enzymatic fragmentation of NOTCH3 that occurs in affected blood vessels of the cerebral small-vessel disorder CADASIL. Characterization of the cleavage event both in cells and with purified proteins suggests that the reaction occurs in cells and is also influenced by the redox states of NOTCH3.
UMI-D and UMI-F antigen localization
UMI-D and UMI-F broadly stain the medial layer of CADASIL vessels while sparing normal-appearing age-matched control vessels. Compared with a NOTCH3 antibody designed against a different part of the ectodomain (1E4; EMD Millipore), UMI-D and UMI-F appear to be more specific for protein from pathological vessels, suggesting that NOTCH3 fragmentation is a process localized to diseased vessels (Fig. 2).
At the ultrastructural level, both UMI-D– and UMI-F–reactive antigens localize to extracellular regions, including the basement membrane, collagen fibers, and GOMs (Fig. 3 and Fig. S1). GOMs are ultrastructural features that are highly specific to CADASIL. Classically, GOMs are extracellular, electron-dense, granular deposits that are visible on EM, often found between neighboring vascular smooth muscle cells. The exact composition of GOMs remains unclear, but it is thought that the ectodomain of NOTCH3 is a major component (17). Our immunolabeling studies suggest that in addition to the full ectodomain of NOTCH3, an N-terminal fragment of the NOTCH3 ectodomain also accumulates within GOMs. We occasionally found increased immunolabeling in GOMs with smaller cross-sectional areas (Fig. S1D) and closely within the perimeter of larger GOM cross-sections. Future studies will be required to see whether accumulation of this UMI-D– and UMI-F–reactive NOTCH3 fragment prefers the surface of GOMS and whether GOMs contribute to CADASIL pathophysiology.
One model of CADASIL proposes that matrisome changes play an important role in disease. The matrisome refers to proteins that make up or interact with the extracellular matrix. Recent studies demonstrated that mutant NOTCH3 multimerizes and interacts with several key extracellular matrix proteins that abnormally accumulate in CADASIL, such as tissue inhibitor of metalloproteinase 3 and vitronectin (18). Additional extracellular matrix proteins found to accumulate in CADASIL vessels include clusterin, endostatin, and LTBP1 (19, 33). UMI-D– and UMI-F–reactive antigens localize to structures of the extracellular matrix, such as the basement membrane, collagen fibers, and GOMs. Further studies will be required to determine whether matrisome proteins interact with the UMI-D– and UMI-F–reactive truncated fragment of NOTCH3 and whether such matrisome changes affect and/or contribute to the cerebrovascular dysfunction seen in CADASIL patients.
NOTCH3 fragmentation
Juxtamembrane NOTCH3 protein cleavage has been described as a key step in ligand-dependent signaling (34, 35). However, cleavage of the protein in the extracellular EGF-like domains has yet to be described until now, to our knowledge. By use of specific monoclonal antibodies that recognize a NOTCH3 cleavage product (UMI-D and UMI-F), we now demonstrate the first example of in situ NOTCH3 post-translational modification that is localized to the regions of the cerebrovasculature that are affected in CADASIL. The co-registration of cellular pathology and NOTCH3 fragmentation in post-mortem human tissue from CADASIL patients is consistent with a pathological role of NOTCH3 cleavage in CADASIL. Additional studies will need to be performed to clarify the role of the NOTCH3 cleavage product in disease progression, in particular whether the fragmentation is, in addition to a marker of disease, also a participant in pathogenesis.
Mechanism of fragmentation
In an in vitro NOTCH3 overexpression system, NOTCH3 cleavage product was found both intracellularly (in the cell lysate) and extracellularly (in the conditioned medium; Fig. 5, A and B). However, ultrastructural localization using immunogold labeling demonstrated accumulation of the NOTCH3 cleavage product primarily in the extracellular matrix compared with inside vascular smooth muscle cells (Fig. 3 and Fig. S1). This difference could be explained by rapid degradation of intracellular NOTCH3 cleavage product in human tissue and/or efficient secretion of cleavage product into the extracellular matrix.
Although our experiments cannot exclude enzymatic processing of NOTCH3, our studies focusing on biochemical characterization of N-terminal NOTCH3 cleavage show that protein cleavage occurs in the absence of enzymes. In these experiments, cleavage of purified Fc-NOTCH3 protein was observed. In further support of non-enzymatic NOTCH3 cleavage, a systematic search of the proteolytic database MEROPS does not reveal a known peptidase capable of fragmentation of NOTCH3 at the defined target sequence. In addition, the use of multiple broad-spectrum protease inhibitors failed to demonstrate prevention of fragmentation. Extensive mutagenesis of the NOTCH3 sequence at the cleavage site demonstrated that the proline is very important; only cysteine substitution was able to sustain a similar amount of cleavage. Drawing upon studies of other proteins, it appears that several independent molecules from multiple species are non-enzymatically cleaved at Asp–Pro bonds, including several isoforms of mucin (28, 29), endothelial nitric-oxide synthase (27), pre-α-inhibitor (32), green mamba toxin MTX2 (30), and herpes simplex virus type 1 (HSV-1) glycoprotein D (gD) (31). Moreover, the Asp–Pro bond has been described as chemically labile, particularly in mildly acidic conditions (27–30, 32). The most parsimonious explanation for NOTCH3 fragmentation is thus chemical lability of Asp–Pro bond within the context of the larger NOTCH3 polypeptide. Of interest, with the exception of endothelial nitric-oxide synthase, all of these proteins are secreted, and it appears that some of the cleaved proteins are cut in the acidic environment of the secretory pathway (29). Further, like NOTCH3, the mucins are cysteine-rich proteins, and in the case of MUC5AC, the cleavage occurs in a cysteine-enriched domain (28).
Enhancement of fragmentation and cysteine alterations
The apparent non-enzymatic processing of NOTCH3 can be accelerated in purified protein preparations by at least three factors. First, acidic conditions increase the degree of fragmentation. The pH transition point corresponds to the pKa of aspartic acid, potentially implicating the protonation state of Asp80 as a determining factor; however, it is recognized that many additional pH-dependent changes outside of aspartates could influence protein chemistry. Second, fragmentation of NOTCH3 appears to be facilitated by reducing conditions, as both thiol and non-thiol reducing agents increase purified NOTCH3 cleavage. Third, cysteine mutations in NOTCH3 regulate the fragmentation of the protein. This was not evident when a single cysteine mutation was introduced; however, the addition of multiple cysteine mutations yielded even more NOTCH3 fragmentation, suggesting that higher degrees of protein structure alteration achieved by elimination of more than one disulfide bond are required to accelerate fragmentation.
The ability of reducing agents and multiple cysteine mutations to induce site-specific cleavage is highly relevant to CADASIL, a disorder that features mutations involving cysteine residues. A clear increase in fragmentation at residue 80 is noted in NOTCH3 proteins that are treated with reducing agents, and this is cooperatively enhanced by mutation of multiple cysteine residues. These data support a role of disruption of secondary structure of NOTCH3 in the promotion of NOTCH3 fragmentation. Interestingly, we found that conformations of NOTCH3 that result from multiple disulfide bond disruptions (26) are present in CADASIL vessels. Whereas single mutations in NOTCH3 failed to result in disease-related epitope formation, chemical reduction that potentially results in multiple disulfide reductions was sufficient to produce protein with disease structure. Moreover, it has recently been proposed by others that several forms of NOTCH3 could exist in mouse tissues engineered to express CADASIL mutant protein (36). We have proposed a model in which NOTCH3 mutations, which include changes in cysteine number, lead to the presence of multiple disruptions of cysteine disulfide bonds, which is a prerequisite for pathology of vessels. One model consistent with these studies is that fragmentation is a key pathological event that occurs following multiple disulfide disruptions.
NOTCH3 fragmentation and the spectrum of mutations in CADASIL
When we examined the fragmentation of NOTCH3 in a cohort of individuals with NOTCH3 mutations dispersed over the extent of the protein, there was a very good correlation between the location of the mutation and the generation of the neo-epitope (Fig. 9). This correlation suggests that CADASIL mutations close to the cleavage site could increase the probability of protein fragmentation. In fact, recent data suggest that mutations of the N-terminal regions of the protein confer more serious disease than mutations in the C-terminal regions (16). Further investigations using models of protein fragmentation that are increased in efficiency are needed to understand the influence of mutations in the N terminus (compared with the C terminus); of particular interest are the precise details about how mutations that occur at a distance away from the cleavage site may propagate structure perturbations that influence chemical fragmentation of NOTCH3 at the N terminus.
Figure 9.

Relationship between NOTCH3 protein fragmentation in cerebral vessels and location of cysteine-altering mutations. The reactivity strength to UMI-D (A) or UMI-F (B) was assessed in a blinded fashion. Slides were then rank-ordered and plotted against the location of the mutations in the NOTCH3 ORF found in each sample.
Future investigations and key questions
Fragmentation of a disease-related protein into peptides in dementing disorders is most notable in Alzheimer's disease, which features a multiple-step enzymatic cleavage of the APP into Aβ and a growing group of other fragmentation products (23). Aβ has emerged as a key therapeutic target of Alzheimer's disease by virtue of its impressive accumulation in plaques of Alzheimer's brains (37–39). However, Aβ has not yet yielded therapeutic advances, and several clinical trials aimed at clearance of Aβ have not been successful (40).
As such, reflection on a potential parallel to AD should stimulate caution in ascribing the predicted NOTCH3 fragmentation product as a key molecule in cerebral small-vessel disease. Several key questions remain. First, does the NOTCH3 fragmentation result in vascular changes in vivo? (In AD, it has been proposed by some that Aβ is simply a consequence of protein fragmentation and partitioning of the protein.) Second, what is the prevalence of the NOTCH3 cleavage product in non-CADASIL vessels? (In AD, doubt has been cast because a large proportion of individuals without AD have accumulation of plaque containing Aβ.) Third, is another form of NOTCH3 that is linked to fragmentation (e.g. the C-terminal remnant of NOTCH3) important for CADASIL pathology? (In AD, failure of antibody therapy suggests the possibility that other parts of APP may be important in disease.) Fourth, it will be important to isolate and sequence the protein that accumulates in CADASIL. This has been done in AD; for cerebral small-vessel diseases, the precise molecular sequence of the fragmentation product will be important for experimental modeling of the disorder.
In contrast to Aβ, which is generated from APP by a sequential enzymatic process, the predicted NOTCH3 fragment does not appear to result from a catalytic process. Therefore, design of drugs that interfere with production of the NOTCH3 cleavage fragment may be challenging because an enzyme cannot be targeted. On the other hand, a more comprehensive understanding the structural elements of NOTCH3 that favor cleavage, such as reducing conditions, could lead to strategies that may have an impact on disease.
The discovery of antibodies to NOTCH3 fragments as described here is a positive development toward therapy. Recent studies have demonstrated the promise of anti-NOTCH3 mouse mAb therapies in preclinical mouse models (36). The new reagents described here thus could be refashioned for diagnostic and therapeutic purposes in the future.
Conclusions
We show that the protein mutated in CADASIL, NOTCH3, undergoes a novel spontaneous cleavage that is independent of proteases. The cleavage event co-registers with degenerating vascular medium, is ultrastructurally localized to the extracellular matrix, and is accelerated by reducing conditions and mutations affecting cysteine residues of NOTCH3. This newly described post-translational alteration of a small-vessel disease protein presents new molecular insight into an increasingly important site of human brain disease and expands the role of pathological protein alterations in neurodegeneration.
Experimental procedures
Chemicals and reagents
Unless noted, chemicals were obtained from Sigma, and cell culture reagents were purchased from Invitrogen.
Monoclonal antibodies against NOTCH3
Rabbits and rats were used to generate NOTCH3 antibodies. As described in a prior study that used polyclonal antibodies to NOTCH3 (26), animals were immunized with peptides from the N-terminal EGF-like repeat of human NOTCH3: ACLCPPGWGERCQLED. Splenic lymphocytes from immunized rabbits were fused to form hybridomas that were cloned and screened for secretion of antibodies exhibiting binding to the immunizing peptides (performed by Epitomics). Avidly binding antibodies were further selected based on their ability to detect protein in the medium of vessels from CADASIL patients. Two of these antibodies, UMI-D and UMI-F, exhibited similar characteristics and were used extensively in this study (see “Results”). Rat monoclonal 1E7 was generated by immunization of rats with the peptide ACLCPPGWGERCQLEDP followed by selection of hybridoma clones that recognize NOTCH3 peptides, CADASIL arteries, and NOTCH3 protein on immunoblotting. These procedures were performed by Genscript.
Immunohistochemistry
Formalin-fixed control frontal lobe sections were obtained from the Alzheimer's Disease Center at the University of Michigan and the Brain Bank of the National Institute for Developmental and Childhood Disorders at the University of Maryland. CADASIL frontal lobe tissue obtained at autopsy has been described previously (41–43). 5-μm sections were analyzed using chromogenic immunohistochemical staining using rabbit monoclonal antibodies after citrate-induced antigen retrieval, as described previously (41–43). Staining was followed by hematoxylin counterstaining. Human vascular antigen integrity in control tissue blocks was confirmed by analysis with the mouse mAb BRIC231 (anti-H; Santa Cruz Biotechnology, Inc.).
TEM and immunogold labeling
Brain tissues previously fixed in 10% buffered formalin were cut into 1 mm × 2-mm pieces and fixed overnight in 2.5% glutaraldehyde, 4% formaldehyde in 0.1 m Sorenson's buffer (Electron Microscopy Sciences). The following day, the tissues were rinsed in 0.1 m Sorenson's buffer and fixed in 1% osmium tetroxide and 2% uranyl acetate for 2 h each. The samples were then dehydrated in a graded ethanol series (50, 70, 85, 95, and 100% × 2) for 10 min each. Tissues were washed with acetone three times for 15 min each and infiltrated with Embed resin (Epon812, DDSA, NMA, DMP30). After incubating at 60 °C for 48 h, the blocks were submitted to the Microscopy and Imaging Core Facility at the University of Michigan, where samples were sectioned on an ultramicrotome (Leica) to obtain 70-nm sections. Grids were stained with uranyl acetate/lead citrate, and high-resolution images were acquired with a JEOL 1400-plus EM.
For immunogold labeling, brain tissues were dehydrated in ethanol (50 and 70%) for 10 min each and infiltrated with LR-White resin according to the manufacturer's instructions (Electron Microscopy Sciences). Polymerization was performed at 50 °C for 24 h. Sections of ∼70-nm thickness were collected on Formvar-coated nickel grids (Electron Microscopy Sciences). Samples were hydrated with TBS buffer (150 mm NaCl and 20 mm Tris, pH 7.6) and treated with 20 mm glycine to quench any remaining formaldehyde. The tissues were then incubated with blocking solution (1% BSA, 0.1% Tween 20 in TBS buffer) for 30 min and incubated overnight at 4 °C with 1:25 (v/v) monoclonal rabbit UMI-F or 1:50 (v/v) monoclonal rabbit UMI-D antibody in blocking solution. The next day, the grids were washed on drops of TBS three times at 5 min each and then incubated with 1 h with a goat anti-rabbit IgG antibody conjugated to UltraSmall 0.2-nm gold nanoparticles (Electron Microscopy Sciences). Grids were washed three times in TBS buffer and three times in double-distilled H2O. Silver enhancement was done according to the manufacturer's protocol (Aurion). Grids were washed in H2O three times for 5 min each. Sections were stained for 10 min in 2% uranyl acetate and 10 min with lead citrate (Electron Microscopy Sciences). Images were acquired using a JEOL 1400-plus electron microscope.
DNA constructs
Fragments of NOTCH3 cDNA were fused to either GST, mouse Fc (IgG), or EGFP in frame to enable production of recombinant NOTCH3 proteins; vectors utilized include, respectively, pGEX (GE Healthcare), pSecTag (Invitrogen), and pEGFP (Clontech). NOTCH3 fragments, derived from full-length NOTCH3 (44), were generated using PCR with restriction recognition sequences engineering into primers to enable standard ligation-mediated cloning. Mutants were also generated by PCR, but for some mutations, nested PCR was required. All plasmids were sequenced to confirm the expected coding sequences.
Transfections, mammalian cell line generation, and recombinant NOTCH3 protein generation
Human HEK293 cells were grown to over 70% confluence and then transfected using Lipofectamine Plus (Life Technologies, Inc.) or PolyJet (SignaGen) according to the manufacturer's instructions (43, 44). DNA was mixed with plasmids that encode puromycin resistance, and the transfected pool of cells was selected by supplementing growth medium with the antibiotic; this process generally yielded dozens of clearly separated cell colonies, which were transferred into individual wells (26).
Colonies were transferred to larger wells, and the conditioned medium was screened for production of appropriate recombinant protein by Western blotting for the Fc portion on the target protein. Productive colonies were expanded by splitting, and prior to medium collection, the supernatant was switched to Opti-MEM, which allowed accumulation of the target proteins in an antibody-free medium (Life Technologies) (26).
Pooled medium was then passed over protein A–agarose columns for single-step purification of recombinant Fc-NOTCH3 fragment fusions. The proteins were eluted at pH 4.5 and immediately neutralized in Tris buffer. All proteins were dialyzed against PBS, and their purity was validated by silver staining (26).
Small-scale recombinant protein expression and purification was performed by transfection of Fc-tagged NOTCH3 deletion constructs. After an overnight incubation with serum containing Dulbecco's modified Eagle's medium growth medium, Opti-MEM was placed over cells. The conditioned medium was then mixed with protein A–agarose beads to purify Fc fusion proteins.
GST fusions were produced in Escherichia coli strain BL21. Induction by isopropyl 1-thio-β-d-galactopyranoside was used to enhance protein production. E. coli cell pellets were sonicated, and the lysates were then mixed with GSH-conjugated magnetic beads. After washing the loaded beads, proteins were eluted with GSH. The proteins were then dialyzed against PBS, and successful purification was validated by silver staining.
Protein analysis
All protein treatments were performed in PBS with supplemental hydrochloric acid or TCEP, as noted for specific experiments. At the end of each treatment, proteins were denatured in sample buffer containing β-mercaptoethanol and boiled at 100 °C for 5 min. All samples were separated on standard SDS-polyacrylamide gels and electroblotted to nitrocellulose. Western blot analysis was performed with antibodies as indicated, followed by incubation with infrared fluorophore–labeled secondary antibodies (Rockland) (45). Bands were detected using a LI-COR Odyssey infrared scanner. For relative analysis of cleavage of Fc proteins, expression levels were normalized to signals obtained using secondary antibodies against mouse Fc.
Author contributions
K. Z. Y., S. J. L., M. T., L. Q., and M. M. W. conceptualization; K. Z. Y., S. J. L., X. Z., N. M. P. C., M. T., and S. R. G. data curation; K. Z. Y., S. J. L., X. Z., N. M. P. C., M. T., S. G. K., S. R. G., J. L. F., and M. M. W. formal analysis; K. Z. Y., S. J. L., X. Z., N. M. P. C., M. T., S. G. K., S. R. G., J. L. F., L. Q., and M. M. W. investigation; K. Z. Y., S. J. L., X. Z., N. M. P. C., M. T., S. G. K., S. R. G., J. L. F., L. Q., and M. M. W. methodology; K. Z. Y. and M. M. W. writing-original draft; K. Z. Y. and M. M. W. project administration; K. Z. Y., S. J. L., X. Z., N. M. P. C., M. T., S. G. K., S. R. G., J. L. F., L. Q., and M. M. W. writing-review and editing; S. J. L., X. Z., N. M. P. C., S. G. K., and M. M. W. validation; L. Q. and M. M. W. resources; L. Q. and M. M. W. supervision; L. Q. and M. M. W. funding acquisition.
Supplementary Material
Acknowledgments
We thank the Alzheimer's Disease Center and Cancer Histology Core at the University of Michigan for tissue and services performed. We thank the Microscopy and Imaging Core at the University of Michigan for services performed and scientific discussions.
This study was supported by National Institutes of Health Grants NS099783 and NS099160 (to M. M. W.), R35GM130292 (to L. Q.), HL108842 (to S. G. K.; awarded to Dr. Jimo Borjigin), and T32-HL125242 and T32 GM007863 (to K. Z. Y.) and United States Department of Veterans Affairs Grants BX003855 and BX003824 (to M. M. W.). This work was also supported by the Frankel Cardiovascular Center Summer Undergraduate Fellowship Program (to J. L. F. and S. R. G.), CADASIL Together We Have Hope, and members of the international CADASIL community who made donations used in this study. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Fig. S1.
- CADASIL
- cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy
- EGF
- epidermal growth factor
- GOM
- granular osmiophilic material
- AD
- Alzheimer's disease
- APP
- amyloid precursor protein
- EGFP
- enhanced GFP
- TEM
- transmission EM
- TCEP
- tris(2-carboxyethyl)phosphine.
References
- 1. Caplan L. R. (2015) Lacunar infarction and small vessel disease: pathology and pathophysiology. J. Stroke 17, 2–6 10.5853/jos.2015.17.1.2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Jackson C., and Sudlow C. (2005) Comparing risks of death and recurrent vascular events between lacunar and non-lacunar infarction. Brain 128, 2507–2517 10.1093/brain/awh636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Norrving B. (2003) Long-term prognosis after lacunar infarction. Lancet Neurol. 2, 238–245 10.1016/S1474-4422(03)00352-1 [DOI] [PubMed] [Google Scholar]
- 4. Tuszynski M. H., Petito C. K., and Levy D. E. (1989) Risk factors and clinical manifestations of pathologically verified lacunar infarctions. Stroke 20, 990–999 10.1161/01.STR.20.8.990 [DOI] [PubMed] [Google Scholar]
- 5. de Groot J. C., de Leeuw F. E., Oudkerk M., van Gijn J., Hofman A., Jolles J., and Breteler M. M. (2000) Cerebral white matter lesions and cognitive function: the Rotterdam Scan Study. Ann. Neurol. 47, 145–151 [DOI] [PubMed] [Google Scholar]
- 6. Jokinen H., Gouw A. A., Madureira S., Ylikoski R., van Straaten E. C., van der Flier W. M., Barkhof F., Scheltens P., Fazekas F., Schmidt R., Verdelho A., Ferro J. M., Pantoni L., Inzitari D., Erkinjuntti T., and LADIS Study Group (2011) Incident lacunes influence cognitive decline: the LADIS study. Neurology 76, 1872–1878 10.1212/WNL.0b013e31821d752f [DOI] [PubMed] [Google Scholar]
- 7. van Dijk E. J., Prins N. D., Vrooman H. A., Hofman A., Koudstaal P. J., and Breteler M. M. (2008) Progression of cerebral small vessel disease in relation to risk factors and cognitive consequences: Rotterdam Scan study. Stroke 39, 2712–2719 10.1161/STROKEAHA.107.513176 [DOI] [PubMed] [Google Scholar]
- 8. Wen H. M., Mok V. C., Fan Y. H., Lam W. W., Tang W. K., Wong A., Huang R. X., and Wong K. S. (2004) Effect of white matter changes on cognitive impairment in patients with lacunar infarcts. Stroke 35, 1826–1830 10.1161/01.STR.0000133686.29320.58 [DOI] [PubMed] [Google Scholar]
- 9. Joutel A., Corpechot C., Ducros A., Vahedi K., Chabriat H., Mouton P., Alamowitch S., Domenga V., Cécillion M., Marechal E., Maciazek J., Vayssiere C., Cruaud C., Cabanis E. A., Ruchoux M. M., Weissenbach J., Bach J. F., Bousser M. G., and Tournier-Lasserve E. (1996) Notch3 mutations in CADASIL, a hereditary adult-onset condition causing stroke and dementia. Nature 383, 707–710 10.1038/383707a0 [DOI] [PubMed] [Google Scholar]
- 10. Tournier-Lasserve E., Joutel A., Melki J., Weissenbach J., Lathrop G. M., Chabriat H., Mas J. L., Cabanis E. A., Baudrimont M., Maciazek J., and et al. (1993) Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy maps to chromosome 19q12. Nat. Genet. 3, 256–259 10.1038/ng0393-256 [DOI] [PubMed] [Google Scholar]
- 11. Joutel A., Vahedi K., Corpechot C., Troesch A., Chabriat H., Vayssière C., Cruaud C., Maciazek J., Weissenbach J., Bousser M. G., Bach J. F., and Tournier-Lasserve E. (1997) Strong clustering and stereotyped nature of Notch3 mutations in CADASIL patients. Lancet 350, 1511–1515 10.1016/S0140-6736(97)08083-5 [DOI] [PubMed] [Google Scholar]
- 12. Joutel A., Andreux F., Gaulis S., Domenga V., Cecillon M., Battail N., Piga N., Chapon F., Godfrain C., and Tournier-Lasserve E. (2000) The ectodomain of the Notch3 receptor accumulates within the cerebrovasculature of CADASIL patients. J. Clin. Invest. 105, 597–605 10.1172/JCI8047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kopan R., and Ilagan M. X. (2009) The canonical Notch signaling pathway: unfolding the activation mechanism. Cell 137, 216–233 10.1016/j.cell.2009.03.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Groot A. J., Habets R., Yahyanejad S., Hodin C. M., Reiss K., Saftig P., Theys J., and Vooijs M. (2014) Regulated proteolysis of NOTCH2 and NOTCH3 receptors by ADAM10 and presenilins. Mol. Cell. Biol. 34, 2822–2832 10.1128/MCB.00206-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lovendahl K. N., Blacklow S. C., and Gordon W. R. (2018) The molecular mechanism of Notch activation. Adv. Exp. Med. Biol. 1066, 47–58 10.1007/978-3-319-89512-3_3 [DOI] [PubMed] [Google Scholar]
- 16. Rutten J. W., Dauwerse H. G., Gravesteijn G., van Belzen M. J., van der Grond J., Polke J. M., Bernal-Quiros M., and Lesnik Oberstein S. A. (2016) Archetypal NOTCH3 mutations frequent in public exome: implications for CADASIL. Ann. Clin. Transl. Neurol. 3, 844–853 10.1002/acn3.344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ishiko A., Shimizu A., Nagata E., Takahashi K., Tabira T., and Suzuki N. (2006) Notch3 ectodomain is a major component of granular osmiophilic material (GOM) in CADASIL. Acta Neuropathol. 112, 333–339 10.1007/s00401-006-0116-2 [DOI] [PubMed] [Google Scholar]
- 18. Monet-Leprêtre M., Haddad I., Baron-Menguy C., Fouillot-Panchal M., Riani M., Domenga-Denier V., Dussaule C., Cognat E., Vinh J., and Joutel A. (2013) Abnormal recruitment of extracellular matrix proteins by excess Notch3 ECD: a new pathomechanism in CADASIL. Brain 136, 1830–1845 10.1093/brain/awt092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Arboleda-Velasquez J. F., Manent J., Lee J. H., Tikka S., Ospina C., Vanderburg C. R., Frosch M. P., Rodríguez-Falcon M., Villen J., Gygi S., Lopera F., Kalimo H., Moskowitz M. A., Ayata C., Louvi A., and Artavanis-Tsakonas S. (2011) Hypomorphic Notch 3 alleles link Notch signaling to ischemic cerebral small-vessel disease. Proc. Natl. Acad. Sci. U.S.A. 108, E128–E135 10.1073/pnas.1101964108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Peters N., Opherk C., Zacherle S., Capell A., Gempel P., and Dichgans M. (2004) CADASIL-associated Notch3 mutations have differential effects both on ligand binding and ligand-induced Notch3 receptor signaling through RBP-Jk. Exp. Cell Res. 299, 454–464 10.1016/j.yexcr.2004.06.004 [DOI] [PubMed] [Google Scholar]
- 21. Joutel A., Monet M., Domenga V., Riant F., and Tournier-Lasserve E. (2004) Pathogenic mutations associated with cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy differently affect Jagged1 binding and Notch3 activity via the RBP/JK signaling Pathway. Am. J. Hum. Genet. 74, 338–347 10.1086/381506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Duering M., Karpinska A., Rosner S., Hopfner F., Zechmeister M., Peters N., Kremmer E., Haffner C., Giese A., Dichgans M., and Opherk C. (2011) Co-aggregate formation of CADASIL-mutant NOTCH3: a single-particle analysis. Hum. Mol. Genet. 20, 3256–3265 10.1093/hmg/ddr237 [DOI] [PubMed] [Google Scholar]
- 23. Andrew R. J., Kellett K. A., Thinakaran G., and Hooper N. M. (2016) A Greek tragedy: the growing complexity of Alzheimer amyloid precursor protein proteolysis. J. Biol. Chem. 291, 19235–19244 10.1074/jbc.R116.746032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jarosz-Griffiths H. H., Noble E., Rushworth J. V., and Hooper N. M. (2016) Amyloid-β receptors: the good, the bad, and the prion protein. J. Biol. Chem. 291, 3174–3183 10.1074/jbc.R115.702704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. De Strooper B., Annaert W., Cupers P., Saftig P., Craessaerts K., Mumm J. S., Schroeter E. H., Schrijvers V., Wolfe M. S., Ray W. J., Goate A., and Kopan R. (1999) A presenilin-1-dependent γ-secretase-like protease mediates release of Notch intracellular domain. Nature 398, 518–522 10.1038/19083 [DOI] [PubMed] [Google Scholar]
- 26. Zhang X., Lee S. J., Young K. Z., Josephson D. A., Geschwind M. D., and Wang M. M. (2014) Latent NOTCH3 epitopes unmasked in CADASIL and regulated by protein redox state. Brain Res. 1583, 230–236 10.1016/j.brainres.2014.08.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fairchild T. A., Fulton D., Fontana J. T., Gratton J. P., McCabe T. J., and Sessa W. C. (2001) Acidic hydrolysis as a mechanism for the cleavage of the Glu298 → Asp variant of human endothelial nitric-oxide synthase. J. Biol. Chem. 276, 26674–26679 10.1074/jbc.M103647200 [DOI] [PubMed] [Google Scholar]
- 28. Lidell M. E., and Hansson G. C. (2006) Cleavage in the GDPH sequence of the C-terminal cysteine-rich part of the human MUC5AC mucin. Biochem. J. 399, 121–129 10.1042/BJ20060443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lidell M. E., Johansson M. E., and Hansson G. C. (2003) An autocatalytic cleavage in the C terminus of the human MUC2 mucin occurs at the low pH of the late secretory pathway. J. Biol. Chem. 278, 13944–13951 10.1074/jbc.M210069200 [DOI] [PubMed] [Google Scholar]
- 30. Ségalas I., Thai R., Ménez R., and Vita C. (1995) A particularly labile Asp–Pro bond in the green mamba muscarinic toxin MTX2. Effect of protein conformation on the rate of cleavage. FEBS Lett. 371, 171–175 10.1016/0014-5793(95)00844-Y [DOI] [PubMed] [Google Scholar]
- 31. Skribanek Z., Mezo G., Mák M., and Hudecz F. (2002) Mass spectrometric and chemical stability of the Asp–Pro bond in herpes simplex virus epitope peptides compared with X-Pro bonds of related sequences. J. Pept. Sci. 8, 398–406 10.1002/psc.395 [DOI] [PubMed] [Google Scholar]
- 32. Thuveson M., and Fries E. (2000) The low pH in trans-Golgi triggers autocatalytic cleavage of pre-α-inhibitor heavy chain precursor. J. Biol. Chem. 275, 30996–31000 10.1074/jbc.M002399200 [DOI] [PubMed] [Google Scholar]
- 33. Kast J., Hanecker P., Beaufort N., Giese A., Joutel A., Dichgans M., Opherk C., and Haffner C. (2014) Sequestration of latent TGF-β binding protein 1 into CADASIL-related Notch3-ECD deposits. Acta Neuropathol. Commun. 2, 96 10.1186/s40478-014-0096-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Aster J. C., Pear W. S., and Blacklow S. C. (2017) The varied roles of Notch in cancer. Annu. Rev. Pathol. 12, 245–275 10.1146/annurev-pathol-052016-100127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kovall R. A., Gebelein B., Sprinzak D., and Kopan R. (2017) The canonical Notch signaling pathway: structural and biochemical insights into shape, sugar, and force. Dev. Cell 41, 228–241 10.1016/j.devcel.2017.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ghezali L., Capone C., Baron-Menguy C., Ratelade J., Christensen S., Østergaard Pedersen L., Domenga-Denier V., Pedersen J. T., and Joutel A. (2018) Notch3(ECD) immunotherapy improves cerebrovascular responses in CADASIL mice. Ann. Neurol. 84, 246–259 10.1002/ana.25284 [DOI] [PubMed] [Google Scholar]
- 37. Mirra S. S., Heyman A., McKeel D., Sumi S. M., Crain B. J., Brownlee L. M., Vogel F. S., Hughes J. P., van Belle G., and Berg L. (1991) The Consortium to Establish a Registry for Alzheimer's Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer's disease. Neurology 41, 479–486 10.1212/WNL.41.4.479 [DOI] [PubMed] [Google Scholar]
- 38. Montine T. J., Phelps C. H., Beach T. G., Bigio E. H., Cairns N. J., Dickson D. W., Duyckaerts C., Frosch M. P., Masliah E., Mirra S. S., Nelson P. T., Schneider J. A., Thal D. R., Trojanowski J. Q., Vinters H. V., et al. (2012) National Institute on Aging–Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease: a practical approach. Acta Neuropathol. 123, 1–11 10.1007/s00401-011-0910-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Thal D. R., Rüb U., Schultz C., Sassin I., Ghebremedhin E., Del Tredici K., Braak E., and Braak H. (2000) Sequence of Aβ-protein deposition in the human medial temporal lobe. J. Neuropathol. Exp. Neurol. 59, 733–748 10.1093/jnen/59.8.733 [DOI] [PubMed] [Google Scholar]
- 40. Morris G. P., Clark I. A., and Vissel B. (2018) Questions concerning the role of amyloid-β in the definition, aetiology and diagnosis of Alzheimer's disease. Acta Neuropathol. 136, 663–689 10.1007/s00401-018-1918-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Dong H., Blaivas M., and Wang M. M. (2012) Bidirectional encroachment of collagen into the tunica media in cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. Brain Res. 1456, 64–71 10.1016/j.brainres.2012.03.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Dong H., Ding H., Young K., Blaivas M., Christensen P. J., and Wang M. M. (2013) Advanced intimal hyperplasia without luminal narrowing of leptomeningeal arteries in CADASIL. Stroke 44, 1456–1458 10.1161/STROKEAHA.111.000721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhang X., Lee S. J., Young M. F., and Wang M. M. (2015) The small leucine-rich proteoglycan BGN accumulates in CADASIL and binds to NOTCH3. Transl. Stroke Res. 6, 148–155 10.1007/s12975-014-0379-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Meng H., Zhang X., Yu G., Lee S. J., Chen Y. E., Prudovsky I., and Wang M. M. (2012) Biochemical characterization and cellular effects of CADASIL mutants of NOTCH3. PLoS One 7, e44964 10.1371/journal.pone.0044964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Meng H., Zhang X., Hankenson K. D., and Wang M. M. (2009) Thrombospondin 2 potentiates Notch3/Jagged1 signaling. J. Biol. Chem. 284, 7866–7874 10.1074/jbc.M803650200 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.