

Abstract
The cystic fibrosis transmembrane conductance regulator (CFTR) is an ion channel protein that is defective in individuals with cystic fibrosis (CF). To advance the rational design of CF therapies, it is important to elucidate how mutational defects in CFTR lead to its impairment and how pharmacological compounds interact with and alter CFTR. Here, using a helical-hairpin construct derived from CFTR's transmembrane (TM) helices 3 and 4 (TM3/4) and their intervening loop, we investigated the structural effects of a patient-derived CF-phenotypic mutation, E217G, located in the loop region of CFTR's membrane-spanning domain. Employing a single-molecule FRET assay to probe the folding status of reconstituted hairpins in lipid bilayers, we found that the E217G hairpin exhibits an altered adaptive packing behavior stemming from an additional GXXXG helix–helix interaction motif created in the mutant hairpin. This observation suggested that the misfolding and functional defects caused by the E217G mutation arise from an impaired conformational adaptability of TM helical segments in CFTR. The addition of the small-molecule corrector Lumacaftor exerts a helix stabilization effect not only on the E217G mutant hairpin, but also on WT TM3/4 and other mutations in the hairpin. This finding suggests a general mode of action for Lumacaftor through which this corrector efficiently improves maturation of various CFTR mutants.
Keywords: cystic fibrosis transmembrane conductance regulator (CFTR), cystic fibrosis, single-molecule biophysics, membrane protein, protein misfolding, helical packing, lumacaftor, pharmacological corrector, single-molecule Förster resonance energy transfer (FRET)
Introduction
The cystic fibrosis transmembrane conductance regulator (CFTR)6 is an ion channel protein that is defective in patients suffering from cystic fibrosis (CF). This inherited disease affects ∼1 of every 3,000 newborns among people of European ancestry and therefore represents the most common life-limiting genetic disorder in the Western world (1). >2,000 CFTR mutations have been reported to date (Cystic Fibrosis Mutation Database; http://www.genet.sickkids.on.ca/cftr/app;7 accessed June 20, 2018), a majority of which impair maturation of CFTR (e.g. at the endoplasmic reticulum (ER)) or alter its channel activity at the cell surface. In addition to the most common phenotypic mutation, a deletion of Phe-508 in the first nucleotide-binding domain, the membrane-associated portions of CFTR, comprising two six-strand transmembrane (TM) domains with adjacent intervening intra- and extracellular loop regions, represent particularly vulnerable hot spots and frequent targets of CF mutations. Strikingly, ∼33% of CFTR's disease-causing mutations are found in these segments, which themselves cover only <20% of CFTR's total residues (3).
Despite considerable progress in understanding CFTR pathology on a cellular level (4–7), the mechanisms by which mutations trigger misfolding and cause channel dysfunction remain largely obscure. In particular, there is very limited information on the underlying structures and conformational states that lead to an altered topology or dysfunctional state. Moreover, enormous efforts are currently being made in developing small-molecule compounds that correct the underlying misfolding or functional defect to increase the amount of matured protein at the cell surface or modulate CFTR activity (8–11). Recently, a binding site for two CFTR potentiators, ivacaftor and GLPG1837, has been found by cryo-EM (12). However, the mechanisms of action of many CFTR modulators still remain largely elusive. This lack of knowledge is mainly rooted in the challenges of studying folding of full-length CFTR. On the one hand, the WT protein is already notoriously difficult to obtain in sufficient quantities and purities for in vitro scrutiny, and proteins carrying destabilizing mutations are even less available. On the other hand, CFTR with its 1,480 amino acid residues is too large and too complex to pinpoint the local structural effects of a single point mutation, particularly for classical ensemble biochemical and biophysical techniques, which are often limited in their ability to resolve the structural heterogeneities of misfolded states.
To overcome these difficulties, we recently introduced a single-molecule approach that exploits helical-hairpin constructs derived from full-length CFTR to gain insights into the structural effects of misfolding and drug rescue (13). Helical hairpins, comprising two TM helices and their intervening loop region, are readily prepared in sufficient amounts for biophysical analysis. They constitute the smallest units that can be inserted autonomously by the translocon, since CFTR topogenesis in the ER is based on the pairwise integration of helical segments (6), and therefore represent minimal in vitro folding units of tertiary contacts between two helices in a membrane (14, 15). In tandem with single-molecule FRET (16), which serves as a spectroscopic ruler (17) to probe the end-to-end distances of hairpins reconstituted in lipid bilayers, these minimalistic folding units thus constitute versatile platforms to characterize the molecular events that link CF disease to structural effects of mutations and drug rescue, mimicking in vivo processes of CFTR misfolding and fold recovery. We have recently applied this approach to study misfolding of the CF-phenotypic TM mutation V232D in TM helix 4 (TM4) and the impact of the pharmacological corrector VX-809 (also known as Lumacaftor) (18) on hairpin misfolding by exploiting the TM3/4 hairpin construct, a helix-loop-helix hairpin comprising CFTR's third and fourth TM helices (human CFTR residues 194–241) and their intervening extracellular loop region 2 (ECL2) (13).
Herein, we exploit the TM3/4 hairpin construct to delineate structural effects of a pathogenic loop mutation and the impact of Lumacaftor on helical packing. Extramembranous loop regions represent key folding determinants (3, 19) and are crucial for the normal functioning of membrane proteins. Mutations in these regions can decrease the stability of a protein and alter topogenesis and are even capable of inducing a change in the secondary structure of TM segments (20–23). They have also been shown to impair the functionality of membrane proteins (24, 25) and membrane channels in particular (26), yet how the removal of a single amino acid in CFTR's loop region compromises the structure/function so extensively that a disease state ensues is still largely unclear. Among the two CFTR mutations found in the extracellular loop region connecting TM3 and TM4, we focus here on the disease-causing loop mutation E217G (Fig. 1a), which causes a mild form of the disease and drastically alters net charge and hydrophobicity of the loop region. Previous cell-based experiments have shown that E217G drastically impairs the maturation to the fully glycosylated, full-length protein (27, 28) and alters CFTR's activity at the cell surface (22). More recently, biophysical studies have yielded insights into secondary structure changes upon mutation and found that the helical content increases upon mutation (27). Yet the influence of the E217G mutation on tertiary structure changes (i.e. helical packing) has received limited study. Moreover, we and others have previously shown that Lumacaftor efficiently targets the first membrane-spanning domain of CFTR, including the TM3/4 hairpin, to rescue misfolding of mutations located in TM helices (13, 29–33), yet the potential effects of Lumacaftor on misfolding mutations located in loop regions, such as E217G, are unexplored.
Figure 1.

a, structure of human CFTR (44) (Protein Data Bank code 5UAK) highlighting the position of the E217G mutation in the interconnecting loop ECL2 (blue) of TM3/4 (yellow/red). Visualization was generated using Visual Molecular Dynamics (VMD) (47). b, schematic representation of the E217G (left) and WT (right) TM3/4 helical-hairpin motifs comprising CFTR's TM helices TM3 (yellow) and TM4 (red) and the intervening extracellular loop ECL2 (blue). The residues at position 217 are represented as van der Waals surfaces. The lengths of TM3 and TM4 are indicated for WT TM3/4, estimated from the cryo-EM structure (44) (Protein Data Bank code 5UAK). Visualization was generated using VMD (47). c, schematic of the single-molecule FRET approach for investigating hairpin conformations. Shown are single fluorescently labeled TM3/4 hairpin molecules reconstituted into phospholipid vesicles (not to scale) freely diffusing through the observation volume of the confocal microscope.
Results and discussion
To probe the effects of the E217G mutation and Lumacaftor action on TM3/4 hairpin folding, we made use of our single-molecule FRET approach and engineered a hairpin construct carrying the E217G mutation and labeled the hairpin with FRET donor and acceptor dyes at its N- and C-terminal ends (Fig. 1b). For reference, we prepared a WT hairpin construct as described previously (13). We reconstituted both variants into phosphatidylcholine (PC) lipid vesicles and employed confocal fluorescence spectroscopy to probe end-to-end distance changes of hairpins by monitoring FRET efficiencies from individual hairpin molecules (Fig. 1c). We first probed hairpin conformations in bilayers composed of 1-palmitoyl-2-oleoyl-sn-glycero-3-PC (i.e. POPC, 16:0–18:1 PC) (Fig. 2a), whose thickness (dh = 29.2 Å (34)) mimics that of the ER membrane (35) and matches well the length of TM3/4's helical segments (i.e. ∼30 Å for TM3 and ∼28 Å for TM4; see Fig. 1b). The FRET efficiency histograms of both hairpins exhibited a bimodal distribution with a major high-FRET and a minor low-FRET population, indicating that both hairpins exist in an equilibrium between a compact, folded structure and an open-state conformation with the equilibrium lying on the side of the compact conformation determined by tight helix–helix interactions. Introduction of the E217G mutation did not cause a change in the occupancy of the two states compared with the WT hairpin. Quantification of open-state and closed-state fractions using probability-distribution analysis (PDA) (Fig. 2a, red cityscapes) showed that the closed state in the E217G hairpin is, within error, indeed equally populated as in the WT hairpin (fC,WT,POPC = 0.76 ± 0.01; fC,E217G,POPC = 0.76 ± 0.004). Accordingly, closed hairpin stability in POPC, as reflected in the Gibbs free-energy change of hairpin closing, is favorable in both WT and E217G TM3/4 (ΔG°WT,POPC = −2.85 kJ/mol; ΔG°E217G,POPC = −2.85 kJ/mol). This suggests that the Glu-to-Gly exchange at position 217 in the loop region of CFTR does not affect the open/closed state equilibrium of the TM3/4 hairpin in POPC membranes.
Figure 2.

a–c, FRET efficiency histograms of E217G TM3/4 (pink) and WT TM3/4 (cyan) in POPC (16:0–18:1 PC) (a), DLPC (12:0 PC) (b), and DEiPC (20:1 PC) (c) lipid vesicles at room temperature. d, further experiments were performed in DEiPC lipid vesicles at 37 °C. PDA fits to the histograms are shown as red cityscapes. Right panels, fraction of closed hairpin (fC) as function of hydrophobic thickness (dh) for E217G TM3/4 (pink) and WT TM3/4 (cyan) as determined by PDA fits. dh represents the thicknesses of the entire hydrocarbon region, as determined at 20 °C (POPC and DLPC) (34) or 30 °C (DEiPC) (39). Errors are S.D. of the PDA χ2 minimization algorithm calculated from 10 iterations.
Whereas the E217G loop mutation might not alter packing stability per se, it might change the conformational adaptability of the helical segments, which is an important factor in the topogenesis and functioning of membrane proteins (36–38). Segmental malleability, for example, is required during co-translational folding and insertion when newly synthesized TM segments seek contacts with neighboring helices, but also for conformational cycling between the different functional states of a membrane protein. The adaptations required in these processes typically also depend on the given bilayer environment. For example, CFTR is embedded in bilayers of increasing thickness on its way from the ER membrane to the plasma membrane. Because loop regions represent hinges connecting TM segments, mutations in these regions may therefore alter the responsiveness of helical segments to changes in bilayer properties.
To test this hypothesis, we probed hairpin conformations in lipid bilayers composed of PCs of different acyl chain lengths and thus of varying hydrophobic thickness. We reconstituted hairpins into vesicles composed of thin 1,2-dilauroyl-sn-glycero-3-phosphopcholine (DLPC; 12:0 PC) and thick 1,2-dieicosenoyl-sn-glycero-3-phosphocholine (DEiPC; 20:1 PC) lipid bilayers to create a positive and negative hydrophobic mismatch situation between the hydrophobic stretches of the TM3/4 hairpins and the thickness of the lipid bilayers, respectively. In particular, the hydrophobic thickness of DLPC is ∼6–8 Å smaller (dh = 21.9 Å (34)), whereas the hydrophobic thickness of DEiPC is ∼3–5 Å larger (dh = 32.6 Å (39)) than the length of TM3/4's α-helices. FRET efficiency histograms in DLPC yielded almost identical equilibrium distributions for the open and closed states of both hairpins (Fig. 2b), indicating that both hairpins respond in a similar fashion to a decrease in membrane thickness and, thus, positive hydrophobic mismatch. By contrast, a marked difference in the adaptability was discernable between the two hairpins in DEiPC (Fig. 2c). Upon increasing bilayer thickness, the WT hairpin responded with a more pronounced opening behavior (fC,WT,DEiPC = 0.30 ± 0.01) than the E217G hairpin, in which the open and closed state were almost equally populated (fC,E217G,DEiPC = 0.48 ± 0.01). Accordingly, hairpin stability is more favorable in E217G TM3/4 (ΔG°E217G,DEiPC = 0.20 kJ/mol) than in WT TM3/4 (ΔG°WT,DEiPC = 2.09 kJ/mol). The same stabilization effect is also observed at physiological temperatures (Fig. 2d). At 37 °C, the E217G TM3/4 hairpin remains relatively more stabilized (ΔG°E217G,DEiPC,37 °C = 1.21 kJ/mol) than WT TM3/4 (ΔG°WT,DEiPC,37 °C = 3.42 kJ/mol), despite the shift in equilibrium of both hairpins toward the open state at 37 °C compared with measurements at room temperature (fC,E217G,DEiPC,37 °C = 0.38 ± 0.02; fC,WT,DEiPC,37 °C = 0.20 ± 0.01). This indicates that the increase in temperature affects the tertiary interactions between TM3 and TM4, but does not specifically impact the altered behavior in the E217G mutant. In summary, the strain imposed by negative hydrophobic mismatch created in DEiPC is more relaxed for the E217G mutant than for the WT hairpin. This suggests an altered responsiveness and conformational adaptability of the E217G hairpin.
The observed altered TM packing behavior likely stems from a difference in the interactions that are formed in the E217G TM3/4 hairpin. Gly residues in TM helices are known to facilitate interhelical interactions, especially in the form of the Gly motif GXXXG (40). Indeed, introduction of the Gly residue at position E217G creates an additional GXXXG motif at the C-terminal end of TM3 by which an extended alanine/glycine-zipper motif emerges (AXXXGXXXG) (41) that has corresponding knobs-into-holes partners in TM4 (Fig. 3a). In contrast to WT TM3/4 (Fig. 3b), this zipper motif might therefore prolong the membrane-spanning helical segments to form additional helix–helix interactions, particularly in situations of adaptive packing within the membrane, as also occurs during folding or channel functioning. Support for this scenario comes from CD spectroscopic studies that have shown that the helical content of the E217G mutant hairpin is increased upon mutation compared with the WT hairpin (27). Hence, our results together with the increased helical content suggest that the mutant hairpin's conformational adaptability is altered due to an additional TM zipper interaction motif that arises upon exchange of a glutamate for a glycine residue.
Figure 3.
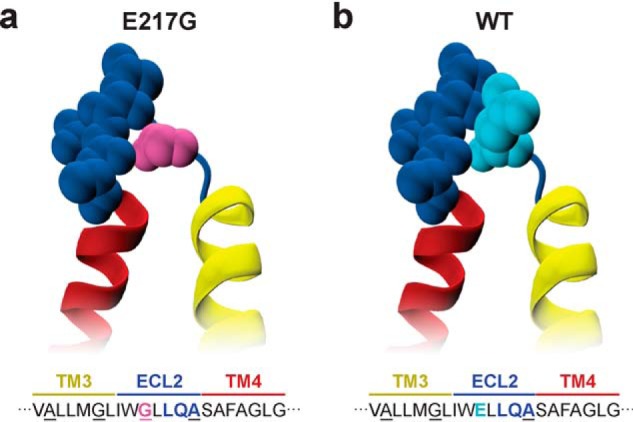
Close-up view of the ECL2 loop region in E217G (a) and WT TM3/4 (b) (yellow/red). Loop residues Gly-217 (pink) in E217G TM3/4 and Glu-217 (cyan) in WT TM3/4 as well as flanking loop residues (blue) are represented as van der Waals surfaces. The alanine/glycine-zipper motifs are indicated in the corresponding TM3/4 amino acid sequence. Visualization was generated using VMD (47).
Mechanistically, the observed changes in the adaptive packing behavior of E217G TM3/4 may help in rationalizing the susceptibility of the E217G full-length mutant protein to misfolding during biogenesis (27, 28). Formation of an additional GXXXG motif may restrict the conformational freedom and adaptability of the helical segments during co-translational folding and insertion of CFTR and impair the formation of interhelical interactions with other TM segments. This may inhibit maturation of CFTR because the protein is trapped in a partially folded, intermediate state at the ER, where it may be targeted for degradation by the ER's quality control machinery (6, 42).
Changes in the conformational plasticity may also help in rationalizing the impaired functioning of the E217G CFTR channel at the cell membrane. Previous electrophysiological experiments have shown that the E217G CFTR mutant exhibits a decreased Cl− efflux compared with WT CFTR upon stimulation, which has been attributed to transient rather than stable openings of E217G CFTR compared with WT CFTR (22). Because the extracellular loop connecting TM3 and TM4 seems to be involved in the open-state stabilization of the CFTR pore (22), our observation of reduced conformational/dynamic adaptability of the E217G containing TM3/4 segment may provide a mechanistic basis for the open-state destabilization of E217G CFTR causing channel dysfunction. Moreover, channel opening of CFTR is linked to a conformational change upon phosphorylation and ATP binding (43), and recent cryo-EM structures of human CFTR have provided further structural details of this opening mechanism (44, 45). Upon pore opening, the TM domains of CFTR rearrange such that the TM3/4 hairpin is in a more compact conformation (45), which appears functionally important. This conformational switch to the closed state may be disturbed in the mutant protein, trapping the CFTR pore in another closed intermediate state or leading to the disfavoring of the conformational change from closed to open state in general.
After having explored the influence of the E217G mutation on TM3/4 hairpin folding, we tested the effect of Lumacaftor on helical packing. Lumacaftor (Fig. 4a) is a corrector drug that has been developed to rescue misfolding of the most common CFTR mutation ΔF508 (18), which is located in the nucleotide-binding domain of CFTR. Yet the promiscuity of Lumacaftor for correcting various CF-causing mutants at different locations within CFTR implies that there may be multiple mechanisms or binding sites at play. To date, various groups have narrowed down the interaction of Lumacaftor to the first nucleotide-binding domain and/or the first TM domain of CFTR. We and others have recently reported that Lumacaftor targets the TM3/4 hairpin in TM domain 1 to rescue misfolding of the TM V232D mutation (13, 29–31), yet Lumacaftor also has a stabilizing effect on the WT hairpin (13, 18). Whereas compaction of WT TM3/4 is already favored in POPC (ΔG°WT,w/o Lumacaftor = −2.85 kJ/mol), the addition of Lumacaftor further increases hairpin stability by ΔΔG° = ΔG°Lumacaftor − ΔG°w/o Lumacaftor = −1.24 kJ/mol to ΔG°WT,Lumacaftor = −4.09 kJ/mol (Fig. 4b). We thus reasoned that this small-molecule corrector may also have a stabilization effect on the TM3/4 hairpin carrying loop mutations.
Figure 4.
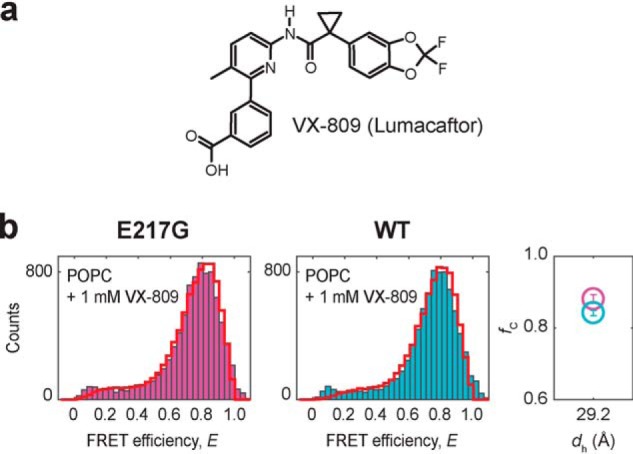
a, structure of the pharmacological corrector VX-809 (Lumacaftor). b, FRET efficiency histograms of E217G (pink) and WT TM3/4 (cyan) in POPC vesicles at 1 mm VX-809 and at room temperature. PDA fits are shown as red cityscapes. Right, fractions of closed hairpin (fC) as function of hydrophobic thickness (dh) for E217G TM3/4 (pink) and WT TM3/4 (blue) at 1 mm VX-809 as determined by PDA fits. dh of POPC represents the thickness of the entire hydrocarbon region, as determined at 20 °C (34). Errors are S.D. of the PDA χ2 minimization algorithm calculated from 10 iterations.
To test this hypothesis, we reconstituted the E217G hairpin into vesicles composed of POPC (Fig. 4b). In the absence of the corrector, the mutant hairpin exhibited a closed-state fraction of fC,E217G = 0.76 ± 0.004, which translates into a net stabilization of ΔG°E217G,w/o Lumacaftor = −2.85 kJ/mol. The addition of Lumacaftor led to an increase of the closed-state fraction (fC,E217G,Lumacaftor = 0.88 ± 0.01). This amounts to a decrease of the open-state fraction of 12% and, thus, an increase in hairpin stability of ΔΔG° = ΔG°Lumacaftor − ΔG°w/o Lumacaftor = −2.07 kJ/mol to ΔG°E217G,Lumacaftor = −4.92 kJ/mol. We also tested Lumacaftor action on another loop mutation in TM3/4, namely the pathogenic Q220R mutation, and observed a similar stabilization effect (ΔG°Q220R,w/o Lumacaftor = −2.21 kJ/mol, ΔG°Q220R,Lumacaftor = −5.43 kJ/mol, ΔΔG° = −3.22 kJ/mol) (Fig. S1). Thus, Lumacaftor exerts its effect also on TM3/4 carrying loop mutations, which, together with our previous results on the WT hairpin (13) and the V232D mutation, supports the idea that the corrector has a rather general mode of action and, thus, specificity for domains (i.e. TM domain segments) rather than for specific mutations.
The stabilization effect of Lumacaftor on the TM3/4 hairpin likely stems from its ability to reside in a phospholipid-bilayer membrane to modulate helical packing of TM domains. It has been proposed that Lumacaftor either interacts with the membrane-spanning stretches of CFTR and promotes tertiary interactions for tighter helix–helix packing or, through its membrane-destabilizing properties (46), aids the adaptation of a more compact fold by decreasing the hydrophobic mismatch between TM segments and the membrane. This could originate from a change in lateral pressure profiles and/or a more disordered hydrocarbon core layer, promoting more dynamic interhelical interactions and allowing polar solvent molecules (e.g. water) to penetrate deeper into the membrane, thereby locally decreasing bilayer hydrophobic thickness.
Indeed, we observed that Lumacaftor imposes changes on the stability of the phospholipid bilayer. Using a terbium(III)/dipicolinic acid fluorescence assay that sensitively reports on ion leakage from terbium-loaded liposomes, we found that Lumacaftor efficiently permeabilizes the membrane yet keeps the vesicles intact and causes a release of ion content in a dose-dependent manner up to 20 μm (Fig. 5a), at which point the disruptive properties of Lumacaftor reach a plateau at ∼40%. This suggests that the corrector perturbs the bilayer order and alters membrane permeability but does not disrupt the vesicles. Furthermore, we observed an increased dynamic behavior of TM segments for all hairpin variants after the addition of Lumacaftor, as judged from correlative analysis of relative donor fluorescence lifetime versus FRET efficiency (Fig. 5b). Accordingly, the hairpin molecules appear less constrained and more dynamic in the presence of Lumacaftor, while still being in a closed conformation. Overall, our results create a picture in which Lumacaftor disorders the membrane in such a way that, on the one hand, it supports native interactions between the two helices of TM3/4 and, on the other hand, intensifies dynamics between closely related compact conformations. This mode of action may be the dominant factor in the observed stabilization of WT TM3/4, E217G TM3/4, and Q220R TM3/4.
Figure 5.

a, terbium(III)/dipicolinic acid (DPA) fluorescence assay with increasing concentrations of VX-809 (Lumacaftor) in POPC (16:0–18:1 PC) liposomes. Measurements were performed at room temperature, and data were normalized to treatment with 0.1% Triton-lysed control. Significance at all doses is compared with the central point of 20 μm (****, p < 0.0001; n.s., not significant). Error bars, S.D. b, contour plots of relative donor fluorescence lifetime versus FRET efficiency of E217G TM3/4 (magenta), Q220R TM3/4 (orange), and WT TM3/4 (cyan) in POPC vesicles without (top row) and with the addition (bottom row) of 1 mm VX-809. All measurements were performed at room temperature. Models for “static FRET” (48, 49) (black line) and reconfiguration dynamics of a Gaussian chain (2) (red line) were fitted. In the presence of VX-809, all TM3/4 variants follow the reconfiguration dynamics of a Gaussian chain more prominently, which indicates that the corrector disorders the lipid bilayer and intensifies dynamics within the compact state. The position of the folded state is indicated by a black arrow.
Conclusion
Taken together, our findings suggest that both the folding defect and the channel dysfunction caused by the E217G mutation stem from an altered adaptive packing behavior of the mutant TM3/4 hairpin. The additional GXXXG helix-interaction motif created by the loop mutation at the C-terminal end of TM3 may restrict the conformational freedom of the TM3/4 helical segments during their folding and insertion and alter the conformational plasticity during ion channeling. The stabilization effect exerted by Lumacaftor on the WT TM3/4, V232D TM mutant, as well as E217G and Q220R loop mutant hairpins, support the idea that this corrector has a rather broad mode of action likely connected to its membrane-destabilizing properties, where it efficiently improves CFTR folding stability to recover misfolding from various CF mutations.
Experimental procedures
Details on experimental procedures including hairpin design (Fig. S2) and protein purification (Fig. S3) are given in the supporting information.
Author contributions
G. K., C. M. D., and M. Schlierf conceptualization; G. K., M. Schenkel, A. H., and D. R.-L. formal analysis; G. K. and M. Schlierf supervision; G. K., M. Schenkel, and A. H. validation; G. K., M. Schenkel, A. H., and M. Schlierf investigation; G. K., M. Schenkel, A. H., and C. M. D. methodology; G. K., M. Schenkel, and M. Schlierf writing-original draft; G. K., M. Schenkel, D. R.-L., C. M. D., and M. Schlierf writing-review and editing; M. Schenkel and D. R.-L. data curation; M. Schenkel visualization; A. H. software; C. M. D. resources; M. Schlierf funding acquisition; M. Schlierf project administration.
Supplementary Material
This work was supported by a scholarship of the European Social Fund and the Free State of Saxony (to M. Schenkel), an Exploration Grant of the Boehringer Ingelheim Foundation (BIS) (to M. Schlierf), Deutsche Forschungsgemeinschaft (DFG) Grant SCHL 1896/3-1 (to M. Schlierf), and a grant from Cystic Fibrosis Canada (to C. M. D.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains supporting Experimental procedures and Figs. S1–S3.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- CFTR
- cystic fibrosis transmembrane conductance regulator
- CF
- cystic fibrosis
- ER
- endoplasmic reticulum
- TM
- transmembrane
- ECL2
- extracellular loop region 2
- PC
- phosphatidylcholine
- PDA
- probability-distribution analysis
- DLPC
- 1,2-dilauroyl-sn-glycero-3-phosphopcholine
- DEiPC
- 1,2-dieicosenoyl-sn-glycero-3-phosphocholine
- w/o
- without.
References
- 1. O'Sullivan B. P., and Freedman S. D. (2009) Cystic fibrosis. Lancet 373, 1891–1904 10.1016/S0140-6736(09)60327-5 [DOI] [PubMed] [Google Scholar]
- 2. Soranno A., Buchli B., Nettels D., Cheng R. R., Müller-Späth S., Pfeil S. H., Hoffmann A., Lipman E. A., Makarov D. E., and Schuler B. (2012) Quantifying internal friction in unfolded and intrinsically disordered proteins with single-molecule spectroscopy. Proc. Natl. Acad. Sci. U.S.A. 109, 17800–17806 10.1073/pnas.1117368109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cheung J. C., and Deber C. M. (2008) Misfolding of the cystic fibrosis transmembrane conductance regulator and disease. Biochemistry 47, 1465–1473 10.1021/bi702209s [DOI] [PubMed] [Google Scholar]
- 4. Lukacs G. L., and Verkman A. S. (2012) CFTR: folding, misfolding and correcting the ΔF508 conformational defect. Trends Mol. Med. 18, 81–91 10.1016/j.molmed.2011.10.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Farinha C. M., and Canato S. (2017) From the endoplasmic reticulum to the plasma membrane: mechanisms of CFTR folding and trafficking. Cell Mol. Life Sci. 74, 39–55 10.1007/s00018-016-2387-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kim S. J., and Skach W. R. (2012) Mechanisms of CFTR folding at the endoplasmic reticulum. Front. Pharmacol. 3, 201 10.3389/fphar.2012.00201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Schlebach J. P., and Sanders C. R. (2015) The safety dance: biophysics of membrane protein folding and misfolding in a cellular context. Q. Rev. Biophys. 48, 1–34 10.1017/S0033583514000110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mijnders M., Kleizen B., and Braakman I. (2017) Correcting CFTR folding defects by small-molecule correctors to cure cystic fibrosis. Curr. Opin. Pharmacol. 34, 83–90 10.1016/j.coph.2017.09.014 [DOI] [PubMed] [Google Scholar]
- 9. Molinski S., Eckford P. D. W., Pasyk S., Ahmadi S., Chin S., and Bear C. E. (2012) Functional rescue of F508del-CFTR using small molecule correctors. Front. Pharmacol. 3, 160 10.3389/fphar.2012.00160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pedemonte N., and Galietta L. J. V. (2012) Pharmacological correctors of mutant CFTR mistrafficking. Front. Pharmacol. 3, 175 10.3389/fphar.2012.00175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rowe S. M., and Verkman A. S. (2013) Cystic fibrosis transmembrane regulator correctors and potentiators. Cold Spring Harb. Perspect. Med. 3, a009761 10.1101/cshperspect.a009761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Liu F., Zhang Z., Levit A., Levring J., Touhara K. K., Shoichet B. K., and Chen J. (2019) Structural identification of a hotspot on CFTR for potentiation. Science 364, 1184–1188 10.1126/science.aaw7611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Krainer G., Treff A., Hartmann A., Stone T. A., Schenkel M., Keller S., Deber C. M., and Schlierf M. (2018) A minimal helical-hairpin motif provides molecular-level insights into misfolding and pharmacological rescue of CFTR. Commun. Biol. 1, 154 10.1038/s42003-018-0153-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Therien A. G., Glibowicka M., and Deber C. M. (2002) Expression and purification of two hydrophobic double-spanning membrane proteins derived from the cystic fibrosis transmembrane conductance regulator. Protein Expr. Purif. 25, 81–86 10.1006/prep.2001.1612 [DOI] [PubMed] [Google Scholar]
- 15. Rath A., Tulumello D. V., and Deber C. M. (2009) Peptide models of membrane protein folding. Biochemistry 48, 3036–3045 10.1021/bi900184j [DOI] [PubMed] [Google Scholar]
- 16. Krainer G., Keller S., and Schlierf M. (2019) Structural dynamics of membrane-protein folding from single-molecule FRET. Curr. Opin. Struct. Biol. 58, 124–137 10.1016/j.sbi.2019.05.025 [DOI] [PubMed] [Google Scholar]
- 17. Hellenkamp B., Schmid S., Doroshenko O., Opanasyuk O., Kühnemuth R., Rezaei Adariani S., Ambrose B., Aznauryan M., Barth A., Birkedal V., Bowen M. E., Chen H., Cordes T., Eilert T., Fijen C., et al. (2018) Precision and accuracy of single-molecule FRET measurements-a multi-laboratory benchmark study. Nat. Methods 15, 669–676 10.1038/s41592-018-0085-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Van Goor F., Hadida S., Grootenhuis P. D. J., Burton B., Stack J. H., Straley K. S., Decker C. J., Miller M., McCartney J., Olson E. R., Wine J. J., Frizzell R. A., Ashlock M., and Negulescu P. A. (2011) Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc. Natl. Acad. Sci. U.S.A. 108, 18843–18848 10.1073/pnas.1105787108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tastan O., Klein-Seetharaman J., and Meirovitch H. (2009) The effect of loops on the structural organization of α-helical membrane proteins. Biophys. J. 96, 2299–2312 10.1016/j.bpj.2008.12.3894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nadeau V. G., and Deber C. M. (2013) Loop sequence dictates the secondary structure of a human membrane protein hairpin. Biochemistry 52, 2419–2426 10.1021/bi400187w [DOI] [PubMed] [Google Scholar]
- 21. Nadeau V. G., and Deber C. M. (2016) Structural impact of proline mutations in the loop region of an ancestral membrane protein. Biopolymers 106, 37–42 10.1002/bip.22765 [DOI] [PubMed] [Google Scholar]
- 22. Hämmerle M. M., Aleksandrov A. A., and Riordan J. R. (2001) Disease-associated mutations in the extracytoplasmic loops of cystic fibrosis transmembrane conductance regulator do not impede biosynthetic processing but impair chloride channel stability. J. Biol. Chem. 276, 14848–14854 10.1074/jbc.M011017200 [DOI] [PubMed] [Google Scholar]
- 23. Kim J. M., Booth P. J., Allen S. J., and Khorana H. G. (2001) Structure and function in bacteriorhodopsin: the role of the interhelical loops in the folding and stability of bacteriorhodopsin. J. Mol. Biol. 308, 409–422 10.1006/jmbi.2001.4603 [DOI] [PubMed] [Google Scholar]
- 24. Kim J., Jiang Q., Glashofer M., Yehle S., Wess J., and Jacobson K. A. (1996) Glutamate residues in the second extracellular loop of the human A2a adenosine receptor are required for ligand recognition. Mol. Pharmacol. 49, 683–691 [PMC free article] [PubMed] [Google Scholar]
- 25. Du K., Nicole P., Couvineau A., and Laburthe M. (1997) Aspartate 196 in the first extracellular loop of the human VIP1 receptor is essential for VIP binding and VIP-stimulated cAMP production. Biochem. Biophys. Res. Commun. 230, 289–292 10.1006/bbrc.1996.5949 [DOI] [PubMed] [Google Scholar]
- 26. Seibert F. S., Jia Y., Mathews C. J., Hanrahan J. W., Riordan J. R., Loo T. W., and Clarke D. M. (1997) Disease-associated mutations in cytoplasmic loops 1 and 2 of cystic fibrosis transmembrane conductance regulator impede processing or opening of the channel. Biochemistry 36, 11966–11974 10.1021/bi9712652 [DOI] [PubMed] [Google Scholar]
- 27. Chang Y.-H., Stone T. A., Chin S., Glibowicka M., Bear C. E., and Deber C. M. (2018) Structural effects of extracellular loop mutations in CFTR helical hairpins. Biochim. Biophys. Acta Biomembr. 1860, 1092–1098 10.1016/j.bbamem.2018.01.003 [DOI] [PubMed] [Google Scholar]
- 28. Lee J. H., Choi J. H., Namkung W., Hanrahan J. W., Chang J., Song S. Y., Park S. W., Kim D. S., Yoon J.-H., Suh Y., Jang I.-J., Nam J. H., Kim S. J., Cho M.-O., Lee J.-E., et al. (2003) A haplotype-based molecular analysis of CFTR mutations associated with respiratory and pancreatic diseases. Hum. Mol. Genet. 12, 2321–2332 10.1093/hmg/ddg243 [DOI] [PubMed] [Google Scholar]
- 29. Ren H. Y., Grove D. E., De La Rosa O., Houck S. A., Sopha P., Van Goor F., Hoffman B. J., and Cyr D. M. (2013) VX-809 corrects folding defects in cystic fibrosis transmembrane conductance regulator protein through action on membrane-spanning domain 1. Mol. Biol. Cell 24, 3016–3024 10.1091/mbc.e13-05-0240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Loo T. W., Bartlett M. C., and Clarke D. M. (2013) Bithiazole correctors rescue CFTR mutants by two different mechanisms. Biochemistry 52, 5161–5163 10.1021/bi4008758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Loo T. W., and Clarke D. M. (2014) The cystic fibrosis V232D mutation inhibits CFTR maturation by disrupting a hydrophobic pocket rather than formation of aberrant interhelical hydrogen bonds. Biochem. Pharmacol. 88, 46–57 10.1016/j.bcp.2013.12.027 [DOI] [PubMed] [Google Scholar]
- 32. Loo T. W., Bartlett M. C., and Clarke D. M. (2013) Corrector VX-809 stabilizes the first transmembrane domain of CFTR. Biochem. Pharmacol. 86, 612–619 10.1016/j.bcp.2013.06.028 [DOI] [PubMed] [Google Scholar]
- 33. Laselva O., Molinski S., Casavola V., and Bear C. E. (2018) Correctors of the major cystic fibrosis mutant interact through membrane-spanning domains. Mol. Pharmacol. 93, 612–618 10.1124/mol.118.111799 [DOI] [PubMed] [Google Scholar]
- 34. Kučerka N., Nieh M.-P., and Katsaras J. (2011) Fluid phase lipid areas and bilayer thicknesses of commonly used phosphatidylcholines as a function of temperature. Biochim. Biophys. Acta 1808, 2761–2771 10.1016/j.bbamem.2011.07.022 [DOI] [PubMed] [Google Scholar]
- 35. Mitra K., Ubarretxena-Belandia I., Taguchi T., Warren G., and Engelman D. M. (2004) Modulation of the bilayer thickness of exocytic pathway membranes by membrane proteins rather than cholesterol. Proc. Natl. Acad. Sci. U.S.A. 101, 4083–4088 10.1073/pnas.0307332101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Langosch D., and Arkin I. T. (2009) Interaction and conformational dynamics of membrane-spanning protein helices: transmembrane domain assembly and dynamics. Protein Sci. 18, 1343–1358 10.1002/pro.154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Frotscher E., Krainer G., Hartmann A., Schlierf M., and Keller S. (2018) Conformational dynamics govern the free-energy landscape of a membrane-interacting protein. ACS Omega 3, 12026–12032 10.1021/acsomega.8b01609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hildebrand P. W., Rother K., Goede A., Preissner R., and Frömmel C. (2005) Molecular packing and packing defects in helical membrane proteins. Biophys. J. 88, 1970–1977 10.1529/biophysj.104.049585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kučerka N., Gallová J., Uhríková D., Balgavý P., Bulacu M., Marrink S.-J., and Katsaras J. (2009) Areas of monounsaturated diacylphosphatidylcholines. Biophys. J. 97, 1926–1932 10.1016/j.bpj.2009.06.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Teese M. G., and Langosch D. (2015) Role of GxxxG motifs in transmembrane domain interactions. Biochemistry 54, 5125–5135 10.1021/acs.biochem.5b00495 [DOI] [PubMed] [Google Scholar]
- 41. Kim S., Jeon T.-J., Oberai A., Yang D., Schmidt J. J., and Bowie J. U. (2005) Transmembrane glycine zippers: physiological and pathological roles in membrane proteins. Proc. Natl. Acad. Sci. U.S.A. 102, 14278–14283 10.1073/pnas.0501234102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sanders C. R., and Myers J. K. (2004) Disease-related misassembly of membrane proteins. Annu. Rev. Biophys. Biomol. Struct. 33, 25–51 10.1146/annurev.biophys.33.110502.140348 [DOI] [PubMed] [Google Scholar]
- 43. Gadsby D. C., Vergani P., and Csanády L. (2006) The ABC protein turned chloride channel whose failure causes cystic fibrosis. Nature 440, 477–483 10.1038/nature04712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Liu F., Zhang Z., Csanády L., Gadsby D. C., and Chen J. (2017) Molecular structure of the human CFTR ion channel. Cell 169, 85–95.e8 10.1016/j.cell.2017.02.024 [DOI] [PubMed] [Google Scholar]
- 45. Zhang Z., Liu F., and Chen J. (2018) Molecular structure of the ATP-bound, phosphorylated human CFTR. Proc. Natl. Acad. Sci. U.S.A. 115, 12757–12762 10.1073/pnas.1815287115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Baroni D., Zegarra-Moran O., Svensson A., and Moran O. (2014) Direct interaction of a CFTR potentiator and a CFTR corrector with phospholipid bilayers. Eur. Biophys. J. 43, 341–346 10.1007/s00249-014-0956-y [DOI] [PubMed] [Google Scholar]
- 47. Humphrey W., Dalke A., and Schulten K. (1996) Visual molecular dynamics. J. Mol. Graph. 14, 33–38, 27–28 10.1016/0263-7855(96)00018-5 [DOI] [PubMed] [Google Scholar]
- 48. Kudryavtsev V., Sikor M., Kalinin S., Mokranjac D., Seidel C. A. M., and Lamb D. C. (2012) Combining MFD and PIE for accurate single-pair Förster resonance energy transfer measurements. Chemphyschem 13, 1060–1078 10.1002/cphc.201100822 [DOI] [PubMed] [Google Scholar]
- 49. Sisamakis E., Valeri A., Kalinin S., Rothwell P. J., and Seidel C. A. M. (2010) Accurate single-molecule FRET studies using multiparameter fluorescence detection. Methods Enzymol. 475, 455–514 10.1016/S0076-6879(10)75018-7 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.