

Abstract
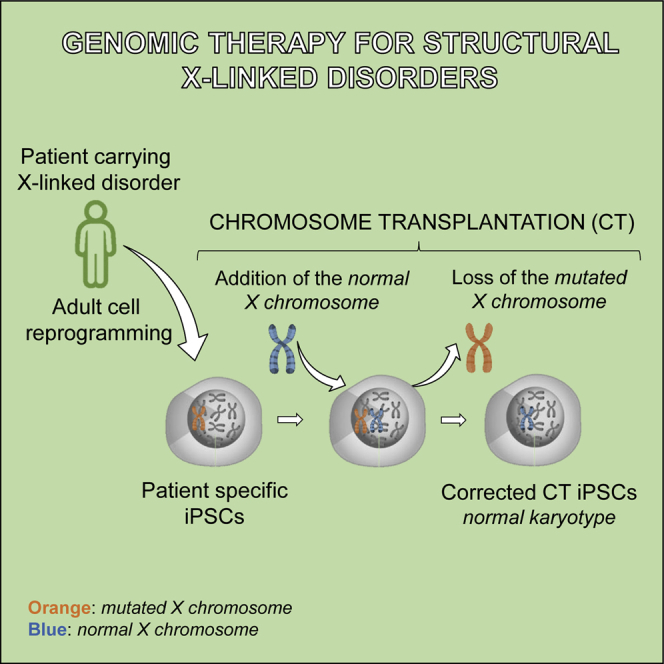
Many human genetic diseases are associated with gross mutations such as aneuploidies, deletions, duplications, or inversions. For these “structural” disorders, conventional gene therapy, based on viral vectors and/or on programmable nuclease-mediated homologous recombination, is still unsatisfactory. To correct such disorders, chromosome transplantation (CT), defined as the perfect substitution of an endogenous defective chromosome with an exogenous normal one, could be applied. CT re-establishes a normal diploid cell, leaving no marker of the procedure, as we have recently shown in mouse pluripotent stem cells. To prove the feasibility of the CT approach in human cells, we used human induced pluripotent stem cells (hiPSCs) reprogrammed from Lesch-Nyhan (LN) disease patients, taking advantage of their mutation in the X-linked HPRT gene, making the LN cells selectable and distinguishable from the resistant corrected normal cells. In this study, we demonstrate, for the first time, that CT is feasible in hiPSCs: the normal exogenous X chromosome was first transferred using an improved chromosome transfer system, and the extra sex chromosome was spontaneously lost. These CT cells were functionally corrected and maintained their pluripotency and differentiation capability. By inactivation of the autologous HPRT gene, CT paves the way to the correction of hiPSCs from several X-linked disorders.
Keywords: genomic disorders, induced pluripotent stem cells, genetic therapy, chromosome transfer, chromosome transplantation
Graphical Abstract
Introduction
The introduction of programmable nucleases such as CRISPR/Cas9 in the arsenal of geneticists trying to edit the human genome has clearly generated great hope of correcting the defects associated with several hereditary diseases.1, 2, 3 In spite of the great potential of this approach, some structural abnormalities involving large chromosome sections remain difficult to amend. These include large inversions, deletions or duplications, complex chromosomal rearrangements, and aneuploidies.4 CRISPR/Cas9 has been tested even in some of these large abnormalities,5 but the results achieved are still unsatisfactory. Indeed, the approach undertaken in Duchenne muscular dystrophy (DMD) animal models and patients, 80% of whom have large deletions, focused on the production of in-frame, albeit abnormal, transcripts by exon skipping, thus coding for an internally deleted protein, which is only partially functional.6, 7, 8, 9, 10, 11, 12 Likewise, the mutation in fragile X patients is due to an abnormal expansion of triplets in the Xq28 region, which cannot be completely restored by canonical approaches.13, 14, 15, 16 In addition, aneuploidies involving sex chromosomes are not manageable with any of these methods.
In previous work, we have introduced the concept of chromosome transplantation (CT) as a means to achieve correction in the selected cases that cannot benefit by classical gene therapy methods.17 CT refers to the precise substitution of an endogenous defective chromosome with a normal exogenous one. CT generates euploid cells and is different from chromosome transfer in which the addition of one or a few chromosomes gives rise to aneuploid cells, which cannot be of clinical utility. Likewise, CT is different from human artificial chromosome (HAC) transfer, in which an artificial vector is added. CT is a new approach that is potentially useful in the new field of “genomic or chromosome therapy,”18 an area that has been long neglected. We showed the feasibility of this approach in mice by performing CT in Hprt-deficient embryonic stem cells (ESCs),17 a model for the human Lesch-Nyhan (LN) disease, and in induced pluripotent stem cells (iPSCs) from a mouse model of chronic granulomatous disease (CGD).19 However, to our knowledge, human pluripotent stem cells have so far been refractory to CT, although recently an artificial chromosome has been successfully transferred to human iPSCs.20
In this study, we report the achievement for the first time of CT in human iPSCs using as proof of concept reprogrammed cells from a LN patient carrying a mutation in the X-linked HPRT gene useful for the selection of the corrected cells. By inactivation of the HPRT gene in patient-derived iPSCs, this approach could be used for the correction of several X-linked disorders.
Results
Experimental Design
The procedure used follows a step-by-step protocol exploiting the HPRT system that allows the selection of corrected cells: LN HPRT− cells die in hypoxanthine-aminopterin-thymidine (HAT) medium, whereas only the corrected HPRT+ cells survive (Figure 1A). (1) iPSCs are generated from a LN patient (LN-iPSCs) with a defect in the HPRT gene. (2) Microcells, carrying a single chromosome, produced from a human HPRT+ X chromosome-containing donor cell line, are fused with LN-iPSCs and selected in HAT medium. (3) HAT-resistant 47,XXY cells are isolated, and clones losing one of the sex chromosomes are amplified and characterized. The transplanted cells maintain their pluripotent status and remain perfectly diploid.
Figure 1.

Overview of the Chromosome Transplantation Protocol Developed to Correct Human iPSCs
(A) Scheme illustrating the CT protocol followed to generate corrected LN-iPSCs in which an endogenous sex chromosome is replaced with an exogenous normal X chromosome (HSAX-HPRT+) to rescue the genetic defect. (1) A donor cell line containing a normal human X chromosome (HSAX-HPRT+) was infected with the EnvΔR lentivirus. (2) Microcells from the EnvΔR donor cells were obtained and (3) co-cultured with the recipient LN-iPSCs to mediate cell-microcell fusion. (4) The resulting fused cells were selected in HAT medium (5) to identify those in which the normal HSAX has been acquired. (6) Finally, corrected diploid clones in which an endogenous sex chromosome has been lost were identified. HSAX, Homo sapiens X chromosome; HPRT, hypoxanthine phosphoribosyltransferase; EnvΔR, lentivirus expressing the envelope protein of the murine leukemia viruses (MLVs); HAT medium, hypoxanthine-aminopterin-thymidine medium. (B) Schematic representation showing the procedure followed to generate the donor cell line (CHO/HSAX) containing a normal X chromosome suitable for the retro-MMCT protocol. WCF, whole-cell fusion; HFC, human fibroblast cell; HSAX, Homo sapiens X chromosome; CHO, Chinese hamster ovary; HPRT, hypoxanthine phosphoribosyltransferase; HAT medium, hypoxanthine-aminopterin-thymidine medium. (C) Representative metaphase spreads after FISH on an HAT-resistant CHO/HSAX clone using the X chromosome painting (red, left) and human genomic DNA (hDNA in red, right) as probes, respectively. (D) Time lines of the two retro-MMCT protocols here exploited (protocols A and B). (E) Efficiencies of different experiments in the generation of HAT-resistant (HATR) clones in various experiments. In protocol A, 1 × 106 recipient human iPSCs were used for every experiment; in protocol B, 1.5 × 105 recipient human iPSCs were used for every experiment.
Generation of LN-iPSCs
Fibroblasts from a male LN patient (46,XY), carrying a deletion of the HPRT exon 1, which was confirmed by PCR analysis (Figure S1A), were reprogrammed21 and LN-iPSC clones were identified and characterized. The clones showed an iPSC-like colony morphology, a normal 46,XY karyotype, and stemness markers and were able to differentiate in vitro, as shown by positivity to all three germ-layer markers (Figures S1B–S1D). Clone LN-iPSC #1 was chosen for CT experiments.
X. Chromosome Transfer
We initially performed several trials of the classical protocol of chromosome transfer by microcell-mediated chromosome transfer (MMCT) without success. Therefore, we tried several modifications, including a strategy recently reported by us for mouse pluripotent cells,22 which was adapted to human iPSC lines. With this approach, we were able to obtain several clones in different cell fusion experiments. We report this achievement in detail below.
MMCT requires donor cells able to generate microcells with high efficiency. Not all cell lines have this peculiarity, and the most suitable ones are mouse fibroblast-derived A9 and Chinese hamster ovary (CHO) cells.23 We initially generated a hybrid A9 mouse cell line (A9/human) containing several human chromosomes, including the X one (Homo sapiens X chromosome [HSAX]), which was used for the initial trials of classical MMCT. However, we were unable to obtain stable clones acquiring the exogenous X chromosome. For this reason, we decided to adapt a strategy recently described by us (retro-MMCT), which exploits the fusogenic properties of the R peptide-deleted envelope (Env) protein (EnvΔR) from the amphotropic murine leukemia virus (MLV).22
In order to use this system, it is important to take into consideration some aspects. The Env protein here exploited is fusogenic for human and mouse cells since they express the receptor for the Env protein; hence, after Env infection, human or mouse cells fuse with each other, forming syncytia. For this reason, we could not use our previously generated donor A9/human cell line for retro-MMCT. We investigated a system to generate a non-auto-fusogenic donor cell line containing the desired HSAX to transplant. To do this, we chose an Hprt− CHO cell line able to form microcells that do not express the receptor for the Env protein. Therefore, we introduced in the CHO genome by MMCT only a single HSAX derived from the A9/human cell line and we isolated by HAT selection some resistant CHO/HSAX hybrid clones (Figure 1B). After fluorescence in situ hybridization (FISH) confirmation of the presence of the HSAX, one positive clone (CHO/HSAX #1) was selected for further use (Figure 1C).
To correct the LN-iPSCs by CT, we first transferred a normal HSAX into LN-iPSCs by retro-MMCT. Microcells were prepared from CHO/HSAX cells expressing the EnvΔR and then co-cultured with LN-iPSCs. In order to maximize the generation of single-chromosome microcells, the preparation was filtered twice. HSAX transferred LN-iPSC clones were selected in 1× HAT medium. In the first five successful experiments, 12 clones initially survived (protocol A).
Considering that human iPSCs are more difficult to manipulate than mouse cells, in subsequent experiments (protocol B), we slightly modified some conditions (Figure 1D). Essentially, we increased the fusion ratio between microcells and recipient cells from 1:1 to 20:1 to improve the fusion rate, and we reduced the HAT concentration to 0.1× to minimize the cell suffering under selection. We obtained a higher number of HAT-resistant clones (41 and 27, respectively), representing a chromosome transfer efficiency on the order of 10−4 clones per recipient cell, comparable to other mouse cell lines previously tested22 (Figure 1E).
Isolation and Characterization of CT Clones
A total of nine HAT-resistant clones obtained were characterized for the presence of the extra HSAX by karyotype, FISH, and PCR analysis of the wild-type (WT) HPRT allele, and all of them exhibited the expected karyotype of 47,XXY (Figure 2; Figure S2). Interestingly, clones with the 47,XXY karyotype showed the tendency to lose in culture the extra sex chromosome: in some clones this loss occurred very rapidly. Three of these clones (LNX-N11.2, LNX-N12.3, and LNX-D) were chosen for deeper analysis. LNX-N11.2 and LNX-N12.3 early lost the Y chromosome, becoming 46,XX (Figures 2C and 2D). The third clone (LNX-D) stably maintained all three sex chromosomes, but after a few passages in culture it started to differentiate spontaneously, even in selective culture medium. For this reason, the clone underwent a second turn of reprogramming with Sendai virus. After reprogramming, the 47,XXY clone, renamed LNX-D1, started to lose one X chromosome and after six passages (from p4 to p10), and 23% of cells showed a normal 46,XY karyotype (Figures 3A and 3B). From this pool, after subcloning, we isolated two subclones (LNX-D1B and LNX-D1T) with a homogeneous corrected 46,XY cell population (Figure 3C).
Figure 2.

Chromosome Transplantation in Human LN-iPSCs
(A) Representative karyotype (left) and metaphase spread (right) after FISH with a probe (green) for the human X chromosome of the HAT-resistant clones (HATR) after retro-MMCT. Chromosomes were counterstained with DAPI (blue). (B) PCR analysis of the HPRT locus in four CT-corrected clones (LNX-D1B, LNX-N9.5, LNX11.2, and LNX12.3) in the HPRT mutated LN-iPSCs and in the HPRT WT control cells, respectively. Neg, negative control. 1 kb, DNA ladder. The 461-bp band, specific for the exon1, is seen only in the HPRT WT control and in the CT clones. (C and D) Analysis of LNX-N11.2 (C) and LNX-N12.3 (D) clones. Left, histogram showing the chromosome distribution; middle, representative DAPI-banding karyotype; and right, metaphase spread after FISH with a human X chromosome painting probe (red). Chromosomes were counterstained with DAPI (blue). p, passage number.
Figure 3.

Cytogenetic Characterization of the HATR LNX-D1 Clone after the Second Round of Reprogramming
(A and B) Analysis of the LNX-D1 clone at p4 (A) and p10 (B). Left, histograms showing the karyotype distribution; and right, representative metaphase spreads after FISH using a human X chromosome painting probe (red). Loss of one of the X chromosomes occurred after few passages in culture (p10). p, passage number. (C) Analysis of the CT 46,XY LNX-D1B subclone. Left, histogram showing the chromosome distribution; middle, representative DAPI-banding karyotype; and right: metaphase spread after FISH with a human X chromosome painting probe (red). Chromosomes were counterstained with DAPI (blue). p, passage number.
Informative SNP analysis of the parental 47,XXY (LNX-D1) and the two 46,XY CT clones confirmed that the remaining X chromosome was of donor origin (Figure 4A).
Figure 4.

Molecular Characterization of the Corrected 46,XY Clones
(A) Informative SNP analysis in the donor and in recipient cell lines, and in the parental HATR 47,XXY clone and its derivative CT-corrected 46,XY clones. (B) Chromosome distribution of the discordant variants. High-quality variants obtained by WES of the LNX-D1B and LNX-D1T CT clones were compared with data from the parental LN-iPSC #1 clone.
The three CT clones maintained their pluripotency features, as shown by the expression of stemness markers (Figure S3A) and the capacity to form embryoid bodies (EBs) and differentiate into the three germ layers (Figure S3B). Although the fusion procedure is not expected to be mutagenic, we performed whole-exome sequencing (WES) on two LNX-D1 46,XY subclones (LNX-D1B and LNX-D1T) and their parental pre-fusion cell line (LN-iPSC #1) to detect putative abnormalities at the nucleotide level. Variant calling was performing using hg19 as the reference genome. Among the selected variants, we detected only one (LNX-D1B) or two discrepancies (LNX-D1T) between the parental cell line and its 46,XY CT corrected clones at the level of all the autosomes. Conversely, 20% of variants on the X chromosome were discordant, in agreement with the different origin of the donor and endogenous X chromosomes (Figure 4B). Taken together, these data confirm the CT event, also suggesting that the fusion process is not particularly damaging to the human genome.
Discussion
In this study, we demonstrate for the first time that human iPSCs represent a suitable substrate for CT, paving the way for personalized medicine also for genomic disorders. Our data allow us to draw several conclusions in this regard. First, we have generated a donor cell line with a normal HSAX that could be used for the correction of iPSCs derived from any patient affected by X-linked “structural” disorders. Second, we have set up a high efficient retro-MMCT protocol for human iPSCs. Third, we observed that pluripotent cells acquiring an extra chromosome by MMCT are prone to spontaneously lose the additional one, probably due to abnormalities in nuclear morphology and decreased cellular fitness associated with trisomic aneuploidy, as recently reported.24,25 This loss has also been shown to occur in human triploid pluripotent cell lines.25,26 Any of the three sex chromosomes can be lost, since both XY and XX diploid clones were obtained, but only cells maintaining the normal (donor) X chromosome would survive selection; however, the specific loss of either endogenous X or Y chromosomes cannot be anticipated. Fourth, the CT method in itself is not mutagenic, although some mutations could arise during prolonged periods of culture, as occurs with any long term-cultured pluripotent stem cells.
In the approach used in the present study, we first generated iPSCs from patient fibroblasts and subsequently performed MMCT. Alternatively, MMCT could be performed first in fibroblasts or other cells, followed by reprogramming to iPSCs. Although it is generally thought that MMCT is easier to perform in pluripotent stem cells than in primary fibroblasts, some authors have been successful with the second strategy.27 In our opinion, both strategies are equivalent, since they use both the MMCT and the reprogramming steps, only changing their time sequence.
CT by MMCT could affect genome integrity, whose maintenance must be tested by whole-genome sequencing. A further limitation of the CT is linked to the epigenetic landscape of the transplanted chromosome, which could differ from the endogenous counterpart. This is particularly important in the case of the X chromosome, due to the phenomenon of X inactivation in corrected 46,XX cells. Although we did not investigate this point, we noticed that the exogenous chromosome was functional and was not inactivated, since the HPRT gene was obviously expressed to resist HAT selection.
The genetic defect of several diseases located on the X chromosome can be corrected by the CT approach. DMD, a severe muscle disease due to large deletions in about 80% of patients, looks like the most obvious candidate. Due to the large dystrophin gene size, spread on about 2 Mb of the X chromosome, and a transcript of more than 10 kb, classical gene therapy approaches have only partially been successful. More recently, results with subsets of mutations have been reported.12,28, 29, 30 In any case, the clinical improvement is modest,31 and novel approaches are eagerly awaited.32 Transfer of an HAC vector containing a complete dystrophin gene has also been shown to correct the DMD phenotype.27,33 However, HACs are sometimes silenced and/or are lost from the cells,20 although other studies did show appropriate transgene maintenance and expression.34,35 Correction of mutations in DMD cells could be achieved by CT followed by differentiation to muscle cells and their injection in the muscles, an option that has been used by several researchers in muscle pathology.27,33,36, 37, 38 The only needed additional step to the protocol used herein is the inactivation, before retro-MMCT fusion, of the HPRT gene on the endogenous X chromosome by CRISPR technology, allowing the use of HAT selection after retro-MMCT. We have recently shown the feasibility of this step by correcting the genetic defect in iPSCs from a mouse model of the X-linked CGD. Finally, this technique might not only lead to sex modification of pluripotent cells but also opens the way to the engineering of a synthetic human X chromosome, which could be modified ad libitum in a host cell line and subsequently introduced in normal pluripotent cells.39
Materials and Methods
Ethics Approval and Consent to Participate
This study was approved by the Independent Ethics Committee of the Humanitas Clinical Institute (approval reference no. 2172).
Cell Cultures
Fibroblasts from the LN patient were obtained from the NIGMS Human Genetic Cell Repository at the Coriell Institute for Medical Research (GM 20394). Human dermal fibroblasts (HDFs) from a normal donor (Thermo Fisher Scientific, Waltham, MA, USA) and the LN fibroblasts were maintained in high-glucose DMEM medium, supplemented with 10% fetal bovine serum (FBS; Lonza, Basel, Switzerland).
The A9 mouse fibroblast line, Hprt− derivative of strain L (Sigma-Aldrich),40 was grown in high-glucose DMEM (Lonza) supplemented with 10% FBS (Lonza). The A9/human hybrid cell line was obtained by whole-cell fusion between A9 cells and HDF cells; clones with a normal human X chromosome were selected in 1× HAT medium (Sigma-Aldrich, Munich, Germany).
Hprt-defective CHO cells were kindly provided by Dr. E. Raimondi (University of Pavia, Pavia, Italy). The CHO cells were cultured in RPMI 1640 medium supplemented with 10% FBS (Lonza), and their microcell-fused derivative cells were selected in 1× HAT medium (Sigma-Aldrich).
Human iPSCs were cultured in feeder-free conditions, using Essential 8 medium (Thermo Fisher Scientific) on human ESC (hESC)-qualified matrix Matrigel-coated six-well plates (BD Biosciences, San Jose, CA, USA) and their microcell-fused derivative cells were selected in HAT medium (Sigma-Aldrich).
Cell Reprogramming
Fibroblasts were reprogrammed by an overnight infection at an MOI of 1 in the presence of 4 ng/mL Polybrene (Sigma-Aldrich) with a third-generation lentiviral vector described by Mostoslavsky and colleagues,21 carrying the Oct4, Sox2, and Klf4 reprogramming factors. The following day, the medium was replaced, and after 48 h cells were seeded onto mouse embryonic fibroblast (MEF) feeder layer with ES medium, which was changed daily. iPSC-like colonies were individually picked 4 weeks later, with a reprogramming efficiency of 0.01%. In the case of clone LNX-D, which was reprogrammed a second time after cell fusion, an integration-free CytoTune-iPS 2.0 Sendai reprogramming kit (Thermo Fisher Scientific), containing Sendai virus particles of the four Yamanaka factors, was used according to the manufacturer’s instructions and cultured as specified above.
Generation of a CHO Cell Line Containing a Human Normal X Chromosome
To generate a CHO/HSAX cell line containing a HPRT+ normal human chromosome, microcells from a hybrid A9 cell line containing a normal human X chromosome (A9/human), obtained by whole-cell fusion (WCF) between A9 cells and human dermal fibroblasts, were fused with Hprt− CHO cells according to standard procedures, and HAT-resistant clones were isolated. Cells were kept in RPMI 1640 supplemented with 10% FBS and 1× HAT medium (Sigma-Aldrich).
MMCT
Classical MMCT was performed as previously described, with some modifications.17,22 After centrifugation at 160 × g, 1 mL of a pre-warmed solution of 50% PEG 1500 (Roche, Mannheim, Germany) was poured onto the cell pellet during 1 min, followed by extensive washing in serum-free RPMI 1640. 2.5 × 106 iPSCs were plated in Matrigel in hESC-qualified matrix p60 dishes, and after 48 h 1× HAT selection was added.
Retro-MMCT
For retro-MMCT, CHO/HSAX cells containing the normal human X chromosome were infected with the vesicular stomatitis virus G protein (VSV-G) pseudotyped lentiviral vector (MOI of 5) encoding EnvΔR, essentially as previously described.22 To analyze the efficiency of retro-MMCT, 5 × 105 CHO/HSAX cells were plated on six gelatin-coated high-speed polycarbonate centrifuge tubes (Nalgene, Rochester, NY, USA). On the next day, to promote micronuclei formation, the medium was changed to Colcemid medium (RPMI 1640 medium containing 20% FBS, penicillin/streptomycin, and 0.1 μg/mL Colcemid [KaryoMAX], Thermo Fisher Scientific). Microcells were prepared after 48 h (protocol A) or after 72 h (protocol B). After Colcemid incubation, the medium was replaced with DMEM medium containing latrunculin B41 (0.2 μM; Enzo Life Sciences, Farmingdale, NY, USA), and the tubes were centrifuged at 16,000 × g for 70 min at 37°C. The pellets containing extruded microcells were resuspended in 10 mL of serum-free RPMI 1640 and purified by two consecutive filtrations using membranes with 8-μm pores followed by one filtration using membranes with 5-μm pores (Millipore, Darmstadt, Germany). For protocol A, about 1 × 106 purified microcells were co-cultured with 1 × 106 recipient cells, with a 1:1 fusion ratio, into a well of a hESC-qualified matrix Matrigel-coated six-well plates with Essential 8 medium supplemented with 5 μM Y-27632 ROCK inhibitor (Selleckchem, Houston, TX, USA). After 2 days, 1× HAT medium supplemented with 5 μM Y-27632 ROCK inhibitor (Selleckchem) was added. For protocol B, about 3 × 106 purified microcells were co-cultured with 1.5 × 105 recipient cells, with a 20:1 fusion ratio, into a well of a hESC-qualified matrix Matrigel-coated six-well plates with Essential 8 medium supplemented with 5 μM Y-27632 ROCK inhibitor (Selleckchem). A killing curve of HAT was established on the parental LN-iPSCs showing that the lowest concentration at which all cells die was 0.1×. Therefore, after 4 days of co-culture, 0.1× HAT medium supplemented with 5 μM Y-27632 ROCK inhibitor (Selleckchem) was added. For both protocols, emerging resistant clones were picked and expanded for further analysis.
Chromosome Preparation and Analysis
Chromosome analysis was done on slide preparations of cells grown on coverslips. Briefly, cell cultures were treated with KaryoMAX Colcemid solution (Thermo Fisher Scientific) at a final concentration of 0.1 μg/mL for 2 h at 37°C. After hypotonic treatment with 0.075 M KCl and fixation in methanol/acetic acid (3:1 v/v), the slides were air-dried and mounted in Eukitt (Sigma-Aldrich). Chromosome counts and karyotype analyses were done on metaphases stained with Vectashield mounting medium with DAPI (Vector Laboratories, Burlingame, CA, USA) for G banding.
Images were captured using an Olympus BX61 research microscope equipped with a cooled CCD (charge-coupled device) camera and analyzed with Applied Imaging software CytoVision (CytoVision master system with mouse karyotyping). At least 10 karyotypes for each iPSC line were analyzed.
FISH
Green or orange whole-chromosome paint (WCP) specific for the human X chromosome (MetaSystems, Milan, Italy) and human genomic DNA were used as DNA probes. In situ hybridization was performed as previously described.42
In brief, slides were treated with pepsin (0.004%) at 37°C for 30 s and dehydrated through the ethanol series before denaturation in 70% formamide/2× SSC. The probes were denaturated at 80°C for 10 min and after a pre-annealing of 20 min at 37°C were applied on the slides. Hybridization was completed overnight at 37°C. Stringent washes were carried out in 0.4% SSC at 72°C for 5 min followed by 4% SSC/0.1% Tween 20 at room temperature (RT) for 5 min. Slides were mounted in Vectashield mounting medium with DAPI and then were scored under an Olympus BX61 research microscope equipped with a cooled CCD camera. Images were captured and analyzed with Applied Imaging software CytoVision (CytoVision master system with karyotyping and FISH).
EB Formation
Semi-confluent human iPSCs in one well of a six-well plate were dissociated by incubation with 2 mg/mL dispase (STEMCELL Technologies, Vancouver, BC, Canada) at 37°C for 2–3 min, or until the colonies began to lift off of the plate. The plates were gently shaken to help dislodge the colonies. The dispase solution was diluted in PBS and the suspension was transferred to a 15-mL centrifuge tube. The cell aggregates were allowed to settle by gravity, and the dispase solution was gently aspirated. The aggregates were washed three more times in PBS before being resuspended in 4 mL of Essential 6 medium supplemented with 5 μM Y-27632 ROCK inhibitor. The suspension was gently transferred to each well of a six-well ultra-low attachment plate (Corning Life Sciences, Acton, MA, USA). After 1 week in suspension, EBs were transferred to Matrigel-coated dishes and cultured in Essential 6 medium (Thermo Fisher Scientific) for an additional 2–3 weeks. Then, the cells were stained with antibodies against markers of all three embryonic germline layers and analyzed by immunofluorescence.
Immunofluorescence
For immunostaining, human iPSCs and EBs were fixed in 4% paraformaldehyde (PFA) for 10 min at room temperature, washed with PBS, and permeabilized in 0.3% Triton X-100 in PBS for 10 min at room temperature. Primary antibodies used were anti-Oct4 (ab18976, Abcam, Cambridge, UK), anti-Nanog (ab62734, Abcam), anti-SOX2 (ab97959, Abcam), anti-TRA-1-60 (ab16288, Abcam), anti-Nestin (ab22035, Abcam), anti-α-SMA (ab5694, Abcam), anti-Brachyury (AF2085, R&D Systems, Minneapolis, MN, USA), and anti-GATA4 (ab134057, Abcam). After primary antibody incubation, samples were washed with PBS and incubated with secondary Alexa Fluor 488-conjugated antibodies (Life Technologies), diluted 1:2,000. Samples were also counterstained with DAPI (200 μg/mL). Slides were observed using an Olympus BX61 research microscope equipped with a cooled CCD camera. Images were captured and analyzed with Applied Imaging software CytoVision.
Sample Preparation and Genomic PCR Analysis
Genomic DNA was extracted from cell lines using a QIAamp DNA kit (QIAGEN, Hilden, Germany) according to the manufacturer’s recommendations. Primer pairs for the analysis of WT HPRT allele and informative SNPs on the X chromosome are listed in Table S1. PCRs were performed under the following conditions: initial denaturing for 5 min at 94°C; denaturing for 30 s at 94°C, 58°C for 30 s, extension for 30 s at 72°C, repeated 35 times; final extension for 5 min at 72°C. The PCR products were recovered using NucleoSpin gel and PCR Clean-up (Macherey-Nagel) and sequenced with specific primers.
WES and Analysis
WES was performed starting from 50 ng of genomic DNA using the Nextera Rapid Capture Exome Library kit (Illumina, San Diego, CA, USA) and the Illumina NextSeq500 platform, according to the manufacturers’ instructions. Reads were aligned against the human reference genome (hg19) using BWA.43 Variant calling was performed with GATK.44 The analyzed target was restricted to the regions with an average coverage >30× in the parental cell line sample. Variants found in the parental cell line, selected to have a minimum sequencing depth of 30x and a Phred Q score >30, were compared to the genotypes at the same genomic positions in the two corrected clones.
Author Contributions
P.V., A.V., and M.P. conceived and designed the experimental plan. L. Susani, A.C., E.C., D.S., L.S.S., S.M., and M.P. performed experiments and analyzed data; T.S. and T.H. designed the retro-MMCT technique; M.P. and L. Susani collected and assembled data, performed data analysis and interpretation; L. Straniero and S.D. performed WES analysis; P.V. and M.P. wrote the paper with support from all of the authors.
Acknowledgments
The research leading to these results has received funding from the PNR-CNR Aging Program 2012–2014 to P.V. A.C. is the recipient of a fellowship from Fondazione Nicola del Roscio.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtm.2020.01.003.
Supplemental Information
References
- 1.Doudna J.A., Charpentier E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science. 2014;346:1258096. doi: 10.1126/science.1258096. [DOI] [PubMed] [Google Scholar]
- 2.Cong L., Ran F.A., Cox D., Lin S., Barretto R., Habib N., Hsu P.D., Wu X., Jiang W., Marraffini L.A., Zhang F. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Urnov F.D. Genome editing B.C. (before CRISPR): lasting lessons from the “Old Testament”. CRISPR J. 2018;1:34–46. doi: 10.1089/crispr.2018.29007.fyu. [DOI] [PubMed] [Google Scholar]
- 4.Lupski J.R. Genomic disorders: structural features of the genome can lead to DNA rearrangements and human disease traits. Trends Genet. 1998;14:417–422. doi: 10.1016/s0168-9525(98)01555-8. [DOI] [PubMed] [Google Scholar]
- 5.Park C.Y., Sung J.J., Choi S.H., Lee D.R., Park I.H., Kim D.W. Modeling and correction of structural variations in patient-derived iPSCs using CRISPR/Cas9. Nat. Protoc. 2016;11:2154–2169. doi: 10.1038/nprot.2016.129. [DOI] [PubMed] [Google Scholar]
- 6.Long C., Li H., Tiburcy M., Rodriguez-Caycedo C., Kyrychenko V., Zhou H., Zhang Y., Min Y.L., Shelton J.M., Mammen P.P.A. Correction of diverse muscular dystrophy mutations in human engineered heart muscle by single-site genome editing. Sci. Adv. 2018;4 doi: 10.1126/sciadv.aap9004. eaap9004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nelson C.E., Hakim C.H., Ousterout D.G., Thakore P.I., Moreb E.A., Castellanos Rivera R.M., Madhavan S., Pan X., Ran F.A., Yan W.X. In vivo genome editing improves muscle function in a mouse model of Duchenne muscular dystrophy. Science. 2016;351:403–407. doi: 10.1126/science.aad5143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xu L., Park K.H., Zhao L., Xu J., El Refaey M., Gao Y., Zhu H., Ma J., Han R. CRISPR-mediated genome editing restores dystrophin expression and function in mdx mice. Mol. Ther. 2016;24:564–569. doi: 10.1038/mt.2015.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Komaki H., Nagata T., Saito T., Masuda S., Takeshita E., Sasaki M., Tachimori H., Nakamura H., Aoki Y., Takeda S. Systemic administration of the antisense oligonucleotide NS-065/NCNP-01 for skipping of exon 53 in patients with Duchenne muscular dystrophy. Sci. Transl. Med. 2018;10:eaan0713. doi: 10.1126/scitranslmed.aan0713. [DOI] [PubMed] [Google Scholar]
- 10.Tabebordbar M., Zhu K., Cheng J.K.W., Chew W.L., Widrick J.J., Yan W.X., Maesner C., Wu E.Y., Xiao R., Ran F.A. In vivo gene editing in dystrophic mouse muscle and muscle stem cells. Science. 2016;351:407–411. doi: 10.1126/science.aad5177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Amoasii L., Long C., Li H., Mireault A.A., Shelton J.M., Sanchez-Ortiz E., McAnally J.R., Bhattacharyya S., Schmidt F., Grimm D. Single-cut genome editing restores dystrophin expression in a new mouse model of muscular dystrophy. Sci. Transl. Med. 2017;9:eaan8081. doi: 10.1126/scitranslmed.aan8081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Min Y.L., Li H., Rodriguez-Caycedo C., Mireault A.A., Huang J., Shelton J.M., McAnally J.R., Amoasii L., Mammen P.P.A., Bassel-Duby R. CRISPR-Cas9 corrects Duchenne muscular dystrophy exon 44 deletion mutations in mice and human cells. Sci. Adv. 2019;5 doi: 10.1126/sciadv.aav4324. eaav4324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu X.S., Wu H., Krzisch M., Wu X., Graef J., Muffat J., Hnisz D., Li C.H., Yuan B., Xu C. Rescue of fragile X syndrome neurons by DNA methylation editing of the FMR1 gene. Cell. 2018;172:979–992.e6. doi: 10.1016/j.cell.2018.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Park C.Y., Halevy T., Lee D.R., Sung J.J., Lee J.S., Yanuka O., Benvenisty N., Kim D.W. Reversion of FMR1 methylation and silencing by editing the triplet repeats in fragile X iPSC-derived neurons. Cell Rep. 2015;13:234–241. doi: 10.1016/j.celrep.2015.08.084. [DOI] [PubMed] [Google Scholar]
- 15.Vershkov D., Fainstein N., Suissa S., Golan-Lev T., Ben-Hur T., Benvenisty N. FMR1 reactivating treatments in fragile X iPSC-derived neural progenitors in vitro and in vivo. Cell Rep. 2019;26:2531–2539.e4. doi: 10.1016/j.celrep.2019.02.026. [DOI] [PubMed] [Google Scholar]
- 16.Gholizadeh S., Arsenault J., Xuan I.C., Pacey L.K., Hampson D.R. Reduced phenotypic severity following adeno-associated virus-mediated Fmr1 gene delivery in fragile X mice. Neuropsychopharmacology. 2014;39:3100–3111. doi: 10.1038/npp.2014.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Paulis M., Castelli A., Susani L., Lizier M., Lagutina I., Focarelli M.L., Recordati C., Uva P., Faggioli F., Neri T. Chromosome transplantation as a novel approach for correcting complex genomic disorders. Oncotarget. 2015;6:35218–35230. doi: 10.18632/oncotarget.6143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Plona K., Kim T., Halloran K., Wynshaw-Boris A. Chromosome therapy: potential strategies for the correction of severe chromosome aberrations. Am. J. Med. Genet. C. Semin. Med. Genet. 2016;172:422–430. doi: 10.1002/ajmg.c.31530. [DOI] [PubMed] [Google Scholar]
- 19.Castelli A., Susani L., Menale C., Muggeo S., Caldana E., Strina D., Cassani B., Recordati C., Scanziani E., Ficara F. Chromosome transplantation: correction of the chronic granulomatous disease defect in mouse induced pluripotent stem cells. Stem Cells. 2019;37:876–887. doi: 10.1002/stem.3006. [DOI] [PubMed] [Google Scholar]
- 20.Sinenko S.A., Skvortsova E.V., Liskovykh M.A., Ponomartsev S.V., Kuzmin A.A., Khudiakov A.A., Malashicheva A.B., Alenina N., Larionov V., Kouprina N., Tomilin A.N. Transfer of synthetic human chromosome into human induced pluripotent stem cells for biomedical applications. Cells. 2018;7:E261. doi: 10.3390/cells7120261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sommer C.A., Stadtfeld M., Murphy G.J., Hochedlinger K., Kotton D.N., Mostoslavsky G. Induced pluripotent stem cell generation using a single lentiviral stem cell cassette. Stem Cells. 2009;27:543–549. doi: 10.1634/stemcells.2008-1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Suzuki T., Kazuki Y., Oshimura M., Hara T. Highly efficient transfer of chromosomes to a broad range of target cells using chinese hamster ovary cells expressing murine leukemia virus-derived envelope proteins. PLoS ONE. 2016;11:e0157187. doi: 10.1371/journal.pone.0157187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Paulis M. Chromosome transfer via cell fusion. Methods Mol. Biol. 2011;738:57–67. doi: 10.1007/978-1-61779-099-7_4. [DOI] [PubMed] [Google Scholar]
- 24.Hwang S., Williams J.F., Kneissig M., Lioudyno M., Rivera I., Helguera P., Busciglio J., Storchova Z., King M.C., Torres E.M. Suppressing aneuploidy-associated phenotypes improves the fitness of trisomy 21 cells. Cell Rep. 2019;29:2473–2488.e5. doi: 10.1016/j.celrep.2019.10.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li T., Zhao H., Han X., Yao J., Zhang L., Guo Y., Shao Z., Jin Y., Lai D. The spontaneous differentiation and chromosome loss in iPSCs of human trisomy 18 syndrome. Cell Death Dis. 2017;8:e3149. doi: 10.1038/cddis.2017.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maclean G.A., Menne T.F., Guo G., Sanchez D.J., Park I.H., Daley G.Q., Orkin S.H. Altered hematopoiesis in trisomy 21 as revealed through in vitro differentiation of isogenic human pluripotent cells. Proc. Natl. Acad. Sci. USA. 2012;109:17567–17572. doi: 10.1073/pnas.1215468109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kazuki Y., Hiratsuka M., Takiguchi M., Osaki M., Kajitani N., Hoshiya H., Hiramatsu K., Yoshino T., Kazuki K., Ishihara C. Complete genetic correction of iPS cells from Duchenne muscular dystrophy. Mol. Ther. 2010;18:386–393. doi: 10.1038/mt.2009.274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Min Y.L., Bassel-Duby R., Olson E.N. CRISPR correction of Duchenne muscular dystrophy. Annu. Rev. Med. 2019;70:239–255. doi: 10.1146/annurev-med-081117-010451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Amoasii L., Hildyard J.C.W., Li H., Sanchez-Ortiz E., Mireault A., Caballero D., Harron R., Stathopoulou T.R., Massey C., Shelton J.M. Gene editing restores dystrophin expression in a canine model of Duchenne muscular dystrophy. Science. 2018;362:86–91. doi: 10.1126/science.aau1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aartsma-Rus A., Krieg A.M. FDA approves eteplirsen for Duchenne muscular dystrophy: the next chapter in the eteplirsen saga. Nucleic Acid Ther. 2017;27:1–3. doi: 10.1089/nat.2016.0657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Levin A.A. Treating disease at the RNA level with oligonucleotides. N. Engl. J. Med. 2019;380:57–70. doi: 10.1056/NEJMra1705346. [DOI] [PubMed] [Google Scholar]
- 32.Jones D. Duchenne muscular dystrophy awaits gene therapy. Nat. Biotechnol. 2019;37:335–337. doi: 10.1038/s41587-019-0103-5. [DOI] [PubMed] [Google Scholar]
- 33.Tedesco F.S., Hoshiya H., D’Antona G., Gerli M.F., Messina G., Antonini S., Tonlorenzi R., Benedetti S., Berghella L., Torrente Y. Stem cell-mediated transfer of a human artificial chromosome ameliorates muscular dystrophy. Sci. Transl. Med. 2011;3:96ra78. doi: 10.1126/scitranslmed.3002342. [DOI] [PubMed] [Google Scholar]
- 34.Benedetti S., Uno N., Hoshiya H., Ragazzi M., Ferrari G., Kazuki Y., Moyle L.A., Tonlorenzi R., Lombardo A., Chaouch S. Reversible immortalisation enables genetic correction of human muscle progenitors and engineering of next-generation human artificial chromosomes for Duchenne muscular dystrophy. EMBO Mol. Med. 2018;10:254–275. doi: 10.15252/emmm.201607284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hoshiya H., Kazuki Y., Abe S., Takiguchi M., Kajitani N., Watanabe Y., Yoshino T., Shirayoshi Y., Higaki K., Messina G. A highly stable and nonintegrated human artificial chromosome (HAC) containing the 2.4 Mb entire human dystrophin gene. Mol. Ther. 2009;17:309–317. doi: 10.1038/mt.2008.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.van der Wal E., Herrero-Hernandez P., Wan R., Broeders M., In ’t Groen S.L.M., van Gestel T.J.M., van IJcken W.F.J., Cheung T.H., van der Ploeg A.T., Schaaf G.J. Large-scale expansion of human iPSC-derived skeletal muscle cells for disease modeling and cell-based therapeutic strategies. Stem Cell Reports. 2018;10:1975–1990. doi: 10.1016/j.stemcr.2018.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Maffioletti S.M., Sarcar S., Henderson A.B.H., Mannhardt I., Pinton L., Moyle L.A., Steele-Stallard H., Cappellari O., Wells K.E., Ferrari G. Three-dimensional human iPSC-derived artificial skeletal muscles model muscular dystrophies and enable multilineage tissue engineering. Cell Rep. 2018;23:899–908. doi: 10.1016/j.celrep.2018.03.091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yuan J., Ma Y., Huang T., Chen Y., Peng Y., Li B., Li J., Zhang Y., Song B., Sun X. Genetic modulation of RNA splicing with a CRISPR-guided cytidine deaminase. Mol. Cell. 2018;72:380–394.e7. doi: 10.1016/j.molcel.2018.09.002. [DOI] [PubMed] [Google Scholar]
- 39.Chari R., Church G.M. Beyond editing to writing large genomes. Nat. Rev. Genet. 2017;18:749–760. doi: 10.1038/nrg.2017.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Littlefield J.W. The use of drug-resistant markers to study the hybridization of mouse fibroblasts. Exp. Cell Res. 1966;41:190–196. doi: 10.1016/0014-4827(66)90558-1. [DOI] [PubMed] [Google Scholar]
- 41.Liskovykh M., Lee N.C., Larionov V., Kouprina N. Moving toward a higher efficiency of microcell-mediated chromosome transfer. Mol. Ther. Methods Clin. Dev. 2016;3:16043. doi: 10.1038/mtm.2016.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Paulis M., Castelli A., Lizier M., Susani L., Lucchini F., Villa A., Vezzoni P. A pre-screening FISH-based method to detect CRISPR/Cas9 off-targets in mouse embryonic stem cells. Sci. Rep. 2015;5:12327. doi: 10.1038/srep12327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., DePristo M.A. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.