

Abstract
Quinolinic acid (QA) is a common intermediate in the biosynthesis of nicotinamide adenine dinucleotide (NAD+) and its derivatives in all organisms that synthesize the molecule de novo. In most prokaryotes, it is formed from the condensation of dihydroxyacetone phosphate (DHAP) and iminoaspartate (IA) by the action of quinolinate synthase (NadA). NadA contains a [4Fe–4S] cluster cofactor with a unique non-cysteinyl-ligated iron ion (Fea), which is proposed to bind the hydroxyl group of an intermediate in its reaction to facilitate a dehydration step. However, direct evidence for this role in catalysis has yet to be provided, and the exact chemical mechanism that underlies this transformation remains elusive. Herein, we present a structure of NadA from Pyrococcus horikoshii (PhNadA) in complex with IA and show that a carboxylate group of the molecule is ligated to Fea of the iron-sulfur cluster, occupying the site to which DHAP has been proposed to bind during catalysis. When crystals of PhNadA in complex with IA are soaked briefly in DHAP before freezing, electron density for a new molecule is observed, which we suggest is related to an intermediate in the reaction. Similar, but slightly different “intermediates” are observed when crystals of a PhNadA Glu198Gln variant are incubated with DHAP, oxaloacetate, and ammonium chloride, conditions under which IA is formed chemically. Continuous-wave and pulse electron paramagnetic resonance techniques are used to verify the binding mode of substrates and proposed intermediates in frozen solution.
Graphical Abstract
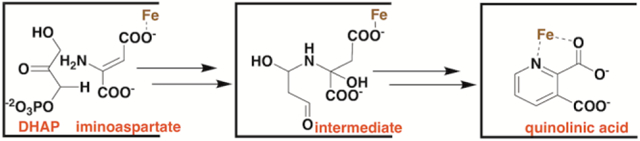
INTRODUCTION
Nicotinamide adenine dinucleotide (NAD) is a ubiquitous metabolite used most often in oxidation-reduction reactions as the donor or acceptor of a hydride ion. It also has many other uses where it becomes irreversibly degraded, as in adenylation, ADP-ribosylation, and histone deacetylation [1–3]. In some enzymes, such as urocanase, its electrophilic C4 position becomes transiently modified by a substrate intermediate during catalysis [4].
Quinolinic acid (QA) is an intermediate in the biosynthesis of NAD. However, the pathway for formation of QA differs in eukaryotes versus prokaryotes, although some eukaryotes are known to use the prokaryotic pathway [5, 6]. In eukaryotes, QA is generated from L-tryptophan via the kynurenine pathway, which requires the participation of molecular oxygen in several steps. In prokaryotes, QA is synthesized from dihydroxyacetone phosphate (DHAP) and iminoaspartate (IA) in a reaction catalyzed by quinolinate synthase (NadA), giving orthophosphate (Pi) and 2 mol of water as additional products (Figure 1) [7–9]. In Escherichia coli and many other organisms, IA is generated in vivo by L-aspartate oxidase (NadB), a flavin-dependent enzyme [10, 11]. During each turnover, the flavin (FAD) becomes reduced at the expense of L-aspartate to afford FADH2, and therefore must be re-oxidized to enable subsequent turnovers. In the presence of oxygen, these reducing equivalents are passed on to molecular oxygen, affording peroxide. In the absence of oxygen, the reducing equivalents are passed on to fumarate, affording succinate. In some organisms, such as Thermotoga maritima, IA is thought to be generated by an NADP+-dependent enzyme [12]. In vitro, IA can be formed chemically by including oxaloacetate (OA) in reaction mixtures that contain a source of ammonium ions [8].
Figure 1.
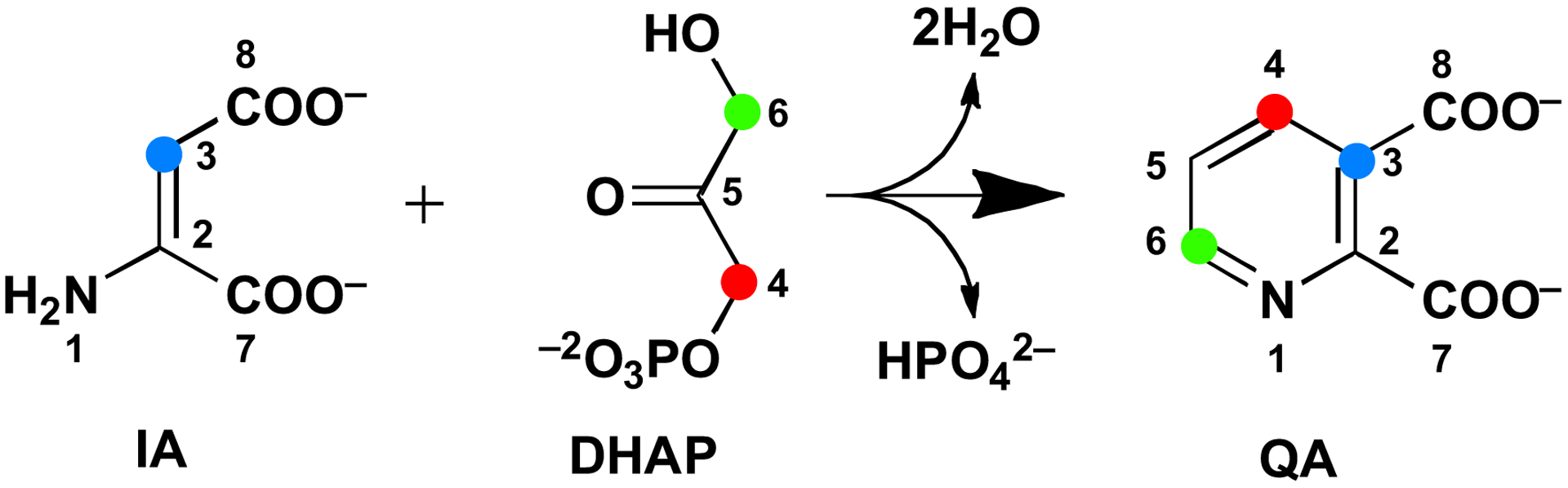
NadA catalyzed condensation of IA and DHAP to give QA. The colored dots indicate known connectivity between atoms. QA is numbered according to IUPAC rules, and this numbering is maintained in the DHAP and IA structures for clarity.
Although the enzymes that catalyze formation of QA in E. coli were discovered in the late 1960s and early 1970s, the exact mechanism by which NadA catalyzes its reaction has not been fully established. However, labeling studies have shown that C3 of IA (shown as C3 in Figure 1) becomes connected to C1 of DHAP (shown as C4 in Figure 1) and that the nitrogen of IA becomes connected to C3 of DHAP (shown as C6 in Figure 1) [13]. NadA contains one [4Fe–4S] cluster [14, 15], which is absolutely required for its activity, and belongs to the iron-sulfur (Fe/S)-dependent hydro-lyase family of enzymes [16, 17]. Enzymes in this family, of which aconitase is the paradigm, ligate [4Fe–4S] clusters using only three cysteines. The fourth noncysteinyl-ligated iron ion, termed Fea, is used as a Lewis acid to facilitate hydration/dehydration reactions [17]. In the prevailing mechanism for NadA, a C3 carbanion of the enamine of IA attacks C4 of DHAP (see note on numbering in legend of Figure 1), eliminating orthophosphate and forming the nascent C–C bond of QA. Subsequent tautomerizations and Schiff-base formation between the amine of the formerly IA and the C6 aldehyde of the formerly DHAP lead to production of the QA precursor (QP), which has recently been renamed intermediate Y [9, 18]. The C5 hydroxyl of the QP is proposed to ligate to Fea to facilitate a dehydration reaction that is initiated by proton removal at C4 (Figure S1) [17].
Recently, a number of structures of NadAs from multiple organisms in the presence of substrates, substrate analogs, products, and potential intermediates have been published. The first structure containing a mechanistically relevant ligand was that of NadA in the presence of its product, QA. In this structure, QA is shown to bind to Fea via its N1 amine and C7 carboxylate groups [19]. Although this structure was reproduced by two additional groups [20, 21], it has been suggested that the structure does not represent the product complex that is generated via catalysis [18]. Additional structures include the enzyme in complex with DHAP (which binds to Fea via its C6 hydroxyl and C5 keto groups (numbering according to Figure 1), in complex with various analogs of IA, and in complex with potential intermediates [18, 20, 21].
Most recently, structures of NadA in complex with two potential reaction intermediates were reported, which supports the mechanism shown in Figure S1 [18, 20]. These intermediates include the condensation product between C1 of DHAP and C3 of IA (termed W), a resulting keto-aldo isomerization to yield intermediate X, and Schiff-base formation between N1 and the C3 aldehyde of formerly DHAP to yield intermediate Y (i.e., QP) (Figure S1).
In an effort to shed additional light on the catalytic mechanism of NadA, and in particular the role of the Fe/S cluster in catalysis, we solved x-ray crystal structures of wild-type (WT) NadAs from Pyrococcus horikoshii (Ph) in the presence of the substrates DHAP or IA, as well as in the presence of both substrates while undergoing turnover. We report structures of species that may reflect unexpected intermediates in the reaction, and use hyperfine-sublevel correlation (HYSCORE) spectroscopy to support the binding mode of these species to the unique iron ion of the FeS cluster. Lastly, we provide a putative mechanism for the NadA reaction that incorporates these species and that is consistent with previous mechanistic studies on the enzyme.
MATERIALS AND METHODS
All general procedures have been previously described [19, 22]. PhNadA and PhNadB were overproduced in E. coli and isolated as previously described [22]. Overproduction of 15N-labeled PhNadA was conducted in E. coli cultured in M9 minimal medium supplemented with 15NH4Cl. PhNadA variants were constructed using the QuikChange II Site–directed Mutagenesis Kit (Agilent; Santa Clara, CA) according to the manufacturer’s specifications and as described previously [22]. Sequences of primers used for mutagenesis are as followed: PhNadA E198Q forward (5’- GCA AAG CTA ATG ATC CAT CCT CAG TGC ATC CCA GAG G −3’); E198Q reverse (5’- C CTC TGG GAT GCA CTG AGG ATG GAT CAT TAG CTT TGC −3’); PhNadA Y109F forward (5’- GCT CCA GTT GTT CTC TTC GTG AAT AGT ACG GCT GAA GC −3’); Y109F reverse (5’- GC TTC AGC CGT ACT ATT CAC GAA GAG AAC AAC TGG AGC −3’).
Anaerobic Screening for Crystallography.
PhNadA was prepared for crystal screening by reconstituting its Fe/S cluster as previously described [22], with the exception that the reconstitution mixture was maintained at room temperature (RT) throughout the procedure. Following concentration of the reconstituted (RCN) protein, it was loaded onto a prepacked HiPrep 16/60 Sephacryl S-200 HR column equilibrated in 50 mM HEPES, pH 7.5, 0.1 M KCl, 10 mM DTT and 10% glycerol that was connected to an ÅKTA chromatography system housed in a Coy (Grass Lake, MI) anaerobic glove box. Fractions containing NadA were pooled and further concentrated. The protein was then treated with thrombin overnight at RT to cleave the N-terminal hexahistidine tag using the Thrombin Cleavage Capture Kit from MilliporeSigma (Burlington, MA, Cat 69022–3FRZ). The protein was then exchanged into 10 mM HEPES, pH 7.5, 0.1 M KCl, 5 mM DTT by gel-filtration chromatography and concentrated to 10 mg/mL. In the structure of PhNadA in complex with DHAP, the protein was diluted to 3 mg/mL in a solution of 10 mM HEPES, pH 7.5, 10 mM aspartate, and 2 mM DHAP. The resulting protein solutions were passed through an EMD Millipore 0.22 μm sterile filter (Ultrafree-MC centrifugal filter units). The filtered protein was screened for suitable crystallization conditions using the NeXtal Classics Suite from Qiagen (Germantown, MD) by the hanging drop vapor diffusion method in the anaerobic chamber at RT. 2 μL of protein (3 to 10 mg/mL) was mixed with 2 μL of the appropriate screen, and the mixtures were equilibrated against a 0.5 M LiCl well solution. Optimal conditions for crystallization of the various complexes and for cryoprotection of the corresponding crystals are shown in Table S2.
Data collection and structure determination.
All data sets were collected at the x-ray Operations and Research beam line 23-ID-B at the Advanced Photon source, Argonne National Laboratory (Lemont, IL, USA). Data collection was remotely controlled using the GM/CA-XSD (Table S1). HKL2000 and iMosflm software were used for the indexing, integration and scaling [23, 24]. Each of the structures was solved by molecular replacement using the Phenix implementation of Phaser [25] with the coordinates of the apo PhNadA structure (PDB ID: 1WZU) as the search model. The PhNadA structures in complex with IA, DHAP and wild-type (WT) intermediate had a monomer in the asymmetric unit; however, the structure of the E198Q variant showed a dimer (Table S1). Structure refinement was executed with Phenix [26], and manual electron density fitting was carried out using COOT [27, 28]. The density of the ligands was interpreted after iterative modeling and refinement of protein residues, iron-sulfur clusters, and water molecules other than those in the active site using difference Fourier maps. The interpretation of the exact chemical species was achieved using a number of standard tools, including 2Fo-Fc maps, Fo-Fc maps, B-factors, R-factors, steric clashes, and hydrogen bonding. Structure analysis was carried out using Pymol software (The PyMOL Molecular Graphics System, Version 1.7.4 Schrödinger, LLC). Data-collection and refinement statistics for the crystal structures solved are shown in Table S1. The atomic coordinates and structure factors are deposited in the protein data bank (PDB) along with their associated PDB IDs (Table S1).
Generation of IA.
The substrate IA was generated enzymatically via the NadB reaction using fumarate as the electron acceptor or chemically by inclusion of OA and ammonium chloride in reaction mixtures. For the generation of IA from L-Asp, or 4-13C-L-Asp, PhNadA (0.7 – 1 mM) was incubated in the presence of NadB (0.3 – 0.5 mM) in a solution containing 100 mM HEPES, pH 7.5, 7.5 mM L-aspartate, 3 mM fumarate, 25 μM FAD, 0.1 M KCl, and 3 mM DHAP when present. When IA was generated chemically, the aspartate/fumarate solution, FAD, and NadB were replaced with 3–10 mM OA and 100 mM ammonium chloride.
Generation of labeled DHAP.
[1,3-13C]DHAP was synthesized from [1,3-13C]glycerol using a coupled enzymatic system containing glycerol kinase (GK), pyruvate kinase (PK), glycerol-3-phosphate oxidase G3PO, and catalase. In the first step, GK adds a phosphate group from ATP to glycerol to afford glycerol-3-phosphate, which is oxidized by dioxygen to DHAP by the flavin dependent enzyme G3PO. PK drives the first step to completion by converting ADP to ATP at the expense of phosphoenolpyruvate, while catalase converts the peroxide generated in the second step back to dioxygen. Reactions were carried out in a final total volume of 0.5 L and contained the following components: 50 mM Tris–HCl, pH 7.8, 2 mM ATP, 20 mM MgCl2, 4 mM phosphoenolpyruvate, 4 mM [1,3-13C]glycerol, 20 mM β-mercaptoethanol, and 10 μM FAD. All of the enzymes were exchanged into buffer that lacked glycerol by gel-filtration chromatography before adding PK (300 U), G3O (75 U), catalase (8500 U), and GK (150 U). The reaction was stirred overnight at room temperature in an aluminum foil-covered large vessel, and production of DHAP was monitored spectrophotometrically in an assay containing α-glycerolphosphate dehydrogenase and NADH. After completion, the reaction was diluted with 3 L of chilled 0.16 mM HCl and loaded onto a BioRad AG1-X2 column (2.5 cm × 20 cm) that had been charged with 0.5 L of 1 M HCl before being equilibrated with 0.16 mM HCl at 4 °C. The column was then washed with 1 L of 0.16 mM HCl followed by 0.25 L of 5 mM HCl before a 5 mM to 35 mM HCl linear gradient was applied over a total volume of 1.5 L. 18 mL fractions were collected, and DHAP was identified by the α-glycerophosphate dehydrogenase assay as described above. The presence of contaminating nucleotides (e.g. ATP and ADP) was monitored by UV-absorbance at 260 nm. Fractions containing DHAP but not contaminating nucleotides were pooled, dried by rotary evaporation, and stored at −20 °C until needed.
Preparation of HYSCORE samples.
PhNadA in all HYSCORE experiments had the N-terminal His-tag. Samples were prepared inside the anaerobic chamber and contained 1–2 mM PhNadA, 200 mM HEPES, pH 7.5, 50 mM KCl, 3 mM DTT and 10% glycerol, in addition to labeled DHAP or IA. They were incubated with 3 mM dithionite at room temperature for 5 min before placing the solutions in EPR tubes (clear-fused quartz tubes, 4 mm OD, 3 mm ID, QSI Quartz) and freezing them in liquid N2,. Detailed experimental conditions are presented in the figure legends. HYSCORE spectra were acquired on a Bruker Elexsys E580 spectrometer equipped with a SuperX-FT microwave bridge and split-ring resonators ER 4118X-MS5. Cryogenic temperature was maintained using an ER 4112-HV Oxford Instruments liquid helium flow cryostat. Data processing was performed using Kazan viewer, a home-written suite of utilities in MATLAB [29].
RESULTS
Structure of WT PhNadA in complex with iminoaspartate.
A number of structures of Fe/S-containing NadAs have recently been published, including several structures of PhNadA [19, 21, 30]. These structures include, among others, the enzyme in complex with its product (QA) [19–21], one of its substrates (DHAP) [20, 21], and a number of compounds that structurally mimic the second substrate (IA) [21]. However, no structure in the presence of IA itself has been published. Crystals of the WT PhNadA/IA complex were generated by including NadB (~1.5 equiv with respect to NadA), FAD, and Asp in the crystallization media. Given the absence of both fumarate and oxygen in the crystallization solution, the NadB present could catalyze maximally only a little more than one turnover, producing IA in concentrations that are at most stoichiometric with that of NadB and in slight excess with that of NadA. The structure of WT PhNadA/IA, determined to a resolution of 2.15 Å, recapitulates some of the features of the previously published structure of apo (i.e. no Fe/S cluster) PhNadA with malate bound, an IA mimic [30], but also shows a number of important distinguishing characteristics (Figure 2A, 2C, Figure S2 A and B). In the structure with IA, one of the oxygen atoms of the C8 carboxylate is bound to Fea, while the remaining oxygen atom forms hydrogen bonds (H-bonds) with the side chains of Asn111 and Glu198, two strictly conserved amino acids. In the apo PhNadA structure, the C4 carboxylate group of malate (equivalent to C8 of IA) is rotated about the C1–C2 bond by 112° and the C2–C3 bond by 150°, and forms H-bonds with the side chains of Tyr109 and Ser126 as well as with the backbone amide protons of Ser38 and Ser126 [30]. The C7 carboxylate of IA forms H-bonds with the side chains of His21, Asn111 and Thr213, as well as with the backbone amide proton of Thr213, while the amino group of IA forms H-bonds with the side chains of Tyr109 and His196. These binding modes for the C7 carboxylate and the amino groups of IA are identical to those of the C1 carboxylate and the C2 hydroxyl groups of malate in the apo-PhNadA structure. However, the important distinction is that the C8 oxygen of IA is within coordinating distance to Fea, while in the aforementioned apo-PhNadA/malate structure, the corresponding C4 position of malate would be the farthest from the same site (Figure S2A). Given this discrepancy and the presumption that the C5 oxygen atom of DHAP would coordinate to the cluster, as has been indicated earlier [20, 21], this observed binding mode for IA is unexpected [17, 30, 31]. However, a more recently published structure of holo-PhNadA in complex with L-malate shows that malate binds similarly to IA in the active site of holo-PhNadA (Figure S2B) [21]. It should also be noted that in the structure of holo-PhNadA with L-malate bound, L-malate was co-crystallized with the protein, while all other IA analogs were soaked into preformed crystals.
Figure 2.
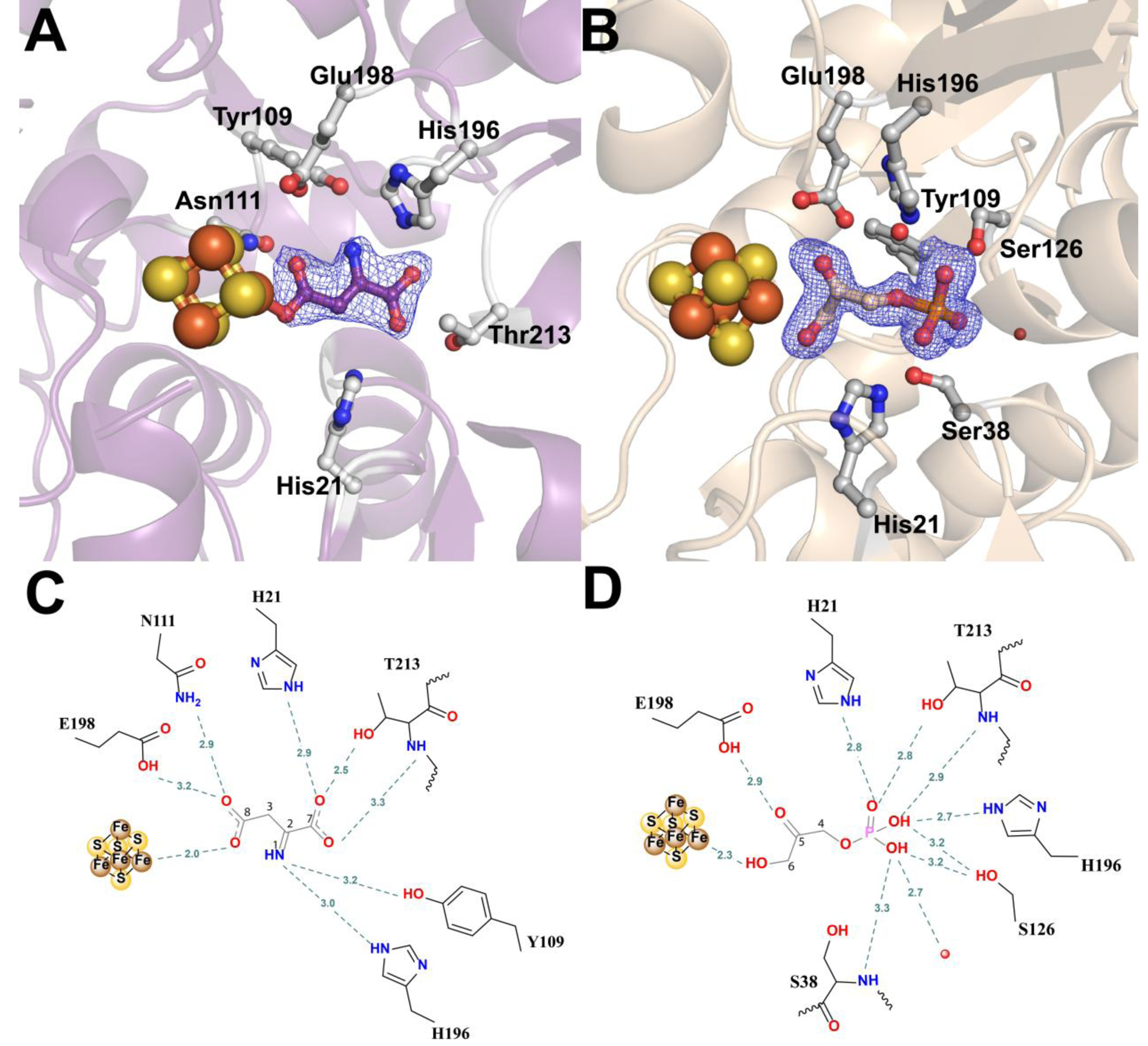
Active site of PhNadA in complex with: (A, C) IA in the active site of PhNadA, (B, D) DHAP in the active site of PhNadA (A, B) Omit electron density map of substrates in the active site of PhNadA, Fo-Fc electron density is contoured at 2.5σ (blue mesh). (C, D) Schematic diagram of substrates bound in the active site of PhNadA. Atom colors: Fe – orange, S – yellow, N - blue, C – grey and O – red.
To verify that the positioning of IA with respect to the open coordination site of the [4Fe-4S] cluster in crystallo is the same as that in solution, a study using continuous-wave (CW) and pulse electron paramagnetic resonance (EPR) spectroscopies was carried out. X-band CW EPR spectra of the [4Fe-4S]+ cluster in the reduced PhNadA/IA complex are significantly different from the spectrum of the substrate-free PhNadA control (g=[2.100, 1.937, 1.913]), the spectrum of the reduced PhNadA/DHAP complex (g=[2.076, 1.918, 1.904]) (see Figure 3), as well as the spectrum of the reduced PhNadA/QA complex [19]. Interestingly, the EPR spectrum of the PhNadA/IA complex consists of two contributions with different g-values (see simulated spectral components in Figure 3). However, given that the average g-value and temperature behavior for these two signals are very similar, we conclude that both signals correspond to a [4Fe-4S]+ cluster. After careful examination of the residual density in the crystallographic data of the PhNadA/IA complex, we identify a minor contribution (~30% occupancy) from a somewhat differently positioned IA in the binding pocket, with the C7 carboxylate group moved away from Thr213 and establishing a weaker H-bond with Ser38 (Figure S2C). From this analysis, we propose that IA has two somewhat different orientations in the pocket, both of which maintain binding with the [4Fe-4S]1+ cluster, resulting in two different EPR spectra. Based on the simulations performed, we conclude that there is roughly a 1:1 ratio of the two EPR-spectral components. The difference in the occupancy determined between EPR and crystallographic experiments is attributed to crystal packing effects on the overall structure of the protein.
Figure 3.
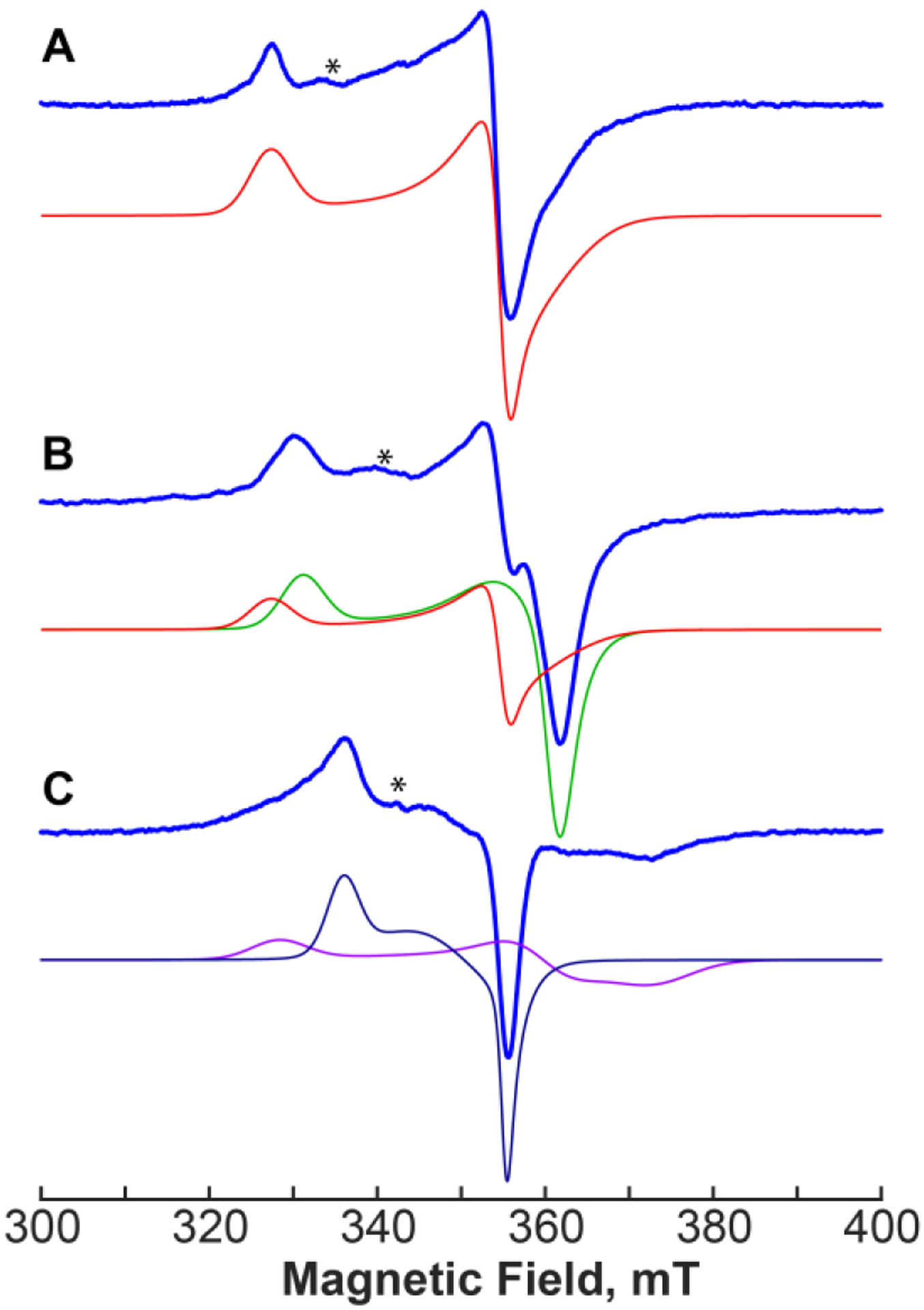
X-band CW EPR spectra (blue) of PhNadA as reduced (A), PhNadA in the presence of DHAP (B) and PhNadA in the presence of IA (C) and simulated spectral components with the following principal g-values: red in A and B, g=[2.100, 1.937, 1.913]; green in B, g=[2.076, 1.918, 1.904]; magenta in C, g=[2.094, 1.911, 1.842]; dark blue in C, g=[2.045, 1.966, 1.936]. Experimental conditions: temperature 15 K (A, B) 8 K (C); MW frequency, 9.623 GHz; MW power, 2 mW; modulation amplitude, 10 G. Asterisks indicate spectral artifacts.
In the previously studied PhNadA/QA complex, we observed a Hyperfine Sublevel Correlation (HYSCORE) signature of an 14N nucleus of QA at a bonding distance to Fea of the [4Fe–4S]+ cluster (Figure S3B) [19]. No such 14N signals are observed in the reduced PhNadA/IA complex (Figures S3C, S3E), suggesting that the amine group of IA is not bound to the Fe/S cluster. To clarify which carboxylate group in IA binds to Fea, we used a site-specifically labeled form of IA containing 13C at the C8 position. This isotopolog was synthesized enzymatically from 4-13C Asp using PhNadB (see Materials and Methods). The resulting X-band HYSCORE spectra of the PhNadA/IA complex show clear evidence for the 13C signals (Figure S3D); however, these signals are partially overlapping with signals of weakly coupled 14N nuclei originating from the protein matrix (Figure S3D). These “contaminating signals” are routinely observed in HYSCORE spectra of [4Fe-4S]-containing proteins and make the analysis of 13C HF coupling difficult. To isolate the 13C signals from the “matrix” 14N signals, we performed uniform 15N isotope labeling of PhNadA by culturing E. coli cells expressing the gene for PhNadA in M9 minimal media in which NH4Cl at natural abundance was replaced with 15NH4Cl (see Materials and Methods).
HYSCORE spectra of 15N-labeled PhNadA in complex with [8-13C]-IA obtained at different field positions across the EPR spectral envelope are shown in Figure 4. All spectra show well-defined ridges that are present if [8-13C]-IA is used and absent if Asp at natural abundance is used to synthesize IA (Figure S4 panel I). The 13C signals can be seen in all HYSCORE measurements performed, with no apparent drop in 13C signal intensity at any particular field position, suggesting that both EPR species identified (Figure 3A and C, and Figure S4 panel II) show that the C8 carboxylate group of IA coordinates to Fea of the cluster.
Figure 4.
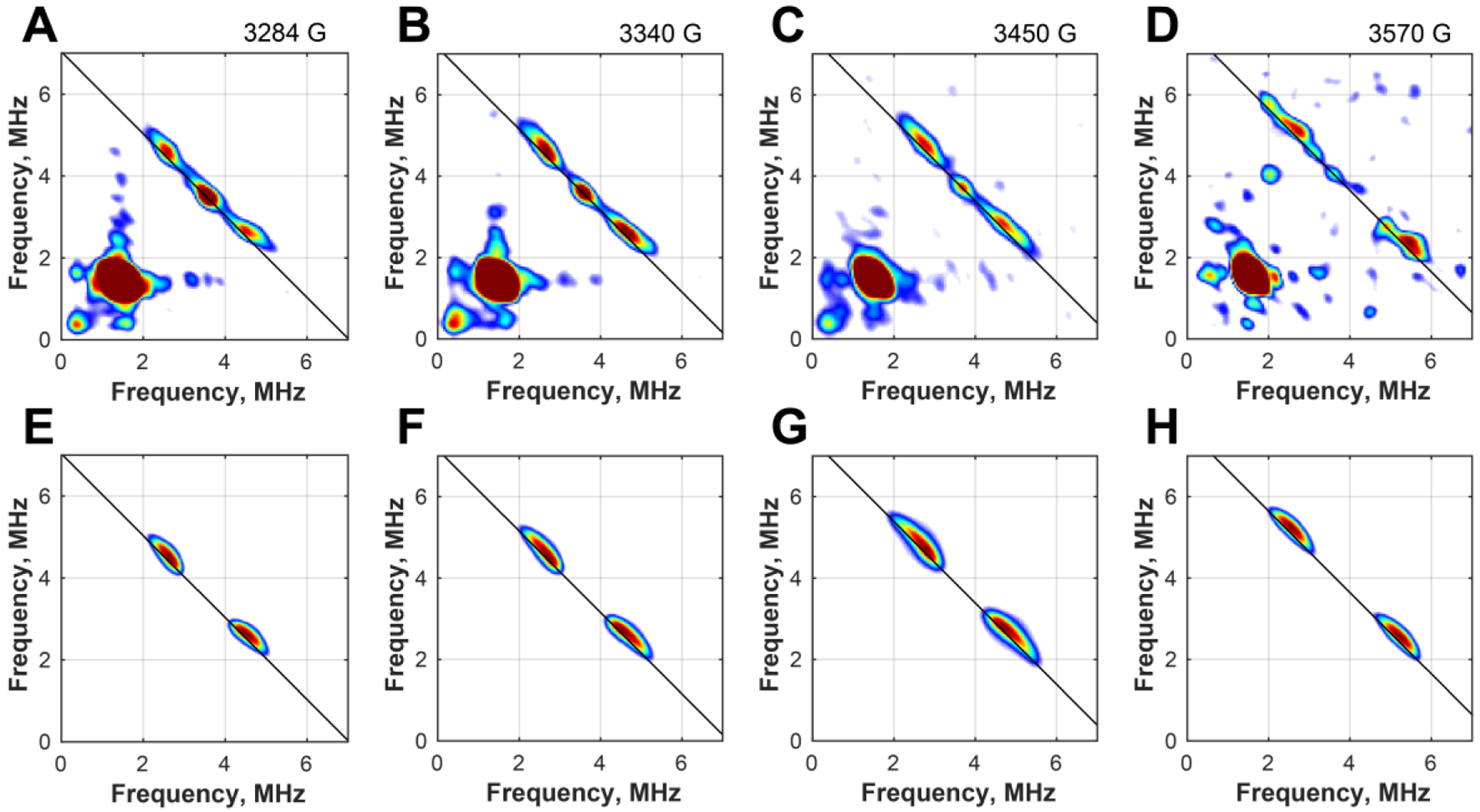
Field-dependent X-band HYSCORE spectra of 15N PhNadA/8-13C IA (A-D) and corresponding simulations using 13C hyperfine coupling of A(13C) = [1.3, 1.0, 4.0] MHz (E-F, see main text for details). Experimental conditions: temperature, 8 K; MW frequency, 9.4011 GHz; τ, 132 ns; t(π/2), 8 ns; magnetic field, A) 328.4 mT; B) 334.0 mT; C) 345.0 mT; D) 357.0 mT.
The position and character of the 13C signals indicate predominant isotropic HF coupling. Due to the two species in the EPR spectrum of PhNadA/IA, it is difficult to determine precisely the 13C HF coupling constants. However, assuming that both species exhibit similar 13C HF coupling, we can simulate the field dependence of the HYSCORE spectra using the following principal constants: A(13C) = [1.3, 1.0, 4.0] MHz. It is worth noting that the isotropic HF coupling constant of Aiso(13C)=2 ± 0.3 MHz is larger than the one observed in the case of the 13C-carboxylate of SAM bound to the unique iron of the [4Fe–4S] cluster in PFL-AE (A(13C)~0.75MHz) [32] and only slightly smaller than that observed for 13CN bound to the auxiliary [4Fe-4S] cluster of HydG (Aiso(13C)= 2.7 MHz) [33]. These spectroscopic experiments unambiguously support the observation made by x-ray crystallography that the C8 carboxylate group of IA binds to Fea of the [4Fe–4S] cluster in the PhNadA/IA complex.
A comparison of the modes of binding of IA and QA to the [4Fe-4S] cluster of PhNadA (Figure S2D) provides interesting insight into a possible new hypothesis for the protein’s catalytic mechanism, especially with respect to the role of the Fe/S cluster. In both structures, a carboxylate group is coordinated to Fea: the C8-carboxylate in the structure with bound IA and the C7-carboxylate in the structure with bound QA. If these binding modes are on the catalytic pathway, they suggest that as the reaction proceeds from substrates to products, there must be an overall rearrangement of the ligand in the active site. Modeling of bond changes in going from the IA-bound structure to the QA-bound structure suggests that there is a rotation of 160° around the C2–C3 bond of IA. As a result of this rotation, the coordination of the Fe/S cluster presumably changes from the C8 carboxylate group of IA to the C7 carboxylate and N1 atom of IA based on the structure with bound QA. The C7 carboxylate would now interact with Asn111 and His21, as is observed in the structure with QA bound. Rotation around the C2–C3 bond places the oxygen atoms of the C8 carboxylate in H-bonding distance to the side chains of His21, Ser38, and Tyr109, as is observed in the structure with QA. The position of the nitrogen atom moves by 2.9 Å in the PhNadA/IA and PhNadA/QA structures, while the C8 carboxylate of QA is bound by two sodium ions in the PhNadA/QA structure (Figure S5E).
Although significant changes in the polypeptide backbone are not observed between the PhNadA/IA and PhNadA/QA structures, minor changes are observed for loop2 (Asp37, Ser38) with adjusted helix α3 (domain 1), loop3 (Tyr109-Asn111) between β5-α6 (domain 2), α7 (domain 2), and the C-terminus of α14 (Figure S5F). Although α4 of domain 2 does not move, Phe60 of α4 undergoes a significant conformational change (Figure S2F), which has also been observed for TmNadA for the complexes with QA and phosphoglycolohydroxamate [20]. In the structure of PhNadA/IA, the side-chain of Phe60 is oriented toward the active site, but undergoes a 180° rotation in the PhNadA/QA structure. This significant change in the orientation of Phe60, which is also accompanied by movement of Asn111 and Tyr109, suggests that these amino acids are involved in supporting the suggested rotation necessary for catalysis and/or release of the product QA (Figure S2F).
Structure of the holo-PhNadA/DHAP.
The structure of holo-PhNadA in complex with DHAP was determined to a resolution of 1.6 Å. This structure was obtained by incubating holo-PhNadA in the presence of DHAP and Asp during crystallization (Figure 2B). In this structure, the C6 hydroxyl group of DHAP coordinates with Fea, and the C5 carbonyl oxygen forms H-bonds with the side chain of Glu198. Although the oxygen atom that bridges C4 and the phosphorous atom of DHAP does not form H-bonds with any amino acid, the remaining oxygen atoms of the phosphate moiety are well anchored and form H-bonds with the side chains of His196, Ser126, Thr213, and His21, as well as with the amide nitrogen of Ser38 and Thr213. A comparison of the structure of PhNadA/DHAP with a recently published structure of PhNadA/DHAP (PDB ID: 5KTN) shows that the phosphate group has the same residue interactions in the structure; however, the three-carbon chain freely rotates by 80° about P-O, 24° about O-C4, 118° about C4–C5, and 126° about the C6-OH bonds. In all of the structures, the C6-OH group coordinates with the unique iron of the Fe/S cluster (Figure S5C).
A superposition of both PhNadA/IA and PhNadA/DHAP structures shows that both substrates occupy roughly the same position in the active site (Figure S5A). A variant analysis of residues that coordinate to DHAP in the active site (His21Gln, His196Gln, and Ser126Ala) shows that none of the substitutions substantially alter the KM for DHAP in the PhNadA reaction [19].
Structure of a possible reaction intermediate in complex with wt PhNadA.
To determine whether structural evidence for any intermediates during catalysis could be provided, PhNadA was crystallized in the presence of PhNadB (~1.5 equiv), FAD, and L-Asp, as described for the PhNadA/IA structure; however, the resulting crystals were cryoprotected by dipping them in a solution containing FAD and DHAP for 15 to 60 s. The resulting structure, solved to a resolution of 1.65 Å and only containing PhNadA (i.e. no PhNadB), reveals electron density consistent with a linkage between the nitrogen atom of IA and C6 of DHAP, but with no phosphate at C4 (Figure 5). Unexpectedly, the C5 oxygen atom of DHAP, proposed to be lost in the last step of catalysis, is also not present. The structure shows one oxygen atom of the C8 carboxylate of the intermediate ligated to Fea, while the second oxygen atom forms H-bonds with the side chain of Glu198 (Figure 5B). The oxygen atoms of the C7 carboxylate group form H-bonds with the side chains of His21 and Thr213 and are also H-bonded to the amide proton of Thr213 (Figure 5B). This arrangement of the carboxylate groups of the intermediate is quite similar to that seen in the structure of PhNadA/IA. The structure also shows electron density at C2 of formerly IA that is consistent with a hydroxyl group, which forms H-bonds with the side chains of Tyr109, Asn111, and His196, while the nitrogen atom of the intermediate interacts with the side chain of Asn111. The emergence of the hydroxyl group at C2 of the wt-intermediate with the conversion of a double to a single bond between N1 and C2 may facilitate rotation of this bond for subsequent ring closure. Interestingly, the C6 carbon of formerly DHAP also bears electron density consistent with a hydroxyl group, suggestive of a carbinolamine. This hydroxyl group forms H-bonds with the side chain and amide groups of Ser38 as well as with a water molecule that is further H-bonded to the side chains of Asp37 and Ser38. Most notably, C4 of formerly DHAP contains electron density that is consistent with an aldehyde. The oxygen of the C4 aldehyde group is in H-bonding distance with the side chain of Tyr109 and a molecule of water.
Figure 5.
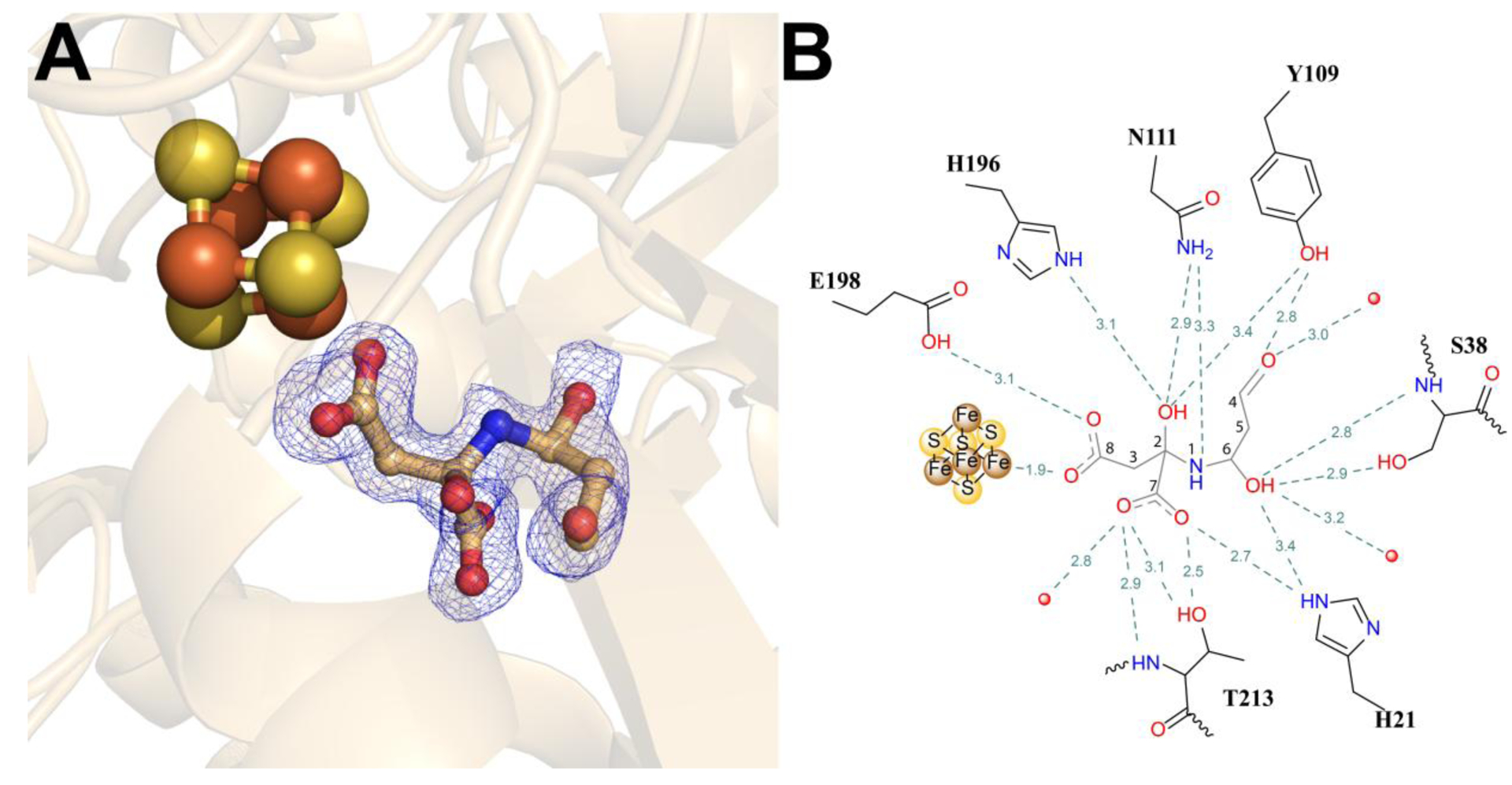
Structure of PhNadA in complex with a possible intermediate in the active site. (A) Omit electron density map of the intermediate in the active site of PhNadA, Fo-Fc electron density is contoured at 2.0 σ (blue mesh). (B) Schematic diagram of binding intermediate in the active site of PhNadA. Atom colors: Fe – orange, S – yellow, N - blue, C – grey and O – red.
A similar intermediate is observed in a structure of the inactive E198Q variant (Table S1 and Figure S6). This complex was crystallized under turnover conditions, but in the presence of OA and ammonium chloride as the source of IA rather than in the presence of NadB and Asp as described for the wt PhNadA/intermediate structure. The structure, solved to 2.2 Å resolution, has two monomers in the asymmetric unit, each of which contains a slightly different intermediate species. The chemical structure of the intermediate in subunit B is similar to that of the wt intermediate (Figures S6B and S6D), but exhibits a different conformation (Figure S6F). The conformation of the wt intermediate resembles an S-shape, while the conformation of the E198Q intermediate is more linear, with changes in the H-bonding network. One oxygen atom of the C8 carboxylate of the E198Q intermediate ligates to Fea of the cluster, while the second oxygen atom forms H-bonds with the side chains of Asn111 and Gln198. The orientation of the C7 carboxylate group places the oxygen atoms in H-bonding distance with the side chains of His21, His196, Thr213 and the amide group of Thr213. The C2-OH group is coordinated by the side chain of His196. The OH-group attached to the C6-carbon is within H-bonding distance of the side chain of Ser38 and the amide group of Ser126. The aldehyde group at C4 is interacting with the side chain Asp37 and Thr125 (Figure S6D).
The chemical structure of the intermediate in subunit A (Figures S6A, S6C and S6E) is slightly different from that of the intermediate in subunit B and from that of the intermediate in the wt enzyme. The intermediate in subunit A lacks an OH-group at C2. We suspect that the intermediate in subunit A precedes that of the wt and variant subunit B intermediates during catalysis, because the latter two have the C2 OH-group that might enable rotation of the ligand. Although the chemical structures of intermediates in the A and B subunits are not identical, the conformation and manner of binding are quite similar (Figures S6C and S6D). Only minor changes are seen in the binding of the OH-group at C6. In subunit A, the hydroxyl group is in H-bonding distance with the backbone amide of Ser126. In subunit B, it is H-bonded to the side chains of Ser38 and the amide group of Ser126.
HYSCORE analysis of the intermediate formed in the active site of the PhNadA E198Q and Y109F variants.
Recently, a structure of a postulated intermediate produced by an inactive variant (Y107F) of Thermotoga maritima NadA (TmNadA) was published, which corresponds to the Y109F variant of PhNadA [20]. This structure was generated by co-crystallizing the protein with DHAP and IA (generated from NH4Cl and OA in the crystallization medium). The authors of the study proposed that the DHAP-part of the intermediate molecule contains the C6-OH group coordinated to the unique Fe of Fe/S-cluster. This result is in contrast to the observed structure of the PhNadA wt-intermediate and the E198Q variant intermediates in this work, in which the C8-carboxylate group of IA coordinates to the cluster. To verify the crystallographically observed intermediate, we analyzed inactive PhNadA E198Q and Y109F variants by HYSCORE using 8-13C-IA (obtained as described earlier) and studied how the presence of DHAP affects IA binding. First, in the absence of DHAP, our experiments confirm that IA binds to the [4Fe-4S] cluster in the same manner both in wt PhNadA (Figure 4) and in the two studied variants (Figures 6 and S7).
Figure 6.
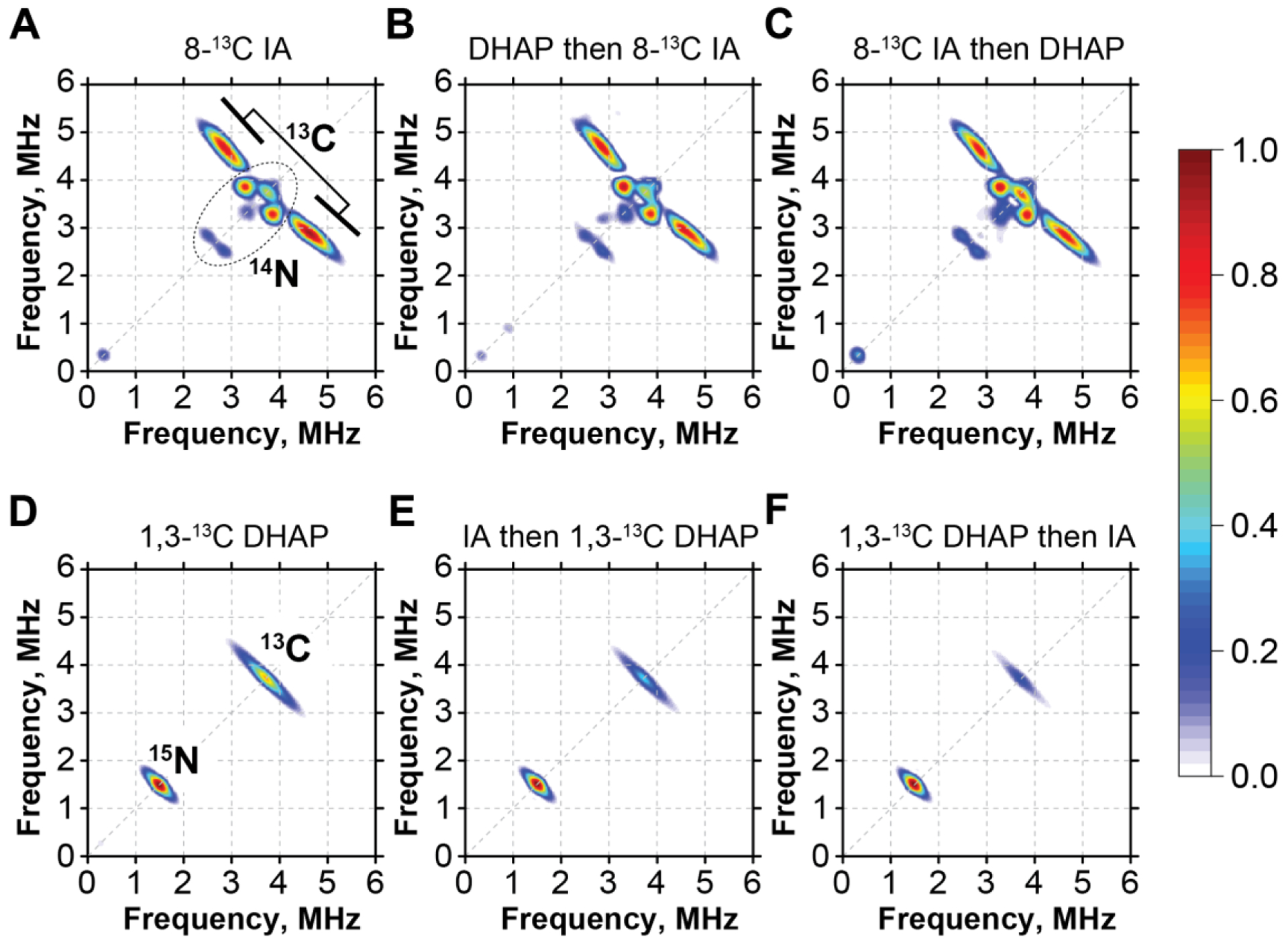
A-C: X-band HYSCORE spectra of PhNadAY109F in the presence of 8-13C IA alone (A) and with an addition of DHAP before (B) or after (C) adding 8-13C IA; D-F 15N-PhNadAY109F in the presence of 1,3-13C DHAP alone (D) and with an addition of IA before (B) or after (C) addition of DHAP. Experimental conditions: temperature, 8K; MW frequency, 9.41 GHz (A-C), 9.38 GHz (D-F); τ, 132 ns; t(π/2), 8ns; Magnetic field, 346.0 mT (A-C), 344.0 mT (D-F).
We then performed two experiments using roughly equimolar (~5 mM) concentrations of both substrates. In one experiment, PhNadA variants were incubated with DHAP first (for 5 h) in the presence of NadB, and then the 8-13C Asp was added to the reaction to generate 8-13C IA (Figure 6B and Figure S7B). In a second experiment, the PhNadA/8-13C IA complex was produced first by incubating PhNadA, 8-13C Asp, and NadB for ~8 h. DHAP was then added to the solution, which was allowed to incubate for 5 h prior to freezing for EPR analysis (Figure 6C and Figure S7C). Regardless of the order of addition of DHAP and IA, we find that the shape and the intensity of the 13C signals in the corresponding HYSCORE spectra remain roughly the same (see Figure S9). Next, we performed a reverse labeling experiment with 1,3-13C-DHAP and unlabeled IA using methodology identical to that described above. Similar to the previous experiment, we alternated the order in which DHAP and IA were incubated with the PhNadA E198Q and Y109F variants. In line with previous reports and our own crystallographic data [21], when no IA is present, we observe a definitive 13C HYSCORE signal, indicating that DHAP indeed binds to the FeS cluster. Because the observed 13C signal overlaps with the matrix 14N signals, we performed these experiments using PhNadA uniformly labeled with 15N. Similar binding is observed with both the E198Q and Y109F variants, although we note that the overall intensity of the 13C-DHAP signal as compared to the 15N matrix signal is lower in the case of 15N-PhNadA E198Q (see Figure S8, and Figures S10 and S11). When IA was generated using NadB and Asp after preincubating the PhNadA Y109F variant with 1,3-13C-DHAP, we observe about a 70 – 80% decrease in the 13C HYSCORE signal, indicating that IA displaces DHAP from binding to the cluster (see Figures 6D, 6F and Figure S10).
A reverse experiment of first incubating PhNadA with NadB-produced IA then adding 1,3-13C DHAP also shows significantly lower levels of 13C signal in both studied variants as compared to those of 1,3-13C DHAP/PhNadA samples (~50 – 60% decrease) (Figures 6D, 6E and Figures S8, S10–S11). We attribute the observation that the reduction of the 13C-DHAP signal is not complete to the inefficiency of in vitro enzymatic synthesis of IA by PhNadB and the instability of IA over the prolonged period of time required for this experiment. Altogether, these experiments indicate that the IA-part of the intermediate is bound to the Fea site. In addition, the modeling of our intermediate from the PhNadA E198Q structure into the density map of the TmNadA/Intermediate W complex (PDB ID: 5F3D) demonstrates a better fit into the electron density with smaller difference Fourier peaks than that observed previously (Figure S12) [20].
DISCUSSION
The mechanism by which NadA catalyzes its reaction has been enigmatic since its discovery, especially with respect to the role of its [4Fe–4S] cluster in catalysis. In the original working hypothesis advanced by Gholson and coworkers, the enamine of IA is proposed to attack C1 of DHAP to eliminate phosphate and create the nascent C–C bond of QA. Subsequent tautomerizations result in the intramolecular formation of an aldehyde and an enamine, which condense to eliminate the first molecule of water and form the nascent C–N bond of QA. In the last step, the second molecule of water is eliminated from QP (intermediate Y), which results in aromatization of the pyridine ring and formation of QA. As shown or postulated in the reactions of all FeS-containing enzymes of the hydro-lyase family, the expected role of the FeS cluster in the NadA reaction is to facilitate dehydration of QP through a coordination with QP’s hydroxyl group. However, in all other known members of the hydro-lyase family, the eliminated water molecule is from a stable precursor. By contrast, the eliminated water molecule is from an unstable intermediate in the proposed NadA reaction mechanism, suggesting that Lewis-acid assistance may not be needed to facilitate catalysis. Nevertheless, this strong paradigm of Lewis acid-mediated catalysis has dominated thinking in the mechanistic understanding of the NadA reaction.
A number of structures of NadAs from multiple organisms have been solved both in the presence and in the absence of ligands; however, no structure has been reported of a NadA–IA complex until the work described herein. Although structures of DHAP or DHAP analogs bound to the unique iron site of NadA have been reported, including a structure reported herein, our studies suggest that IA binding to the unique iron site may be more mechanistically relevant. Consistent with this premise, we observe that an IA-derived fragment of a predicted intermediate in the NadA reaction is bound to Fea, and we observe binding of the QA product to Fea through its N1 and C7 carboxylate, which are both derived from IA. In fact, in all structures that contain IA or remnants of the molecule, a carboxylate group from IA is always coordinated to Fea, suggesting that this coordination is mechanistically important. However, in the IA- and intermediate-bound structures, the C8 carboxylate is bound to Fea, while in the QA-bound structure, the C7 carboxylate is bound to Fea. These observations suggest that in the progression from the “intermediate” to product, rotations around both the N1–C2 and C2–C3 bonds take place, and in the intermediate species, the C8 carboxylate dissociates and the intermediate’s C7 carboxylate and N1 atoms rebind to the Fe/S cluster. This suggested conformational change is strikingly reminiscent of catalysis by aconitase, the paradigm enzyme for the Fe/S-dependent hydrolyase family of enzymes, which catalyzes the isomerization of citrate to isocitrate via the intermediate, cis-aconitate. When citrate is bound to aconitase, its C3 carboxylate and C3 hydroxyl group coordinate to Fea in a bidentate fashion. Deprotonation at C2 of the pro-R arm of citrate initiates an anti-elimination of a molecule of water to afford the intermediate, cis-aconitate. In order to add a hydroxyl group at C2 of cis-aconitate to generate isocitrate, the molecule undergoes a flip in the active site, which involves the release of the C3 carboxylate from Fea with concomitant coordination of the C2 carboxylate. This conformational change positions the hydroxyl group attached to Fea in the proper orientation to add to C2 of the molecule (Figure S13).
In a similar manner, we propose that the observed intermediate species (8) undergoes an analogous flip about the N1–C2 and C2–C3 bonds to generate (9) (Figure 7). Loss of a molecule of water affords an imine (10), which facilitates two deprotonation events at C3 that lead to ring closure (11) and dehydration (12). The last step is loss of the C6 hydroxyl group to afford QA (13), which is initiated by proton abstraction at C5. These steps could also occur in other sequences. For example, dehydration of the C5–C6 bond could occur before ring closure and dehydration of the C3–C4 bond.
Figure 7.
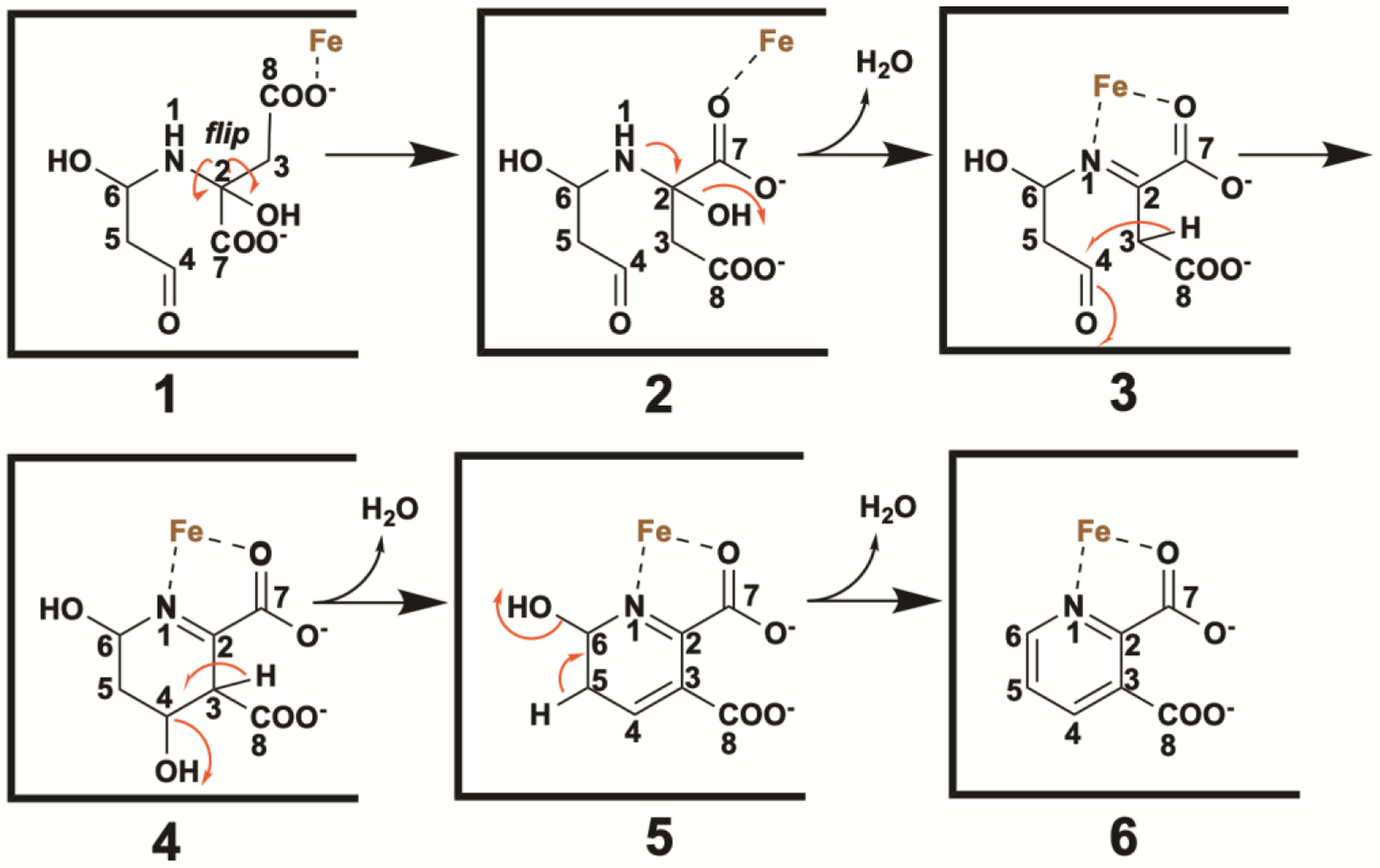
Working hypothesis for the PhNadA-dependent conversion of the “intermediate” to the final product.
The structure of PhNadA/QA provides some insight into residues that might be involved in conversion of the intermediate species to product. Our site-directed mutagenesis studies [19] show that Tyr23, Tyr109, and Glu198 are absolutely essential in catalysis, given that changing them to Phe23, Phe109 and Gln198 caused the activity of the corresponding variant protein to be undetectable in our assays. Tyr109 is juxtaposed equidistant between C3 and C4 of QA at 3.3 Å from each of the atoms and appears to be perfectly situated to abstract the C3 proton of species 10 and deliver the proton to the nascent hydroxyl group in species 11. A second round of proton abstraction and oxygen-atom protonation by the same residue would result in the elimination of water to yield species 12. The last step, conversion of 12 to QA (13) could be mediated by Tyr23 as the general base that removes the proton from C5, and Glu198 as the general acid that protonates the C6 hydroxyl group, which leaves as a molecule of water.
As detailed above, the electron density for the unexpected species obtained by x-ray crystallography is robust, and given the resolution of 1.65 Å, determining the structure of the species was straightforward. However, the mechanism shown in Figure 7 begs the question, “how is this intermediate species formed.” Indeed, the absence of the C5 hydroxyl group of DHAP in the intermediate species is unexpected, given that all previously suggested mechanisms for the NadA reaction show loss of this hydroxyl group in the very last step in a reaction that is facilitated by association of the hydroxyl group with the Fe/S cluster. In Figure 8, we outline a plausible mechanism for formation of the intermediate species (8) shown in Figure 7. Tautomerization of DHAP affords an enol-phosphate (2), which is attacked at phosphorous by water, eliminating water from C6 of DHAP and setting up a reactive Michael acceptor. The Michael acceptor is trapped by the nitrogen of the enamine of IA to afford species 4, which tautomerizes to give species 5. Proton abstraction from species 5 allows for the elimination of the hydroxyl group at C5 as a molecule of water. Water then attacks at C2 and C6 to give species 8. We hypothesize that the predicted ensuing conformational change allows this species to build up in the crystal. Alternatively, species 7 might proceed directly through the catalytic sequence, bypassing species 8. In this instance, the last step in the mechanism described in Figure 7 would be dehydration at C4, given that the C5–C6 dehydration would have already occurred.
Figure 8.
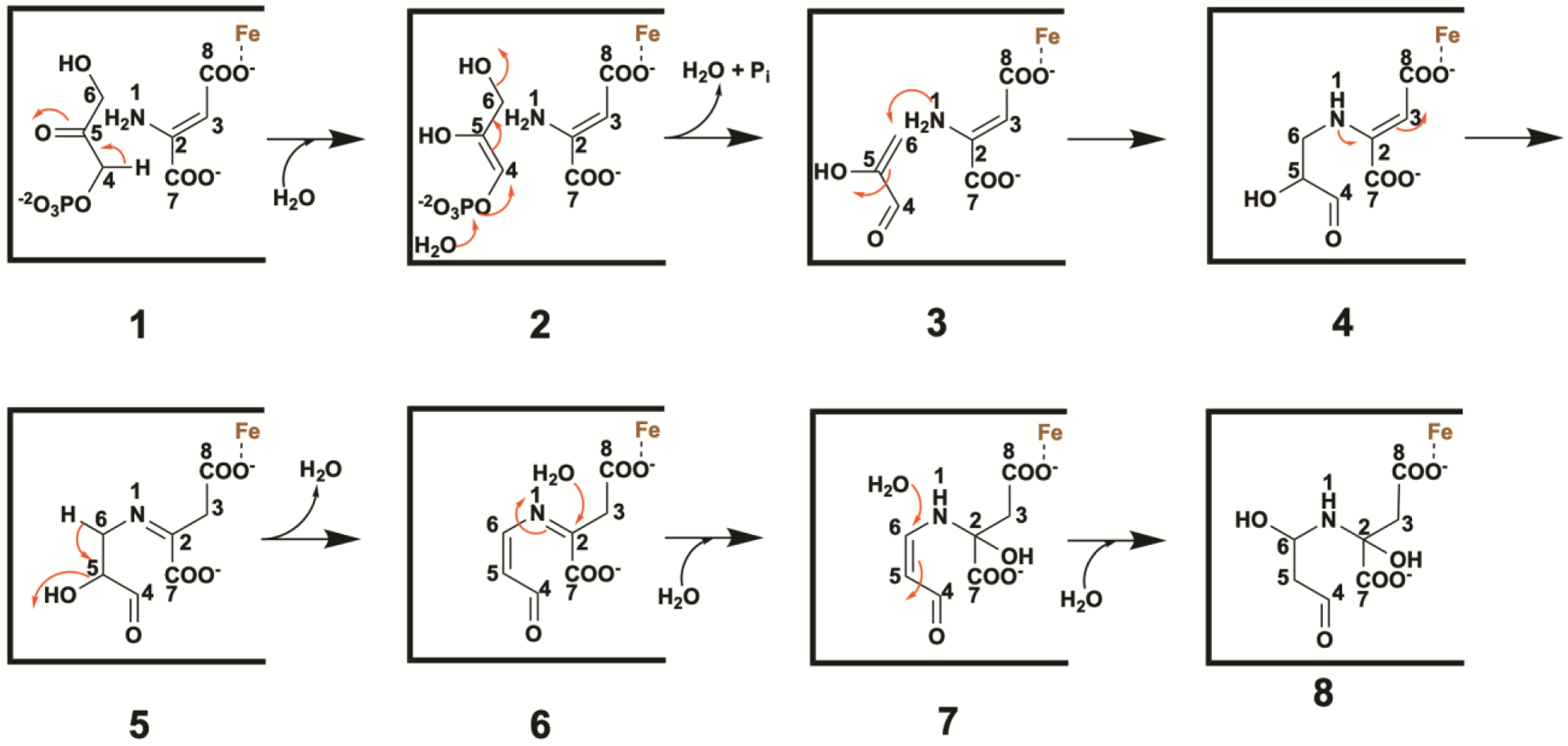
Working hypothesis for the PhNadA-dependent conversion of DHAP and IA to the “intermediate” species (8) observed by x-ray crystallography.
In conclusion, we provide evidence for an unexpected species in the NadA reaction that may in fact be an intermediate, and a plausible mechanism for formation of this species from DHAP and IA, as well as formation of QA from this species. This species was obtained by soaking crystals of wt PhNadA/IA with DHAP for only short period of time or by co-crystallizing a PhNadA Glu198Gln variant with DHAP and chemically generated IA. Strikingly, in both instances, the hydroxyl group thought to be eliminated in the last step in the reaction is missing in this species. Moreover, our ability to generate this species with stoichiometric concentrations of IA (at most) and with very short incubation times with DHAP, suggests that it is not formed from aberrant chemistry or steps that are off the normal reaction cycle. The proposed working hypothesis shown in Figures 7 and 8 suggests that the major role of the Fe/S cluster in catalysis is for substrate and product orientation, but that it may also facilitate some of the proton transfer steps by lowering the pKa values of relevant carbon acids.
Supplementary Material
ACKNOWLEDGMENT
This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357.
Funding Sources
This work was supported by NIH (GM-122595 to S.J.B.), NSF (MCB-1158486 and 1716686 to S.J.B.), the Eberly Family Distinguished Chair in Science (S.J.B.), and The Pennsylvania State University Huck Institute for Life Sciences (N.H.Y). SJB is an investigator of the Howard Hughes Medical Institute.
ABBREVIATIONS
- Asp
aspartate
- DHAP
dihydroxyacetone phosphate
- DTT
dithiothreitol
- CW
continuous wave
- EPR
electron paramagnetic resonance
- FAD
flavin adenine dinucleotide
- FADH2
reduced form of FAD
- FeS
iron–sulfur
- G3P
glyceraldehyde 3-phosphate
- HF
hyperfine
- HYSCORE
hyperfine sublevel correlation
- IA
iminoaspartate
- NadA
quinolinate synthase
- NadB
L-aspartate oxidase
- OA
oxaloacetate
- Ph
Pyrococcus horikoshii
- QA
quinolinic acid
- QP
quinolinate precursor
- Tm
Thermotoga maritima
- wt
wild-type
Footnotes
Supporting Information
Tables S1–S2 and Figures S1-S13. This material is available free of charge via the internet at http://pubs.acs.org.
REFERENCES
- 1.Belenky P, Bogan KL, and Brenner C, NAD+ metabolism in health and disease. Trends Biochem. Sci, 2007. 32: p. 12–19. [DOI] [PubMed] [Google Scholar]
- 2.Frey PA and Hegeman AD, Enzymatic Reaction Mechanisms. 2007, New York: Oxford University Press. [Google Scholar]
- 3.Pollak N, Dölle C, and Ziegler M, The power to reduce: pyridine nucleotides–small molecules with a multitude of functions. Biochem. J, 2007. 402: p. 205–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kessler D, Rétey J, and Schulz GE, Structure and action of urocanase. J. Mol. Biol, 2004. 342: p. 183–194. [DOI] [PubMed] [Google Scholar]
- 5.Foster JW and Moat AG, Nicotinamide adenine dinucleotide biosynthesis and pyridine nucleotide cycle metabolism in microbial systems. Microbiol. Rev, 1980. 44: p. 83–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gazzaniga F, Stebbins R, Chang SZ, McPeek MA, and Brenner C, Microbial NAD Metablism: lessons from comparative genomics. Microbiol. Mol. Biol. Rev, 2009. 73: p. 529–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chandler JL and Gholson RK, Studies on the de novo biosynthesis of NAD in Escherichia coli. Anal. Biochem, 1972. 48: p. 529–535. [DOI] [PubMed] [Google Scholar]
- 8.Nasu S and Gholson RK, Replacement of the B protein requirement of the E. coli quinolinate synthetase system by chemically-generated iminoaspartate. Biochem. Biophys. Res. Commun, 1981. 101(2): p. 533–539. [DOI] [PubMed] [Google Scholar]
- 9.Nasu S, Wicks FD, and Gholson RK, L-Aspartate oxidase, a newly discovered enzyme of Escherichia coli, is the B protein of quinolinate synthetase. J. Biol. Chem, 1982. 257(2): p. 626–632. [PubMed] [Google Scholar]
- 10.Mortarino M, Negri A, Tedeschi G, Simonic T, Duga S, Gassen HG, and Ronchi S, L-Aspartate oxidase from Escherichia coli. I. Characterization of coenzyme binding and product inhibition. Eur. J. Biochem, 1996. 239: p. 418–426. [DOI] [PubMed] [Google Scholar]
- 11.Tedeschi G, Negri A, Mortarino M, Ceciliani F, Simonic T, Faotto L, and Ronchi S, L-Aspartate oxidase from Escherichia coli. II. Interaction with C4 dicarboxylic acids and identification of a novel L-aspartate:fumarate oxidoreductase activity. Eur. J. Biochem, 1996. 239: p. 427–433. [DOI] [PubMed] [Google Scholar]
- 12.Yang Z, Savchenko A, Yakunin A, Zhang R, Edwards A, Arrowsmith C, and Tong L, Aspartate dehydrogenase, a novel enzyme identified from structural and functional studies of TM1643. J. Biol. Chem, 2003. 278: p. 8804–8808. [DOI] [PubMed] [Google Scholar]
- 13.Wicks FD, Sakakibara S, Gholson RK, and Scott TA, The mode of condensation of aspartic acid and dihydroxyacetone phosphate in quinolinate synthesis in Escherichia coli. Biochim. Biophys. Acta, 1977. 500: p. 213–216. [DOI] [PubMed] [Google Scholar]
- 14.Cicchillo RM, Tu L, Stromberg JA, Hoffart LM, Krebs C, and Booker SJ, Escherichia coli quinolinate synthetase does indeed harbor a [4Fe–4S] cluster. J Am Chem Soc, 2005. 127: p. 7310–7311. [DOI] [PubMed] [Google Scholar]
- 15.Ollagnier-de Choudens S, Loiseau L, Sanakis Y, Barras F, and Fontecave M, Quinolinate synthetase, an iron–sulfur enzyme in NAD biosynthesis. FEBS Lett, 2005. 579: p. 3737–3743. [DOI] [PubMed] [Google Scholar]
- 16.Beinert H, Kennedy MC, and Stout CD, Aconitase as iron-sulfur protein, enzyme, and iron-regulatory protein. Chem. Rev, 1996. 96: p. 2335–2373. [DOI] [PubMed] [Google Scholar]
- 17.Flint DH and Allen RM, Iron–sulfur proteins with nonredox functions. Chem Rev, 1996. 96(7): p. 2315–2334. [DOI] [PubMed] [Google Scholar]
- 18.Volbeda A, Cabodevilla JS, Darnault C, Gigarel O, Han T-H-L, Renoux O, Hamelin O, Ollagnier-de Choudens S, Amara P, and Fontecilla-Camps JC, Crystallographic trapping of reaction intermediates in quinolinic acid synthesis by NadA. ACS Chem. Biol, 2018. 13: p. 1209–1217. [DOI] [PubMed] [Google Scholar]
- 19.Esakova OA, Silakov A, Grove TL, Saunders AH, McLaughlin MI, Yennawar NH, and Booker SJ, Structure of quinolinate synthase from Pyrococcus horikoshii in the presence of its product, quinolinic acid. J. Am. Chem. Soc, 2016. 138: p. 7224–7227. [DOI] [PubMed] [Google Scholar]
- 20.Volbeda A, Darnault C, Renoux O, Reichmann D, Amara P, Ollagnier-de Choudens S, and Fontecilla-Camps JC, Crystal structures of quinolinate synthase in complex with a substrate analogue, the condensation intermediate, and substrate-derived product. J. Am. Chem. Soc, 2016. 138: p. 11802–11809. [DOI] [PubMed] [Google Scholar]
- 21.Fenwick MK and Ealick SE, Crystal structures of the iron-sulfur cluster-dependent quinolinate synthase in complex with dihydroxyacetone phosphate, iminoaspartate analogues, and quinolinate. Biochemistry, 2016. 55: p. 4135–4139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Saunders AH, Griffiths AE, Lee K-H, Cicchillo RM, Tu L, Stromberg JA, Krebs C, and Booker SJ, Characterization of quinolinate synthases from Escherichia coli, Mycobacterium tubculosis, and Pyrococcus horikoshii indicates that [4Fe–4S] clusters are common cofactors throughout this class of enzymes. Biochemistry, 2008. 47: p. 10999–11012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Otwinowski Z and Minor W, Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol, 1997. 2765: p. 307–326. [DOI] [PubMed] [Google Scholar]
- 24.Battye TGG, Kontogiannis L, Johnson O, Powell HR, and Leslie AGW, iMosflm: a new graphical interface for diffraction image processing with MOSFLM. Acta Crystallogr. D Biol. Crystallogr, 2011. D67: p. 271–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McCoy AJ, Grosse–Kunstleve RW, Adams PD, Winn MD, Storoni LC, and Read RJ, Phaser crystallographic software. J. Appl. Crystallogr, 2007. 40: p. 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse–Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, and Zwart PH, PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr, 2010. 66: p. 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Emsley P and Cowtan K, Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr, 2004. 60: p. 2126–2132. [DOI] [PubMed] [Google Scholar]
- 28.Emsley P, Lohkamp B, Scott WG, and Cowtan K, Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr, 2010. 66: p. 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Silakov A, Kazan Viewer. https://sites.google.com/site/silakovalexey/kazan-viewer, 2011.
- 30.Sakuraba H, Tsuge H, Yoneda K, Katunuma N, and Ohshima T, Crystal structure of the NAD biosynthetic enzyme quinolinate synthase. J. Biol. Chem, 2005. 280: p. 26645–26648. [DOI] [PubMed] [Google Scholar]
- 31.Chan A, Clémancey M, Mouesca J-M, Amara P, Hamelin O, Latour J-M, and Ollagnier-de Choudens S, Studies of inhibitor binding to the [4Fe–4S] cluster of quinolinate synthase. Angew. Chem. Int. Ed, 2012. 124: p. 1–5. [DOI] [PubMed] [Google Scholar]
- 32.Walsby CJ, Ortillo D, B.W. E., J.B. Broderick, and B.M. Hoffman, An anchoring role for FeS clusters: chelation of the amino acid moiety of S-adenosylmethionine to the unique iron site of the [4Fe–4S] cluster of pyruvate formate–lyase activating enzyme. J. Am. Chem. Soc, 2002. 124: p. 11270–11271. [DOI] [PubMed] [Google Scholar]
- 33.Dinis P, Suess DLM, Fox SJ, Harmer JE, Driesener RC, De La Paz L, Swartz JR, Essex JW, Britt RD, and Roach PL, X-ray crystallographic and EPR spectroscopic analysis of HydG, a maturase in [FeFe]-hydrogenase H-cluster assembly. Proc. Natl. Acad. Sci. U S A, 2015. 112: p. 1362–1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.