

Abstract
Converging evidence indicates the dysregulation of unique cytosolic compartments called stress granules (SGs) might facilitate the accumulation of toxic protein aggregates that underlie many age-related neurodegenerative pathologies (ANPs). SG dynamics are particularly susceptible to the cellular conditions that are commonly induced by aging, including the elevation in reactive oxygen species (ROS) and increased concentration of aggregate-prone proteins. In turn, the persistent formation of these compartments is hypothesized to serve as a seed for subsequent protein aggregation. Notably, the protein quality control (PQC) machinery responsible for inhibiting persistent SGs (e.g. Hsc70-BAG3) can become compromised with age, suggesting that the modulation of such PQC mechanisms could reliably inhibit pathological processes of ANPs. As exemplified in the context of accelerated aging syndromes (i.e. Hutchinson–Gilford progeria), PQC enhancement is emerging as a potential therapeutic strategy, indicating similar techniques might be applied to ANPs. Collectively, these recent findings advance our understanding of how the processes that might facilitate protein aggregation are particularly susceptible to aging conditions, and present investigators with an opportunity to develop novel targets for ANPs.
Keywords: Neurodegeneration, stress granules, BAG3
Introduction
Age-related neurodegenerative pathologies (ANPs), such as Alzheimer’s Disease (AD), and Parkinson’s Disease (PD), are a growing public health concern, while effective treatments remain scarce. The mounting burden of such pathologies is due to an unparalleled shift in aging demographics: by 2050, the percentage of aged individuals (i.e. >60 years old) will exceed 21% (an increase from 10% in 2010), and, for the first time in human history, the number of aged persons in the world will exceed the number of young (World Health Organization, 2006, 2015). Furthermore, unlike other public health challenges, where advances in treatment have reduced pathological manifestations and decreased mortality rates (e.g. cancer, HIV/AIDS), therapies to combat ANPs remain ineffective. For instance, in the case of AD, no FDA approved therapy can stop or reverse its progression (Cummings, Morstorf, & Zhong, 2014; Mangialasche, Solomon, Winblad, Mecocci, & Kivipelto, 2010). Therefore, it is increasingly urgent to advance treatment options for ANPs by targeting those processes which drive pathological consequences.
The causal mechanisms underlying ANPs may be related to a common characteristic amongst their pathogeneses, namely the failure to maintain intercellular protein homeostasis (i.e. proteostasis) and ensuing accumulation of so-called aggregates (Kurtishi, Rosen, Patil, Alves, & Moller, 2018; Ross & Poirier, 2004). Aggregate formation is initiated following anomalous interactions between polypeptides that are induced by distinct intermolecular properties in said proteins, including aberrant post-translational modifications (PTMs) and variation in amino acid sequences (Fink, 1998). Subsequent interactions amongst proximal counterparts, as well as nascent bonding with distal species, facilitates the polymerization of polypeptides into amorphous assemblies or highly ordered structures (i.e. amyloid) (Chiti & Dobson, 2006; Invernizzi, Papaleo, Sabate, & Ventura, 2012). Thus, although the structural relevance of certain aggregates remain debated (e.g. pre-fibril, proto-fibril, mature oligomer etc.), it is increasingly clear that initial interactions within a limited quantity of proteins can further precipitate the assembly of those aberrant proteinaceous compartments common amongst ANPs (i.e. “seeding”) (Walker, Diamond, Duff, & Hyman, 2013). In turn, the persistence of such aggregates can hinder a multitude of cellular processes and homeostatic mechanisms that ultimately result in a net loss of function (Brundin, Melki, & Kopito, 2010; Stefani & Dobson, 2003).
In support of their causal contribution to pathogenesis, not only is each ANP associated with a distinct aggregating proteomic profile, but genomic interrogations demonstrate a robust association between ANPs and those variations in polypeptide sequences that facilitate abnormal protein accumulation (J. P. Taylor, Hardy, & Fischbeck, 2002). For instance, numerous mutations within the amyloid beta (Aβ) sequence in AD patients result in protein conformations that increase its propensity to aggregate (Hatami, Monjazeb, Milton, & Glabe, 2017). Similarly, deleterious mutations amongst those protein quality control (PQC) mechanisms responsible for maintaining proteostasis also result in increased propensities for aggregation (Ciechanover & Brundin, 2003; Levine & Kroemer, 2019; Macario, Grippo, & Conway de Macario, 2005). Curiously, as aging can compromise proteostasis and facilitate protein aggregation, while proteome maintenance has been linked to increased fitness across mammalian and non-mammalian species, the consequences of age-dependent alterations in cellular conditions appear to be particularly relevant to underlying ANP mechanisms (D. C. David et al., 2010; Pérez et al., 2009; R. C. Taylor & Dillin, 2011; Treaster et al., 2014).
Intercellular conditions commonly associated with aging include elevated oxidative stress as well as increased concentrations of aggregate-prone polypeptides (Finkel & Holbrook, 2000; Huang et al., 2018). A cellular component that is especially susceptible to such factors is stress granules (SGs) (Alberti, Mateju, Mediani, & Carra, 2017; Lechler et al., 2017). SGs are membrane-less cytoplasmic compartments composed of mRNA and RNA-binding proteins (RBPs) whose formation ensues following the detection of cellular stress (e.g. ROS) and resulting inhbition of translation (Protter & Parker, 2016). PQC factors, specifically the Hsc70-BAG3 complex, are otherwise responsible for maintaining SG dynamics and facilitating their disassembly (Ganassi et al., 2016). However, given their unique molecular composition and physical characteristics, SG dynamics can readily be effected by such intercellular conditions commonly associated with aging (Emara et al., 2012; Patel et al., 2015). Consequently, persistent SG formation and the accumulation of polypeptides in such compartments might serve as a seed for the aberrant protein aggregation that is common amongst ANPs.
Given that PQC components, including Hsc70-BAG3 functioning, can be compromised in age, enhancing SG-relevant PQC could represent a novel strategy to inhibit pathological processes underlying ANPs (Dahlmann, 2007; Rubinsztein, Mariño, & Kroemer, 2011). Indeed, investigations of accelerated aging syndromes, particularly Hutchinson–Gilford progeria, demonstrate how enhancing PQC can mitigate protein aggregation as well as its pathological consequences (K. Cao et al., 2011; Cenni et al., 2011). Thus, it behooves researchers to consider how aging conditions might precipitate protein aggregation through disrupted SG regulation, as well as how the methods used to ameliorate similar pathological processes in accelerated aging conditions could be applied to those mechanisms underlying ANPs.
Aging conditions: ROS/RNS and Aggregate-Prone Proteins
Aging is associated with accumulating impairment in various homeostatic processes that ultimately compromise efficent cellular functioning (Lopez-Otin, Blasco, Partridge, Serrano, & Kroemer, 2013). Such impairment is accompanied by an elevation in concentrations of reactive oxygen species (ROS) and reactive nitrogen species (RNS) across tissue types (Finkel & Holbrook, 2000). Broadly, these molecules consist of the more reactive superoxide, peroxynitrate and hydroxyl radicals as well as the less potent hydrogen peroxide and nitric oxide (Murphy et al., 2011). The inherent properties of the CNS render it particularly susceptible to the accumulation of such species. Along with the post-mitotic origin of neurons, the brain maintains less antioxidant defenses compared to other organs and its increased energetic demand, in conjunction with decreased energetic reserve, is compensated by a high degree of vascular turnover, which itself can generate ROS/RNS due to increased heavy metal exposure (e.g. Fe, Mg) (Halliwell, 2006). In aging, such accumulation is exacerbated due to a number of age-dependent variables, including chronic microglial activation, decreased mitochondrial efficiency, increased permeability of the blood-brain-barrier (BBB) and further compromised antioxidant mechanisms (Balaban, Nemoto, & Finkel, 2005; Finkel & Holbrook, 2000; von Bernhardi, Eugenín-von Bernhardi, & Eugenín, 2015).
Aging can also be characterized by an increased population of aggregate-prone proteins (D. David, 2012; Klaips, Jayaraj, & Hartl, 2018). This elevation in polypeptides that maintain increased aggregation propensities is sometimes referred to as supersaturation (Ciryam, Kundra, Morimoto, Dobson, & Vendruscolo, 2015; Ciryam, Tartaglia, Morimoto, Dobson, & Vendruscolo, 2013). Although the age-dependent mechanisms that generate such proteins are not fully elucidated, current postulations consider the elevated ROS/RNS in aging as a primary causal factor (Hohn, Konig, & Grune, 2013; Santos & Lindner, 2017). This is due to the fact that the inherent electrostatic properties of ROS/RNS allow them to induce modifications in proteins that are known to increase their propensity to aggregate (Reichmann, Voth, & Jakob, 2018). For instance, through deamidation, α-amidation, β-scission and hydrogen abstraction, ROS/RNS can facilitate the cleavage of polypeptide backbones and expose hydrophobic residues that are highly aggregate prone (Chiti & Dobson, 2006; Stadtman & Levine, 2006). In addition, ROS/RNS modifications to amino acid sidechains, including oxidation, carbonylation and carbonmylation, decrease the activation energy required for crosslinking between proteins and increase the likelihood for ensuing polymerization into proteinaceous assemblies (Gorisse et al., 2016; Mirzaei & Regnier, 2008; Stadtman & Levine, 2006; Tanase et al., 2016). In addition to the increased generation of aggregate-prone polypeptides, aging decreases the efficacy of PQC (see below for detailed PQC description), leading to impaired clearance of aggregate-prone proteins (Klaips et al., 2018; Vilchez, Saez, & Dillin, 2014). As a result, the increased concentrations of ROS/RNS and aggregate-prone proteins in aging are capable of disrupting processes that are dependent on such factors, including the regulation of SGs.
Stress Granules (SGs)
SGs are membrane-less cytosolic compartments whose dynamics are increasingly relevant to the underlying mechanisms of ANPs (Benarroch, 2018; Gan, Cookson, Petrucelli, & La Spada, 2018). Under normal physiological conditions, transcripts are bound to RNA binding proteins (RBPs) in ribonucleoprotein (RNP) complexes, which function to translocate mRNA to sites of protein translation and modify protein synthesis (Kiebler & Bassell, 2006). Upon the detection of a stress factor, RNPs are disassembled, while mRNA-bound RBPs are rapidly reformed into SGs in order to sequester transcripts which might otherwise be degraded due to the molecular consequences of the stressor itself or the host cell’s non-specific defense mechanisms (Buchan & Parker, 2009; Protter & Parker, 2016). Although SG assembly has been observed in response to various types of stress factors, a physiologically relevant factor that can induce SG formation and is known to increase with age is elevated concentrations of ROS/RNS (Aulas, Lyons, Fay, Anderson, & Ivanov, 2018; Brown et al., 2011; J. G. Daigle et al., 2016; Emara et al., 2012). Subsequently, when stressful conditions are ameliorated, SG disassembly is facilitated by specific PQC machinery (Anderson & Kedersha, 2008; Kedersha & Anderson, 2002; Panas, Ivanov, & Anderson, 2016). However, SG dynamics can readily be disrupted, due in part to their distinctive molecular composition (i.e. RBPs) as well as the unique physical state in which they exist (i.e. Liquid Like Phase Separated; LLPS).
SGs are composed of RBPs, which are themselves characterized by intrinsically disorders regions (IDRs): repeated, specific orders of a limited number amino acids, with high concentrations of glycine and proline residues, as well as polar amino acids (e.g. asparagine, glutamine, and tyrosine) (Uversky, 2017; Uversky & Dunker, 2010). IDRs can allow proteins to engage in a multitude of multivalent, low affinity interactions with proximal polypeptides, that over time can crosslink and promote polymerization into higher order assemblies (Li et al., 2012; Lin, Protter, Rosen, & Parker, 2015; Nott et al., 2015). Thus, RBPs inherently maintain similar functional properties as those aggregate-prone proteins known to increase with age (see above). Notably, genomic interrogations indicate a compelling association between deleterious mutations in RBPs (e.g. TDP-43, FUS) and ensuing protein aggregation observed in ANPs (Conlon & Manley, 2017; Maziuk, Ballance, & Wolozin, 2017).
RBPs are also responsible for the unique physical state in which SGs exist in the cytoplasm, namely LLPS (Kato et al., 2012; Molliex et al., 2015). This state is characterized by a dynamic, fluid-like structure that, although initially homogenous, separates into two liquid phases that coexist: a slightly denser core encased by an outer shell that is in direct contact with surrounding cytosolic elements (Jain et al., 2016; Shin & Brangwynne, 2017). Given the capacity for aggregate-prone polypeptides to accumulate in SGs, (i.e. due in part to protein-protein interactions involving RBPs), in conjunction with the lack of membrane encapsulation that might otherwise inhibit such contacts, SGs can condense into persistent, dense foci (Jain et al., 2016; Mateju et al., 2017). SG persistence can then hinder disassembly processes by PQC mechanisms and contribute to further protein accumulation into aggregates that resemble those observed in ANPs (J. G. Daigle et al., 2016; Ganassi et al., 2016; Lechler et al., 2017; Patel et al., 2015).
Taken together, evidence suggests the chronic elevation of ROS/RNS in aging is capable of inducing SG formation, while the concurrent elevation of aggregate-prone proteins observed in aging is capable of facilitating their accumulation into SGs. Such accumulation (i.e. due in part to the unique composition and physical state of SGs) then inhibits SG disassembly by distinct PCQ components. In turn, persistent SG formation might serve as a “seed” for further protein aggregation common in ANPs (Figure 1). In support of this hypothesis, the persistent assembly of SGs, due to the optogenetically-induced dimerization of the RBP G3BP, has recently been shown to emulate pathological characteristics of ANPs (e.g. ALS, FTD), including the condensation into compartments that harbor amyloid oligomers (P. Zhang et al., 2019). Furthermore, aggregate-prone polypeptides and RBPs implicated in various ANPs are known to colocalize with SGs (Ash, Vanderweyde, Youmans, Apicco, & Wolozin, 2014; Liu-Yesucevitz et al., 2010; S. Waelter et al., 2001) (Table 1).
Figure 1.
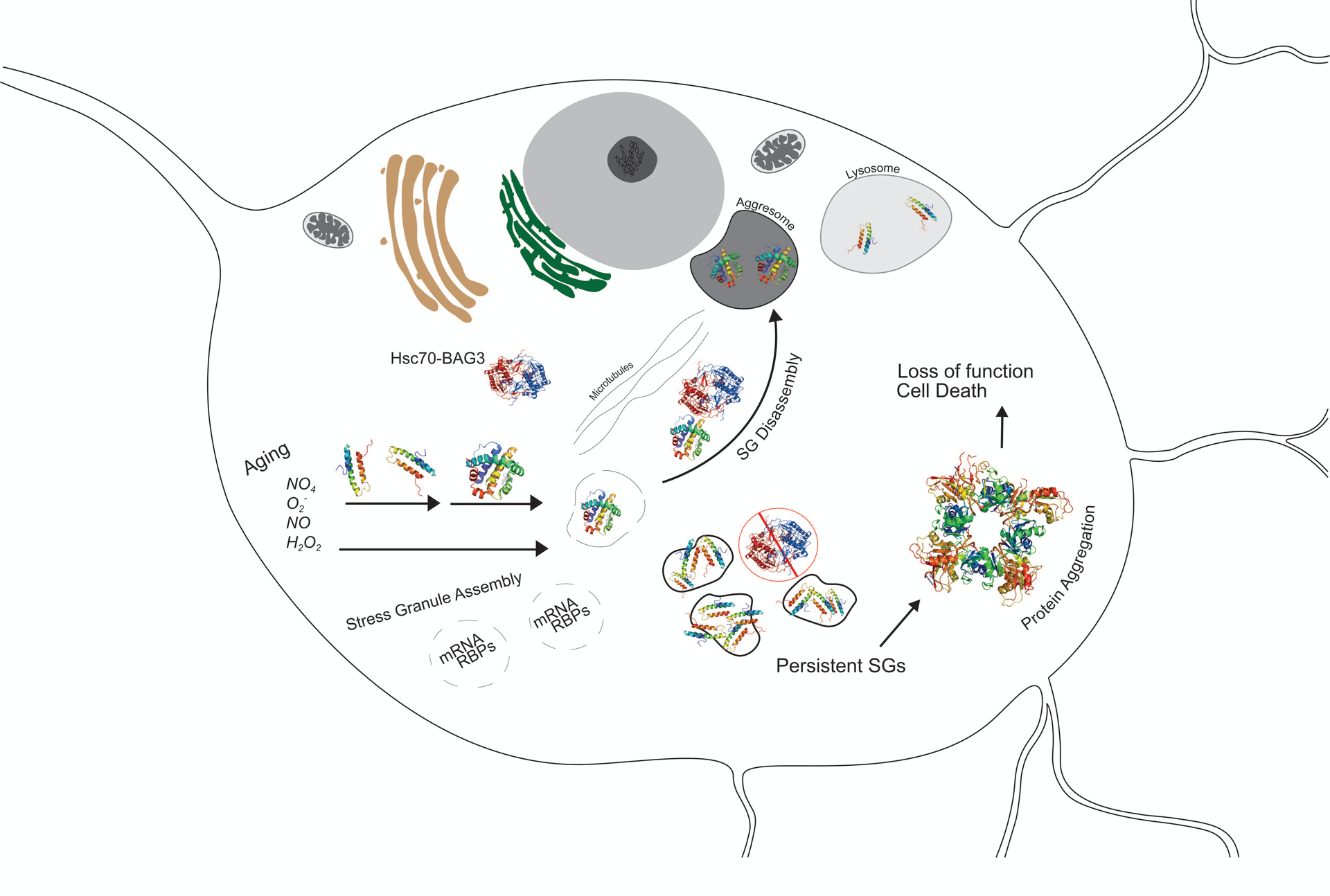
Proposed mechanism by which age-dependent SG dysregulation facilitates pathological aggregation of proteins. SGs are transient, cytosolic compartments that sequester mRNA upon cellular detection stress factors, such as ROS/RNS; upon the removal of a transient stressor, SG disassembly is facilitated by certain PQC mechanisms, specifically the Hsc70-BAG3 complex. Aging is associated with increased concentrations of ROS/RNS as well as aggregate-prone polypeptides; such conditions are capable of inducing SG formation and inhibiting SG disassembly, respectively. In attempts to maintain SG regulation, the Hsc70-BAG3 complex transports aggregate-prone polypeptides along microtubules to the aggresome for subsequent lysosomal degradation. The age-dependent tonic elevation in ROS/RNS and aggregate prone proteins, in conjunction with decreased Hsc70-BAG3 functional capacities in aging, might therefore facilitate the formation of persistent SGs. The prolonged formation of SGs could then serve as a “seed” for further protein accumulation, which leads to cytotoxic aggregates that perturb homeostasis, compromise cellular functioning, and precipitate cell death. SG, Stress Granules; ROS/RNS, Reactive Oxygen Species/Reactive Nitrogen Species; PQC, Protein Quality Control.
Table 1.
Evidence for SG colocalization with polypeptides implicated in ANPs
| Protein | Primary ANPs | Colocalization with SGs [Model (Marker)] | |
|---|---|---|---|
| Tau | AD, FTD | In vitro | HT22 (TIA, PABP) (Vanderweyde et al., 2016) |
| Primary Mouse hippocampal neurons (TIA) (Vanderweyde et al., 2016) | |||
| In vivo | Mice, frontal cortex (TIA, PABP) (Apicco et al., 2018) | ||
| Mice, fontal cortex (TIA) (Vanderweyde et al., 2016) | |||
| Mice, cortex, hippocampus (TIA, eIF3) (Vanderweyde et al., 2012) | |||
| Human, frontal cortex (TIA) (Vanderweyde et al., 2012) | |||
| FUS | ALS, FTD | In vitro | HeLa (TIA, TIAR, G3BP, PABP) (Dormann et al., 2010) |
| HeLa (TIA) (Bentmann et al., 2012) | |||
| Hek293 (TIAR) (Bosco et al., 2010) | |||
| Hek293 (TIA, PABP) (Gal et al., 2011) | |||
| Primary rat cortical neurons & CV1 fibroblasts (TIA, PABP) (Vance et al., 2013) | |||
| N2a (TIAR) (J. Gavin Daigle et al., 2012) | |||
| SH-SY5Y(TIAR, G3BP) (Shelkovnikova, Robinson, Connor-Robson, & Buchman, 2013) | |||
| iPSCs (TIAR) (Lenzi et al., 2015) | |||
| Primary Zebrafish cells (eIF3) (Acosta et al., 2014) | |||
| Fibroblasts (TIAR) (Lo Bello et al., 2017) | |||
| In vivo | Human, dentate gyrus (PABP) (Dormann et al., 2010) | ||
| Human, spinal cord (PABP) (Vance et al., 2013) | |||
| TDP43 | AD, FTD, ALS | In vitro | Hek293 & BeM17 (TIA, TIAR, eIF3, PABP) (Liu-Yesucevitz et al., 2010) |
| Hek293T (HuR, TIAR) (Dewey et al., 2011) | |||
| HeLa (TIA) (Bentmann et al., 2012) | |||
| HeLa (TIA) (McDonald et al., 2011) | |||
| HeLa (TIAR, HuR) (Parker et al., 2012) | |||
| Nsc34 (HuR, TIAR) (Colombrita et al., 2009) | |||
| In vivo | Human, frontal cortex (TIA, eIF3) (Liu-Yesucevitz et al., 2010) | ||
| PolyQ | HD | In vitro | Hek 293T (TIA) (Stephanie Waelter et al., 2001) |
| Repeat | N2a (TIA) (Furukawa, Kaneko, Matsumoto, Kurosawa, & Nukina, 2009) | ||
| In vivo | Mouse, hippocampus, cortex, striatum (TIA) (Furukawa et al., 2009) | ||
| C9orf72 | ALS, FTD | In vitro | HeLa (G3BP) (Boeynaems et al., 2017) |
| Repeat | HeLa (G3BP) (Lee et al., 2016) | ||
| HeLa & U2OS (eIF4) (Lee et al., 2016) | |||
| Hek293T (eIF3, TIA) (Y. J. Zhang et al., 2018) | |||
| In vivo | Mouse, cortex, hippocampus (TIA) (Chew et al., 2019) | ||
| Human, frontal cortex (eIF3) (Y. J. Zhang et al., 2018) | |||
SG-Relevant Protein Quality Control (PQC)
Given the abundant diversity in an organism’s proteome, complimentary degradation pathways are necessary to maintain efficient proteostasis. The specific PQC systems utilized to clear a given protein are dictated by its inherent properties (e.g. quaternary structure), in conjunction with an array of enzymes (e.g. ligases), transport complexes (e.g. chaperones) and disposal mechanisms. One facet, the ubiquitin-protease-system (UPS), is responsible for clearing short lived protein species, whereby ubiquitin-bound substrates are shuttled to the 26s proteasome via specific chaperone complexes (e.g. Hsc70-BAG1-CHIP), and undergo degradation following incorporation into catalytic barrels (Ciechanover, 2005; Ravid & Hochstrasser, 2008). Conversely, peptide assemblies not cleared by the proteasome and its subunits (i.e. 19S, 20S components), are degraded via autophagy; here, double membrane structures called autophagosomes sequester unwanted proteins and subsequently fuse with lysosomes to ensure catabolism of their contents (Glick, Barth, & Macleod, 2010). If a polypeptide contains certain recognition motifs (e.g. KREFQ), substrates can be directly transported (i.e. by Hsc70-BAG1) to the lysosome for degradation in a process referred to as chaperone-mediated autophagy (CMA) (Massey, Zhang, & Cuervo, 2006). A chaperone complex that mediates multiple clearance mechanisms, Hsc70-BAG3, is also emerging as a necessary regulator of SG dynamics (Ganassi et al., 2016; Kampinga & Craig, 2010; Kim, Hipp, Bracher, Hayer-Hartl, & Hartl, 2013).
In its ADP-bound conformation, Hsc70’s substrate binding domain (SBD) can recognize specific hydrophobic amino acid motifs, recruit complimentary PQC modulators, and mediate ensuing protein degradation (Hartl, Bracher, & Hayer-Hartl, 2011; Rüdiger, Germeroth, Schneider-Mergener, & Bukau, 1997). BAG3, a member of the BCL2-associated athanogene family, functions as a nuclear exchange factor (NEF) by hydrolyzing Hsc70-bound-ATP and thus ensuring efficient Hsc70 functioning (Rosati, Graziano, De Laurenzi, Pascale, & Turco, 2011). If a polypeptide is accompanied by autophagy adapters (e.g. p62/SQSTM1, NRB1), the Hsc70-BAG3 complex can facilitate clearance via autophagosome formation, regardless of the substrate’s ubiquitin moieties (Behl, 2016). In a more selective processes, recent findings also demonstrate Hsc70-BAG3’s capacity to regulate SGs by removing aggregate-prone peptides in a process generally referred to as the BAG-3 mediated selective macroautophagy pathway (Gamerdinger, Kaya, Wolfrum, Clement, & Behl, 2011; Ganassi et al., 2016).
With the assistance of co-chaperones and other HSPs (e.g. HSPB8), the Hsc70-BAG3 complex is recruited to SG loci upon the application of cellular stress and subsequently binds to polypeptides, particularly those that are aggregate-prone (Crippa et al., 2010; Ganassi et al., 2016). Utilizing BAG3’s proline rich domain, along with the 14–3–3 adapter protein, substrate bound complexes are then associated with the microtubule motor dynein (Gamerdinger et al., 2011; Tyedmers, Mogk, & Bukau, 2010; Xu et al., 2013). In conjunction with HDAC6, substrates are then subject to retrograde transport along cytoskeletal elements to perinuclear sites called aggresomes (Gamerdinger et al., 2009; Gamerdinger et al., 2011; Kawaguchi et al., 2003; Mateju et al., 2017). Although their precise mechanisms are not fully elucidated, such vimentin encased juxtanuclear compartments are thought to be storage locations for aggregate-prone peptides and stimulate subsequent degradation via autophagy (Kopito, 2000). By removing the aggregate-prone substrates that are recruited to SGs upon the induction of cellular stress, Hsc70-BAG3 machinery is necessary to prevent the condensation of SGs into insoluble, aberrant entities that can precipitate further protein aggregation (Ganassi et al., 2016; Mateju et al., 2017). Therefore, as aging conditions maintain the capacity to induce persistent SGs, in conjunction Hsc70-BAG3’s ability mitigate aberrant SGs that might otherwise facilitate protein accumulation, the functional augmentation of Hsc70-BAG3 in aging represents an attractive target for inhibiting mechanisms underlying ANPs.
The enhancement of Hsc70-BAG3 could be particularly effective in hindering the protein aggregation underlying many ANPs due to the fact that Hsc7-BAG3 functioning is particularly vulnerable in aging. For instance, mitochondrial functioning and oxidative phosphorylation are hindered with age, which inherently limits the ability of the ATP-dependent chaperones, including Hsc70-BAG3, to fulfill its functions (Beal, 2005; Brehme et al., 2014; Jain et al., 2016). Potentially due to the complimentary relationship between the UPS and autophagy, aged cells also maintain mitigated levels of BAG1 and elevated levels of BAG3, indicative of a compensatory yet disproportionate demand for Hsc70-BAG3 functioning in age (Behl, 2016; Gamerdinger et al., 2009). In addition, evidence gathered from preclinical and clinical samples illustrates the aged brain exhibits decreased levels of LC3, a protein that is necessary for phagophore elongation and ensuing autophagosome formation (Guebel & Torres, 2016; Kaushik et al., 2012; Yu et al., 2017). Similarly, both aged rodents as well as elderly individuals maintain decreased levels of Hsc70 in the CNS (Gleixner et al., 2014; Loeffler, Klaver, Coffey, Aasly, & LeWitt, 2016). Interestingly, clinical evidence also suggests the aged brain maintains decreased expression for HDA6, the adapter protein that ensures efficient transport of Hsc70-BAG3 bound substrates to the aggresome (Guebel & Torres, 2016). Thus, not only does aging induce conditions that are capable of hindering SG dynamics, but the capacity to maintain SG regulation and clear SG contents via Hsc70-BAG3 is concomitantly reduced.
Future investigations are encouraged to validate current evidence suggesting the benefits of enhanced Hsc70-BAG3 functioning, particularly with respect to those aggregate-prone polypeptides that are commonly found in aberrant SGs and are associated with ANPs. For instance, increasing Hsc70’s capacity to recognize aggregate-prone substrates, via the stabilization of its ADP-bound conformation, inhibits the formation insoluble cytosolic aggregates by promoting the clearance of α-synuclein as well as polyQ repeats (Roodveldt et al., 2009; Wang et al., 2013). In addition, recent evidence demonstrated adequate Hsc70 levels are necessary to ensure the clearance of persistent proteinaceous aggregates (Hjerpe et al., 2016). Consistent with such findings, BAG3 overexpression mitigates α-synuclein concentrations and increases its translocation to perinuclear compartments in conjunction with Hsc70 and p62/SQSTM1 (Y. L. Cao et al., 2017). Similarly, BAG3 overexpression results in significantly reduced levels of polyQ repeats, and is capable of significantly decreasing levels of hyperphosphorylated tau in primary neurons (Carra, Seguin, Lambert, & Landry, 2008; Lei, Brizzee, & Johnson, 2015). The validation of such results, along with promising investigations of accelerated aging disorders, would encourage the examination of SG-relevant PQC as a treatment option for ANPs.
Enhancing PQC: insights from HGPS
Often grouped under the nomenclature of progeria syndromes, accelerated aging disorders are characterized by their abnormal protein aggregation and rapidly induce cellular conditions commonly associated with normal aging (Ghosh & Zhou, 2014). One of the most well studied syndromes, Hutchinson–Gilford progeria (HGPS), is caused by a single, de novo C to T point mutation in the LMNA gene, which encodes the protein Lamin A (De Sandre-Giovannoli et al., 2003; Eriksson et al., 2003). As a result, a truncated form of prelamin A, termed progerin, inhibits the removal of its farnesylated C-terminal residue by ZMPSTE24/FACE-1 (Davies, Fong, Yang, Coffinier, & Young, 2009). Similar to the aggregate-prone proteins that can hinder SG disassembly and are common amongst ANPs, the electrostatic properties of progerin’s farnesylated moiety render it more capable of engaging in aberrant protein-protein interactions (Kalinowski et al., 2014). As a result, farnesylated progerin accumulates along the rim of the inner nuclear membrane, where it compromises the integrity of the nuclear envelope and renders the nucleus more susceptible to damaging conditions emanating from the cytosol (Goldman et al., 2004).
As exemplified by emerging data from HGPS research, the augmentation of PQC processes represent a reliable method for effectively inhibiting aberrant protein aggregation. Striking results from cultured HGPS cells demonstrate the direct enhancement of autophagy mechanisms, via the application of rapamycin and ensuing inhibition of mTOR1, significantly reduces concentrations of progerin (particularly its insoluble conformation), inhibits the accumulation of progerin puncta in the nucleoplasm, and significantly enhances cell viability (K. Cao et al., 2011). Similar results were obtained in a separate cohort of HGPS fibroblasts, where the induction of autophagy machinery (i.e. via rapamycin treatment) lead to significantly decreased progerin aggregation, and inhibited the deleterious consequences of progerin accumulation (i.e. damage to chromatin from a comprised nuclear envelope) (Cenni et al., 2011). Moreover, given the reliable mitigation of pathological mechanisms through the enhancement of PQC processes, Phase 2 clinical trials are currently underway at Boston Children’s Hospital in order to examine the efficacy of a direct mTOR1 inhibitor amongst a range of HGPS patients. Thus, provided that both ANPs and HGPS are fundamentally characterized by their abnormal aggregation of toxic protein species, further investigations are encouraged to determine if the targeting of PQC mechanisms in accelerated aging disorders might serve as a broader model in advancing treatment options for ANPs (Burtner & Kennedy, 2010).
Conclusion.
In the context of an unprecedented demand for effective ANP therapies due to shifting demographics as well as limited treatment options, researchers have gained a significantly greater understanding of the mechanisms which might underlie such conditions. The elucidation of SG dynamics has enabled investigators to demonstrate how the intracellular consequences of aging, namely the elevation in ROS/RNS and aggregate-prone polypeptides, have the potential to both induce SG formation and disrupt SG disassembly, respectively. In turn, the prolonged formation of these otherwise transient compartments allows persistent SGs to facilitate further polypeptide accumulation and potentially serve as a “seed” for subsequent protein aggregation. However, insights from accelerated aging syndromes, particularly HGPS, suggest the augmentation of particular PQC processes might serve as an effective therapeutic strategy to mitigate cytotoxic protein aggregation. Indeed, given their function in SG disassembly, along with their compromised capacities in aging, enhancing SG-relevant PQC mechanisms (i.e. Hsc70-BAG3) represent an attractive opportunity to combat ANPs. Thus, it behooves future investigators to determine if the modulation of such PQC components are capable of effectively ameliorating the pathological protein aggregation common in ANPs.
Although recent evidence has illuminated a number of crucial mechanisms, several confounds remain, including the limited external validity with respect to in vivo models. Whereas many current studies have utilized in vitro techniques and non-mammalian models to establish reliable causal precedence amongst aging conditions (e.g. ROS/RNS, aggregate-prone polypeptides), perturbed SG dynamics and ensuring protein aggregation, further examinations are necessary to determine if such relationships are maintained in animal models as well as clinical samples. Consistent with such a limitation, future investigations are also encouraged to establish the direct association between SG aberrations and behavioral outcomes across preclinical and clinical samples, provided that performance on behavioral diagnostic assessments are a common pathological manifestation in individuals living with ANPs. Such considerations will provide more accurate and reliable means for assessing if the augmentation of SG-relevant PQC is efficacious in combatting ANPs.
ACKNOWLEDGMENTS
The authors wish to thank past and present members of the Department of Neuroscience and Center for Neurovirolgy for their support, and sharing of ideas and reagents. We thank C. Papaleo for editorial assistance. This work was made possible by grant number R01AG060962 awarded by the NIH to JG and P30MH092177 awarded by NIH to KK. In addition, this investigation was supported by the National Institutes of Health under Ruth L. Kirchstein National Research Service Award (1T32MH079785) providing support to Michael Duggan.
Footnotes
CONFLICT OF INTERESTS
The authors have no financial conflicts of interest to declare.
REFERENCES
- Acosta JR, Goldsbury C, Winnick C, Badrock AP, Fraser ST, Laird AS, … Cole NJ (2014). Mutant human FUS Is ubiquitously mislocalized and generates persistent stress granules in primary cultured transgenic zebrafish cells. PLoS ONE, 9(6), e90572. doi: 10.1371/journal.pone.0090572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alberti S, Mateju D, Mediani L, & Carra S (2017). Granulostasis: Protein Quality Control of RNP Granules. Front Mol Neurosci, 10, 84. doi: 10.3389/fnmol.2017.00084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson P, & Kedersha N (2008). Stress granules: the Tao of RNA triage. Trends Biochem Sci, 33(3), 141–150. doi: 10.1016/j.tibs.2007.12.003 [DOI] [PubMed] [Google Scholar]
- Apicco DJ, Ash PEA, Maziuk B, LeBlang C, Medalla M, Al Abdullatif A, … Wolozin B (2018). Reducing the RNA binding protein TIA1 protects against tau-mediated neurodegeneration in vivo. Nature neuroscience, 21(1), 72–80. doi: 10.1038/s41593-017-0022-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ash PEA, Vanderweyde TE, Youmans KL, Apicco DJ, & Wolozin B (2014). Pathological Stress Granules in Alzheimer’s Disease. Brain Res, 1584, 52–58. doi: 10.1016/j.brainres.2014.05.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aulas A, Lyons SM, Fay MM, Anderson P, & Ivanov P (2018). Nitric oxide triggers the assembly of “type II” stress granules linked to decreased cell viability. Cell Death & Disease, 9(11), 1129. doi: 10.1038/s41419-018-1173-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balaban RS, Nemoto S, & Finkel T (2005). Mitochondria, oxidants, and aging. Cell, 120(4), 483–495. doi: 10.1016/j.cell.2005.02.001 [DOI] [PubMed] [Google Scholar]
- Beal MF (2005). Mitochondria take center stage in aging and neurodegeneration. Ann Neurol, 58(4), 495–505. doi: 10.1002/ana.20624 [DOI] [PubMed] [Google Scholar]
- Behl C (2016). Breaking BAG: The Co-Chaperone BAG3 in Health and Disease. Trends in Pharmacological Sciences, 37(8), 672–688. doi: 10.1016/j.tips.2016.04.007 [DOI] [PubMed] [Google Scholar]
- Benarroch EE (2018). Cytoplasmic RNA granules, ribostasis, and neurodegeneration. Neurology. doi: 10.1212/WNL.0000000000005172 [DOI] [PubMed] [Google Scholar]
- Bentmann E, Neumann M, Tahirovic S, Rodde R, Dormann D, & Haass C (2012). Requirements for stress granule recruitment of fused in sarcoma (FUS) and TAR DNA-binding protein of 43 kDa (TDP-43). J Biol Chem, 287(27), 23079–23094. doi: 10.1074/jbc.M111.328757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeynaems S, Bogaert E, Kovacs D, Konijnenberg A, Timmerman E, Volkov A, … Van Den Bosch L (2017). Phase Separation of C9orf72 Dipeptide Repeats Perturbs Stress Granule Dynamics. Mol Cell, 65(6), 1044–1055 e1045. doi: 10.1016/j.molcel.2017.02.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosco DA, Lemay N, Ko HK, Zhou H, Burke C, Kwiatkowski TJ Jr., … Hayward LJ (2010). Mutant FUS proteins that cause amyotrophic lateral sclerosis incorporate into stress granules. Hum Mol Genet, 19(21), 4160–4175. doi: 10.1093/hmg/ddq335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brehme M, Voisine C, Rolland T, Wachi S, Soper JH, Zhu Y, … Morimoto RI (2014). A chaperome subnetwork safeguards proteostasis in aging and neurodegenerative disease. Cell Rep, 9(3), 1135–1150. doi: 10.1016/j.celrep.2014.09.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JA, Roberts TL, Richards R, Woods R, Birrell G, Lim YC, … Lavin MF (2011). A novel role for hSMG-1 in stress granule formation. Mol Cell Biol, 31(22), 4417–4429. doi: 10.1128/mcb.05987-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brundin P, Melki R, & Kopito R (2010). Prion-like transmission of protein aggregates in neurodegenerative diseases. Nat Rev Mol Cell Biol, 11(4), 301–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchan J, & Parker R (2009). Eukaryotic stress granules: the ins and outs of translation. Mol Cell, 36(6), 932–941. doi: 10.1016/j.molcel.2009.11.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burtner CR, & Kennedy BK (2010). Progeria syndromes and ageing: what is the connection? Nat Rev Mol Cell Biol, 11(8), 567–578. doi: 10.1038/nrm2944 [DOI] [PubMed] [Google Scholar]
- Cao K, Graziotto JJ, Blair CD, Mazzulli JR, Erdos MR, Krainc D, & Collins FS (2011). Rapamycin reverses cellular phenotypes and enhances mutant protein clearance in Hutchinson-Gilford progeria syndrome cells. Sci Transl Med, 3(89), 89ra58. doi: 10.1126/scitranslmed.3002346 [DOI] [PubMed] [Google Scholar]
- Cao YL, Yang YP, Mao CJ, Zhang XQ, Wang CT, Yang J, … Liu CF (2017). A role of BAG3 in regulating SNCA/alpha-synuclein clearance via selective macroautophagy. Neurobiol Aging, 60, 104–115. doi: 10.1016/j.neurobiolaging.2017.08.023 [DOI] [PubMed] [Google Scholar]
- Carra S, Seguin SJ, Lambert H, & Landry J (2008). HspB8 chaperone activity toward poly(Q)-containing proteins depends on its association with Bag3, a stimulator of macroautophagy. J Biol Chem, 283(3), 1437–1444. doi: 10.1074/jbc.M706304200 [DOI] [PubMed] [Google Scholar]
- Cenni V, Capanni C, Columbaro M, Ortolani M, D’Apice MR, Novelli G, … Lattanzi G (2011). Autophagic degradation of farnesylated prelamin A as a therapeutic approach to lamin-linked progeria. Eur J Histochem, 55(4), e36. doi: 10.4081/ejh.2011.e36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chew J, Cook C, Gendron TF, Jansen-West K, Del Rosso G, Daughrity LM, … Petrucelli L (2019). Aberrant deposition of stress granule-resident proteins linked to C9orf72-associated TDP-43 proteinopathy. Mol Neurodegener, 14(1), 9. doi: 10.1186/s13024-019-0310-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiti F, & Dobson CM (2006). Protein Misfolding, Functional Amyloid, and Human Disease. Annual Review of Biochemistry, 75(1), 333–366. doi: 10.1146/annurev.biochem.75.101304.123901 [DOI] [PubMed] [Google Scholar]
- Ciechanover A (2005). Proteolysis: from the lysosome to ubiquitin and the proteasome. Nat Rev Mol Cell Biol, 6(1), 79–87. doi: 10.1038/nrm1552 [DOI] [PubMed] [Google Scholar]
- Ciechanover A, & Brundin P (2003). The Ubiquitin Proteasome System in Neurodegenerative Diseases: Sometimes the Chicken, Sometimes the Egg. Neuron, 40(2), 427–446. doi: 10.1016/S0896-6273(03)00606-8 [DOI] [PubMed] [Google Scholar]
- Ciryam P, Kundra R, Morimoto RI, Dobson CM, & Vendruscolo M (2015). Supersaturation is a major driving force for protein aggregation in neurodegenerative diseases. Trends Pharmacol Sci, 36(2), 72–77. doi: 10.1016/j.tips.2014.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciryam P, Tartaglia G, Morimoto RI, Dobson CM, & Vendruscolo M (2013). Widespread Aggregation and Neurodegenerative Diseases Are Associated with Supersaturated Proteins. Cell Rep, 5(3), 781–790. doi: 10.1016/j.celrep.2013.09.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombrita C, Zennaro E, Fallini C, Weber M, Sommacal A, Buratti E, … Ratti A (2009). TDP-43 is recruited to stress granules in conditions of oxidative insult. J Neurochem, 111(4), 1051–1061. doi: 10.1111/j.1471-4159.2009.06383.x [DOI] [PubMed] [Google Scholar]
- Conlon EG, & Manley JL (2017). RNA-binding proteins in neurodegeneration: mechanisms in aggregate. Genes Dev, 31(15), 1509–1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crippa V, Sau D, Rusmini P, Boncoraglio A, Onesto E, Bolzoni E, … Poletti A (2010). The small heat shock protein B8 (HspB8) promotes autophagic removal of misfolded proteins involved in amyotrophic lateral sclerosis (ALS). Hum Mol Genet, 19(17), 3440–3456. doi: 10.1093/hmg/ddq257 [DOI] [PubMed] [Google Scholar]
- Cummings JL, Morstorf T, & Zhong K (2014). Alzheimer’s disease drug-development pipeline: few candidates, frequent failures. Alzheimers Res Ther, 6(4), 37. doi: 10.1186/alzrt269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlmann B (2007). Role of proteasomes in disease. BMC biochemistry, 8 Suppl 1(Suppl 1), S3–S3. doi: 10.1186/1471-2091-8-S1-S3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daigle JG, Krishnamurthy K, Ramesh N, Casci I, Monaghan J, McAvoy K, … Pandey UB (2016). Pur-alpha regulates cytoplasmic stress granule dynamics and ameliorates FUS toxicity. Acta Neuropathol, 131(4), 605–620. doi: 10.1007/s00401-015-1530-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daigle JG, Lanson NA Jr, Smith RB, Casci I, Maltare A, Monaghan J, … Pandey UB (2012). RNA-binding ability of FUS regulates neurodegeneration, cytoplasmic mislocalization and incorporation into stress granules associated with FUS carrying ALS-linked mutations. Hum Mol Genet, 22(6), 1193–1205. doi: 10.1093/hmg/dds526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- David D (2012). Aging and the aggregating proteome. Frontiers in Genetics, 3(247). doi: 10.3389/fgene.2012.00247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- David DC, Ollikainen N, Trinidad JC, Cary MP, Burlingame AL, & Kenyon C (2010). Widespread Protein Aggregation as an Inherent Part of Aging in C. elegans. PLoS biology, 8(8). doi: 10.1371/journal.pbio.1000450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies BS, Fong LG, Yang SH, Coffinier C, & Young SG (2009). The posttranslational processing of prelamin A and disease. Annu Rev Genomics Hum Genet, 10, 153–174. doi: 10.1146/annurev-genom-082908-150150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Sandre-Giovannoli A, Bernard R, Cau P, Navarro C, Amiel J, Boccaccio I, … Levy N (2003). Lamin a truncation in Hutchinson-Gilford progeria. Science, 300(5628), 2055. doi: 10.1126/science.1084125 [DOI] [PubMed] [Google Scholar]
- Dewey CM, Cenik B, Sephton CF, Dries DR, Mayer P 3rd, Good SK, … Yu G (2011). TDP-43 is directed to stress granules by sorbitol, a novel physiological osmotic and oxidative stressor. Mol Cell Biol, 31(5), 1098–1108. doi: 10.1128/MCB.01279-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dormann D, Rodde R, Edbauer D, Bentmann E, Fischer I, Hruscha A, … Haass C (2010). ALS-associated fused in sarcoma (FUS) mutations disrupt Transportin-mediated nuclear import. Embo j, 29(16), 2841–2857. doi: 10.1038/emboj.2010.143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emara MM, Fujimura K, Sciaranghella D, Ivanova V, Ivanov P, & Anderson P (2012). Hydrogen peroxide induces stress granule formation independent of eIF2alpha phosphorylation. Biochem Biophys Res Commun, 423(4), 763–769. doi: 10.1016/j.bbrc.2012.06.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksson M, Brown WT, Gordon LB, Glynn MW, Singer J, Scott L, … Collins FS (2003). Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature, 423(6937), 293–298. doi: 10.1038/nature01629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fink AL (1998). Protein aggregation: folding aggregates, inclusion bodies and amyloid. Folding and Design, 3(1), R9–R23. doi: 10.1016/S1359-0278(98)00002-9 [DOI] [PubMed] [Google Scholar]
- Finkel T, & Holbrook NJ (2000). Oxidants, oxidative stress and the biology of ageing. Nature, 408(6809), 239–247. doi: 10.1038/35041687 [DOI] [PubMed] [Google Scholar]
- Furukawa Y, Kaneko K, Matsumoto G, Kurosawa M, & Nukina N (2009). Cross-Seeding Fibrillation of Q/N-Rich Proteins Offers New Pathomechanism of Polyglutamine Diseases. The Journal of Neuroscience, 29(16), 5153. doi: 10.1523/JNEUROSCI.0783-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gal J, Zhang J, Kwinter DM, Zhai J, Jia H, Jia J, & Zhu H (2011). Nuclear localization sequence of FUS and induction of stress granules by ALS mutants. Neurobiol Aging, 32(12), 2323.e2327–2323.e2340. doi: 10.1016/j.neurobiolaging.2010.06.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamerdinger M, Hajieva P, Kaya AM, Wolfrum U, Hartl FU, & Behl C (2009). Protein quality control during aging involves recruitment of the macroautophagy pathway by BAG3. Embo j, 28(7), 889–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamerdinger M, Kaya AM, Wolfrum U, Clement AM, & Behl C (2011). BAG3 mediates chaperone‐based aggresome-targeting and selective autophagy of misfolded proteins. EMBO reports, 12(2), 149–156. doi: 10.1038/embor.2010.203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan L, Cookson MR, Petrucelli L, & La Spada AR (2018). Converging pathways in neurodegeneration, from genetics to mechanisms. Nature neuroscience, 21(10), 1300–1309. doi: 10.1038/s41593-018-0237-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganassi M, Mateju D, Bigi I, Mediani L, Poser I, Lee HO, … Carra S (2016). A Surveillance Function of the HSPB8-BAG3-HSP70 Chaperone Complex Ensures Stress Granule Integrity and Dynamism. Mol Cell, 63(5), 796–810. doi: 10.1016/j.molcel.2016.07.021 [DOI] [PubMed] [Google Scholar]
- Ghosh S, & Zhou Z (2014). Genetics of aging, progeria and lamin disorders. Current Opinion in Genetics & Development, 26, 41–46. doi: 10.1016/j.gde.2014.05.003 [DOI] [PubMed] [Google Scholar]
- Gleixner A, Pulugulla S, Pant D, Posimo J, Crum T, & Leak R (2014). Impact of aging on heat shock protein expression in the substantia nigra and striatum of the female rat. Cell and tissue research, 357(1), 43–54. [DOI] [PubMed] [Google Scholar]
- Glick D, Barth S, & Macleod KF (2010). Autophagy: cellular and molecular mechanisms. The Journal of pathology, 221(1), 3–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman RD, Shumaker DK, Erdos MR, Eriksson M, Goldman AE, Gordon LB, … Collins FS (2004). Accumulation of mutant lamin A causes progressive changes in nuclear architecture in Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci U S A, 101(24), 8963–8968. doi: 10.1073/pnas.0402943101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorisse L, Pietrement C, Vuiblet V, Schmelzer CE, Köhler M, Duca L, … Gillery P (2016). Protein carbamylation is a hallmark of aging. Proc Natl Acad Sci U S A, 113(5), 1191–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guebel DV, & Torres NV (2016). Sexual dimorphism and aging in the human hyppocampus: identification, validation, and impact of differentially expressed genes by factorial microarray and network analysis. Frontiers in Aging Neuroscience, 8, 229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halliwell B (2006). Oxidative stress and neurodegeneration: where are we now? J Neurochem, 97(6), 1634–1658. doi: 10.1111/j.1471-4159.2006.03907.x [DOI] [PubMed] [Google Scholar]
- Hartl FU, Bracher A, & Hayer-Hartl M (2011). Molecular chaperones in protein folding and proteostasis. Nature, 475, 324. doi: 10.1038/nature10317 [DOI] [PubMed] [Google Scholar]
- Hatami A, Monjazeb S, Milton S, & Glabe CG (2017). Familial Alzheimer’s Disease Mutations within the Amyloid Precursor Protein Alter the Aggregation and Conformation of the Amyloid-beta Peptide. J Biol Chem, 292(8), 3172–3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hjerpe R, Bett JS, Keuss MJ, Solovyova A, McWilliams TG, Johnson C, … Kurz T (2016). UBQLN2 Mediates Autophagy-Independent Protein Aggregate Clearance by the Proteasome. Cell, 166(4), 935–949. doi: 10.1016/j.cell.2016.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohn A, Konig J, & Grune T (2013). Protein oxidation in aging and the removal of oxidized proteins. J Proteomics, 92, 132–159. doi: 10.1016/j.jprot.2013.01.004 [DOI] [PubMed] [Google Scholar]
- Huang C, Wagner-Valladolid S, Stephens AD, Jung R, Poudel C, Sinnige T, … David DC (2018). Intrinsically aggregation-prone proteins form amyloid-like aggregates and contribute to tissue aging in C. elegans. bioRxiv [DOI] [PMC free article] [PubMed] [Google Scholar]
- Invernizzi G, Papaleo E, Sabate R, & Ventura S (2012). Protein aggregation: Mechanisms and functional consequences. The International Journal of Biochemistry & Cell Biology, 44(9), 1541–1554. doi: 10.1016/j.biocel.2012.05.023 [DOI] [PubMed] [Google Scholar]
- Jain S, Wheeler JR, Walters RW, Agrawal A, Barsic A, & Parker R (2016). ATPase-modulated stress granules contain a diverse proteome and substructure. Cell, 164(3), 487–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalinowski A, Yaron PN, Qin Z, Shenoy S, Buehler MJ, Losche M, & Dahl KN (2014). Interfacial binding and aggregation of lamin A tail domains associated with Hutchinson-Gilford progeria syndrome. Biophys Chem, 195, 43–48. doi: 10.1016/j.bpc.2014.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kampinga HH, & Craig EA (2010). The HSP70 chaperone machinery: J proteins as drivers of functional specificity. Nat Rev Mol Cell Biol, 11(8), 579–592. doi: 10.1038/nrm2941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato M, Han TW, Xie S, Shi K, Du X, Wu LC, … McKnight SL (2012). Cell-free formation of RNA granules: low complexity sequence domains form dynamic fibers within hydrogels. Cell, 149(4), 753–767. doi: 10.1016/j.cell.2012.04.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaushik S, Arias E, Kwon H, Lopez NM, Athonvarangkul D, Sahu S, … Singh R (2012). Loss of autophagy in hypothalamic POMC neurons impairs lipolysis. EMBO reports, 13(3), 258–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi Y, Kovacs JJ, McLaurin A, Vance JM, Ito A, & Yao T-P (2003). The Deacetylase HDAC6 Regulates Aggresome Formation and Cell Viability in Response to Misfolded Protein Stress. Cell, 115(6), 727–738. doi: 10.1016/S0092-8674(03)00939-5 [DOI] [PubMed] [Google Scholar]
- Kedersha N, & Anderson P (2002). Stress granules: sites of mRNA triage that regulate mRNA stability and translatability. Biochem Soc Trans, 30(Pt 6), 963–969. doi:10.1042// [DOI] [PubMed] [Google Scholar]
- Kiebler MA, & Bassell GJ (2006). Neuronal RNA granules: movers and makers. Neuron, 51(6), 685–690. doi: 10.1016/j.neuron.2006.08.021 [DOI] [PubMed] [Google Scholar]
- Kim YE, Hipp MS, Bracher A, Hayer-Hartl M, & Hartl FU (2013). Molecular Chaperone Functions in Protein Folding and Proteostasis In Kornberg RD (Ed.), Annual Review of Biochemistry, Vol 82 (Vol. 82, pp. 323–355). Palo Alto: Annual Reviews. [DOI] [PubMed] [Google Scholar]
- Klaips CL, Jayaraj GG, & Hartl FU (2018). Pathways of cellular proteostasis in aging and disease. J Cell Biol, 217, 51–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopito RR (2000). Aggresomes, inclusion bodies and protein aggregation. Trends in cell biology, 10(12), 524–530. doi: 10.1016/S0962-8924(00)01852-3 [DOI] [PubMed] [Google Scholar]
- Kurtishi A, Rosen B, Patil KS, Alves GW, & Moller SG (2018). Cellular Proteostasis in Neurodegeneration. Mol Neurobiol. doi: 10.1007/s12035-018-1334-z [DOI] [PubMed] [Google Scholar]
- Lechler MC, Crawford ED, Groh N, Widmaier K, Jung R, Kirstein J, … David DC (2017). Reduced Insulin/IGF-1 Signaling Restores the Dynamic Properties of Key Stress Granule Proteins during Aging. Cell Rep, 18(2), 454–467. doi: 10.1016/j.celrep.2016.12.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KH, Zhang P, Kim HJ, Mitrea DM, Sarkar M, Freibaum BD, … Taylor JP (2016). C9orf72 Dipeptide Repeats Impair the Assembly, Dynamics, and Function of Membrane-Less Organelles. Cell, 167(3), 774–788 e717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei Z, Brizzee C, & Johnson GVW (2015). BAG3 facilitates the clearance of endogenous tau in primary neurons. Neurobiol Aging, 36(1), 241–248. doi: 10.1016/j.neurobiolaging.2014.08.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenzi J, De Santis R, de Turris V, Morlando M, Laneve P, Calvo A, … Bozzoni I (2015). ALS mutant FUS proteins are recruited into stress granules in induced pluripotent stem cell-derived motoneurons. Dis Model Mech, 8(7), 755–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine B, & Kroemer G (2019). Biological Functions of Autophagy Genes: A Disease Perspective. Cell, 176(1–2), 11–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Banjade S, Cheng H-C, Kim S, Chen B, Guo L, … Rosen MK (2012). Phase transitions in the assembly of multivalent signalling proteins. Nature, 483, 336. doi: 10.1038/nature10879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y, Protter DS, Rosen MK, & Parker R (2015). Formation and Maturation of Phase-Separated Liquid Droplets by RNA-Binding Proteins. Mol Cell, 60(2), 208–219. doi: 10.1016/j.molcel.2015.08.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu-Yesucevitz L, Bilgutay A, Zhang YJ, Vanderweyde T, Citro A, Mehta T, … Wolozin B (2010). Tar DNA binding protein-43 (TDP-43) associates with stress granules: analysis of cultured cells and pathological brain tissue. PLoS ONE, 5(10), e13250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo Bello M, Di Fini F, Notaro A, Spataro R, Conforti FL, & La Bella V (2017). ALS-Related Mutant FUS Protein Is Mislocalized to Cytoplasm and Is Recruited into Stress Granules of Fibroblasts from Asymptomatic FUS P525L Mutation Carriers. Neurodegener Dis, 17(6), 292–303. doi: 10.1159/000480085 [DOI] [PubMed] [Google Scholar]
- Loeffler DA, Klaver AC, Coffey MP, Aasly JO, & LeWitt PA (2016). Age-related decrease in heat shock 70-kDa protein 8 in cerebrospinal fluid is associated with increased oxidative stress. Frontiers in Aging Neuroscience, 8, 178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Otin C, Blasco MA, Partridge L, Serrano M, & Kroemer G (2013). The Hallmarks of Aging. Cell, 153(6), 1194–1217. doi: 10.1016/j.cell.2013.05.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macario AJ, Grippo TM, & Conway de Macario E (2005). Genetic disorders involving molecular-chaperone genes: a perspective. Genet Med, 7(1), 3–12. doi:10.109701.Gim.0000151351.11876.C3 [DOI] [PubMed] [Google Scholar]
- Mangialasche F, Solomon A, Winblad B, Mecocci P, & Kivipelto M (2010). Alzheimer’s disease: clinical trials and drug development. Lancet neurology, 9(7), 702–716. doi: 10.1016/s1474-4422(10)70119-8 [DOI] [PubMed] [Google Scholar]
- Massey AC, Zhang C, & Cuervo AM (2006). Chaperone-mediated autophagy in aging and disease. Current topics in developmental biology, 73, 205–235. doi: 10.1016/S0070-2153(05)73007-6 [DOI] [PubMed] [Google Scholar]
- Mateju D, Franzmann TM, Patel A, Kopach A, Boczek EE, Maharana S, … Alberti S (2017). An aberrant phase transition of stress granules triggered by misfolded protein and prevented by chaperone function. Embo j, 36(12), 1669–1687. doi: 10.15252/embj.201695957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maziuk B, Ballance HI, & Wolozin B (2017). Dysregulation of RNA Binding Protein Aggregation in Neurodegenerative Disorders. Front Mol Neurosci, 10, 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald KK, Aulas A, Destroismaisons L, Pickles S, Beleac E, Camu W, … Vande Velde C (2011). TAR DNA-binding protein 43 (TDP-43) regulates stress granule dynamics via differential regulation of G3BP and TIA-1. Hum Mol Genet, 20(7), 1400–1410. doi: 10.1093/hmg/ddr021 [DOI] [PubMed] [Google Scholar]
- Mirzaei H, & Regnier F (2008). Protein:protein aggregation induced by protein oxidation. J Chromatogr B Analyt Technol Biomed Life Sci, 873(1), 8–14. doi: 10.1016/j.jchromb.2008.04.025 [DOI] [PubMed] [Google Scholar]
- Molliex A, Temirov J, Lee J, Coughlin M, Kanagaraj AP, Kim HJ, … Taylor JP (2015). Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell, 163(1), 123–133. doi: 10.1016/j.cell.2015.09.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy Michael P., Holmgren A, Larsson N-G, Halliwell B, Chang Christopher J., Kalyanaraman B, … Winterbourn Christine C. (2011). Unraveling the Biological Roles of Reactive Oxygen Species. Cell Metabolism, 13(4), 361–366. doi: 10.1016/j.cmet.2011.03.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nott TJ, Petsalaki E, Farber P, Jervis D, Fussner E, Plochowietz A, … Baldwin AJ (2015). Phase transition of a disordered nuage protein generates environmentally responsive membraneless organelles. Mol Cell, 57(5), 936–947. doi: 10.1016/j.molcel.2015.01.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Organization WH (2006). Neurological disorders: public health challenges: World Health Organization.
- Organization WH (2015). World report on ageing and health: World Health Organization.
- Panas MD, Ivanov P, & Anderson P (2016). Mechanistic insights into mammalian stress granule dynamics. J Cell Biol, 215(3), 313–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker SJ, Meyerowitz J, James JL, Liddell JR, Crouch PJ, Kanninen KM, & White AR (2012). Endogenous TDP-43 localized to stress granules can subsequently form protein aggregates. Neurochemistry International, 60(4), 415–424. doi: 10.1016/j.neuint.2012.01.019 [DOI] [PubMed] [Google Scholar]
- Patel A, Lee HO, Jawerth L, Maharana S, Jahnel M, Hein MY, … Alberti S (2015). A Liquid-to-Solid Phase Transition of the ALS Protein FUS Accelerated by Disease Mutation. Cell, 162(5), 1066–1077. doi: 10.1016/j.cell.2015.07.047 [DOI] [PubMed] [Google Scholar]
- Pérez VI, Buffenstein R, Masamsetti V, Leonard S, Salmon AB, Mele J, … Chaudhuri A (2009). Protein stability and resistance to oxidative stress are determinants of longevity in the longest-living rodent, the naked mole-rat. Proceedings of the National Academy of Sciences, 106, 3059–3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Protter DS, & Parker R (2016). Principles and properties of stress granules. Trends in cell biology, 26(9), 668–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravid T, & Hochstrasser M (2008). Diversity of degradation signals in the ubiquitin-proteasome system. Nat Rev Mol Cell Biol, 9(9), 679–690. doi: 10.1038/nrm2468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichmann D, Voth W, & Jakob U (2018). Maintaining a Healthy Proteome during Oxidative Stress. Mol Cell, 69(2), 203–213. doi: 10.1016/j.molcel.2017.12.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roodveldt C, Bertoncini CW, Andersson A, van der Goot AT, Hsu ST, Fernandez-Montesinos R, … Dobson CM (2009). Chaperone proteostasis in Parkinson’s disease: stabilization of the Hsp70/alpha-synuclein complex by Hip. Embo j, 28(23), 3758–3770. doi: 10.1038/emboj.2009.298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosati A, Graziano V, De Laurenzi V, Pascale M, & Turco MC (2011). BAG3: a multifaceted protein that regulates major cell pathways. Cell Death &Amp; Disease, 2, e141. doi: 10.1038/cddis.2011.24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross CA, & Poirier MA (2004). Protein aggregation and neurodegenerative disease. Nat Med, 10(7), S10–S17. doi: 10.1038/nm1066 [DOI] [PubMed] [Google Scholar]
- Rubinsztein David C., Mariño, G., & Kroemer, G. (2011). Autophagy and Aging. Cell, 146(5), 682–695. doi: 10.1016/j.cell.2011.07.030 [DOI] [PubMed] [Google Scholar]
- Rüdiger S, Germeroth L, Schneider-Mergener J, & Bukau B (1997). Substrate specificity of the DnaK chaperone determined by screening cellulose-bound peptide libraries. Embo j, 16(7), 1501–1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos AL, & Lindner AB (2017). Protein Posttranslational Modifications: Roles in Aging and Age-Related Disease. Oxid Med Cell Longev, 2017, 5716409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelkovnikova TA, Robinson HK, Connor-Robson N, & Buchman VL (2013). Recruitment into stress granules prevents irreversible aggregation of FUS protein mislocalized to the cytoplasm. Cell Cycle, 12(19), 3194–3202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin Y, & Brangwynne CP (2017). Liquid phase condensation in cell physiology and disease. Science, 357(6357). doi: 10.1126/science.aaf4382 [DOI] [PubMed] [Google Scholar]
- Stadtman ER, & Levine RL (2006). Chemical Modification of Proteins by Reactive Oxygen Species. Redox Proteomics. doi:doi: 10.1002/0471973122.ch1 [DOI] [Google Scholar]
- Stefani M, & Dobson CM (2003). Protein aggregation and aggregate toxicity: new insights into protein folding, misfolding diseases and biological evolution. Journal of Molecular Medicine, 81(11), 678–699. doi: 10.1007/s00109-003-0464-5 [DOI] [PubMed] [Google Scholar]
- Tanase M, Urbanska AM, Zolla V, Clement CC, Huang L, Morozova K, … Santambrogio L (2016). Role of Carbonyl Modifications on Aging-Associated Protein Aggregation. Scientific reports, 6, 19311. doi: 10.1038/srep19311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor JP, Hardy J, & Fischbeck KH (2002). Biomedicine - Toxic proteins in neurodegenerative disease. Science, 296(5575), 1991–1995. doi: 10.1126/science.1067122 [DOI] [PubMed] [Google Scholar]
- Taylor RC, & Dillin A (2011). Aging as an event of proteostasis collapse. Cold Spring Harb Perspect Biol, 3(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treaster SB, Ridgway ID, Richardson CA, Gaspar MB, Chaudhuri AR, & Austad SN (2014). Superior proteome stability in the longest lived animal. Age (Dordr), 36(3), 9597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyedmers J, Mogk A, & Bukau B (2010). Cellular strategies for controlling protein aggregation. Nat Rev Mol Cell Biol, 11(11), 777–788. doi: 10.1038/nrm2993 [DOI] [PubMed] [Google Scholar]
- Uversky VN (2017). The roles of intrinsic disorder-based liquid-liquid phase transitions in the “Dr. Jekyll-Mr. Hyde” behavior of proteins involved in amyotrophic lateral sclerosis and frontotemporal lobar degeneration. Autophagy, 13(12), 2115–2162. doi: 10.1080/15548627.2017.1384889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uversky VN, & Dunker AK (2010). Understanding protein non-folding. Biochim Biophys Acta, 1804(6), 1231–1264. doi: 10.1016/j.bbapap.2010.01.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vance C, Scotter EL, Nishimura AL, Troakes C, Mitchell JC, Kathe C, … Shaw CE (2013). ALS mutant FUS disrupts nuclear localization and sequesters wild-type FUS within cytoplasmic stress granules. Hum Mol Genet, 22(13), 2676–2688. doi: 10.1093/hmg/ddt117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderweyde T, Apicco DJ, Youmans-Kidder K, Ash PEA, Cook C, Lummertz da Rocha E, … Wolozin B (2016). Interaction of tau with the RNA-Binding Protein TIA1 Regulates tau Pathophysiology and Toxicity. Cell Rep, 15(7), 1455–1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderweyde T, Yu H, Varnum M, Liu-Yesucevitz L, Citro A, Ikezu T, … Wolozin B (2012). Contrasting pathology of the stress granule proteins TIA-1 and G3BP in tauopathies. Journal of Neuroscience, 32(24), 8270–8283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilchez D, Saez I, & Dillin A (2014). The role of protein clearance mechanisms in organismal ageing and age-related diseases. Nature Communications, 5, 5659. doi: 10.1038/ncomms6659 [DOI] [PubMed] [Google Scholar]
- von Bernhardi R, Eugenín-von Bernhardi L, & Eugenín J (2015). Microglial cell dysregulation in brain aging and neurodegeneration. Front Aging Neurosci, 7. doi: 10.3389/fnagi.2015.00124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waelter S, Boeddrich A, Lurz R, Scherzinger E, Lueder G, Lehrach H, & Wanker EE (2001). Accumulation of mutant huntingtin fragments in aggresome-like inclusion bodies as a result of insufficient protein degradation. Mol Biol Cell, 12(5), 1393–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker LC, Diamond MI, Duff KE, & Hyman BT (2013). Mechanisms of protein seeding in neurodegenerative diseases. JAMA Neurol, 70(3), 304–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang AM, Miyata Y, Klinedinst S, Peng HM, Chua JP, Komiyama T, … Lieberman AP (2013). Activation of Hsp70 reduces neurotoxicity by promoting polyglutamine protein degradation. Nat Chem Biol, 9(2), 112–118. doi: 10.1038/nchembio.1140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z, Graham K, Foote M, Liang F, Rizkallah R, Hurt M, … Zhou Y (2013). 14–3–3 protein targets misfolded chaperone-associated proteins to aggresomes. J Cell Sci, 126(18), 4173–4186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Feng L, Li J, Lan X, Lixiang A, Lv X, … Chen L (2017). The alteration of autophagy and apoptosis in the hippocampus of rats with natural aging-dependent cognitive deficits. Behav Brain Res, 334, 155–162. [DOI] [PubMed] [Google Scholar]
- Zhang P, Fan B, Yang P, Temirov J, Messing J, Kim HJ, & Taylor JP (2019). Chronic optogenetic induction of stress granules is cytotoxic and reveals the evolution of ALS-FTD pathology. Elife, 8. doi: 10.7554/eLife.39578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YJ, Gendron TF, Ebbert MTW, O’Raw AD, Yue M, Jansen-West K, … Petrucelli L (2018). Poly(GR) impairs protein translation and stress granule dynamics in C9orf72-associated frontotemporal dementia and amyotrophic lateral sclerosis. Nat Med, 24(8), 1136–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]