

Abstract
A new non-cytotoxic [(+)-17β-hydroxystrebloside (1)] and two known cytotoxic [(+)-3′-de-O-methylkamaloside (2) and (+)-strebloside (3)] cardiac glycosides were isolated and identified from the combined flowers, leaves, and twigs of Streblus asper collected in Vietnam, with the absolute configuration of 1 established from analysis of its ECD and NMR spectroscopic data and confirmed by computational ECD calculations. A new 14,21-epoxycardanolide (3a) was synthesized from 3 that was treated with base. A preliminary structure-activity relationship study indicated that the C-14 hydroxy group and the C-17 lactone unit and the established conformation are important for the mediation of the cytotoxicity of 3. Molecular docking profiles showed that the cytotoxic 3 and its non-cytotoxic analogue 1 bind differentially to Na+/K+-ATPase. Compound 3 docks deeply in the Na+/K+-ATPase pocket with a sole pose, and its C-10 formyl and C-5, C-14, and C-4’ hydroxy groups may form hydrogen bonds with the side-chains of Glu111, Glu117, Thr797, and Arg880 of Na+/K+-ATPase, respectively. However, 1 fits the cation binding sites with at least three different poses, which all depotentiate the binding between 1 and Na+/K+-ATPase. Thus, 3 was found to inhibit Na+/K+-ATPase, but 1 did not. In addition, the cytotoxic and Na+/K+-ATPase inhibitory 3 did not affect glucose uptake in human lung cancer cells, against which 3 showed potent activity, indicating that this cardiac glycoside mediates its cytotoxicity by targeting Na+/K+-ATPase but not by interacting with glucose transporters.
Keywords: Streblus asper, cardiac glycosides, cytotoxicity, docking profiles, Na+/K+-ATPase inhibition, glucose transport inhibition
Graphic Abstract
1. Introduction
The genus Streblus (Moraceae) contains about 25 species,1 of which Streblus asper Lour., a medium-sized tree, is used in several systems of traditional medicine.2 The potential antitumor activity of S. asper has been investigated in both in vitro and in vivo studies,3–5 and cardiac glycosides have been characterized as the main active components, of which their content varies among the different plant parts of S. asper. 6‒9
Cardiac glycosides are well-known Na+/K+-ATPase inhibitors that contribute to the treatment of congestive heart failure and also have been investigated extensively for their potential anticancer activities.10,11 Recently, Na+/K+-ATPase has been studied as a promising molecular target for the discovery of new anticancer drugs,12 and its ion-transport function but not the receptor function was found to be involved in the inhibition of MDA-MB-231 human breast cancer cell migration by cardiac glycosides.13
In a continuing search for anticancer agents,14,15 several new and known cytotoxic cardiac glycosides were obtained from an earlier investigation on the stem bark of S. asper, of which (+)-strebloside was found to show in vivo antitumor efficacy mediated potentially by inhibiting Na+/K+-ATPase, NF-κB, and mutant p53 expression through induction of the ERK pathways.4,5 Following this prior work, the cardiac glycosides of the combined flowers, leaves, and twigs of S. asper collected in Vietnam have been investigated, leading to the isolation and characterization of a new compound, (+)-17β-hydroxystrebloside (1). Also, obtained were two known analogues, (+)-3′-de-O-methylkamaloside (2) and (+)-strebloside (3). Preliminary structure-activity relationships in terms of Na+/K+-ATPase and glucose transport inhibitory effects of (+)-strebloside (3) and binding profiles between 1 or 3 and Na+/K+-ATPase were investigated.
2. Results and discussion
2.1. Isolation and bioactivity of cardiac glycosides from Streblus asper
The combined flowers, leaves, and twigs of S. asper collected in Vietnam were extracted with MeOH, and then the extract was partitioned with n-hexane and CHCl3. When the cytotoxic CHCl3 partition was subjected to chromatographic separation guided by inhibitory activity against the HT-29 human colon cancer cell line, a new cardiac glycoside, (+)-17β-hydroxystrebloside (1), and two known analogues, (+)-3′-de-O-methylkamaloside (2)4 and (+)-strebloside (3),4 were isolated (Fig. 1).
Fig. 1.

Structures of cardiac glycosides (1–3) isolated from the combined flowers, leaves, and twigs of S. asper and a reference compound, ouabain, for the docking studies.
Compound 1 was purified as an amorphous colorless powder with a molecular formula of C31H46O11, 16 Da (one oxygen atom) more than 3, as shown by a sodiated molecular ion peak at m/z 617.2936 (calcd 617.2932) observed in its HRESIMS, in conjunction with the 1H and 13C NMR spectroscopic data (Table 1).16 The IR spectrum showed hydroxy group absorption at νmax 3479 cm−1, and the 1H and 13C NMR data of 1 (Table 1) exhibited characteristic resonances for a cardiac glycoside.4 Comparison of these NMR spectroscopic data with those of (+)-strebloside (3) indicated that 1 and 3 contain an identical saccharide moiety and a similar steroid core, but the substitution at the C-17 position was found to be different (Fig. 1). Previously, the conformation and α- and β-configured protons of (+)-digoxin and (+)-strebloside (3) have been reported.4,17 Based on these determinations, the 1H and 13C NMR spectroscopic data of 1 have been assigned completely, with its α- or β-configured protons being proposed (Table 1).
Table 1.
1H and 13C NMR spectroscopic data of 1 and 3aa.
| position | 1b | 1c | 3ab | 3ac |
|---|---|---|---|---|
| 1 | 23.2 CH2 | α 1.25 m | 23.8 CH2 | α 1.18 m |
| β 2.03 m | β 2.17 m | |||
| 2 | 25.2 CH2 | α 1.51 m | 25.3 CH2 | α 1.50 m |
| β 2.03 m | β 1.97 m | |||
| 3 | 73.1 CH | α 4.23 br s | 72.8 CH | α 4.23 t (2.9) |
| 4 | 34.3 CH2 | α 1.75 m | 34.4 CH2 | α 1.74 m |
| β 1.97 m | β 1.99 m | |||
| 5 | 73.0 C | 73.3 C | ||
| 6 | 36.4 CH2 | β 1.72 m | 36.4 CH2 | β 1.69 m |
| α 2.00 m | α 2.11 dt (14.1, 4.5) | |||
| 7 | 18.2 CH2 | α 1.67m | 17.8 CH2 | α 1.72 m |
| β 2.34 m | β 2.21m | |||
| 8 | 41.7 CH | β 2.08 m | 37.9 CH | β 1.96 m |
| 9 | 39.4 CH | α 1.44 m | 38.7 CH | α 1.59 m |
| 10 | 54.4 C | 54.9 C | ||
| 11 | 21.1 CH2 | α 1.32 m | 20.6 CH2 | α 1.41 m |
| β 1.55 m | β 1.49 m | |||
| 12 | 33.9 CH2 | α 1.00 m | 31.6 CH2 | 1.29 m |
| β 1.25 m | 1.28 m | |||
| 13 | 51.7 C | 43.7 C | ||
| 14 | 88.5 C | 87.7 C | ||
| 15 | 29.9 CH2 | α 1.88 m | 27.4 CH2 | α 1.94 m |
| β 2.05 m | β 2.01 m | |||
| 16 | 37.0 CH2 | 2.30 m | 19.7 CH2 | α 1.54 m |
| β 1.83 m | ||||
| 17 | 86.4 C | 46.9 CH | α 1.83 m | |
| 18 | 12.6 CH3 | β 1.06 s | 15.0 CH3 | β 0.98 s |
| 19 | 208.5 CH | β 9.97 s | 208.4 CH | β 10.12 s |
| 20 | 171.5 C | 35.6 CH | β 2.98 m | |
| 21 | 73.3 CH2 | 4.83 d (18.5) | 102.5 CH | β 5.84 d (5.7) |
| 5.02 d (18.4) | ||||
| 22 | 116.6 CH | 5.84 br s | 35.1 CH2 | β 2.79 dd (18.0, 9.2) |
| α 2.39 d (18.0) | ||||
| 23 | 173.8 C | 174.4 C | ||
| 1′ | 100.1 CH | α 4.35 d (6.8) | 99.9 CH | α 4.35 m |
| 2′ | 80.1 CH | β 3.25 m | 80.1 CH | β 3.23 m |
| 3′ | 84.0 CH | α 3.23 m | 84.0 CH | α 3.22 m |
| 4′ | 68.5 CH | α 3.82 m | 68.6 CH | α 3.82 m |
| 5′ | 70.5 CH | α 3.55 m | 70.5 CH | α 3.54 m |
| 6′ | 16.5 CH3 | β 1.36 (6.4) | 16.5 CH3 | β 1.35 d (6.5) |
| OH-5 | 4.45 br s | |||
| OH-17 | 4.78 br s | |||
| OCH3-2′ | 61.4 CH3 | α 3.56 s | 61.4 CH3 | α 3.55 s |
| OCH3-3′ | 57.7 CH3 | β 3.48 s | 57.8 CH3 | β 3.48 s |
Assignments of chemical shifts are based on the analysis of 1D- and 2D-NMR spectra. CH3, CH2, CH, and C multiplicities were determined by DEPT 90, DEPT 135, and HSQC experiments.
13C NMR spectroscopic data (δ) measured in CDCl3 at 100.61 MHz and referenced to the solvent residual peak at δ 77.16.16
1H NMR spectroscopic data (δ) measured in CDCl3 at 400.13 MHz and referenced to the solvent residual peak at δ 7.26.16
A 13C NMR spectroscopic resonance for C-17 of 1 was observed at δC 86.4 rather than at δC 50.6 for 3,4 indicating an additional hydroxy group to be substituted at the C-17 position, as supported by the HMBC correlations observed between H-16, H-18, and H-22 and C-17 (Fig. 2). This inference was also evidenced by the 13C NMR spectroscopic resonances at δC 51.7 (C-13), δC 88.5 (C-14), δC 29.9 (C-15), δC 37.0 (C-16), δC 12.6 (C-18), and δC 171.5 (C-20), which were different from those at δC 49.6, 85.4, 32.1, 27.0, 15.7, and 174.3 for these respective carbons of 3 (Table 1).4
Fig. 2.

COSY (▬, 1H→1H) and key HMBC (↷,1H→13C) (upper) and selected NOESY ( , 1H→1H) (below) correlations of 1.
, 1H→1H) (below) correlations of 1.
Comparison of the specific rotation value and 1D and 2D NMR spectroscopic data of 1 with those of 3 indicated that both 1 and 3 have the same configuration for their respective glycose moiety and the A-, B-, and C-ring system, but that for their D-ring proved to be different, as implied by their NOESY correlations (Fig. 2). The 1H NMR resonances at δ 4.45 and δ 4.78 for the respective OH-5 and OH-17 of 1 were assigned by the HMBC correlations observed between OH-5 and C-4, C-5, and C-6, and between OH-17 and C-20. NOESY correlations between H-8 and OH-5 and OH-17 of 1 (Fig. 2) indicated that a β-configured hydroxy group is connected at C-17 of 1. This was also supported by the 13C NMR spectroscopic resonances at δC 21.1, 33.9, 51.7, and 88.5 for C-11–C-14 of 1, respectively, rather than those proposed at δC 22.1, 40.0, 49.6, and 85.4 for these same carbon atoms of 3 (Table 1),4 owing to effects from the different orientation of the C-17 lactone unit.18,19 A pronounced difference was observed for the NMR resonances for C-12 and H-12 at δC 33.9 and δH 1.00, 1.25 in 1 when compared with those at δC 40.0 and δH 1.38 and 1.51 in 3 (Table 1),4 which could result from the anisotropic effects of the C-23 carbonyl group.20 Thus, a 3S, 5S, 8R, 9S, 10S, 13S, 14S, 17R, 1’R, 2’R, 3’S, 4’S, 5’R absolute configuration could be defined for 1, with the structure being assigned as (3β)-3-[(2,3-di-O-methyl-β-D-fucopyranosyl)oxy]-5β,14β,17β-trihydroxy-19-oxo-17-epi-card-20(22)-enolide, namely, (+)-17β-hydroxystrebloside.
This absolute configuration determination for 1 was also confirmed by computational TDDFT ECD calculations. Six and seven conformers for the respective 17β- and 17α-hydroxystrebloside have been yielded from a conformer search, and one conformer with 100% of population for the β-epimer and two conformers with respective 63% and 32% of populations for the α-epimer have been characterized by facilitating a Boltzmann distribution. An overview of all of the calculated ECD spectra of 17β-hydroxystrebloside from the M062X, CAM-B3LYP, and wB97XD functionals was consistent with its experimental ECD spectrum (Fig.3A), and the best fit M062X was selected for the ECD calculations. Compound 1 was confirmed as 17β-hydroxystrebloside from its consistent experimental ECD with the computational ECD calculated for 17β-hydroxystrebloside but not for its 17α epimer (Fig.3B).
Fig. 3.

Overlay of the experimental ECD spectrum of (+)-17β-hydroxystrebloside (1) (black, obtained in MeOH and corrected by subtracting a spectrum of the appropriate solution in the absence of the samples recorded under identical conditions) and (A) three TDDFT calculated ECD spectra of 1 with functionals M062X (green), CAM-B3LYP (blue), and wB97XD (red) on the Def2TZVP basis set and (B) the TDDFT calculated ECD spectra of 17β-hydroxystrebloside (blue) at the M062X/Def2TZVP level of theory and the Boltzmann weighted ECD spectrum of 17α-hydroxystrebloside (red).
As characterized previously, the known cardiac glycosides, (+)-3′-de-O-methylkamaloside (2) and (+)-strebloside (3), were identified by comparison of their spectroscopic data with literature values.4
All chromatographically and spectroscopically pure compounds 1‒3 (Figs. S1‒S3, supplementary data) isolated from S. asper were evaluated for their cytotoxicity against the human HT-29 colon cancer, OVCAR3 ovarian cancer, and MDA-MB-435 melanoma cell lines, using paclitaxel as the positive control.5 The known compounds 2 and 3 showed potent cytotoxicity, with IC50 values in the range 0.079‒0.93 μM, but the new compound 1 did not exhibit any discernible activities at the concentrations used (Table 2). This indicated that C-17 isomerization accompanied by introducing a hydroxy group resulted in activity being abolished, and thus the C-17 position may be inferred as important for mediation of the cytotoxicity of 3.
Table 2.
Cytotoxicity against human cancer cells of 1–3 and 3aa.
| compound | HT-29b | MDA-MB-435c | OVCAR3d |
|---|---|---|---|
| 1 | >20 | >20 | >20 |
| 2 | 0.93 | 0.64 | 0.62 |
| 3 | 0.17 | 0.079 | 0.13 |
| 3a | >20 | >20 | >20 |
| Paclitaxele | 0.0008 | 0.0015 | 0.0015 |
IC50 values are the concentration (μM) required for 50% inhibition of cell viability for a given test compound after a 72 h treatment and were calculated using nonlinear regression analysis with measurements performed in triplicate and representative of three independent experiments, where the values generally agreed within 10%.
IC50 value toward the HT-29 human colon cancer cells.
IC50 value toward the MDA-MB-435 human melanoma cells.
IC50 value toward the OVCAR3 human ovarian cancer cells.
Positive control.
2.2. Synthesis and cytotoxicity of (+)-20,22-dihydro-14,21-epoxystrebloside
Previously, several cardiac glycosides have been isolated from the stem bark of S. asper, with some of their derivatives being synthesized. A preliminary structure-activity relationship study indicated that the C-3 saccharide residue and the C-5 and C-14 hydroxy and C-10 formyl groups were all important for (+)-strebloside (3) to mediate its cytotoxicity toward HT-29 cells.4 To test the importance of the C-17 lactone unit of this compound, a new derivative (3a) containing a saturated C-17 lactone ring and an additional cyclic 14,21-epoxy unit was synthesized herein from 3 (Scheme 1).
Scheme 1.

Synthesis of compound 3a from 3. Reagents and conditions: (a) MeOH saturated with NaOH, 65 °C, overnight.
The new product (3a) showed a sodiated molecular ion peak at m/z 601.2979 for a molecular formula of C31H46O10 (calcd 601.2983), which is the same as that of 3. Closely similar NMR resonances for the saccharide moiety were observed for 3 and 3a, but those for their steroid core were found to be different (Table 1 and Figs. S3 and S4, Supplementary Data). The 13C NMR resonances at δC 174.3 and 118.0 assigned for the C-20 and C-22 double bond of 3 were found to be replaced by those at δC 35.6 and 35.1 for these respective carbons of 3a (Table 1), and that at δC 73.6 for C-21 of 3 changed to δC 102.5 in 3a, indicating 3a as being a 20,22-dihydro cardiac glycoside hemiacetal. Both C-14 and C-21 of 3a were found to be connected by an oxygen atom, as supported by the HMBC correlation observed between H-21 and C-14 and its 13C NMR spectroscopic resonances at δC 37.9 (C-8), 31.6 (C-12), 43.7 (C-13), 87.7 (C-14), 27.4 (C-15), 19.7 (C-16), and 46.9 (C-17). These values varied from those at δC 41.8, 40.0, 49.6, 85.4, 32.1, 27.0, and 50.6 observed for C-8 and C-12–C-17, respectively, in 3,4 owing to the formation of a cyclic hemiacetal, as substantiated by the HMBC correlations between H-17 and C-12–C-16 and C-20–C-22, between H-21 and C-14, C-17, C-20, C-22, and C-23, and between H-7, H-12, and H-18 and C-14 of 3a (Fig. S6, Supplementary Data).
A positive Cotton effect (CE) around 235 nm arising from the n→π* transition of the α,β-unsaturated lactone unit to indicate the 17R configuration of 3 was missing in the ECD spectrum of 3a,4 owing to the loss of the α,β-unsaturated lactone unit. Comparison of the specific rotation value and 1D and 2D NMR spectroscopic data of 3a with those of 3 demonstrated that the configuration of the glycose moiety and the A-, B-, and C-ring system of both compounds is the same, but that for their D ring and lactone unit is different. A 3S, 5S, 8R, 9S, 10S, 13R, 14S, 17R, 20S, 21S, 1’R, 2’R, 3’S, 4’S, 5’R absolute configuration could be defined for 3a, as supported by the NOESY correlations observed between H3-18 and both of H-20 and H-21 of 3a (Fig. S6, Supplementary Data). Thus, 3a was assigned as (+)-20,22-dihydro-14,21-epoxystrebloside.
Several 14,21-epoxycardanolides have been reported in the 1920s, including isostrophanthidin and isodigitoxigenin, which were produced from the treatment of strophanthidin and digitoxigenin, respectively, with base.21–23 Following these investigations, the absolute configuration of 3β-hydroxy-14,21-epoxy-5β,14β-cardanolide and its 12β-hydroxy derivative was established by analysis of their NMR spectroscopic data,24 and the molecular structure of (21R)-3β-acetoxy-14,21-epoxy-5β,14β-card-20(22)-enolide was determined by analysis of its single-crystal X-ray diffraction data.25 The mechanism of production of 14,21-epoxycardanolides has been discussed briefly in literature,22,23 and the details of the formation of 3a from 3 were proposed herein as shown in Scheme 2. This could involve base-mediated isomerization of the C-20 and C-22 double bond followed by opening of the resulting β,γ-unsaturated lactone unit, which produces a formyl group at the C-20 position. Attack of the base-activated C-14 hydroxy group on the resultant aldehyde, followed by re-formation of the lactone unit, would result in the generation of 3a.
Scheme 2.

Plausible mechanism of formation of 3a from 3.
When evaluated for the cytotoxicity toward HT-29, MDA-MB-435, and OVCAR3 human cancer cells, this chromatographically and spectroscopically pure synthetic cardiac glycoside 3a (Fig. S4, Supplementary Data) did not exhibit any evident activities (Table 2), indicating that both the C-14 hydroxy group and the unsaturated C-17 lactone unit and its conformation are important factors for the cytotoxicity of (+)-strebloside (3).
2.3. Docking studies
As discussed above, (+)-strebloside (3) showed cytotoxicity in the nM range toward human cancer cells, but its analogue, (+)-17β-hydroxystrebloside (1), did not, which could result from differential interactions between these cardiac glycosides and their molecular targets. Previously, (+)-strebloside (3) was proposed to mediate its cytotoxicity through inhibition of Na+/K+-ATPase activity.5 It binds to Na+/K+-ATPase, and its docking profile is similar to that published for digitoxin, indicating that both cardiac glycosides share a similar Na+/K+-ATPase-targeted mechanism to mediate their cytotoxicity.5
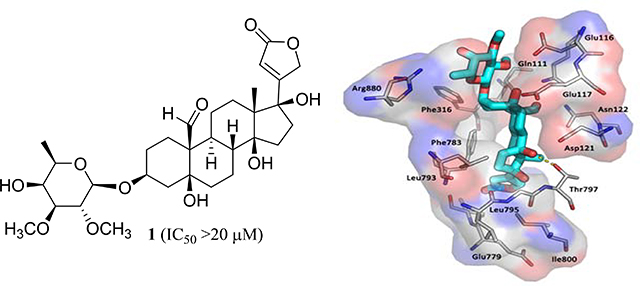
In the present work, computer-simulated docking profiles for the cytotoxic (+)-strebloside (3) and its non-cytotoxic analogue, (+)-17β-hydroxystrebloside (1), were investigated. Compound 3 adopts a binding pose similar to that of ouabain bound to Na+/K+-ATPase in the crystal structure 4HYT (Figs. 4A, 4B, and 5B).26,27 The α-surface of the steroid core of 3 interacts with bulky hydrophobic side chains of αM4–αM5, and its C-10 formyl and C-5 and C-14 hydroxy groups on the β-surface orient toward αM1–αM2 and αM6. These α helices provide several polar residues for the formation of hydrogen bonds between the C-10 formyl and C-5, C-14, and C-4’ hydroxy groups of 3 and the side-chains of Glu111, Glu117, Thr797, and Arg880 of Na+/K+-ATPase, respectively (Fig. 4D).
Fig. 4.

The docking box (A), binding poses of ouabain (B, yellow sticks, from the crystal structure), 1 (C, cyan sticks) and 3 (D, blue sticks) in Na+/K+-ATPase pocket and inhibition of Na+/K+-ATPase by 1 (E) and 3 (F).5
Fig. 5.

The overlaid binding poses of 1 (cyan sticks) and 3 (blue sticks) (A) and 3 (blue sticks) and ouabain (yellow sticks) (B) and different binding poses of 1 (cyan sticks) (C–E) in Na+/K+-ATPase pocket.
However, in contrast to 3, which docks deeply in the Na+/K+-ATPase pocket with a sole pose, compound 1 fits the cation binding sites with at least three different poses (Figs. 4C, 5A, and 5C–5E). In the first binding pose (Figure 5C), the lactone unit of 1 can overlay the ouabain binding pose in the crystal structure 4HYT,26 but the D-ring of its steroid moiety rotates around 30°. This rotation results in the orientational change of the α-surface of its steroid core, from αM4–αM5 to αM4 and αM1, and, in turn, the hydrogen bond between its OH-14 group and the Thr797 side-chain of Na+/K+-ATPase is lost. In the second binding pose (Figure 5D), the C-23 carbonyl group of 1 docks to a small hydrophobic groove on the side of the pocket lined by Ile320, Ala323 (αM4) and Ile780 and Phe783 (αM5) of Na+/K+-ATPase, and this docking breaks the hydrogen bond network between 1 and Na+/K+-ATPase. In the third binding pose (Figure 5E), the steroid moiety of 1 moves towards the entrance of the hydrophobic cavity, owing to the opposite orientation of its lactone unit, which results in breaking of the hydrogen bond network between 1 and Na+/K+-ATPase. The hydrogen bonds between the C-10 formyl and C-5, C-14, and C-4’ hydroxy groups of 3 and the respective side-chains of Glu111, Glu117, Thr797, and Arg880 of Na+/K+-ATPase seem to contribute greatly to the binding between 3 and Na+/K+-ATPase, but these H-bonds are broken in all of three different binding poses of 1. Thus, compound 1 binds to Na+/K+-ATPase less strongly than 3, which may result in the lack of cytotoxicity of 1.
To test the effect of the C-17 configuration on the binding between 1 and Na+/K+-ATPase, a docking profile was generated and depicted for its 17-epimer, 17α-hydroxystrebloside (1a), which showed that the binding between 1a or 3 and Na+/K+-ATPase is similar, but is different from that between 1 and Na+/K+-ATPase (Figs. 4 and 5 and Fig. S7, Supplementary Data). Compounds 1a and 3 share a similar binding pose, but the OH-17 group of 1a was found to be surrounded by several hydrophobic residues within a 5 Å distance, including L125, I800, and T797 sidechains of Na+/K+-ATPase (Fig. S7, Supplementary Data), which may affect the binding between 1a and Na+/K+-ATPase. Also, OH-17 of 1a docks to a hydrophobic pocket, which should result in an energy penalty.28 Thus, 1a could not bind Na+/K+-ATPase as strongly as 3, indicating that the substituent, configuration, and conformation of the C-17 lactone unit are all important for the binding between 3 and Na+/K+-ATPase.
2.4. Na+/K+-ATPase inhibition
In our previous investigation, (+)-strebloside (3) was reported to inhibit potently Na+/K+-ATPase activity (IC50 159.7 nM) (Fig. 4F),5 and the substituents at the C-10, −12, and −17 of the cardiac glycosides evaluated were found to be critically important for their inhibition of Na+/K+-ATPase.29 Thus, the non-cytotoxic cardiac glycoside, (+)-17β-hydroxystrebloside (1), was tested for its ability to inhibit Na+/K+-ATPase activity. The cellular enzyme adenosine 5′-triphosphatase from the porcine cerebral cortex was treated with various concentrations of 1, and, as expected, it did not show any activity at a concentration less than 100 μM (IC50 231.9 μM) (Fig. 4E).
2.5. Glucose transport inhibition
The inhibition of Na+/K+-ATPase may result in a higher intracellular Na+/K+ ratio followed by preventing calcium exit and a resultant higher concentration of cytoplasmic calcium. Thus, calcium uptake into the sarcoplasmic reticulum could be increased by the sarco/endoplasmic reticulum (ER) calcium ATPase2 (SERCA2) transporter, which leads to tumor cell apoptosis through cell cycle arrest and caspase activation.10 In addition, Na+ has been found to be required in extracellular solutions to drive glucose transport, for which the energy is provided by the sodium gradient maintained by the Na+/K+ pump across the brush-border membrane, and Na+/glucose cotransporters (SGLTs) depend critically on the stoichiometry of the Na+ and sugar fluxes to accumulate sugar.30 Accordingly, the sugar pump could be closely related to the sodium pump, as supported by Na+/K+-ATPase inhibitors ouabain and thevetin, of which both compounds inhibited completely the active transport of 3-methylglucose across the intestine of frogs in vitro.31
Cancer cells require an increase in glucose uptake and metabolism to meet the energy and biomass synthesis for their rapid proliferation rates, and these cells take a majority of glucose in via glucose transporters, which thus have become important targets for the development of anticancer agents.32 Using a procedure reported previously,33 the Na+/K+-ATPase inhibitory (+)-strebloside (3) was tested for its effects on glucose uptake in H1299 human lung cancer cells, against which this cardiac glycoside showed potent cytotoxicity. However, the results obtained showed that 3 did not inhibit glucose uptake in H1299 cells (Fig. 6), indicating that (+)-strebloside (3) mediates its cytotoxicity toward H1299 cells through a molecular signaling exclusive of glucose transport.
Fig. 6.

Cytotoxicity of 3 against H1299 cells and glucose transport inhibition in the cells. H1299 human lung cancer cells were treated by vehicle, WZB117 (30 μM, positive control), or 3 (30 μM) for 15 min. After glucose uptake was initiated by 2-deoxy-D-[3H]glucose, cells were lysed and the radioactivity of the cell lysates was measured. The result showed that compound 3 showed cytotoxicity toward H1299 cells (IC50 0.29 μM), but it did not inhibit glucose transport (columns, means, n = 3; bars, SE; **p ≤ 0.01; ***p ≤ 0.001).
The major challenges for the development of cardiac glycoside-like anticancer drugs are their especially narrow therapeutic index, limited selectivity, resistance shown by rodent subunits of the Na+/K+-ATPase, and the limited promise shown in cancer clinical trials to date.15 As concluded previously, such problems may correlate with the inhibition of Na+/K+-ATPase by cardiac glycosides.11 The present investigation indicates that the OH-14β group and the C-17 unsaturated lactone unit play important roles in the mediation of the cytotoxicity of 3 and in the binding between this compound and Na+/K+-ATPase. The inhibition of Na+/K+-ATPase by 3 was also found to be blocked by C-17 isomerization and introduction of a hydroxy group at this position, indicating that further structural modifications of (+)-strebloside (3) could result in the production of modified cardiac glycosides showing an improved antineoplastic activity to combat the problems of the limited clinical trial outcomes.
3. Conclusion
The well-known cardiac glycoside, (+)-strebloside (3), has been characterized as the main cytotoxic component of S. asper, which binds to Na+/K+-ATPase and inhibits the activity of this enzyme. However, (+)-17β-hydroxystrebloside (1), a C-17 hydroxylated (+)-strebloside (3) isolated in the present investigation of S. asper, did not show any activities against human cancer cells and Na+/K+-ATPase. Investigation of the docking profiles of 1 and 3 indicates that the C-17 lactone unit is important, with its substituent, configuration, and conformation all play a key role in the binding between 3 and Na+/K+-ATPase. Thus, it seems that (+)-strebloside (3) targets Na+/K+-ATPase to mediate its cancer cell cytotoxicity, and modification of 3 could produce some new Na+/K+-ATPase-independent antitumor cardiac glycosides to contribute to the development of new anticancer agents from (+)-strebloside (3).
4. Materials and methods
4.1. General
Optical rotations were measured at room temperature on an Anton Paar polarimeter (Anton Paar, Graz, Austria). UV spectra were recorded on a Hitachi U2910 ultraviolet spectrophotometer. ECD measurements were performed using a JASCO J-810 spectropolarimeter. IR spectra were recorded on a Nicolet 6700 FT-IR spectrometer. 1H and 13C, DEPT 90, DEPT 135, HSQC, HMBC, NOESY, and COSY NMR spectra were recorded at room temperature on a Bruker Avance II 400, a Bruker Avance III HD 700, or a Bruker Avance III HD 800 MHz NMR spectrometer. ESIMS or HRESIMS data were collected on a Bruker Maxis 4G Q-TOF mass spectrometer in the positive-ion mode. Column chromatography was conducted using silica gel (65 × 250 or 230 × 400 mesh, Sorbent Technologies, Atlanta, GA, USA). Analytical thin-layer chromatography (TLC) was performed on precoated silica gel 60 F254 plates (Sorbent Technologies, Atlanta, GA, USA). Sephadex LH-20 was purchased from Amersham Biosciences, Uppsala, Sweden. For visualization of TLC plates, H2SO4 was used as a spray reagent. All procedures were carried out using solvents purchased from commercial sources and employed without further purification. Paclitaxel and reagents for chemical synthesis were purchased from Sigma-Aldrich (St. Louis, MO, USA) (purity ≥ 98%).
4.2. Plant material
A sample of the combined flowers, leaves, and twigs of Streblus asper (acquisition number AA06919) was collected in January 2010 by D.D.S. and T.N.N. (voucher specimen: DDS 14849) from a tree (eight-meters tall) at Dahang Village (11° 40.53’ N; 109° 10.3’ E), Nui Chua National Park, Ninh Hai District, Ninh Thuan Province, Vietnam. The voucher herbarium specimen has been deposited at the John G. Searle Herbarium of the Field Museum of Natural History, Chicago, IL, USA, under the accession number F-2300803.
4.3. Extraction and isolation
The milled air-dried combined flowers, leaves, and twigs of S. asper (sample AA06919, 3400 g) was extracted with MeOH (5 L × 7) at room temperature. The solvent was evaporated in vacuo, and the dried MeOH extract (170.0 g, 5.0%) was resuspended in 10% H2O in MeOH (1000 mL) and partitioned with n-hexane (800, 500, and 500 mL) to yield an n-hexane-soluble residue (31.0 g, 0.9%). Then, 200 mL of H2O were added to the aqueous MeOH layer, and this was partitioned with CHCl3 (800, 500, and 500 mL). The combined CHCl3 partition was washed with a 1% aqueous solution of NaCl, to partially remove plant polyphenols, and the solvent was evaporated to afford a CHCl3-soluble extract (20.0 g, 0.6%). The CHCl3-soluble extract exhibited cytotoxicity toward the HT-29 human colon cancer cell line (IC50 < 5.0 μg/mL). Both the n-hexane- and water-soluble extracts were inactive (IC50 >20.0 μg/mL) in the bioassay system used. The CHCl3-soluble extract (19.5 g) was subjected to silica gel column chromatography (6.0 × 45 cm) and eluted with a gradient of DCM (CH2Cl2)-MeOH. Eluates were pooled by TLC analysis to give 21 combined fractions (D2F1−D2F21), and fraction D2F7 was deemed active against HT-29 cells (IC50 <1 μg/mL).
Fraction D2F7 was chromatographed over a silica gel column (4.5 × 40 cm), eluted with a gradient of DCM-MeOH to yield three pooled subfractions, D2F7F1–D2F7F3. Of these, fraction D2F7F1 was chromatographed over a silica gel column (4.5 × 40 cm), eluted with a gradient of DCM-MeOH, and purified by separation over a Sephadex LH-20 column, eluted with DCM-MeOH (1:1), affording (+)-strebloside4 (3, 150 mg). Fraction D2F7F2 was separated by silica gel chromatography (2.5 × 30 cm), eluted with a gradient of DCM-MeOH, and purified by separation over a Sephadex LH-20 column, eluted with DCM-MeOH (1:1), to yield (+)-17β-hydroxystrebloside (1, 5.0 mg). Fraction D2F7F3 was separated over a series of silica gel columns (1.5 × 20 cm), eluted with a gradient mixture of DCM-MeOH, and purified by separation over Sephadex LH-20 columns, eluted with DCM-MeOH (1:1), to afford (+)-3′-de-O-methylkamaloside4 (2, 1.0 mg).
4.3.1. (+)-17β-Hydroxystrebloside (1)
Amorphous colorless powder; [α]20D +31.0 (c 0.1, MeOH); UV (MeOH) λmax (log ε) 215 (4.09) nm; ECD (MeOH, nm) λmax (Δε) 244.1 (9.06); IR (dried film) νmax 3478, 1737, 1645, 1427, 1171, 1050 cm−1; 1H and 13C NMR data, see Table 1; positive-ion HRESIMS m/z 617.2936 (calcd for C31H46O11Na, 617.2932).
4.4. Base hydrolysis of (+)-strebloside (3)
To a 25 mL glass vial equipped with a magnetic stirrer, containing 17.3 mg (0.03 mmol) of (+)-strebloside (3), 5 mL of NaOH saturated MeOH were added, and the vial was sealed. After the mixture was stirred at 60 °C overnight and then cooled to room temperature, 1 N HCl was added until the pH value being around 7.0. The solution was extracted with DCM (5 mL × 3), and the organic layer was washed with distilled H2O followed by evaporation at reduced pressure. The residue was purified by silica gel column chromatography, using DCM-MeOH (20:1→1:1), to afford 1.0 mg (0.002 mmol) of 3a (5.6%).
4.4.1. (+)-20(22)-Dihydro-14,21-epoxystrebloside (3a)
Amorphous colorless powder; [α]20D +16.0 (c 0.1, MeOH); IR (dried film) νmax 3481, 1713, 1455, 1213, 1060 cm−1; 1H and 13C NMR data, see Table 1; positive-ion HRESIMS m/z 601.2979 (calcd for C31H46O10Na, 601.2983).
4.5. ECD Calculations
The 2D structures of 17α- and 17β-hydroxystreblosides were converted to 3D coordinates with the MMFF9434 force field in OpenBabel 2.3.1.35 The confab function within OpenBabel was then used to sample at most one-million conformers with at least a 0.5 Å RMSD cut-off. The conformers were re-ranked after optimizations, followed by a frequency calculation in a self-consistent reaction field (SCRF) model of methanol at the wB97XD/6–31G(d,p)36,37 level calculated with Gaussian 16.38 A Boltzmann distribution using thermal free energy was then applied to the conformers to obtain the populations. Gaussian 16 was then used to calculate excited state structures of the most populated conformers. TDDFT was used to calculate 8 N-states at three separate levels of theory, wB97XD, CAM-B3LYP,39 and M062X,40 and all of TDDFT calculations used the Def2TZVP basis set.41,42 Functionals and basis sets were selected based on previous investigations,43,44 and the calculated spectra were weighted based on their Boltzmann populations and translated by plus of 15 nm for those from wB97XD and CAM-B3LYP and 10 nm for M062X on the x-axis.
4.6. Cell Lines
All cell lines were purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA) and cultured at 37 °C in 5% CO2. The human HT-29 colon and OVCAR3 ovarian cancer and MDA-MB-435 melanoma cell lines were cultured in RPMI 1640 medium, supplemented with FBS (10%), penicillin (100 units/mL), and streptomycin (100 μg/mL). The H1299 human non-small cell lung cancer cell line was maintained in Dulbecco’s Modified Eagle Medium (DMEM containing 25 mM glucose) supplemented with FBS (10%), penicillin (100 units/mL), and streptomycin (100 μg/mL).
4.7. Cytotoxicity Assays
The cytotoxicity of all the cardiac glycosides presented was screened against the cancer cell lines selected, using a procedure reported previously.4,5 Briefly, for cytotoxicity tested against HT-29, MDA-MB-435, or OVCAR3 cells, after log-phase-growth, cells were seeded in 96-well clear flat-bottomed plates (Microtest 96, Falcon) and treated with the samples or paclitaxel (the positive control) (both dissolved in DMSO and diluted to different concentrations required) or the vehicle (DMSO) for 72 h. Viability of cells was evaluated by a commercial absorbance assay (CellTiter 96 AQueous One Solution Cell Proliferation Assay, Promega Corp., Madison, WI, USA), with the IC50 values calculated from the vehicle control. For cytotoxicity tested against the H1299 human non-small cell lung cancer cell line,4 cell proliferation was assessed using the MTT proliferation assay kit (Cayman Chemical, Ann Arbor, MI, USA). After cells were seeded in each well of a 96-well plate and treated with the samples for 24 h, they were treated with MTT for 4 h. Then, the medium was removed, and 100 μL Crystal Dissolving Solution were added to each well. The absorbance of the solution was measured at 570 nm, with IC50 values calculated from the vehicle control.
4.8. Molecular modeling
For the docking profiles presented in Figures 4 and 5, the chain A of the crystal structure 4HYT (ouabain) was used as the receptor, which was prepared by MGLTools45 to add non-polar hydrogens and charges. The 3D structure of (+)-17β-hydroxystrebloside (1) was built in Maestro and prepared by LigPrep from Schrodinger Suite 2018–2 [Schrödinger Suite 2018–2 Protein Preparation Wizard (Schrödinger, LLC, New York, NY, 2018)]. The geometric optimization was performed using the OPLS3 (optimized potentials for liquid simulation 3) force field with all possible ionization states at pH 7.4 ± 0.1 created by Epik [a software program for pK(a) prediction and protonation state generation].46 Sixteen conformations of 1 generated by LigPrep were used for the molecular docking against the receptor by Autodock VINA.47 A rectangular box (25 × 25 × 30 Å3) centered around the center of Leu793, Ile787, Glu117 and Gln111 defines the region that the ligands can explore, which could cover nearly all of the α-M1–M6 of Na+/K+-ATPase. (+)-Strebloside (3) was also docked to the receptor, with ouabain from 4HYT used as the reference.26
For molecular modeling presented in Figure S7 (Supplementary Data), molecular docking profiles were produced following a protocol reported previously.5 Briefly, protein and ligand were prepared from the Schrödinger Release 2016–1, and the Protein Preparation Wizard was used to optimize the crystal structure of the Na+/K+-ATPase in complex with (+)-digoxin (PDB code 4RET).48 Restrained minimization was performed on the hydrogens, with RMSD heavy atoms being converged to less than 0.30 Å, using the OPLS3 force field. The 2D structure of 17α-hydroxystrebloside (1a) with defined chiral atoms was built in Maestro, and the 3D structure was created by LigPrep. OPLS3 was used for ligand geometric optimization, with all possible ionization states created at pH 7.4 by Epik. All the chiral atoms were kept during LigPrep, and default values were utilized for other parameters for protein and ligand preparations. Molecular docking was performed using GOLD v5.2.2,49 with the above prepared protein and ligands. The active site for Na+/K+-ATPase was defined as being within 10 Å around the catalytic site of in the prepared protein. The best scoring pose for each compound was selected for further analysis, and illustrations were made using Chimera.50
4.9. Na+/K+-ATPase activity assay
Na+/K+-ATPase activity was assessed using a luminescent ADP detection assay (ADP-Glo Max Assay; Promega) that measures enzymatic activity by quantitating the ADP produced during the enzymatic first half-reaction.5 Specifically, 10 μL of assay buffer containing adenosine 5′-triphosphatase (ATP) from porcine cerebral cortex (Sigma) were added to the wells of a 96-well plate followed by 10 μL of DMSO or the test compounds dissolved in DMSO. After 5.0 μL of ATP were added to each well followed by a 15-min incubation, 25 μL of ADP-Glo Reagent were added. After a 40-min incubation, 50 μL of kinase detection reagent were added to each well followed by a 60-min incubation. Then, ATP was measured via a luciferin/luciferase reaction using a Synergy Mx (BioTek, Winooski, VT, USA) to assess luminescence.
4.10. Glucose uptake assay
Using a protocol reported previously,33,51 after H1299 cells grown in 24-well plates were washed and treated with FBS-free DMEM for 1.5 h, the cell were washed and incubated for 30 min in glucose free KRP medium. WZB117 (used as the positive control) or (+)-strebloside was then added to cells at a final concentration of 30 μM. The cells were incubated for 15 min, and then glucose uptake was initiated by adding 37 MBq/L 2-deoxy-D-[3H]glucose and 1 mM regular glucose as the final concentrations to cells. After 40 min incubation, the glucose uptake was terminated by phosphate-buffered saline. Cells were lysed, and the radioactivity retained in the cell lysates was measured by an LS 6000 series liquid scintillation counter (Beckman Coulter, Inc., Fullerton, CA, USA). The data were analyzed statistically using Student’s t-test, by comparison of the data from the experimental samples with those from the vehicle control, and p < 0.05 was set as the level of significant difference.
4.11. Statistical analysis
The in vitro measurements were performed in triplicate and are representative of three independent experiments, where the values generally agreed within 10%. The dose response curve was calculated for IC50 determinations using non-linear regression analysis (Table Curve2DV4; AISN Software Inc. Mapleton, OR, USA). Differences among samples were assessed by one-way ANOVA followed by Tukey-Kramerʼs test, and the significance level was set at p < 0.05.
Supplementary Material
Acknowledgments
This investigation was supported by grant P01 CA125066 funded by the National Cancer Institute, NIH, Bethesda, MD. The plant sample of Streblus asper was collected under a collaborative arrangement between the University of Illinois at Chicago (USA) and the Institute of Ecology and Biological Resources of the Vietnam Academy of Science and Technology, Hanoi (Vietnam). We thank Emeritus Professor Dr. David J. Hart, Department of Chemistry and Biochemistry, The Ohio State University, for helpful comments and suggestions about the mechanism of the formation of compound 3a. Drs. Arpad Somogy and Nanette M. Kleinholz of the Mass Spectrometry and Proteomics of the Campus Chemical Instrument Center, The Ohio State University, are thanked for access to the mass spectrometer. Drs. Craig A. McElroy and Deepa Krishnan, College of Pharmacy, The Ohio State University, are thanked for access to some of the NMR instrumentation used in this investigation.
Footnotes
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at https://doi.org/10.1016/j.bmc. These include copies of mass and NMR spectra and the structures of compounds 1‒3 and 3a and the COSY, key HMBC, and selective NOESY correlations of 3a, and docking profiles for 1a and 3. Supplementary data associated with this article can be found in the online version.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Datwyler SL, Weiblen GD. On the origin of the fig: Phylogenetic relationships of Moraceae from ndhF sequences. Am. J. Bot 2004;91:767–777. [DOI] [PubMed] [Google Scholar]
- 2.Neekhra S, Awasthi H, Singh DCP. Potential therapeutic use of Streblus asper: A review. Int. J. Res. Dev. Pharm. Life Sci 2017;6:2845–2849. [Google Scholar]
- 3.Kumar RBS, Kar B, Dolai N, et al. Antitumor activity and antioxidant status of Streblus asper bark against Dalton’s ascitic lymphoma in mice. Interdiscip. Toxicol 2015;8:125–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ren Y, Chen WL, Lantvit DD, et al. Cardiac glycoside constituents of Streblus asper with potential antineoplastic activity. J. Nat. Prod 2017;80:648–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen WL, Ren Y, Ren J. et al. (+)-Strebloside-induced cytotoxicity in ovarian cancer cells is mediated through cardiac glycoside signaling networks. J. Nat. Prod 2017;80:659–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Saxena VK, Chaturvedi SK. Cardiac glycosides from the roots of Streblus asper. Planta Med. 1985;51:343–344. [DOI] [PubMed] [Google Scholar]
- 7.Fiebig M, Duh CY, Pezzuto JM, et al. Plant anticancer agents, XLI. Cardiac glycosides from Streblus asper. J. Nat. Prod 1985;48:981–985. [DOI] [PubMed] [Google Scholar]
- 8.Hano Y, Juma P, Abliz Z, et al. Two new cardenolide glycosides from Streblus asper. Heterocycles 2003;59:805–809. [Google Scholar]
- 9.Bai Y, Zhu W, Xu Y, et al. Characterization, quantitation, similarity evaluation and combination with Na+,K+-ATPase of cardiac glycosides from Streblus asper. Bioorg. Chem 2019;87:265–275. [DOI] [PubMed] [Google Scholar]
- 10.Patel S Plant-derived cardiac glycosides: Role in heart ailments and cancer management. Biomed. Pharmacother 2016;84:1036–1041. [DOI] [PubMed] [Google Scholar]
- 11.Diederich M, Muller F, Cerella C. Cardiac glycosides: From molecular targets to immunogenic cell death. Biochem. Pharmacol 2017;125:1–11. [DOI] [PubMed] [Google Scholar]
- 12.Alevizopoulos K, Calogeropoulou T, Lang F, et al. Na+/K+-ATPase inhibitors in cancer. Current Drug Targets 2014;15:988–1000. [PubMed] [Google Scholar]
- 13.Magpusao AN, Omolloh G, Johnson J, et al. Cardiac glycoside activities link Na+/K+-ATPase ion-transport to breast cancer cell migration via correlative SAR. ACS Chem. Biol 2015;10:561–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kinghorn AD, Carcache de Blanco EJ, Lucas DM, et al. Discovery of anticancer agents of diverse natural origin. Anticancer Res. 2016;36:5623–5637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ren Y, Carcache de Blanco EJ, Fuchs JR, et al. Potential anticancer agents characterized from selected tropical plants. J. Nat. Prod 2019;82:657–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gottlieb HE, Kotlyar V, Nudelman A. NMR chemical shifts of common laboratory solvents as trace impurities. J Org Chem. 1997;62:7512–7515. [DOI] [PubMed] [Google Scholar]
- 17.Aulabaugh AE, Crouch RC, Martin GE, et al. The conformational behavior of the cardiac glycoside digoxin as indicated by NMR spectroscopy and molecular dynamics calculations. Carbohydr. Res 1992;230:201–212. [DOI] [PubMed] [Google Scholar]
- 18.Hanna AG, Elgamal MHA, Morsy NAM, et al. Two cardenolides from Calotropis procera. Magn. Reson. Chem 1999;37:754–757. [Google Scholar]
- 19.Araya JJ, Kindscher K, Timmermann BN. Cytotoxic cardiac glycosides and other compounds from Asclepias syriaca. J. Nat. Prod 2012;75:400–407. [DOI] [PubMed] [Google Scholar]
- 20.Crombie L, Lown JW. Proton magnetic studies of rotenone and related compounds. J. Chem. Soc 1962;1962:775–781. [Google Scholar]
- 21.Jacobs WA, Heidelburger M. Strophanthin. I. Strophanthidin. J. Biol. Chem 1922;54:253–261. [Google Scholar]
- 22.Jacobs WA, Gustus EL. Strophanthin. XIII. Isostrophanthidin and its derivatives. J. Biol. Chem 1927;74:811–827. [Google Scholar]
- 23.Jacobs WA, Gustus EL. The digitalis glucosides. I. Digitoxigenin and isodigitoxigenin. J. Biol. Chem 1928;78:573–581. [Google Scholar]
- 24.Krasso AF, Binder M, Tamm Ch. Stereochemistry of epoxycardanolides (isogenins). Helv. Chim. Acta 1972;55:1352–1371. [Google Scholar]
- 25.Pfeiffer D, Reck G, Lindig C, et al. Crystal and molecular structure of (21R)-3β-acetoxy-14,21-epoxy-5β,14β-card-20(22)-enolide. Crystal Res. Technol 1985;20:189–194. [Google Scholar]
- 26.Laursen M, Yatime L, Nissen P, et al. Crystal structure of the high-affinity Na+,K+-ATPase-ouabain complex with Mg2+ bound in the cation binding site. Proc. Natl. Acad. Sci. U. S. A 2013;110:10958–10963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Seeliger D, Groot BL. Ligand docking and binding site analysis with PyMOL and Autodock/Vina. J. Comp. Aided Mol. Des 2010;24:417–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Barratt E, Bronowska A, Vondrášek J, et al. Thermodynamic penalty arising from burial of a ligand polar group within a hydrophobic pocket of a protein receptor. J. Mol. Biol 2006;362:994–1003. [DOI] [PubMed] [Google Scholar]
- 29.Stanton DT, Ankenbauer J, Rothgeb D, et al. Identification and characterization of novel sodium/potassium-ATPase inhibitors by virtual screening of a compound database. Bioorg. Med. Chem 2007;15:6062–6070. [DOI] [PubMed] [Google Scholar]
- 30.Wright EM, Loo DDF, Hirayama BA. Biology of human sodium glucose transporters. Physiol. Rev 2011;91:733–794. [DOI] [PubMed] [Google Scholar]
- 31.Csáky TZ, Hartzog III HG, Fernald GW. Effect of digitalis on active intestinal sugar transport. Am. J. Physiol 1961;200:459–460. [DOI] [PubMed] [Google Scholar]
- 32.Qian Y, Wang X, Chen X. Inhibitors of glucose transport and glycolysis as novel anticancer therapeutics. World J. Transl. Med 2014;3:37–57. [Google Scholar]
- 33.Ren Y, Yuan C, Qian Y, et al. Constituents of an extract of Cryptocarya rubra housed in a repository with cytotoxic and glucose transport inhibitory effects. J. Nat. Prod 2014;77:550–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Halgren TA. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J. Comput. Chem 1996;17:490–519. [Google Scholar]
- 35.O’Boyle NM, Banck M, James CA, et al. Open Babel: An open chemical toolbox. J. Cheminf 2011;3:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chai JD, Head-Gordon M. Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections. Phys. Chem. Chem. Phys 2008;10:6615–6620. [DOI] [PubMed] [Google Scholar]
- 37.Ditchfield R, Hehre WJ, Pople JA. Self-consistent molecular-orbital methods. IX. Extended Gaussian-type basis for molecular-orbital studies of organic molecules. J. Chem. Phys 1971;54:724–728. [Google Scholar]
- 38.Frisch MJ, Trucks GW, Schlegel HB, et al. Gaussian 16 Rev. C.01. Gaussian, Inc, Wallingford CT, 2016. [Google Scholar]
- 39.Yanai T, Tew DP, Handy NC. A new hybrid exchange-correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett 2004;393:51–57. [Google Scholar]
- 40.Zhao Y, Truhlar DG. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Account 2008;120:215–241. [Google Scholar]
- 41.Weigend F, Ahlrichs R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys 2005;7:3297–3305. [DOI] [PubMed] [Google Scholar]
- 42.Weigend F Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys 2006;8:1057–1065. [DOI] [PubMed] [Google Scholar]
- 43.Pescitelli G, Bruhn T. Good computational practice in the assignment of absolute configurations by TDDFT calculations of ECD spectra. Chirality 2016;28:466–474. [DOI] [PubMed] [Google Scholar]
- 44.Agarwal G, Wilson JR, Kurina SJ, et al. Structurally modified cyclopenta[b]benzofuran analogues isolated from Aglaia perviridis. J. Nat. Prod 2019;82:2870–2877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Goodsell DS, Morris GM, Olson AJ. Automated docking of flexible ligands: Applications of AutoDock. J. Mol. Recognit 1996;9:1–5. [DOI] [PubMed] [Google Scholar]
- 46.Shelley JC, Cholleti A, Frye LL, et al. Epik: A software program for pKα prediction and protonation state generation for drug-like molecules. J. Comp. Aided Mol. Des 2007;21:681–691. [DOI] [PubMed] [Google Scholar]
- 47.Trott O, Olson AJ. Software news and update AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comp. Chem 2010;31:455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Laursen M, Gregersen JL, Yatime L, et al. Structures and characterization of digoxin- and bufalin-bound Na+,K+-ATPase compared with the ouabain-bound complex. Proc. Natl. Acad. Sci. U. S. A 2015;112:1755–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Verdonk ML, Cole JC, Hartshorn MJ, et al. Improved protein-ligand docking using GOLD. Proteins Struct. Funct. Gene 2003;52:609–623. [DOI] [PubMed] [Google Scholar]
- 50.Pettersen EF, Goddard TD, Huang CC, et al. UCSF Chimera-A visualization system for exploratory research and analysis. J. Comput. Chem 2004;25:1605–1612. [DOI] [PubMed] [Google Scholar]
- 51.Liu Y, Cao Y, Zhang W, et al. A small-molecule inhibitor of glucose transporter 1 downregulates glycolysis, induces cell-cycle arrest, and inhibits cancer cell growth in vitro and in vivo. Mol. Cancer Ther 2012;11:1672–1682. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.