

Abstract
Background
Little is known about whether mitochondria 8-hydroxy-2′-deoxyguanosine (8-OHdG), a biomarker of mitochondrial DNA (mtDNA) oxidative damage, contributes to the development of coronary artery disease (CAD) in diabetic patients. Here, we explored the associations of mtDNA 8-OHdG in leukocytes with obstructive CAD, coronary stenosis severity, cardiovascular biomarkers, and 1-year adverse outcomes after coronary revascularization in patients with type 2 diabetes mellitus (T2DM).
Methods
In a total of 1920 consecutive patients with T2DM who underwent coronary angiography due to symptoms of angina or angina equivalents, the presence of obstructive CAD, the number of diseased vessels with ≥ 50% stenosis, and modified Gensini score were cross-sectionally evaluated; the level of mtDNA 8-OHdG was quantified by quantitative PCR. Then, 701 of 1920 diabetic patients who further received coronary revascularization completed 1-year prospective follow-up to document major adverse cardiovascular and cerebral events (MACCEs). In vitro experiments were also performed to observe the effects of mtDNA oxidative damage in high glucose-cultured human umbilical vein endothelial cells (HUVECs).
Results
Cross-sectionally, greater mtDNA 8-OHdG was associated with increased odds of obstructive CAD (odds ratio [OR] 1.38, 95% CI confidence interval 1.24–1.52), higher degree of coronary stenosis (number of diseased vessels: OR 1.29, 95% CI 1.19–1.41; modified Gensini scores: OR 1.28, 95% CI 1.18–1.39), and higher levels of C-reactive protein (β 0.18, 95% CI 0.06–0.31) after adjusting for confounders. Sensitivity analyses using propensity score matching yielded similar results. Stratification by smoking status showed that the association between mtDNA 8-OHdG and obstructive CAD was most evident in current smokers (Pinteration < 0.01). Prospectively, the adjusted hazards ratio per 1-SD increase in mtDNA 8-OHdG was 1.59 (95% CI 1.33–1.90) for predicting 1-year MACCEs after revascularization. In HUVECs, exposure to antimycin A, an inducer for mtDNA oxidative damage, led to adverse alterations in markers of mitochondrial and endothelia function.
Conclusion
Greater mtDNA 8-OHdG in leukocytes may serve as an independent risk factor for CAD in patients with T2DM.
Keywords: Mitochondrial 8-OHdG, Coronary artery disease, Type 2 diabetes mellitus, Clinical outcomes after revascularization
Introduction
Patients with type 2 diabetes mellitus (T2DM) exhibit a high prevalence of coronary artery disease (CAD) and cardiac deaths [1, 2]. T2DM-related variables, including long diabetes duration [3], high glycemic variability [4, 5], and elevated glycated hemoglobin (HbA1c) [6], are also considered as potent predictors for cardiovascular events in large-scale prospective studies. The pathogenesis of clinically overt CAD in T2DM, while not fully elucidated, has been related to the excess production of reactive oxygen species (ROS), which may active proinflammatory pathways, reduce nitric oxide bioavailability, and consequently induce diabetic endothelial dysfunction [7, 8]. Mitochondria, as central regulators of energy metabolism, are primary cellular sources and targets of oxidants associated with vascular disease in T2DM [9].
Mitochondrial DNA (mtDNA) is more vulnerable to the accumulation of ROS-mediated damage than nuclear DNA, in part because of its close proximity to the mitochondrial oxidative phosphorylation chain, and because it lacks protective histones and efficient DNA repair mechanisms [10]. Under diabetic conditions, oxidative damage to the mtDNA may be an important consequence of excess ROS production [11], inducing a loss of mitochondrial networks, decreased expression of mitochondrial respiratory complexes, and consequent T2DM-related atherosclerosis [12–14]. Among numerous types of mtDNA oxidative lesions, the formation of 8-hydroxy-2′-deoxyguanosine (8-OHdG) is the most abundant one, reflecting a steady-state level of oxidative damage occurring within mtDNA [15]. In kidney of diabetic rats, levels of 8-OHdG were up-regulated in mtDNA but not in nuclear DNA [16]. In population studies, we and others previously showed that leukocyte mtDNA copy number variation (mtDNA-CN), as an indirect measure of mtDNA damage [17], inversely correlated with CAD risk and severity independent of diabetic status [18, 19]. However, little is known about whether 8-OHdG in mtDNA, a more direct marker of oxidative DNA damage, contributes to the development of CAD in patients with T2DM.
Hence, in this cross-sectional study, we enrolled a total of 1920 consecutive patients with T2DM to assess the associations of mitochondrial 8-OHdG in leukocytes with the presence of obstructive CAD, the severity of coronary artery stenosis, and cardiometabolic phenotypes. Then, 701 of 1920 patients who received percutaneous or surgical revascularization were prospectively followed for 1 year to investigate the prognostic value of mitochondrial 8-OHdG on clinical outcomes after revascularization. Finally, we performed in vitro experiments to assess the effect of antimycin A (AMA)-induced mtDNA oxidative damage on markers of mitochondrial and endothelial dysfunction in high glucose-cultured human umbilical vein endothelial cells (HUVECs).
Methods
Study population
From May 2016 to May 2018, 2381 consecutive patients with T2DM who underwent coronary angiography due to symptoms of angina or angina equivalents were cross-sectionally evaluated in a discovery cohort from The First Affiliated Hospital of Zhengzhou University (at Henan province, northern China) and in a validation cohort from Zhongnan Hospital of Wuhan University (at Hubei province, central China). Patients were excluded if they (1) were younger than 40 years (n = 59); (2) did not complete blood tests (n = 33); (3) had a history of type 1 diabetes (n = 23); (4) had a history of systemic disease including cancer of any type, liver disease, and clinically significant infection (n = 229); (5) had a history of cardiac disease including valvular disease, congenital heart disease, and cardiomyopathy (n = 117). After excluding 461 cases due to exclusion criteria, a total of 1920 patients with T2DM were finally included, involving 1125 cases in a discovery cohort and 795 cases in a validation cohort (Fig. 1). The diagnosis of T2DM was based on the American Diabetes Association 2014 criteria: fasting plasma glucose (FPG) ≥ 7.0 mmol/L and/or HbA1c ≥ 6.5% and/or 2-h post-challenge plasma glucose ≥ 11.1 mmol/L and/or ongoing therapy for T2DM [20]. Obstructive CAD was defined as the maximum luminal stenosis of ≥ 50% in any segment of major coronary arteries [21, 22].
Fig. 1.
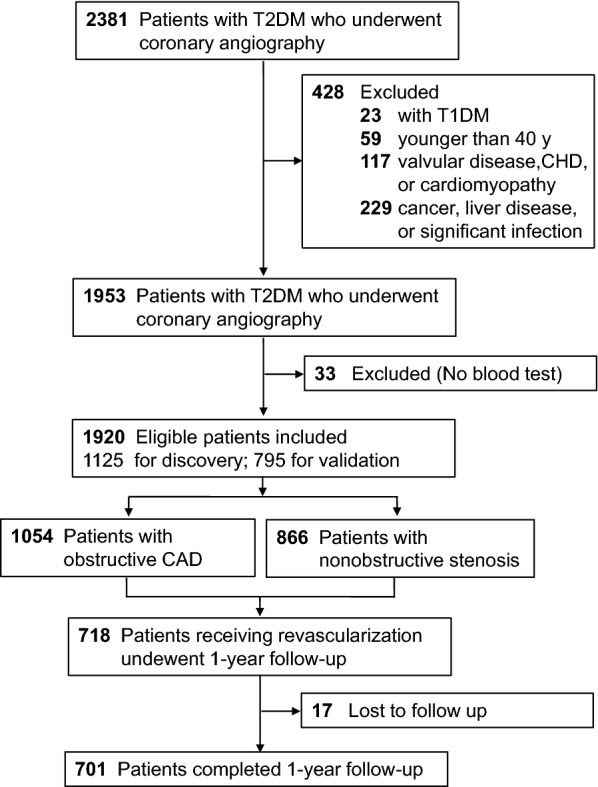
Flow chart of our study
The severity of coronary artery stenosis was quantified by the number of diseased vessels with ≥ 50% stenosis and modified Gensini scores. We first categorized each patient by the number of diseased vessels with ≥ 50% stenosis in a single, double, or triple-vessel distribution. We defined vessel distribution by the left anterior descending artery and its branches, the left circumflex artery and its branches, and the right coronary artery and its branches. Patients with ≥ 50% stenosis of left main coronary artery were classified into the 3-vessel obstructive CAD group [22]. So we finally created four categories of CAD extent: non-obstructive CAD, 1-, 2-, 3-vessel/left main obstructive CAD. In the Gensini scoring system [23], angiographic stenosis of each coronary segment was first scored according to the degree of luminal narrowing: 1 for 0–25%, 2 for 26–50%, 4 for 51–75%, 8 for 76–90%, 16 for 91–99%, and 32 for 100% stenosis. Then a multiplier was assigned to each segment depending on the prognostic importance of the area supplied by that segment: 5 for left main coronary artery, 2.5 for proximal left anterior descending coronary artery (LAD) and proximal left circumflex branch, 1.5 for mid-segment of LAD, 0.5 for second diagonal branch and posterolateral branch, and 1 for other branches. Finally, the weighted score of each lesion was summed to give final Gensini scores.
During the study period, 718 of 1920 patients with T2DM underwent percutaneous coronary intervention (PCI) or coronary artery bypass grafting (CABG) due to the presence of indications for revascularization [24, 25]. These 718 patients were followed up for 12 months after revascularization to track major adverse cardiovascular and cerebrovascular events (MACCEs) as the primary outcome. MACCE was defined as a composite of all-cause death, repeat revascularization, non-fatal myocardial infarction (MI), and non-fatal stroke (Additional file 1: Methods) [26]. The second outcome was any cause of cardiac death. All outcomes of interest were ascertained through telephone interviews with participants and verification by checking medical records and administrative billing codes for readmissions at 30 days and 6, 9, 12 months after revascularization. In total, 701 patients (97.6%) were completely followed for clinical outcomes up to 12 months or deaths. The study protocol followed the Declaration of Helsinki and was approved by the Ethics Committees of participating centers. All participants signed written informed consent.
Collection and definition of clinical variables
Detailed description of clinical variables has been presented in our previous studies [18, 27]. Briefly, demographic factors, including age, sex, smoking status (never, former, and current), alcohol drinking status (ever and never), diabetes duration, and type of diabetes management (lifestyle modification, oral agents, insulin, and oral agents plus insulin), were documented by checking participants’ medical records and questionnaires. Weight and height were measured on the day of enrollments, and body mass index (BMI) was coded into three categories: normal BMI (18–24.9 kg/m2), overweight (25–30 kg/m2), and obese (> 30 kg/m2). Dyslipidemia and hypertension were defined based on the previously published guidelines [28, 29].
Fasting blood samples were drawn on the day of angiography. FPG, low-density lipoprotein cholesterol (LDL-c), high-density lipoprotein cholesterol (HDL-c), total cholesterol (TC), triglycerides (TG), and C-reactive protein (CRP) were measured on a Cobas 8000 Analyzer (Roche Diagnostics, Germany) using standard methods. HbA1c was assayed by high-performance liquid chromatography on a Bio-Rad Hemoglobin Testing System. Plasma D-dimer was detected with latex enhanced immunoassay on an ACL TOP 300 Analyzer (Beckman, USA).
Quantitative of the levels of 8-OHdG in leukocyte mtDNA
Leukocyte DNA was extracted using the QIAamp DNA minikit (Qiagen, MD, USA). The levels of mtDNA 8-OHdG were measured by quantitative PCR (qPCR) to amplify a fragment of mtDNA with or without treatment of human 8-oxoguanine DNA glycosylase 1 (hOGG1), as described by Lin et al. [30, 31]. Briefly, this method is based on the theory that pretreatment of hOGG1 would remove the 8-OHdG residues to form abasic sites that reduce the amplification efficacy [32]. So, the content of 8-OHdG in mtDNA is relative to the differences in amplification efficiency between hOGG1-treated and untreated DNA when the cycling number was in the linear amplification stage [30]. First, 100 ng of DNA sample was treated with or without 1 U of hOGG1 (TREVIGEN, MD, USA) in a mix containing 1.5 μL of 10× buffer NE, 8.5 μL of RNase free water, and 100 μg/L of bovine serum albumin, followed by an incubation of 1 h for 37 °C. Then, the treated and untreated samples were amplified by qPCR using a specific primer pair designed for the mitochondrial ND1 gene in a reaction mixture containing ~ 25 ng of DNA sample, 2 μL of ddH2O, and 10 pM of each primer (F: 5′-CAC CCA AGA ACA GGG TTT GT-3′; R: 5′-TGG CCA TGG GAT TGT TGT TAA-3′). Each sample was assayed in duplicate, and the mean value of the ∆ct (treated ct − untreated ct) was calculated as the level of mtDNA 8-OHdG.
For quality control, four samples were included into each plate to analyze variability. The average CV across plates was 4.58%. For validation of qPCR data, we also measured the levels of 8-OHdG in mtDNA using a previously described ELISA assay based on mitochondria isolation, mtDNA extraction and digestion, and immunosorption [16] in 200 participants randomly selected from the whole cohorts. The bivariate correlation coefficient between qPCR and ELISA data was greater than 0.72 (Additional file 2: Fig. S1), suggesting that the levels of 8-OHdG determined by qPCR were reliable.
In vitro experiment in HUVECs
HUVECs (ATCC Cat# PCS-100-010) were cultured in complete endothelial cell growth medium (EGM-2 Bulletkit, LONZA) containing high levels of glucose (25 mM d-glucose) in a humidified incubator with 5% CO2 at 37 °C. The cells were incubated for 72–96 h until fully confluent, and treated with or without 50 mM of AMA (Sigma, MO, USA), an inducer of mtDNA oxidative damage by specifically inhibiting mitochondrial complex 3 [33]. After 24 h of incubation, cells with or without exposure to AMA were separately collected.
For the measurement of mitochondrial membrane potential (∆ψm), cells were washed twice with PBS, and incubated with 200 μM of JC-1 (Invitrogen, CA, USA) for 20 min, as described by Widlansky et al. [34]. JC-1 is a cationic dye whose mitochondrial uptake is directly correlated with the magnitude of ∆ψm. Mitochondrial membrane potential was expressed as the JC-1 fluorescence intensities detected by the Microplate Reader at Ex/Em(green)/Em(red) of 485/538/590 nm. MtDNA 8-OHdG, mtDNA-CN, and mRNA expression of peroxisome proliferator-activated receptor coactivator 1a (PGC-1α, a master gene of mitochondrial biogenesis [35]) gene were determined using qPCR, as previously described [18, 36, 37]. Culture supernatants of HUVECs were collected to measure the levels of nitric oxide (NO), soluble vascular adhesion molecule 1 (sVCAM-1) and E-selectin using colorimetric kits (for NO) or ELISA (Invitrogen). We also performed a MTT assay (Sigma, MO, USA) to examine cell viability of HUVECs. Optical densities were measured at 562 nm with the results being normalized to untreated cells [33]. All in vitro experiments were performed in triplicate.
Statistical analysis
We applied a Z transformation to standardize the distribution of mtDNA 8-OHdG across cohorts. The association of 8-OHdG in mtDNA with obstructive CAD was analyzed by logistic regression models with three progressive degrees of adjustment. Model 1 was a crude model without any confounders; model 2 was adjusted for age, sex, and cardiovascular risk factors including smoking habit, alcohol drinking habit, overweight/obesity, hypertension, and dyslipidemia; model 3 was additionally adjusted for T2DM-related variables including FPG, HbA1c, diabetes duration, and type of diabetes management. For each model, 8-OHdG in mtDNA was first analyzed as a continuous variable, and then as an ordinal variable based on its quartile distribution. We used the restricted cubic spline (RCS) to analyze the shape of the association between mtDNA 8-OHdG and CAD presence, with four equally spaced knots set at 5th, 35th, 65th, and 95th percentiles as recommended by Harrell et al. [38]. Sensitivity analyses were based on two statistical approaches: (1) stratification analysis assessing the effect modification by clinical covariates, (2) propensity score (PS) matching [39], which matched the distribution of covariates after using a 1:1 nearest neighbor matching (caliper = 0.05) to yield 673 pairs of participants.
We calculated the increases in Harrell’s C-statistic (i.e. ΔC-statistic) to assess the incremental value of adding mtDNA 8-OHdG separately to the 2013 ACC/AHA Pooled Cohorts Equations (PCE) [40] and the Framingham Risk Score (FRS) [41] in discriminating obstructive CAD. We computed the net reclassification improvement (NRI) as an alternative approach to evaluate risk classification ability when the risk estimates of obstructive CAD was categorized as low (< 20%), intermediate (20–50%), and high risk (> 50%). Finally, we calculated the integrated discrimination improvement (IDI), which integrates the continuous NRI over all possible cutoffs of risk estimates and mathematically corresponds to the difference in discrimination slopes of the two models in comparison [42]. The correlations of mtDNA 8-OHdG with coronary stenosis severity and cardiometabolic phenotypes were assessed by ordinal logistic regression and multivariable linear regression, respectively.
When assessing the prognostic value of mtDNA 8-OHdG on 1-year clinical outcomes after revascularization, we first used the time-dependent receiver operator characteristics (ROC) curve to screen the optimal cutoff value of mtDNA 8-OHdG with the highest combined sensitivity and specificity to predict MACCEs. Then, the Cox regression was performed to estimate hazard ratios (HRs) for the association between mtDNA 8-OHdG (as a continuous variable or as a dichotomous variable categorized by the ROC-derived cutoff value) and the incidence of MACCEs after adjusting for all confounders in model 3. For in vitro experiments, the Student t test was used to assess the differences in tested markers between AMA-treated and untreated HUVECs. All above analyses were conducted using STATA 12.0 (StataCorp, TC, USA). A statistical power was estimated using PS software 3.0 (Vanderbilt, TN, USA). A two-sided P < 0.05 was considered statistically significant.
Results
Characteristics of participants
Table 1 presents the clinical characteristics of all 1920 participants. Overall, type 2 diabetic patients with CAD were older, more likely to be smokers, obese, hypertensive, and dyslipidemic, and had higher levels of FPG, HbA1c, TC, LDL-c, and CRP than those without. After PS matching, a sub-cohort of 673 pairs of participants was screened. Within this cohort, all clinical characteristics were similar between CAD and non-CAD groups.
Table 1.
Clinical characteristics of 1920 diabetic patients
| Variables | All participants | After PS matching | ||||
|---|---|---|---|---|---|---|
| Obstructive CAD (N = 1054) |
Non-CAD (N = 866) |
Pa | Obstructive CAD (N = 673) |
Non-CAD (N = 673) |
Pa | |
| Age, years | 62.8 ± 9.6 | 61.1 ± 10.7 | < 0.001 | 62.3 ± 9.5 | 62.3 ± 10.8 | 0.977 |
| Male, n (%) | 583 (55.3) | 480 (55.4) | 0.960 | 360 (53.5) | 373 (55.4) | 0.477 |
| BMI, mean (SD), kg/m2 | 25.2 ± 4.0 | 24.3 ± 2.6 | < 0.001 | 24.7 ± 3.9 | 24.5 ± 2.6 | 0.204 |
| BMI categories | ||||||
| Normal | 540 (51.2) | 494 (57.0) | < 0.001 | 412 (61.2) | 396 (58.8) | 0.613 |
| Overweight | 372 (35.3) | 317 (36.6) | 116 (17.2) | 118 (17.6) | ||
| Obese | 142 (13.5) | 55 (6.4) | 145 (21.6) | 159 (23.6) | ||
| Smoking, n (%) | ||||||
| Never smoker | 582 (55.2) | 555 (64.1) | < 0.001 | 382 (56.8) | 358 (53.2) | 0.157 |
| Former smoker | 168 (15.9) | 130 (15.0) | 231 (34.3) | 264 (39.2) | ||
| Current smoker | 304 (28.9) | 181 (20.9) | 60 (8.9) | 51 (7.6) | ||
| Alcohol drinkers, n (%) | 343 (32.5) | 248 (28.6) | 0.065 | 210 (31.2) | 196 (29.1) | 0.406 |
| FPG, mmol/L | 7.3 ± 2.0 | 6.7 ± 1.8 | < 0.001 | 7.0 ± 1.6 | 6.9 ± 2.0 | 0.386 |
| HbA1c, (%) | 8.1 ± 1.8 | 7.9 ± 1.7 | 0.031 | 8.0 ± 1.8 | 8.0 ± 1.7 | 0.779 |
| Diabetes duration, years | 10.3 ± 5.4 | 9.9 ± 5.7 | 0.110 | 10.1 ± 5.4 | 9.7 ± 5.7 | 0.105 |
| Diabetes management | ||||||
| Lifestyle modification | 291 (27.6) | 255 (29.4) | 0.566 | 200 (29.7) | 199 (29.6) | 0.500 |
| Oral agents only | 332 (31.5) | 283 (32.7) | 206 (30.6) | 229 (34.0) | ||
| Insulin only | 275 (26.1) | 214 (24.7) | 166 (24.7) | 157 (23.3) | ||
| Oral agents and insulin | 156 (14.8) | 114 (13.2) | 101 (15.0) | 88 (13.1) | ||
| Dyslipidemia, n (%) | 334 (31.7) | 166 (19.2) | < 0.001 | 164 (24.4) | 158 (23.5) | 0.701 |
| Cholesterol, mmol/L | ||||||
| TC, mmol/L | 4.95 ± 0.91 | 4.80 ± 0.85 | < 0.001 | 4.88 ± 0.90 | 4.85 ± 0.87 | 0.194 |
| LDL-c, mmol/L | 3.28 ± 0.83 | 3.08 ± 0.68 | < 0.001 | 3.16 ± 0.80 | 3.10 ± 0.67 | 0.123 |
| HDL-c, mmol/L | 1.12 ± 0.15 | 1.16 ± 0.21 | 0.318 | 1.09 ± 0.15 | 1.12 ± 0.21 | 0.756 |
| TG, mmol/L | 1.48 ± 0.85 | 1.36 ± 0.76 | 0.104 | 1.49 ± 0.84 | 1.43 ± 0.80 | 0.114 |
| Hypertension, n (%) | 619 (58.7) | 338 (39.0) | < 0.001 | 330 (49.0) | 318 (47.3) | 0.513 |
| Blood pressure | ||||||
| SBP, mmHG | 138.2 ± 21.4 | 134.8 ± 19.5 | < 0.001 | 136.7 ± 25.0 | 135.4 ± 19.1 | 0.157 |
| DBP, mmHG | 88.0 ± 18.4 | 85.1 ± 12.0 | < 0.001 | 86.4 ± 18.1 | 85.9 ± 11.4 | 0.109 |
| CRP, mg/L | 1.57 ± 1.01 | 1.10 ± 0.86 | < 0.001 | 1.34 ± 0.94 | 1.30 ± 0.87 | 0.400 |
| D-dimer, mg/L | 0.98 ± 0.93 | 0.91 ± 0.82 | 0.077 | 0.93 ± 0.95 | 0.91 ± 0.79 | 0.228 |
Data are presented as the mean ± SD or n (%) as appropriate
BMI body mass index, FPG fasting plasma glucose, HbA1c glycosylated haemoglobin, TC total cholesterol, LDL-c low-density lipoprotein cholesterol, HDL-c high-density lipoprotein cholesterol, TG triglycerides, SBP systolic blood pressure, DBP diastolic blood pressure, CRP C-reactive protein, CAD coronary artery disease, PS propensity score
aP values were obtained from the student t test for continuous variables, and the Pearson χ2 test for categorical variables
mtDNA 8-OHdG and obstructive CAD in diabetic patients
In the whole cohort of 1920 diabetic patients, each 1-SD increase (0.87 ∆ct) in mtDNA 8-OHdG was associated with a 1.41-fold (95 confidence interval [95% CI] 1.28–1.56, Table 2) increased odds of CAD in the crude model. When further adjusted for age, sex, and, cardiovascular risk factors (model 2), the odds ratio (OR) for CAD was attenuated to 1.38 (95% CI 1.25–1.53) per 1-SD increase in mtDNA 8-OHdG. Additional adjustment for FPG, HbA1c, diabetes duration, and type of diabetes management (model 3) did not modify the magnitude of the association (OR = 1.38). Assuming an OR of 1.38 and a type I error of 0.05, the whole population could provide a statistical power of 98.3% to address the association. When analyzed with the bottom quartile of mtDNA 8-OHdG as the reference, the OR for CAD was 0.93 in the second quartile, 1.32 in the third quartile, and 2.41 in the top quartile, with a significant dose–response trend observed (Ptrend < 0.001, Table 2). Similarly, the RCS analysis suggested that mtDNA 8-OHdG yielded much greater and linear-shaped increases in CAD rates across its ranges (Pnon-linearity = 0.745, Fig. 2).
Table 2.
Association of mtDNA 8-OHdG with CAD risk in patients with T2DM
| mtDNA 8-OHdG | Obstructive CAD N (%) | Non-CAD N (%) |
OR (95%CI) | ||
|---|---|---|---|---|---|
| Model 1a | Model 2b | Model 3c | |||
| All participants | |||||
| Each 1-SD increase | 1054 | 866 | 1.41 (1.28–1.56) | 1.38 (1.25–1.53) | 1.38 (1.24–1.52) |
| Quartiles | |||||
| Q1 (< 2.84) | 232 (22.0) | 261 (30.1) | 1 (Reference) | 1 (Reference) | 1 (Reference) |
| Q2 (2.84–3.20) | 224 (21.3) | 251 (29.0) | 1.00 (0.78–1.29) | 0.94 (0.72–1.22) | 0.93 (0.71–1.22) |
| Q3 (3.20–3.53) | 267 (25.3) | 213 (24.6) | 1.41 (1.10–1.82) | 1.33 (1.02–1.73) | 1.32 (1.01–1.72) |
| Q4 (> 3.53) | 331 (31.4) | 141 (16.3) | 2.64 (2.03–3.44) | 2.42 (1.84–3.19) | 2.41 (1.82–3.19) |
| P for trend | < 0.001 | < 0.001 | < 0.001 | ||
| After PS matching | |||||
| Each 1-SD increase | 673 | 673 | 1.39 (1.24–1.56) | 1.39 (1.24–1.55) | 1.39 (1.24–1.55) |
| Quartiles | |||||
| Q1 (< 2.84) | 149 (22.1) | 187 (27.8) | 1 (Reference) | 1 (Reference) | 1 (Reference) |
| Q2 (2.84–3.17) | 148 (22.0) | 190 (28.2) | 0.98 (0.72–1.33) | 0.97 (0.71–1.31) | 0.94 (0.69–1.28) |
| Q3 (3.17–3.52) | 163 (24.2) | 176 (26.2) | 1.16 (0.86–1.57) | 1.16 (0.86–1.57) | 1.17 (0.86–1.58) |
| Q4 (> 3.52) | 213 (31.7) | 120 (17.8) | 2.22 (1.63–3.04) | 2.19 (1.60–3.00) | 2.18 (1.60–2.99) |
| P for trend | < 0.001 | < 0.001 | < 0.001 | ||
PS propensity score, 8-OHdG 8-hydroxy-2′-deoxyguanosine, CAD coronary artery disease
aCrude model without any confounders
bModel 2 was adjusted for age, sex, smoking habit, alcohol drinking habit, overweight/obesity, hypertension, and dyslipidemia
cModel 3 was adjusted for age, sex, smoking habit, alcohol drinking habit, overweight/obesity, hypertension, dyslipidemia., FPG, HbA1c, diabetes duration, and type of diabetes management
Fig. 2.

Restricted cubic spline analysis for assessing the shape of the association between mtDNA 8-OHdG and CAD presence in patients with T2DM after adjusting for age, sex, smoking habit, alcohol drinking habit, overweight/obesity, hypertension, dyslipidemia., FPG, HbA1c, diabetes duration, and type of diabetes management
When stratified by smoking status, the OR for CAD per 1-SD increase of mtDNA 8-OHdG gradually increased from 1.10 in never smokers, to 1.81 in former smokers, and ultimately to 2.15 in current smokers (Pinteraction < 0.001, Fig. 3). When stratified by study cohorts and other clinical covariates, no effect modification was observed. In the PS-matched cohort of 673 pairs of participants with balanced distributions of clinical covariates, mtDNA 8OHdG, either as a continuous variable or as an ordinal variable, continued to be positively associated with an increased odds of CAD, to a similar degree as in the whole cohort (Table 2).
Fig. 3.

Association between continuous mtDNA 8-OHdG and CAD presence stratified by different clinical covariates in patients with T2DM. Pinteraction was obtained from the χ2-based Q test
mtDNA 8-OHdG and risk discrimination and reclassification for CAD in diabetic patients
Adding mtDNA 8-OHdG to the 2013 ACC/AHA PCE model significantly improved risk discrimination and reclassification for CAD events, measured by ΔC-statistic (0.024), continuous NRI (0.299) and IDI (0.020, all P < 0.001). When categorizing the overall NRI with cutoffs at 20% and 50%, the addition of mtDNA 8-OHdG properly upclassified 45 individuals with CAD events, and appropriately downclassified 52 individuals without CAD (categorical NRI = 0.103, P < 0.001, Table 3). Similarly, when mtDNA 8-OHdG was added to the FRS, there were significant improvements in discrimination (ΔC-statistic: 0.010, P = 0.003) and risk reclassification (categorical NRI = 0.059, continuous NRI = 0.196, IDI = 0.014, all P < 0.001) for CAD events (Table 3). All models including mtDNA 8-OHdG were well-calibrated, with P values of the Hosmer–Lemeshow test of > 0.1.
Table 3.
Discrimination and reclassification improvements of mtDNA 8-OHdG beyond the 2013 ACC/AHA PCE and the FRS in 1920 patients with T2DM
| Risk estimates | ACC/AHA 2013 PCE + mtDNA 8-OHdG | FRS + mtDNA 8-OHdG | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| < 20% | 20–50% | > 50% | Total | Reclassified | < 20% | 20–50% | > 50% | Total | Reclassified | |
| Individuals without obstructive CAD, No. | ||||||||||
| < 20% | 268 | 23 | 0 | 291 | 8% | 352 | 10 | 0 | 362 | 3% |
| 20–50% | 68 | 275 | 41 | 384 | 28% | 59 | 191 | 14 | 264 | 28% |
| > 50% | 0 | 48 | 143 | 191 | 25% | 0 | 27 | 213 | 240 | 11% |
| Total | 336 | 346 | 184 | 866 | 411 | 228 | 227 | 866 | ||
| Individuals with obstructive CAD, No. | ||||||||||
| < 20% | 125 | 30 | 3 | 158 | 21% | 105 | 7 | 0 | 112 | 6% |
| 20–50% | 35 | 288 | 82 | 405 | 29% | 30 | 205 | 41 | 276 | 26% |
| > 50% | 0 | 35 | 456 | 491 | 7% | 0 | 31 | 635 | 666 | 5% |
| Total | 160 | 353 | 541 | 1054 | 135 | 243 | 676 | 1054 | ||
| Estimate (95% CI) | P value | Estimate (95% CI) | P value | |
|---|---|---|---|---|
| Δ C-statistic | 0.024 (0.013–0.035) | < 0.001 | 0.010 (0.003–0.017) | 0.003 |
| IDI | 0.020 (0.014–0.026) | < 0.001 | 0.014 (0.001–0.019) | < 0.001 |
| NRI continuous | 0.299 (0.210–0.388) | < 0.001 | 0.196 (0.107–0.286) | < 0.001 |
| NRI categorical | 0.103 (0.064–0.142) | < 0.001 | 0.059 (0.029–0.090) | < 0.001 |
PCE ACC/AHA Pooled Cohorts Equations, FRS the Framingham Risk Score, IDI integrated discrimination improvement, NRI integrated discrimination improvement
mtDNA 8-OHdG and coronary stenosis severity in diabetic patients
For each diabetic patient, the severity of coronary stenosis was ordinally scored as the number of diseased vessels with stenosis of ≥ 50% and the quartiles of modified Gensini scores. Ordinal logistic regression showed that each 1-SD increase in mtDNA 8-OHdG was associated with a 29% higher odds of having more diseased vessels and a 28% greater odds of being a higher quartile of Gensini scores after model 3 adjustment (both P < 0.001, Table 4). When analyzing mtDNA 8-OHdG as an ordinal variable, the adjusted OR in the top versus bottom quartile of mtDNA 8-OHdG was 1.91 (95% CI 1.50–2.42) for having more diseased vessels, and 1.94 (95% CI 1.54–2.44) for being higher quartiles of Gensini scores. Sensitivity analyses based on PS matching continuously observed the significant associations of mtDNA 8-OHdH with the number of diseased vessels and Gensini scores (Table 4).
Table 4.
Associations of mtDNA 8-OHdG with coronary stenosis severity as determined by the number of disease vessels and quartiles of modified Gensini scores
| mtDNA 8-OHdG | No. of diseased vessels with > 50% stenosis | OR (95% CI)a | Quartiles of modified Gensini scoresb | OR (95% CI)a | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Non-CAD | 1 | 2 | 3/LMCAD | Q1 | Q2 | Q3 | Q4 | |||
| All participants | ||||||||||
| Each 1-SD increase | 866 | 448 | 337 | 269 | 1.29 (1.19–1.41) | 486 | 478 | 482 | 474 | 1.28 (1.18–1.39) |
| Quartiles, N (%) | ||||||||||
| Q1 | 261 (30.1) | 97 (21.7) | 86 (25.5) | 49 (18.2) | 1 (Reference) | 149 (30.6) | 137 (28.7) | 117 (24.3) | 90 (19.0) | 1 (Reference) |
| Q2 | 251 (29.0) | 108 (24.1) | 59 (17.5) | 57 (21.2) | 0.92 (0.72–1.18) | 132 (27.2) | 130 (27.2) | 116 (24.0) | 97 (20.4) | 1.08 (0.86–1.36) |
| Q3 | 213 (24.6) | 104 (23.2) | 85 (25.2) | 78 (29.0) | 1.33 (1.06–1.69) | 115 (23.7) | 110 (23.0) | 131 (27.2) | 124 (26.2) | 1.42 (1.13–1.78) |
| Q4 | 141 (16.3) | 139 (31.0) | 107 (31.8) | 85 (31.6) | 1.91 (1.50–2.42) | 90 (18.5) | 101 (21.1) | 118 (24.5) | 163 (34.4) | 1.94 (1.54–2.44) |
| P for trend | < 0.001 | < 0.001 | ||||||||
| After PS matching | ||||||||||
| Each 1-SD increase | 673 | 295 | 211 | 167 | 1.36 (1.22–1.50) | 358 | 364 | 288 | 336 | 1.33 (1.21–1.47) |
| Quartiles | ||||||||||
| Q1 | 187 (27.8) | 69 (23.4) | 56 (26.5) | 24 (14.4) | 1 (Reference) | 98 (27.4) | 108 (29.7) | 77 (26.8) | 53 (15.8) | 1 (Reference) |
| Q2 | 190 (28.2) | 71 (24.0) | 38 (18.0) | 39 (23.4) | 0.98 (0.73–1.31) | 95 (26.5) | 103 (28.3) | 66 (22.9) | 74 (22.0) | 1.12 (0.85–1.48) |
| Q3 | 176 (26.2) | 63 (21.4) | 54 (25.6) | 46 (27.5) | 1.27 (0.95–1.70) | 87 (24.3) | 80 (22.0) | 85 (29.5) | 87 (25.9) | 1.49 (1.14–1.96) |
| Q4 | 120 (17.8) | 92 (31.2) | 63 (29.9) | 58 (34.7) | 2.04 (1.53–2.72) | 78 (21.8) | 73 (20.0) | 60 (20.8) | 122 (36.3) | 2.01 (1.52–2.65) |
| P for trend | < 0.001 | |||||||||
PS propensity score, 8-OHdG 8-hydroxy-2′-deoxyguanosine, LMCAD left main coronary artery disease
aModels were adjusted for age, sex, smoking habit, alcohol drinking habit, overweight/obesity, hypertension, dyslipidemia., FPG, HbA1c, diabetes duration, and type of diabetes management
bThe median (interquartile range) of modified Gensini scores before and after PS matching was 27 (20–36) and 27 (20–35.5), respectively
mtDNA 8-OHdG and cardiometabolic phenotypes
We totally tested 11 biomarkers covering five categories of cardiometabolic phenotypes, i.e. blood lipids (LDL-C, HDL-c, TC, and TG), blood glucose (FPG, HbA1c), blood pressure (systolic and diastolic), proinflammatory measures (CRP and D-dimer), and adiposity measure (BMI). Overall, greater mtDNA 8-OHdG levels was significantly correlated with higher CRP levels (β = 0.182, P = 0.005), marginally with lower HDL-c levels (β = − 0.010, P = 0.059), but not with other 9 biomarkers after adjustment for confounders (Table 5). In the PS-matched cohort, only the association between mtDNA 8-OHdG and CRP remained significant.
Table 5.
Associations of mtDNA 8-OHdG with 11 cardiovascular biomarkers in patients with T2DM
| Variables | All participants | After PS matching | ||||
|---|---|---|---|---|---|---|
| N | β (95% CI)a | Pa | N | β (95% CI)a | Pa | |
| Blood lipids | ||||||
| TG (mmol/L) | 1920 | 0.035 (− 0.007, 0.078) | 0.100 | 1346 | 0.046 (− 0.006, 0.098) | 0.081 |
| TC (mmol/L) | 1920 | 0.002 (− 0.044, 0.048) | 0.925 | 1346 | − 0.001 (− 0.057, 0.054) | 0.962 |
| LDL-c (mmol/L) | 1920 | 0.014 (− 0.023, 0.050) | 0.462 | 1346 | − 0.008 (− 0.020, 0.005) | 0.251 |
| HDL-c (mmol/L) | 1920 | − 0.010 (− 0.020, 0.000) | 0.059 | 1346 | 0.011 (− 0.032, 0.053) | 0.498 |
| Blood glucose | ||||||
| FPG (mmol/L) | 1920 | 0.040 (− 0.038, 0.118) | 0.319 | 1346 | 0.001 (− 0.085, 0.087) | 0.987 |
| HbA1c (%) | 1920 | 0.000 (− 0.053, 0.053) | 0.996 | 1346 | − 0.038 (− 0.148, 0.071) | 0.492 |
| Blood pressure | ||||||
| SBP (mmHg) | 1920 | 1.971 (− 0.210, 4.043) | 0.077 | 1346 | 1.297 (− 0.186, 2.780) | 0.086 |
| DBP (mmHg) | 1920 | 0.725 (− 0.124, 1.575) | 0.094 | 1346 | 0.718 (− 0.277, 1.714) | 0.157 |
| Inflammation | ||||||
| CRP (mg/L) | 1920 | 0.182 (0.055, 0.309) | 0.005 | 1346 | 0.172 (0.028, 0.315) | 0.019 |
| D-dimer (mg/L) | 1798 | − 0.019 (− 0.067, 0.028) | 0.431 | 1258 | 0.000 (− 0.057, 0.055) | 0.980 |
| Adiposity | ||||||
| BMI (m/kg2) | 1920 | 0.068 (− 0.016, 0.153) | 0.111 | 1346 | 0.045 (− 0.162, − 0.252) | 0.670 |
Italic values indicate significant results with P < 0.05
BMI body mass index, FPG fasting plasma glucose, HbA1c glycosylated haemoglobin, TC total cholesterol, LDL-c low-density lipoprotein cholesterol, HDL-c high-density lipoprotein cholesterol, TG triglycerides, SBP systolic blood pressure, DBP diastolic blood pressure, CRP C-reactive protein
aMultivariable linear regression was performed with the mtDNA 8-OHdG as the independent variable and each cardiovascular biomarker as the dependent variable, with adjustment for age, sex, smoking habit, alcohol drinking habit, overweight/obesity, hypertension, dyslipidemia., diabetes duration, and type of diabetes management
mtDNA 8-OHdG and 1-year clinical outcomes after coronary revascularization
During a median follow-up of 12.4 (IQR: 11.7–12.6) months, 109 (15.5%) MACCEs occurred in 701 diabetic patients who underwent coronary revascularization (PCI: n = 475; CABG: n = 226). After adjustment for confounders, high level of mtDNA 8-OHdG was estimated as an independent predictor for MACCEs (HR = 1.59, 95% CI 1.33–1.90), especially for all-cause death (HR = 2.06) and cardiac death (HR = 1.92, all P < 0.001, Table 6). The time-dependent ROC at 12 months suggested that the optimal cut-off value of mtDNA 8-OHdG was 3.22 ∆ct for predicting MACCEs, with an AUC of 0.66, a sensitivity of 0.72, and a specificity of 0.55. Patients with mtDNA 8-OHdG values of > 3.22 ∆ct had a multivariable-adjusted HR of 2.74 (95% CI 1.80–4.17, P < 0.001, Table 6) for predicting MACCEs, compared with those without. When stratified by revascularization methods, the HRs for MACCE, all-cause death, and cardiac death were associated with increasing mtDNA 8-OHdG in the PCI group; there were positive associations of mtDNA 8-OHdG with MACCEs and all-cause death in the CABG group (Additional file 3: Table S1).
Table 6.
Associations of continuous and dichotomous mtDNA 8-OHdG with 1-year outcomes after coronary revascularization
| Outcomes | Continuous mtDNA 8-OHdG | Dichotomous mtDNA 8-OHdG | ||||
|---|---|---|---|---|---|---|
| HR (95%CI)a | Pa | No. of eventsb | HR (95%CI)a | Pa | ||
| High (n = 344) | Low (n = 357) | |||||
| MACCE | 1.59 (1.33–1.90) | < 0.001 | 78 (22.7) | 31 (8.7) | 2.74 (1.80–4.17) | < 0.001 |
| All-cause death | 2.06 (1.59–2.66) | < 0.001 | 34 (9.9) | 11 (3.1) | 3.36 (1.69–6.69) | < 0.001 |
| Cardiac death | 1.92 (1.33–2.78) | < 0.001 | 19 (5.5) | 5 (1.4) | 3.80 (1.40–10.3) | 0.009 |
| Repeat revascularization | 1.26 (0.98–1.62) | 0.078 | 47 (13.7) | 19 (5.3) | 2.67 (1.55–4.57) | < 0.001 |
| Non-fatal MI | 1.42 (0.91–2.22) | 0.126 | 12 (3.5) | 7 (2.0) | 2.30 (0.89–5.92) | 0.085 |
| Non-fatal stroke | 1.45 (0.59–3.58) | 0.417 | 3 (0.9) | 2 (0.6) | 1.71 (0.27–10.8) | 0.569 |
MACCE major adverse cardiovascular and cerebral events, MI myocardial infarction
aModels were adjusted for age, sex, smoking habit, alcohol drinking habit, overweight/obesity, hypertension, dyslipidemia., FPG, HbA1c, diabetes duration, and type of diabetes management
bThe cutoff that categorized the value of mtDNA 8-OHdG as high or low was 3.22 ∆ct based on the results of time-dependent ROC
In vitro experiments for mtDNA oxidative damage in HUVECs under hyperglycemic conditions
To provide in vitro evidence for the nature of the associations reported herein, we tested the effects of AMA, an inducer of mtDNA oxidative damage, in HUVECs under hyperglycemic conditions. As presented in Fig. 4a, exposure to 50 mM of AMA for 24 h led to a 3.7-times increase in mtDNA 8-OHdG compared with that in untreated cells, supporting the direct effect of AMA on mtDNA oxidative damage. Pretreatment of AMA also resulted in adverse alterations in markers of mitochondrial (i.e. decreased mtDNA-CN and PGC-1α expression, increased ∆ψm magnitude, Fig. 4b–d) and endothelia dysfunction (i.e. decreased NO levels and increased sVCAM-1 levels, Fig. 4e, f). Meanwhile, the cell viability in AMA-treated HUVECs was 37% lower than that in untreated HUVECs (Fig. 4h).
Fig. 4.

Effects of exposure to antimycin A, an inducer for mtDNA oxidative damage, on mtDNA 8-OHdG (a), mtDNA-CN (b), mitochondrial membrane potential (c), PGC1-α mRNA expression (d), sVCAM-1 levels (e), NO levels (f), E-selectin levels (g), and cell viability (h) in glucose-cultured HUVECs. Data are mean ± SD of three independent experiments. *P < 0.05, **P < 0.01 compared with the AMA-untreated cells
Discussion
To our knowledge, this is the first study to explore the association of 8-OHdG in leukocyte mtDNA with CAD in diabetic patients. First, in a cross-sectional design covering 1920 patients with T2DM from two provinces in China, mtDNA 8-OHdG was positively associated with CAD presence, coronary stenosis severity, and CRP levels. Then, in 701 prospectively followed diabetic patients, mtDNA 8-OHdG was suggested as a significant predictor of 1-year MACCEs after coronary revascularization. Finally, in vitro experiments showed that exposure to AMA, an inducer of mtDNA oxidative damage, led to adverse alterations in biomarkers of mitochondrial and endothelia dysfunction in HUVECs.
Previous studies consistently showed excreted levels of 8-OHdG in serum, plasma, or urine, mainly reflecting nuclear DNA damage, in association with coronary atherosclerosis in T2DM [43, 44]. This may be partly explained by the generally high caloric intake of diabetic patients with atherosclerotic cardiovascular disease, leading to an increase in mitochondrial energy synthesis, over-production of mitochondrial ROS, and consequently mtDNA oxidative damage in endothelial cells [45, 46]. However, large population studies linking mtDNA oxidative damage to CAD in T2DM are still sparse. A small case–control study involving 275 patients with diabetes or clinical atherosclerosis observed a significant association of mtDNA adducts in blood mononuclear cells with vascular function [47]. In our previous work, mtDNA-CN in leukocytes, while not a direct marker for mtDNA damage, was suggested as a risk factor for CAD, independent of diabetic status [18]. In the present study, we extended our previous work by directly measuring 8-OHdG in mtDNA, which had the advantage of straightly reflecting a steady-state level of oxidative DNA damage occurring within mitochondria. Besides the observation that the linear association between mtDNA 8-OHdG and CAD presence in patients with T2DM remained significant after adjusting for multiple cardiovascular risk factors and T2DM-related variables, we also found that adding mtDNA 8-OHdG into established risk prediction models improved risk discrimination and reclassification for CAD events. Recently, a case-series study of patients who underwent virtual histology intravascular ultrasound also suggested that elevated mtDNA damage in leukocytes was correlated with increased number of thin-capped fbroatheromas, which is a key feature of high-risk plaques [48]. These findings, combined with further in vivo evidence that mtDNA damage occurred prior to plaque formation in ApoE-null mice [48], suggest a causative role for mtDNA 8-OHdG as a risk factor for CAD in patients with T2DM.
The previous studies may not be large enough to observe the effect modification by smoking status on mtDNA oxidative damage in T2DM. So the current observation that the association between mtDNA 8-OHdG and CAD presence in diabetic patients was most significant for current smokers, but no longer significant for never smokers was meaningful. After all, a major contribution of cigarette smoking in the pathogenesis of diabetic macrovascular complications is the over-production of ROS, which may aggravate mtDNA oxidative damage and greatly increase CAD risk in patients with T2DM [49]. In population studies, smoking has been associated with increased mtDNA-CN and decreased activity of mitochondrial respiratory chain complexes in a dose-dependent manner [50, 51]. In vitro, the exposure to nicotine-derived nitrosamine ketone, a major toxic ingredient of cigarette smoke, may lead to imbalanced cellular redox status, defective mitochondrial biogenesis, and impaired energy production in mitochondria [52]. Taken together, our data support the view that cigarette smoking may modify the effects of mtDNA oxidative damage on the development of CAD in patients with T2DM.
A novel finding of our study is that mtDNA 8-OHdG was a significant predictor for composite MACCEs, especially for all-cause mortality and cardiac mortality after revascularization in patients with T2DM. This result was supported by our further observation that mtDNA 8-OHdG positively correlated with coronary stenosis severity, as determined by the number of diseased vessels and modified Gensini scores. Indeed, functional studies have suggested that mtDNA oxidative damage under diabetic conditions may trigger a series of pathological events including abnormal mitochondrial gene transcription, uncontrolled oxidative stress, and endothelial dysfunction [12, 53], all of which contribute to plaque progression and rupture [54]. In vitro, hyperglycemia may also induce the interaction of mtDNA oxidative damage with inflammatory cytokine release, causing apoptosis of monocytes, T2DM-related endothelial dysfunction, and plaque instability [48]. In line with this evidence, we found that mtDNA 8-OHdG positively correlated with CRP levels, a well established measure of systemic inflammation, in patients with T2DM, and that AMA-induced mtDNA oxidative damage was linked to adverse alterations in markers of endothelial dysfunction and cell viability in high glucose-cultured HUVECs. Together, these findings suggest that mtDNA oxidative damage is on a chain from cardiovascular morbidity to mortality in patients with T2DM.
The pivotal effect of elevated mtDNA 8-OHdG on obstructive CAD in T2DM raises the question of whether this effect is mediated by possible lifestyle or genetic factors. Recent evidence from aged mice showed that increased mitochondrial oxidative stress might induce age-associated vascular wall remodeling, causing aortic stiffening and cardiovascular disease [55, 56], whereas supplementation with cobalamin (i.e. vitamin B12) and folate could improve mitochondrial oxidative capacity to protect the failing heart in mice [57]. There was also ex vivo evidence showing that a dominant negative Ogg1 mutant F460A restored Ogg1 activity, and, consequently reduced mtDNA damage under the hyperglycemic milieus [15]. So, human studies, specifically exploring the mediation of mtDNA 8-OHdG variations by aging, B-vitamin nutrition status, and OGG1 mutations, would be of interest.
Our study has limitations. First, although we used a series of rigorous statistical approaches to minimize the effect of residual confounding, we could not establish a causal relationship between mtDNA 8-OHdG and obstructive CAD owing to our observational design. Second, our study only enrolled high-risk participants who were referred for coronary angiography. This may cause the selection bias that influences the stability of results. Third, further assessment of the dose–response effect of smoking and mtDNA 8-OHdG was not available, due to lack of collection of more detailed information on cigarette smoking such as pack-years of smoking or smoking duration. Fourth, the observation of the significant association between mtDNA 8-OHdG and CRP levels should be interpreted cautiously, because all cardiovascular biomarkers were measured at a single time point, which might bias the results. Finally, 1 year of follow-up was not long enough to assess the association of mtDNA 8-OHdG with long-term outcomes after coronary revascularization.
Conclusions
In summary, our data show that higher mtDNA 8-OHdG was cross-sectionally associated with increased odds of obstructive CAD, higher degree of coronary stenosis, and higher CRP levels, and prospectively with 1-year adverse clinical outcomes after coronary revascularization. Future prospective studies in seeking mtDNA-related biomarkers for incident CAD should focus on direct detection of mtDNA 8-OHdG rather than on other indirect measurements. Our in vitro experiments also suggest that exposure to AMA, an inducer for mtDNA oxidative damage, may correlate with adverse alterations in endothelia function in HUVECs. These results may serve as a basis for future physiological studies to explore whether AMA-induced endothelia dysfunction is directly caused by mtDNA 8-OHdG or mediated by other casual factors.
Supplementary information
Additional file 1: Methods. Assessment of artery stenosis severity and definitions of composites of MACCE.
Additional file 2: Fig. S1. Correlations of the data of mtDNA 8-OHdG quantified by aPCR and ELISA in 200 randomly selected participants. R coefficient and P value were obtained from the Pearson correlation test.
Additional file 3: Table S1. Associations of mtDNA 8-OHdG with 1-year clinical outcomes stratified by revascularization modality.
Additional file 4: Data S1. Original data of all 1920 participants analyzed in this study.
Acknowledgements
Not applicable.
Abbreviations
- 8-OHdG
8-Hydroxy-2′-deoxyguanosine
- CAD
Coronary artery disease
- T2DM
Type 2 diabetes mellitus
- mtDNA
Mitochondrial DNA
- ROS
Reactive oxygen species
- BMI
Body mass index
- FPG
Fasting plasma glucose
- HbA1c
Glycosylated haemoglobin
- CRP
C-reactive protein
- PS
Propensity score
- MACCE
Major adverse cardiovascular and cerebrovascular events
- HUVECs
Human umbilical vein endothelial cells
Authors’ contributions
XW contributed to design the work and draft the manuscript. XW, NC, and XL contributed to acquisition, analysis, or interpretation of data for the work. XL contributed to data presentation and statistical analysis. All authors read and approved the final manuscript.
Funding
This study was supported by the National Basic Research Program of China (81800317; to X.W.) and the Union Program of the Key Scientific and Technological Project of Henan Province (2018020008; to X.W.).
Availability of data and materials
Data generated or analyzed during this study are included in the Additional file 4: Data S1.
Ethics approval and consent to participate
The study protocol followed the Declaration of Helsinki and was approved by the Ethics Committees of participating centers. All participants signed written informed consent.
Consent for publication
All authors consent this manuscript for publication.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary information accompanies this paper at 10.1186/s12933-020-00998-6.
References
- 1.Niccoli G, Giubilato S, Di Vito L, Leo A, Cosentino N, Pitocco D, et al. Severity of coronary atherosclerosis in patients with a first acute coronary event: a diabetes paradox. Eur Heart J. 2013;34(10):729–741. doi: 10.1093/eurheartj/ehs393. [DOI] [PubMed] [Google Scholar]
- 2.Tancredi M, Rosengren A, Svensson AM, Kosiborod M, Pivodic A, Gudbjornsdottir S, et al. Excess mortality among persons with type 2 diabetes. N Engl J Med. 2015;373(18):1720–1732. doi: 10.1056/NEJMoa1504347. [DOI] [PubMed] [Google Scholar]
- 3.Venuraju SM, Lahiri A, Jeevarethinam A, Cohen M, Darko D, Nair D, et al. Duration of type 2 diabetes mellitus and systolic blood pressure as determinants of severity of coronary stenosis and adverse events in an asymptomatic diabetic population: PROCEED study. Cardiovasc Diabetol. 2019;18(1):51. doi: 10.1186/s12933-019-0855-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Takahashi H, Iwahashi N, Kirigaya J, Kataoka S, Minamimoto Y, Gohbara M, et al. Glycemic variability determined with a continuous glucose monitoring system can predict prognosis after acute coronary syndrome. Cardiovasc Diabetol. 2018;17(1):116. doi: 10.1186/s12933-018-0761-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gu J, Pan JA, Fan YQ, Zhang HL, Zhang JF, Wang CQ. Prognostic impact of HbA1c variability on long-term outcomes in patients with heart failure and type 2 diabetes mellitus. Cardiovasc Diabetol. 2018;17(1):96. doi: 10.1186/s12933-018-0739-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schottker B, Rathmann W, Herder C, Thorand B, Wilsgaard T, Njolstad I, et al. HbA1c levels in non-diabetic older adults—No J-shaped associations with primary cardiovascular events, cardiovascular and all-cause mortality after adjustment for confounders in a meta-analysis of individual participant data from six cohort studies. BMC Med. 2016;14:26. doi: 10.1186/s12916-016-0570-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vanhoutte PM, Shimokawa H, Feletou M, Tang EH. Endothelial dysfunction and vascular disease—a 30th anniversary update. Acta Physiol (Oxf) 2017;219(1):22–96. doi: 10.1111/apha.12646. [DOI] [PubMed] [Google Scholar]
- 8.Ravassa S, Beaumont J, Huerta A, Barba J, Coma-Canella I, Gonzalez A, et al. Association of low GLP-1 with oxidative stress is related to cardiac disease and outcome in patients with type 2 diabetes mellitus: a pilot study. Free Radic Biol Med. 2015;81:1–12. doi: 10.1016/j.freeradbiomed.2015.01.002. [DOI] [PubMed] [Google Scholar]
- 9.Chen S, Dong Z, Zhao Y, Sai N, Wang X, Liu H, et al. Homocysteine induces mitochondrial dysfunction involving the crosstalk between oxidative stress and mitochondrial pSTAT3 in rat ischemic brain. Sci Rep. 2017;7(1):6932. doi: 10.1038/s41598-017-07112-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mikhed Y, Daiber A, Steven S. Mitochondrial oxidative stress, mitochondrial DNA damage and their role in age-related vascular dysfunction. Int J Mol Sci. 2015;16(7):15918–15953. doi: 10.3390/ijms160715918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Widlansky ME, Hill RB. Mitochondrial regulation of diabetic vascular disease: an emerging opportunity. Transl Res. 2018;202:83–98. doi: 10.1016/j.trsl.2018.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shenouda SM, Widlansky ME, Chen K, Xu G, Holbrook M, Tabit CE, et al. Altered mitochondrial dynamics contributes to endothelial dysfunction in diabetes mellitus. Circulation. 2011;124(4):444–453. doi: 10.1161/CIRCULATIONAHA.110.014506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mercer JR, Cheng KK, Figg N, Gorenne I, Mahmoudi M, Griffin J, et al. DNA damage links mitochondrial dysfunction to atherosclerosis and the metabolic syndrome. Circ Res. 2010;107(8):1021–1031. doi: 10.1161/CIRCRESAHA.110.218966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kizhakekuttu TJ, Wang J, Dharmashankar K, Ying R, Gutterman DD, Vita JA, et al. Adverse alterations in mitochondrial function contribute to type 2 diabetes mellitus-related endothelial dysfunction in humans. Arterioscler Thromb Vasc Biol. 2012;32(10):2531–2539. doi: 10.1161/ATVBAHA.112.256024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cividini F, Scott BT, Dai A, Han W, Suarez J, Diaz-Juarez J, et al. O-GlcNAcylation of 8-oxoguanine DNA glycosylase (Ogg1) impairs oxidative mitochondrial DNA lesion repair in diabetic hearts. J Biol Chem. 2016;291(51):26515–26528. doi: 10.1074/jbc.M116.754481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kakimoto M, Inoguchi T, Sonta T, Yu HY, Imamura M, Etoh T, et al. Accumulation of 8-hydroxy-2′-deoxyguanosine and mitochondrial DNA deletion in kidney of diabetic rats. Diabetes. 2002;51(5):1588–1595. doi: 10.2337/diabetes.51.5.1588. [DOI] [PubMed] [Google Scholar]
- 17.Ashar FN, Zhang Y, Longchamps RJ, Lane J, Moes A, Grove ML, et al. Association of mitochondrial DNA copy number with cardiovascular disease. JAMA Cardiol. 2017;2(11):1247–1255. doi: 10.1001/jamacardio.2017.3683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang XB, Cui NH, Zhang S, Liu ZJ, Ma JF, Ming L. Leukocyte telomere length, mitochondrial DNA copy number, and coronary artery disease risk and severity: a two-stage case-control study of 3064 Chinese subjects. Atherosclerosis. 2019;284:165–172. doi: 10.1016/j.atherosclerosis.2019.03.010. [DOI] [PubMed] [Google Scholar]
- 19.Liu LP, Cheng K, Ning MA, Li HH, Wang HC, Li F, et al. Association between peripheral blood cells mitochondrial DNA content and severity of coronary heart disease. Atherosclerosis. 2017;261:105–110. doi: 10.1016/j.atherosclerosis.2017.02.013. [DOI] [PubMed] [Google Scholar]
- 20.Association AD. Diagnosis and classification of diabetes mellitus. Diabetes Care. 2014;37(Suppl 1):S81–S90. doi: 10.2337/dc14-S081. [DOI] [PubMed] [Google Scholar]
- 21.Rosenberg S, Elashoff MR, Beineke P, Daniels SE, Wingrove JA, Tingley WG, et al. Multicenter validation of the diagnostic accuracy of a blood-based gene expression test for assessing obstructive coronary artery disease in nondiabetic patients. Ann Intern Med. 2010;153(7):425–434. doi: 10.7326/0003-4819-153-7-201010050-00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee KY, Hwang BH, Kim TH, Kim CJ, Kim JJ, Choo EH, et al. Computed tomography angiography images of coronary artery stenosis provide a better prediction of risk than traditional risk factors in asymptomatic individuals with type 2 diabetes: a long-term study of clinical outcomes. Diabetes Care. 2017;40(9):1241–1248. doi: 10.2337/dc16-1844. [DOI] [PubMed] [Google Scholar]
- 23.Montorsi P, Ravagnani PM, Galli S, Rotatori F, Veglia F, Briganti A, et al. Association between erectile dysfunction and coronary artery disease. Role of coronary clinical presentation and extent of coronary vessels involvement: the COBRA trial. Eur Heart J. 2006;27(22):2632–2639. doi: 10.1093/eurheartj/ehl142. [DOI] [PubMed] [Google Scholar]
- 24.Patel MR, Calhoon JH, Dehmer GJ, Grantham JA, Maddox TM, Maron DJ, et al. ACC/AATS/AHA/ASE/ASNC/SCAI/SCCT/STS 2016 appropriate use criteria for coronary revascularization in patients with acute coronary syndromes: a report of the American College of Cardiology Appropriate Use Criteria Task Force, American Association for Thoracic Surgery, American Heart Association, American Society of Echocardiography, American Society of Nuclear Cardiology, Society for Cardiovascular Angiography and Interventions, Society of Cardiovascular Computed Tomography, and the Society of Thoracic Surgeons. J Am Coll Cardiol. 2017;69(5):570–591. doi: 10.1016/j.jacc.2016.10.034. [DOI] [PubMed] [Google Scholar]
- 25.Patel MR, Calhoon JH, Dehmer GJ, Grantham JA, Maddox TM, Maron DJ, et al. ACC/AATS/AHA/ASE/ASNC/SCAI/SCCT/STS 2017 appropriate use criteria for coronary revascularization in patients with stable ischemic heart disease: a report of the American College of Cardiology Appropriate Use Criteria Task Force, American Association for Thoracic Surgery, American Heart Association, American Society of Echocardiography, American Society of Nuclear Cardiology, Society for Cardiovascular Angiography and Interventions, Society of Cardiovascular Computed Tomography, and Society of Thoracic Surgeons. J Nucl Cardiol. 2017;24(5):1759–1792. doi: 10.1007/s12350-017-0917-9. [DOI] [PubMed] [Google Scholar]
- 26.Valle JA, Glorioso TJ, Bricker R, Barón AE, Armstrong EJ, Bhatt DL, et al. Association of coronary anatomical complexity with clinical outcomes after percutaneous or surgical revascularization in the veterans affairs clinical assessment reporting and tracking program. JAMA Cardiol. 2019;4(8):727–735. doi: 10.1001/jamacardio.2019.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang XB, Cui NH, Liu X, Ming L. Identification of a blood-based 12-gene signature that predicts the severity of coronary artery stenosis: an integrative approach based on gene network construction, Support Vector Machine algorithm, and multi-cohort validation. Atherosclerosis. 2019;291:34–43. doi: 10.1016/j.atherosclerosis.2019.10.001. [DOI] [PubMed] [Google Scholar]
- 28.National Cholesterol Education Program (NCEP) Expert Panel on Detection E, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) Third Report of the National Cholesterol Education Program (NCEP) expert panel on detection, evaluation, and treatment of high blood cholesterol in adults (adult treatment panel III) final report. Circulation. 2002;106(25):3143–3421. doi: 10.1161/circ.106.25.3143. [DOI] [PubMed] [Google Scholar]
- 29.James PA, Oparil S, Carter BL, Cushman WC, Dennison-Himmelfarb C, Handler J, et al. 2014 evidence-based guideline for the management of high blood pressure in adults: report from the panel members appointed to the Eighth Joint National Committee (JNC 8) JAMA. 2014;311(5):507–520. doi: 10.1001/jama.2013.284427. [DOI] [PubMed] [Google Scholar]
- 30.Lin CS, Wang LS, Tsai CM, Wei YH. Low copy number and low oxidative damage of mitochondrial DNA are associated with tumor progression in lung cancer tissues after neoadjuvant chemotherapy. Interact Cardiovasc Thorac Surg. 2008;7(6):954–958. doi: 10.1510/icvts.2008.177006. [DOI] [PubMed] [Google Scholar]
- 31.Lin CS, Wang LS, Chou TY, Hsu WH, Lin HC, Lee SY, et al. Cigarette smoking and hOGG1 Ser326Cys polymorphism are associated with 8-OHdG accumulation on mitochondrial DNA in thoracic esophageal squamous cell carcinoma. Ann Surg Oncol. 2013;20(Suppl 3):S379–S388. doi: 10.1245/s10434-012-2576-z. [DOI] [PubMed] [Google Scholar]
- 32.Sikorsky JA, Primerano DA, Fenger TW, Denvir J. Effect of DNA damage on PCR amplification efficiency with the relative threshold cycle method. Biochem Biophys Res Commun. 2004;323(3):823–830. doi: 10.1016/j.bbrc.2004.08.168. [DOI] [PubMed] [Google Scholar]
- 33.Hytti M, Korhonen E, Hyttinen JMT, Roehrich H, Kaarniranta K, Ferrington DA, et al. Antimycin A-induced mitochondrial damage causes human RPE cell death despite activation of autophagy. Oxid Med Cell Longev. 2019;2019:1583656. doi: 10.1155/2019/1583656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Widlansky ME, Wang J, Shenouda SM, Hagen TM, Smith AR, Kizhakekuttu TJ, et al. Altered mitochondrial membrane potential, mass, and morphology in the mononuclear cells of humans with type 2 diabetes. Transl Res. 2010;156(1):15–25. doi: 10.1016/j.trsl.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Caporarello N, Meridew JA, Jones DL, Tan Q, Haak AJ, Choi KM, et al. PGC1alpha repression in IPF fibroblasts drives a pathologic metabolic, secretory and fibrogenic state. Thorax. 2019;74(8):749–760. doi: 10.1136/thoraxjnl-2019-213064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lien LM, Chiou HY, Yeh HL, Chiu SY, Jeng JS, Lin HJ, et al. Significant association between low mitochondrial DNA content in peripheral blood leukocytes and ischemic stroke. J Am Heart Assoc. 2017;6(11):e006157. doi: 10.1161/JAHA.117.006157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Christiansen D, Murphy RM, Bangsbo J, Stathis CG, Bishop DJ. Increased FXYD1 and PGC-1alpha mRNA after blood flow-restricted running is related to fibre type-specific AMPK signalling and oxidative stress in human muscle. Acta Physiol (Oxf) 2018;223(2):e13045. doi: 10.1111/apha.13045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Harrell FE, Jr, Lee KL, Pollock BG. Regression models in clinical studies: determining relationships between predictors and response. J Natl Cancer Inst. 1988;80(15):1198–1202. doi: 10.1093/jnci/80.15.1198. [DOI] [PubMed] [Google Scholar]
- 39.Deb S, Austin PC, Tu JV, Ko DT, Mazer CD, Kiss A, et al. A review of propensity-score methods and their use in cardiovascular research. Can J Cardiol. 2016;32(2):259–265. doi: 10.1016/j.cjca.2015.05.015. [DOI] [PubMed] [Google Scholar]
- 40.Goff DC, Jr, Lloyd-Jones DM, Bennett G, Coady S, D’Agostino RB, Gibbons R, et al. 2013 ACC/AHA guideline on the assessment of cardiovascular risk: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation. 2014;129(25 Suppl 2):S49–S73. doi: 10.1161/01.cir.0000437741.48606.98. [DOI] [PubMed] [Google Scholar]
- 41.D’Agostino RB, Sr, Vasan RS, Pencina MJ, Wolf PA, Cobain M, Massaro JM, et al. General cardiovascular risk profile for use in primary care: the Framingham Heart Study. Circulation. 2008;117(6):743–753. doi: 10.1161/CIRCULATIONAHA.107.699579. [DOI] [PubMed] [Google Scholar]
- 42.Pencina MJ, D’Agostino RB, Sr, D’Agostino RB, Jr, Vasan RS. Evaluating the added predictive ability of a new marker: from area under the ROC curve to reclassification and beyond. Stat Med. 2008;27(2):157–172. doi: 10.1002/sim.2929. [DOI] [PubMed] [Google Scholar]
- 43.Di Minno A, Turnu L, Porro B, Squellerio I, Cavalca V, Tremoli E, et al. 8-Hydroxy-2-deoxyguanosine levels and cardiovascular disease: a systematic review and meta-analysis of the literature. Antioxid Redox Signal. 2016;24(10):548–555. doi: 10.1089/ars.2015.6508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stephens JW, Khanolkar MP, Bain SC. The biological relevance and measurement of plasma markers of oxidative stress in diabetes and cardiovascular disease. Atherosclerosis. 2009;202(2):321–329. doi: 10.1016/j.atherosclerosis.2008.06.006. [DOI] [PubMed] [Google Scholar]
- 45.Paneni F, Beckman JA, Creager MA, Cosentino F. Diabetes and vascular disease: pathophysiology, clinical consequences, and medical therapy: part I. Eur Heart J. 2013;34(31):2436–2443. doi: 10.1093/eurheartj/eht149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xuan Y, Gao X, Anusruti A, Holleczek B, Jansen E, Muhlack DC, et al. Association of serum markers of oxidative stress with incident major cardiovascular events, cancer incidence, and all-cause mortality in type 2 diabetes patients: pooled results from two cohort studies. Diabetes Care. 2019;42(8):1436–1445. doi: 10.2337/dc19-0292. [DOI] [PubMed] [Google Scholar]
- 47.Fetterman JL, Holbrook M, Westbrook DG, Brown JA, Feeley KP, Breton-Romero R, et al. Mitochondrial DNA damage and vascular function in patients with diabetes mellitus and atherosclerotic cardiovascular disease. Cardiovasc Diabetol. 2016;15:53. doi: 10.1186/s12933-016-0372-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yu E, Calvert PA, Mercer JR, Harrison J, Baker L, Figg NL, et al. Mitochondrial DNA damage can promote atherosclerosis independently of reactive oxygen species through effects on smooth muscle cells and monocytes and correlates with higher-risk plaques in humans. Circulation. 2013;128(7):702–712. doi: 10.1161/CIRCULATIONAHA.113.002271. [DOI] [PubMed] [Google Scholar]
- 49.Niemann B, Rohrbach S, Miller MR, Newby DE, Fuster V, Kovacic JC. Oxidative stress and cardiovascular risk: obesity, diabetes, smoking, and pollution: part 3 of a 3-part series. J Am Coll Cardiol. 2017;70(2):230–251. doi: 10.1016/j.jacc.2017.05.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Masayesva BG, Mambo E, Taylor RJ, Goloubeva OG, Zhou S, Cohen Y, et al. Mitochondrial DNA content increase in response to cigarette smoking. Cancer Epidemiol Biomarkers Prev. 2006;15(1):19–24. doi: 10.1158/1055-9965.EPI-05-0210. [DOI] [PubMed] [Google Scholar]
- 51.Miro O, Alonso JR, Jarreta D, Casademont J, Urbano-Marquez A, Cardellach F. Smoking disturbs mitochondrial respiratory chain function and enhances lipid peroxidation on human circulating lymphocytes. Carcinogenesis. 1999;20(7):1331–1336. doi: 10.1093/carcin/20.7.1331. [DOI] [PubMed] [Google Scholar]
- 52.Wu S, Li X, Meng S, Fung T, Chan AT, Liang G, et al. Fruit and vegetable consumption, cigarette smoke, and leukocyte mitochondrial DNA copy number. Am J Clin Nutr. 2019;109(2):424–432. doi: 10.1093/ajcn/nqy286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chiang SC, Meagher M, Kassouf N, Hafezparast M, McKinnon PJ, Haywood R, et al. Mitochondrial protein-linked DNA breaks perturb mitochondrial gene transcription and trigger free radical-induced DNA damage. Sci Adv. 2017;3(4):e1602506. doi: 10.1126/sciadv.1602506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Forstermann U, Xia N, Li H. Roles of vascular oxidative stress and nitric oxide in the pathogenesis of atherosclerosis. Circ Res. 2017;120(4):713–735. doi: 10.1161/CIRCRESAHA.116.309326. [DOI] [PubMed] [Google Scholar]
- 55.Zhou RH, Vendrov AE, Tchivilev I, Niu XL, Molnar KC, Rojas M, et al. Mitochondrial oxidative stress in aortic stiffening with age: the role of smooth muscle cell function. Arterioscler Thromb Vasc Biol. 2012;32(3):745–755. doi: 10.1161/ATVBAHA.111.243121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vendrov AE, Vendrov KC, Smith A, Yuan J, Sumida A, Robidoux J, et al. NOX4 NADPH oxidase-dependent mitochondrial oxidative stress in aging-associated cardiovascular disease. Antioxid Redox Signal. 2015;23(18):1389–1409. doi: 10.1089/ars.2014.6221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Piquereau J, Moulin M, Zurlo G, Mateo P, Gressette M, Paul JL, et al. Cobalamin and folate protect mitochondrial and contractile functions in a murine model of cardiac pressure overload. J Mol Cell Cardiol. 2017;102:34–44. doi: 10.1016/j.yjmcc.2016.11.010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Methods. Assessment of artery stenosis severity and definitions of composites of MACCE.
Additional file 2: Fig. S1. Correlations of the data of mtDNA 8-OHdG quantified by aPCR and ELISA in 200 randomly selected participants. R coefficient and P value were obtained from the Pearson correlation test.
Additional file 3: Table S1. Associations of mtDNA 8-OHdG with 1-year clinical outcomes stratified by revascularization modality.
Additional file 4: Data S1. Original data of all 1920 participants analyzed in this study.
Data Availability Statement
Data generated or analyzed during this study are included in the Additional file 4: Data S1.