

Abstract
Spinal bulbar muscular atrophy (SBMA) is an adult-onset, slowly progressive motor neuron disease caused by abnormal CAG repeat expansion in the androgen receptor (AR) gene. Although ligand (testosterone)-dependent mutant AR aggregation has been shown to play important roles in motor neuronal degeneration by the analyses of transgenic mice models and in vitro cell culture models, the underlying disease mechanisms remain to be fully elucidated because of the discrepancy between model mice and SBMA patients. Thus, novel human disease models that recapitulate SBMA patients’ pathology more accurately are required for more precise pathophysiological analysis and the development of novel therapeutics. Here, we established disease specific iPSCs from four SBMA patients, and differentiated them into spinal motor neurons. To investigate motor neuron specific pathology, we purified iPSC-derived motor neurons using flow cytometry and cell sorting based on the motor neuron specific reporter, HB9e438::Venus, and proceeded to the genome-wide transcriptome analysis by RNA sequences. The results revealed the involvement of the pathology associated with synapses, epigenetics, and endoplasmic reticulum (ER) in SBMA. Notably, we demonstrated the involvement of the neuromuscular synapse via significant upregulation of Synaptotagmin, R-Spondin2 (RSPO2), and WNT ligands in motor neurons derived from SBMA patients, which are known to be associated with neuromuscular junction (NMJ) formation and acetylcholine receptor (AChR) clustering. These aberrant gene expression in neuromuscular synapses might represent a novel therapeutic target for SBMA.
Keywords: Spinal bulbar muscular atrophy, Induced pluripotent stem cells, iPSC-derived motor neurons, RNA sequencing, Gene set enrichment analysis, Synapse, Neurotransmitter, Neuromuscular junctions, Epigenetics, Endoplasmic reticulum
Introduction
Spinal bulbar muscular atrophy (SBMA) is an adult-onset slowly progressive lower motor neuron (MN) disease caused by abnormal CAG repeat expansion in the androgen receptor (AR) gene. SBMA is characterized by weakness and atrophy of limbs and bulbar muscles caused by the degeneration of spinal and bulbar MNs [1–3]. Through analysis using transgenic mice models harboring mutant AR with expanded polyglutamine tract (AR-97Q) and in vitro cell culture models, mutant AR has been shown to form nuclear aggregation in a ligand (testosterone)-dependent manner, causing neuronal cell death through transcriptional dysregulation, impaired axonal transport, and other mechanisms [4, 5]. However, mechanisms underlying neuronal degeneration in SBMA are not yet fully elucidated, as SBMA model mice exhibit several discrepancies with human SBMA patients. For instance, the number of CAG repeat required for the onset of the disease is 38 or more in patients [6, 7], but more than 90 in transgenic mice models [8]. Moreover, the skeletal muscle degeneration is more prominent in model mice than in human patients [8]. From the aspect of therapeutics, although leuprorelin acetate, a luteinizing hormone-releasing hormone (LHRH) analogue, dramatically improved symptoms of AR-97Q mice [9], it could not ameliorate the symptoms of human SBMA patients, as had been observed in model mice, and induced an antianabolic action of skeletal muscle as an adverse effect [10, 11]. Therefore, a novel human disease model that more accurately recapitulates SBMA patients’ pathology has been expected for more precise pathophysiological analysis and the development of novel therapeutics.
Interestingly, recent analyses have shown that phenotypes and MN pathology of transgenic mice models were unexpectedly rescued by muscle-specific silencing of mutant AR, suggesting the involvement of muscle pathology in motor neuronal degeneration [12, 13]. In addition, impaired transmission of neuromuscular synapses was observed in SBMA mice models [14]. These results strongly indicate muscular pathology, which causes neurodegeneration in SBMA. However, the molecular pathology underlying neuromuscular interactions has not been sufficiently investigated, due to the lack of appropriate disease models.
Disease specific human induced pluripotent stem cells (hiPSCs) provide valuable disease models for neurodegenerative disorders [15, 16]. They are able to give rise to otherwise unavailable neural cells, which more accurately model patients’ pathology and provide us with new platforms for pathophysiological analysis and drug discovery [17]. For instance, α-synuclein accumulation observed in PARK2 iPSC-derived dopaminergic neurons well corresponded to the pathological findings of the Lewy body pathologies observed in autopsied brain specimens of the same patient [18]. In addition, MNs derived from sporadic amyotrophic lateral sclerosis (ALS)-iPSCs exhibited a time course of neuronal degeneration consistent with the clinical course observed in corresponding ALS patients [19].
On the other hand, iPSC-derived disease models still have issues to overcome, including time and labor required for the establishment and differentiation of iPSCs, heterogeneities of differentiated neural cells, and large variations among iPSC clones. In particular, the phenotypes or molecular changes may be masked by variations of the differentiation efficiency among iPSC clones, indicating the necessity of a method to purify target cells. To overcome these issues, we previously established rapid, efficient, and simple MN differentiation system from hiPSCs, in which MN progenitors could be obtained within two weeks with the induction efficiency of HB9+-MNs of approximately 40–50%. Moreover, MNs could be enriched by flow cytometry and cell sorting with MN specific HB9e438::Venus reporter lentivirus [20].
In this study, we established iPSCs from four SBMA patients, differentiated them into MNs, and investigated molecular pathology behind the motor neuronal degeneration in SBMA. We focused on common molecular changes among multiple iPSC clones derived from different patients. Differentiated MNs were purified using the lentiviral reporter system to overcome clonal variations of iPSCs, and to elucidate MN specific pathology. Furthermore, genome-wide transcriptome analyses by RNA sequences revealed the involvement of the key molecular pathology associated with synapses, epigenetics, and endoplasmic reticulum (ER) in SBMA. Pathology associated with neuromuscular synapses such as Synaptotagmin proteins, RSPO2, and WNT ligands were particularly highlighted, which may serve as novel therapeutic targets for SBMA.
Results
Establishment of SBMA disease specific iPSCs
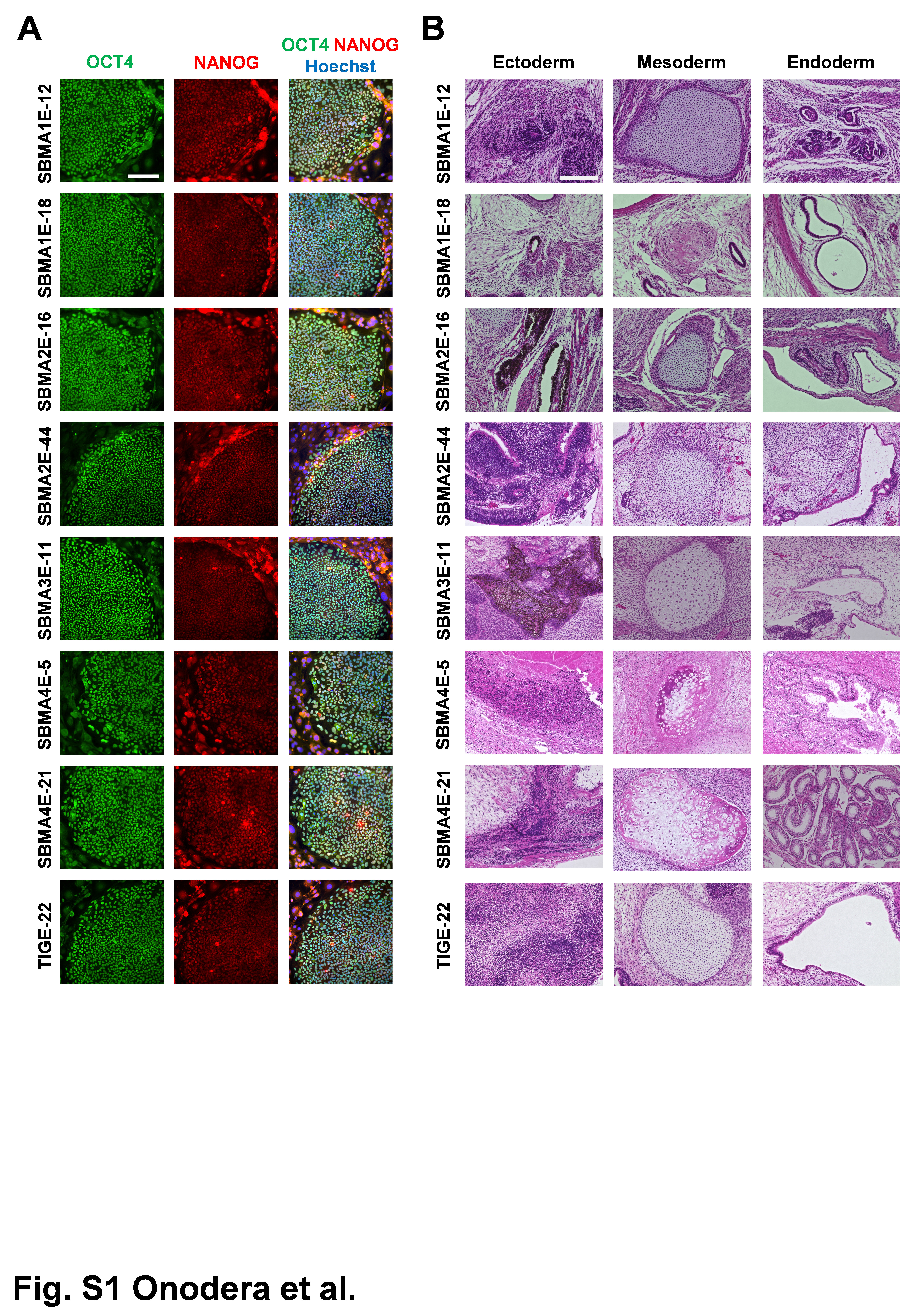
We first established iPSCs from skin fibroblasts isolated from four SBMA patients (SBMA1, 2, 3, and 4), and three healthy age-matched control adults (TIG114, YF, KN) (Table 1), by introducing OCT4, SOX2, KLF4, L-MYC, LIN28, and shTP53 via episomal vectors. Three of the control iPSC clones, TIGE-9, YFE-16, and EKN3, have already been described previously [20, 21] (Table 1). All the SBMA and control iPSC clones showed characteristic human embryonic stem cell-like morphology and expressed pluripotent stem cell markers, Oct4 and Nanog, as revealed by immunocytochemical analysis (Fig. 1a and Additional file 1: Figure S1). The expressions of OCT4 and NANOG were also confirmed by quantitative RT-PCR, which showed comparable expression to KhES1 (human embryonic stem cells) [22] in all the clones examined (Fig. 1b). We also examined teratoma forming capacity, which showed differentiation potentials into all three germ layers (Fig. 1c and Additional file 1: Figure S1), and normal karyotypes by G-banding (Fig. 1d and Additional file 2: Figure S2). These results suggest that we have established iPSC clones with the quality necessary for pathophysiological analysis.
Table 1.
Characterization of iPSCs
| Name | Race | Gender | Age | CAG repeat |
iPSC clones | |
|---|---|---|---|---|---|---|
| SBMA | SBMA1 | Japanese | male | 42 | 52 | SBMA1E-12, 18 |
| SBMA2 | Japanese | male | 46 | 47 | SBMA2E-16, 44 | |
| SBMA3 | Japanese | male | 33 | 50 | SBMA3E-10, 11 | |
| SBMA4 | Japanese | male | 38 | 49 | SBMA4E-5, 21 | |
| Control | TIG114 | Japanese | male | 36 | 24 | TIGE-9a, 22 |
| YF | Japanese | male | 24 | 21 | YFE-16a, 19 | |
| KN | Japanese | male | 39 | 20 | EKN3b |
Fig. 1.

Establishment of iPSCs from SBMA patients and controls. a Immunocytochemical analysis of YFE-19 and SBMA3E-10 iPSCs for pluripotent markers OCT4 (green) and NANOG (red). Scale bar, 100 μm. The established iPSC clones were positive for OCT4 and NANOG. b Quantitative RT-PCR analysis of the expression of pluripotent makers OCT4 and NANOG in the established iPSC clones. Data are normalized to β-ACTIN and presented as the relative expressions to KhES1 (hESC). All established iPSC clones expressed OCT4 and NANOG. c Hematoxylin and eosin staining of teratomas derived from YFE-19 and SBMA3E-10 iPSCs. Scale bar, 200 μm. The established iPSC clones were able to differentiate into 3 germ layers. d Karyotype analysis of YFE-19 and SBMA3E-10 iPSCs via G-banding analysis. The established iPSC clones showed normal karyotypes, 46, XY. See also Additional file 1: Figures S1 and S2, Additional file 2
Stability of CAG repeat numbers during reprogramming
To determine the stability of expanded CAG repeat during reprogramming, we next examined the length of the CAG repeat in the AR gene by fragment analysis. As expected, the length of the CAG repeat in each iPSC clone was stable during reprogramming, except for SBMA1E-18, whose number of CAG repeat increased from 52 to 55 (Fig. 2a). These results were confirmed by direct Sanger sequencing, as shown in Fig. 2b (representative images of YFE-19 and SBMA3E-10 are shown) and Table 1. These results suggest that the CAG repeat number was maintained during the reprogramming process in SBMA.
Fig. 2.

Stability of CAG repeats in the AR gene during reprogramming. a CAG repeat determination by fragment analysis. CAG repeat number in the AR gene was stable during reprogramming in the established iPSC clones, except for SBMA1E-18, which showed 55 CAG repeats. b Direct sequence analysis of CAG repeats in genomic DNA from YFE-19 and SBMA3E-10 iPSCs showed 21 and 50 CAG repeats, respectively. Repeat sequencing was read from the 3′ side
Differentiation of SBMA disease specific iPSCs into MNs
To confirm the differentiation potentials of established iPSC clones, control and SBMA iPSC clones were induced to differentiate into MNs by the previously reported method [20]. After 2 weeks of embryoid body (EB) formation, which contains MN progenitors, the EBs were dissociated into single cells and adherently differentiated and maturated on poly-L-ornithine- and laminin-coated cover glasses or plates for 1–4 weeks (maturation culture) (Fig. 3a). In one week, we confirmed differentiation into HB9+ and ISL-1+ MNs from all SBMA and control iPSC clones by immunocytochemistry (Fig. 3b). According to quantitative RT-PCR analysis, the expressions of HB9 and ISL-1 in SBMA iPSC-derived MNs were higher than those in control iPSC-derived MNs, while mature MN markers ChAT and AR did not show significant differences at 4 weeks of maturation culture (Fig. 3c). These results suggest that all the established clones have the potential to sufficiently differentiate into MNs, and exhibit some clonal variations for the differentiation propensity among hiPSC clones.
Fig. 3.

Differentiation of SBMA disease specific iPSCs into motor neurons. a Schematic presentation of the culture protocol for the differentiation of iPSCs. LSC, LDN-193189 (L), SB4315342 (S), CHIR99021 (C); RA, retinoic acid; PM, purmorphamine. b Immunocytochemical analysis of HB9, ISL-1, and βIII-Tubulin after 1 week of monolayer differentiation with 10 nM DHT. Scale bar, 100 μm. c Quantitative RT-PCR analysis of HB9, ISL-1, ChAT, and AR expressions at 4 weeks (n = 4, mean ± SEM). HB9 and ISL-1 expressions were higher in SBMA than controls in the presence of 10 nM DHT (*p < 0.05; Student’s t-test)
Purification of HB9e438::Venus positive MNs by flow cytometry and cell sorting
For the transcriptome analysis using purified/enriched populations of MNs, MNs were infected with the HB9e438::Venus reporter lentivirus four days after the initiation of the maturation culture, and were cultured for a further four weeks in the presence of 10 nM dihydrotestosterone (DHT), which is a ligand of AR, to detect ligand-dependent pathology of SBMA. The cells were then detached and dissociated into single cells, and purified by flow cytometry and cell sorting based on HB9e438::Venus fluorescence (Fig. 4a). The cells were divided into 3 fractions, a negative fraction (NF: gated by the fluorescence intensity of uninfected controls, gray peak), positive fraction (PF: green peak), where the top 2/3 of PF was determined as high-positive fractions (HPF; purified MN fraction), and others (Fig. 4b), following previously reported protocol with some modifications [20]. We also examined the expression of Venus fluorescent protein from a reporter lentivirus containing the same vector backbone, but without the HB9e438 enhancer element (background reporter lentivirus; blue peak in Fig. 4b) and found that the fluorescence observed in the HPF of HB9e438::Venus reporter-transduced cells overlapped minimally with the fluorescence generated by the background reporter. We further confirmed the expression of the MN markers, HB9, ISL-1 and ChAT, and astrocyte maker, GFAP, by quantitative RT-PCR using NF and HPF. We observed significant increases in the expression of MN markers and a decrease in the astrocyte maker in HPF. The expression levels of HB9, ISL-1, and ChAT in HPF were 7.7 ± 3.1-folds, 5.0 ± 0.7-folds, and 3.4 ± 0.7-folds higher than those in NF (n = 4, p < 0.05) (Fig. 4c), respectively, suggesting HPF was highly purified MN fraction. Although the expressions of HB9 and ISL-1 were higher in SBMA iPSC-derived MNs compared with those of control iPSC-derived MNs before purification, which indicates clonal variation of the differentiation efficiencies (Fig. 3c), they were equally expressed in all the clones after purification, suggesting that the clonal variation was corrected by the purification of MNs by flow cytometry and cell sorting based on HB9e438::Venus. The significant decrease in the expression of GFAP indicated the elimination of non-MNs/astrocytes from MN cultures by purification (0.56 ± 0.12-folds, n = 4, p < 0.05). AR expression was comparable in the NF and the HPF, consistent with the finding that AR expression was not specific to MNs.
Fig. 4.

Purification of HB9e438::Venus positive MNs by flow cytometry. a Schematic presentation of the culture protocol for flow cytometry and cell sorting. b Schematic histogram for determining a negative fraction (NF) and high-positive fraction (HPF). NF consists of uninfected cells (gray). HPF consists of top 2/3 fraction of HB9e438::Venus+ cells (green). NT consists of unpurified cells. Blue, background lentiviral infected cells. c Quantitative RT-PCR analysis of HB9, ISL-1, ChAT, AR, and GFAP expressions at 4 weeks. Data are normalized to β-ACTIN and presented as the relative expressions to controls (n = 4, means ± SEM). Significant increases in the expressions of HB9, ISL-1, and ChAT are observed in the HPF. A significant decrease in the expression of GFAP is observed in the HPF (*p < 0.05; Student’s t-test)
Differentially expressed genes between SBMA- and control-MNs by RNA sequencing
To investigate disease-associated genes in SBMA-MNs, total RNA was extracted from both unpurified MNs (NT) and purified MN fraction (HPF) from MNs derived from 4 SBMA-iPSCs and 4 control-iPSCs, and processed for RNA sequencing analysis. As a result, 119 genes showed adjusted p-values less than 0.05 in NT, and 79 genes in HPF (Fig. 5a). In total, of these differentially expressed genes (DEGs) in NT and HPF, 107 genes were upregulated and 73 genes were downregulated in SBMA-MNs (Fig. 5b). The heatmap of the DEGs showed that the majority of DEGs in NT were upregulated genes in SBMA-MNs, while the majority of DEGs in HPF were downregulated genes (Fig. 5c, d). All the ranked genes were shown in Additional file 3: Tables S1 and S2. Among these DEGs, CXC motif chemokine ligand 14 (CXCL14), Insulin-like growth factor-1 (IGF-1), Neurotrophin-3 (NTF3, NT-3), Glutamate ionotropic receptor delta type subunit 2 (GRID2), Glutamate ionotropic receptor kainate type subunit 1 (GRIK1), Glutamate metabotropic receptor 2 (GRM2), Regulator of G protein signaling 4 (RGS4) and Regulator of G protein signaling 5 (RGS5) are markedly upregulated, and Family with sequence similarity 135 member B (FAM135B) and Membrane-type frizzled-related protein (MFRP) are prominently downregulated, as shown in Fig. 5c, d, and Fig. 6d. As dysregulation of these DEGs are also previously shown to be associated with neurodegeneration, they could also be involved in the pathology of SBMA.
Fig. 5.

Identification of differentially expressed genes (DEGs) by RNA sequencing analysis. a Number of DEGs in the RNA-seq data, adjusted p-value (p-adj) < 0.05. The number of genes with p-adj < 0.05 in NT was 119, and 79 genes in HPF from a total of 58,825 annotated genes. b The Venn diagrams of up-regulated or down-regulated genes in SBMA (p-adj < 0.05). Up-regulation of 107 genes and down-regulation of 73 genes are shown. c The heatmap of the hierarchical clustering analysis based on the DEGs with p-adj < 0.05 in NT. Red represents higher expression and green indicates lower expression. For the list of the genes, see also Additional file 3: Table S1. d The heatmap of hierarchical clustering analysis based on the DEGs with p-adj < 0.05 in HPF. Red represents higher expression and green indicates lower expression. For the list of the genes, see also Additional file 3: Table S2
Fig. 6.

Enrichment of synapse-related pathology by GSEA. a Gene ontology (GO) enrichment analysis. Up-regulated gene sets in SBMA with FDR < 0.2 are shown. Blue indicates enriched in NT, and red in HPF. For negatively enriched GO terms, see also Tables S4 and S5, Additional file 3. b Pathways significantly enriched in SBMA with FDR < 0.2 are shown. Blue indicates enriched in NT, and red in HPF. (R), Reactome is a curated database of pathways and reactions in human biology. For negatively enriched pathway terms, see also Tables S6 and S7, Additional file 3. c Representative enrichment plots. Synapse, neurotransmitter, exocytosis, and epigenetics associated gene sets were involved in enrichment of SBMA. Ribosome and ER were negatively enriched in SBMA. The green curves show the enrichment score and reflect the degree to each black line, which shows a position of a gene in the ranked list of genes. NES, normalized enrichment score. FDR, false discovery rate. ER, endoplasmic reticulum. d The focused gene expression from DEGs and GSEA. The vertical axis represented relative log2 fold change (SBMA / Control) in the gene expression (n = 4, mean ± SEM)
Involvement of synapse pathology in SBMA shown by GSEA
To investigate important pathways and signals involved in SBMA pathology, we next performed gene set enrichment analysis (GSEA) [23]. The dataset has 58,825 native features. After collapsing features into gene symbols, there are 57,238 genes remaining. In the gene ontology (GO) analysis, gene set size filters (minimum = 15, maximum = 500) resulted in filtering out 4652 / 9996 gene sets. The remaining 5344 gene sets were used in the analysis. In the pathway analysis, gene set size filters resulted in filtering out 1406 / 5501 gene sets. The remaining 4095 gene sets were used in the analysis. The numbers of gene sets either up- or down-regulated in SBMA is listed in Additional file 3: Table S3.
GO analysis revealed that the gene sets that were enriched in SBMA-MNs included gene sets related to synapse in both NT and HPF; those related to synaptic vesicles, calcium ions, and exocytosis in HPF (Fig. 6a). Pathway analysis revealed that gene sets related to neurotransmitters were enriched in SBMA-MNs in both NT and HPF (Fig. 6b). Among these gene sets, acetylcholine neurotransmitter release cycle could be closely associated with synaptic pathology in MNs, and all gene sets were associated with the synaptic function and development of NMJs. In addition, the gene sets associated with histone methylation, including H3K4me2 and H3K27me3, were also enriched in SBMA in HPF, and located at higher ranks, suggesting that epigenetics was also involved in the pathogenesis of SBMA, as previously reported [24]. Representative enrichment plots which were significantly enriched in SBMA with regards to synapse, neurotransmitter, exocytosis, and epigenetics are shown in Fig. 6c (upper 4 panels). The members of each gene set (black lines) appear close to SBMA (red zone). This distribution indicates significant enrichment in SBMA.
As for negatively enriched gene sets in SBMA-MNs in GO analysis, chromosome and ribosome related gene sets were occasionally detected in both NT and HPF. Furthermore, in HPF, in addition to the gene sets of mRNA catabolic processes and ribosomes, the gene sets of endoplasmic reticulum (ER) are positioned at higher ranks (Additional file 3: Tables S4 and S5), suggesting that there is decreased protein synthesis in SBMA-MNs and the involvement of ER in SBMA pathology (Additional file 3: Tables S4-S7). Representative enrichment plots which were significantly enriched in control-MNs, including ribosome and ER are shown in Fig. 6c (lower 2 panels). The members of the gene set (black lines) appear close to the control (blue zone).
Finally, we investigated genes that are particularly elevated in SBMA-MNs, among gene sets detected by GO and pathway analyses, using a FDR q-value < 0.2, fold change ≧ 2, and p < 0.05. As a result, 96 genes were extracted (Additional file 3: Table S8), including Calcitonin-related polypeptide α (CALCA), which has previously been reported to be involved in SBMA pathogenesis [25], GRID2, GRIK1, R-spondin 2 (RSPO2), Synaptotagmin6 (SYT6), Synaptotagmin9 (SYT9), Wnt family member 2B (WNT2B) and Wnt family member 3A (WNT3A), most of which are involved in the synaptic functions in NMJs independent of agrin (Fig. 6d). Agrin (AGRN), which induces acetylcholine receptor (AChR) clustering at NMJs, did not show significant alteration (Fig. 6d), suggesting that neuromuscular pathology, independent of agrin, plays crucial roles in the pathogenesis of SBMA (Fig. 6d).
Discussion
Stability of CAG repeat numbers in SBMA disease specific iPSCs
Some of the triplet repeat diseases exhibit trinucleotide repeat instability, which results in the increase of repeat numbers, earlier disease onset, and increased severity of the disease in successive generations, known as anticipation. In SBMA, such an instability of the CAG repeat has not been observed in somatic and germ cells [26], and the anticipation is rare [7]. These observations are consistent with our results, in which CAG repeat expansions in the AR gene were relatively stable during reprogramming to establish iPSCs. Similar results were obtained in the previously established SBMA disease specific iPSCs, in which CAG repeat numbers were unaltered during reprogramming, long-term maintenance of iPSCs, and differentiation [27, 28], except for one report that showed variations in the repeat number, possibly due to the mosaicism of parental fibroblasts [29].
However, the consistency between the trinucleotide repeat instability in patients’ tissues and those in iPSCs is still not clear in other triplet repeat diseases, such as Huntington disease (HD), caused by CAG repeat expansion in the Huntingtin (HTT) gene, and myotonic dystrophy (DM1), caused by CTG repeat expansion in the 3′ UTR of the DMPK gene, both of which exhibit somatic instability of the repeats, and significant anticipation [30]. Although the augmentation of the CAG repeat expansion was not observed during reprograming into iPSCs, the long-term maintenance of iPSCs, and the neural differentiation in HD disease-specific iPSCs [31, 32], all 41 iPSC clones established from DM1 patients’ fibroblasts showed different CTG repeat length as expected [33]. Thus, trinucleotide repeat instability during reprogramming does not completely correspond to that observed in patients’ tissues, and the details remain yet to be elucidated.
Identification of cytokines and neurotrophic factors involved in motor neuron degeneration
Since mutant AR is known to translocate into the nucleus of MNs in a ligand (testosterone)-dependent manner, causing transcriptional dysregulation and MN degeneration in SBMA [5, 8], iPSC-derived MNs were treated with 10 nM DHT for 4 weeks to detect ligand-dependent pathogenesis. A previous report has shown that SBMA iPSC-derived MNs demonstrated relatively mild cellular phenotypes without mutant AR aggregations for up to 30 days of MN differentiation culture in the presence of DHT [34]. Since our SBMA iPSC-derived MNs were also cultured for up to 4 weeks in this study, they may have recapitulated similar molecular changes to those observed in the above-mentioned pathological stages without mutant AR aggregations.
Through RNA sequence analysis of four SBMA-MNs and four control-MNs, we identified 79 and 119 DEGs in purified (HPF) and unpurified (NT) MNs, respectively. Only 18 genes were commonly identified, suggesting the purification of MN enabled the detection of MN-specific pathology (Fig. 5b). More importantly, several molecules which have been previously reported to be associated with SBMA pathology, such as CALCA and FAM135B, were also identified [25, 34], indicating reproducibility of the previous findings in our iPSC-derived SBMA model. Among DEGs, several cytokines and neurotrophic factors were upregulated in SBMA-MNs, including CXCL14, IGF-1, and NTF3. CXCL14 is known to be upregulated in mice exhibiting similar phenotypes to ALS [35], suggesting that it may also be involved in MN degeneration in SBMA. IGF-1 is shown to suppress apoptosis of a mutant AR expressing MN cell line (MN-1) by phosphorylating AR via Akt, which inhibits ligand-dependent translocation of the mutant AR into the nucleus [36]. IGF-1 and Pituitary adenylyl cyclase activating polypeptide (PACAP) rescued the reduction of depolarizing current and electro-physiologically improved MN-1 cells that express mutant AR (AR100Q) [37]. Moreover, muscle specific overexpression of IGF-1 rescued the phenotypes of SBMA model mice, which include mutant AR aggregations through Akt-dependent phosphorylation of the mutant AR [38]. Thus, the upregulation of IGF-1 in SBMA-MNs could be attributable to the compensatory negative feedback of rescuing the pathology of SBMA. NTF3 (NT-3) is primarily known to be expressed and involved in the development of proprioceptive neurons in dorsal root ganglia [39, 40]. NT-3 also promotes survival and regeneration of MNs [41, 42], and formation and function of NMJs, including neuromuscular transmission [43, 44]. Thus, the upregulation of NTF3 in SBMA-MNs could induce increased synapse density or could be the result of compensatory negative feedback, associated with neuronal or neuromuscular synapse degeneration.
GSEA revealed core gene sets involved in the pathogenesis of SBMA
According to GSEA, upregulation of (1) synapse-, (2) neurotransmitter-, (3) calcium-related exocytosis-, and (4) epigenetics-related gene sets, and downregulation of (5) ER-related gene sets were identified as involved in the pathogenesis of SBMA-MNs. Notably, in purified MNs (HPF), gene sets related to activated synapse function or those related to epigenetics were more significantly enriched in SBMA-MNs.
Regarding epigenetics, the gene sets of high-CpG-density promoters (HCP) bearing the methylation mark at K4 and K27 (H3K4me2 and H3K27me3) were enriched in the HPF of the SBMA-MNs. Although abnormal histone modification has never been reported, DNA methyltransferase1 is known to be strongly expressed in SBMA model mice and patients’ spinal cords, which could be rescued by its inhibitor through the suppression of Hes5 [24]. As the role of epigenetics is also reported in the etiology of ALS [45], epigenetics may play important roles in the neurodegeneration present in SBMA.
As negatively enriched gene sets, ER-related gene sets are identified. ER plays crucial roles in the unfolded protein response (UPR), which is the ER protein quality control pathway. The deficiency of the UPR mediator, CHOP, was shown to activate macro-autophagy, which is a lysosomal protein quality control pathway and which accentuated phenotypes of SBMA model mice [46]. Moreover, ER-associated calcium homeostasis was shown to be disturbed in cultured embryonic MNs using SBMA model mice [47]. Therefore, down-regulation of ER-related gene sets suggests dysregulation of UPR in the SBMA-MNs.
Pathology in neuromuscular synapses were highlighted to cause neurodegeneration in SBMA
Most highlighted differences between the SBMA- and control-MNs were within synapse-related gene sets. Recently, the skeletal muscle specific overexpression of IGF-1, or the silencing of mutant AR, was shown to rescue the phenotypes of SBMA model mice, suggesting the involvement of skeletal muscles in the neurodegeneration in SBMA [12, 13, 38]. Moreover, by analyzing NMJs in the SBMA model mice, pathological fragmentation of NMJs and abnormal synaptophysin distribution, as well as defects in the neuromuscular synaptic transmission, were demonstrated, which is consistent with the previously reported insufficient synaptic functions in SBMA [14, 48]. Interestingly, our transcriptome analysis of iPSC-MNs revealed upregulation of synapse-related gene sets in the SBMA-MNs, including activation of intracellular signaling via calcium ions, formation of synaptic vesicles, and the release of neurotransmitters such as acetylcholine. Similarly, analysis of DEGs, synapse-related genes were abundantly enriched and were found to be upregulated in the SBMA-MNs (Figs. 5c, d, and 6d). For instance, glutamate receptors GRID2, GRIK1, and GRM2 could be involved in glutamate toxicity in MNs, a phenomenon reported to occur in ALS. Upregulation of these genes in the SBMA-MNs suggests the involvement of similar glutamate toxicity in SBMA. Another example is that RGS proteins, including RGS4 and RGS5, negatively regulate G protein-coupled receptor (GPCR) signaling, and work in coordination to regulate key aspects of neurotransmitter release, synaptic transmission, and synaptic plasticity at neuronal synapses [49]. Increased expressions of RGS4 and RGS5 inactivate GPCR signaling, which could induce neurodegeneration [50]. Moreover, the synaptotagmin family, including SYT6 and SYT9, plays important roles in calcium-dependent exocytosis of synaptic vesicles, and SYT9 has been shown to be involved in the pathology of ALS [51].
As for the neuromuscular synapse formation, RSPO2 is highly expressed in MNs and directly binds to its receptor, leucine-rich repeat-containing G-protein coupled receptor 5 (Lgr5) at the neuromuscular synapse. RSPO2 not only activates canonical Wnt pathways, but also promotes AChR clustering via muscle-specific tyrosine kinase (MuSK) and low-density lipoprotein receptor-related protein 4 (Lrp4) [52, 53]. Similarly, some of the Wnt ligands (Wnt4, Wnt7a, Wnt7b, Wnt9a, Wnt9b, Wnt10b, Wnt11, and Wnt16) also promote AChR clustering independent of Agrin [54–56], but WNT3A negatively regulate NMJ formation [57]. Thus, increased expression of RSPO2, and Wnt ligands in SBMA-MNs, could induce abnormal neuromuscular synapse formation, or could be caused by compensatory negative feedback of pathological synaptic degeneration.
In this study, SBMA disease-specific iPSCs provided a valuable disease model that enabled elucidation of previously unknown pathologies underlying neurodegeneration. Comprehensive transcriptome analysis using purified MNs revealed MN-specific and patient-specific pathologies that have never been investigated in detail. Notably, this analysis further highlighted synaptic pathologies in SBMA. In contrast to most of the previous studies which focused on muscular pathology causing non-cell autonomous neurodegeneration and NMJ dysfunctions, we demonstrated that neuromuscular synapses and synaptic functions of NMJs could be affected by MN pathology in SBMA, and also identified alteration of gene expressions in MNs, such as Synaptotagmin, RSPO2, and WNT ligands. Through these analysis, identification of novel therapeutic targets focusing on neuromuscular synapses is expected.
Materials and methods
Isolation of human skin fibroblasts and generation of iPSCs
Human dermal fibroblasts (HDFs) were obtained from 4 Japanese SBMA patients and 3 controls: a 36-year-old Japanese male from the Japanese Collection of Research Bioresources (JCRB) Cell Bank (TIG114), a 24-year-old Japanese male (YF), and a 39-year-old Japanese male (KN). HDFs were cultured in DMEM (Sigma-Aldrich, USA), 10% fetal bovine serum (FBS; Sigma-Aldrich, USA), 2 mM L-glutamine, and 1% penicillin/streptomycin. Then, 6 × 105 HDFs were transfected with 1 μg of each of the following: pCXLE-hOCT3/4-shp53 (OCT4 and shTP53), pCXLE-hSK (SOX2 and KLF4), and pCXLE-hUL (L-MYC and LIN28; a gift from Dr. Yamanaka). Plasmid transfection was performed using the Neon transfection system (Thermo Fisher Scientific, USA). After 6 days, the cells were harvested and plated on mitomycin-C-treated SNL murine fibroblast feeder cells in 0.1% gelatin-coated tissue culture dishes in human fibroblast medium. On the next day, the medium was changed to standard human embryonic stem cell (hESC) medium containing DMEM/F-12 (Wako, Japan), 20% knockout serum replacement (KSR) (Thermo Fisher Scientific, USA), 2 mM L-glutamine, 1% non-essential amino acids (NEAA) (Sigma- Aldrich, USA), 0.1 mM 2-mercaptoethanol (2-ME) (Sigma- Aldrich, USA), 0.5% penicillin/streptomycin, and 4 ng/mL recombinant human fibroblast growth factor-2 (FGF-2) (Peprotech, USA). When the colonies had grown to a sufficiently large size, they were picked and expanded in the same way as hESCs and hiPSCs. The properties of the established iPSC clones were evaluated, as described previously (Fig. 1, Additional files 1 and 2: Figure S1 and Figure S2). Control iPSC clones, which were TIGE-9, TIGE-22, YFE-16, YFE-19, and EKN3, and the SBMA iPSC clones, which were SBMA1E-12, SBMA1E-18, SBMA2E-16, SBMA2E-44, SBMA3E-10, SBMA3E-11, SBMA4E-5, and SBMA4E-21, were established. The control iPSC clones (TIGE-9, YE-16, and EKN3) were previously reported [20, 21]. For the RNA sequence analysis, control iPSC clones (EKN3, TIGE-9, YFE-16 and YFE-19) and SBMA iPSC clones (1E-12, 2E-16, 3E-10, and 4E-5) were used and the data are presented as the average of four clones for the controls and SBMA.
iPSC culture and differentiation
iPSCs were differentiated into spinal MNs, as previously described [20]. iPSCs were maintained on mitomycin-C-treated SNL murine fibroblast feeder cells in 0.1% gelatin-coated tissue culture dishes in hESC medium and were used for MN induction. For differentiation, hiPSC colonies were detached using a dissociation solution (0.25% trypsin, 100 μg/ml collagenase IV (Gibco), 1 mM CaCl2, and 20% KSR) and cultured in suspension in bacteriological dishes in standard hESC medium, after the removal of SNL feeder cells, with incubation for 1–2 h in gelatin-coated dishes. On day 1, the medium was changed to human embryoid body (hEB) medium containing DMEM/F-12, 5% KSR, 2 mM L-glutamine, 1% NEAA, and 0.1 mM 2-ME with 300 nM LDN-193189 (Sigma-Aldrich, USA), 3 μM SB431542 (Tocris, UK), and 3 μM CHIR99021 (FCS, USA). On day 2, the medium was changed to fresh hEB medium containing 300 nM LDN-193189, 3 μM SB431542, and 3 μM CHIR99021, and 1 μM retinoic acid (RA) (Sigma-Aldrich, USA). From day 4 to day 14, hEBs were cultured in hEB medium containing 1 μM RA and 1 μM purmorphamine (Calbiochem, Germany), and the medium was changed every 2–3 days. On day 14, hEBs were enzymatically dissociated into single cells using TrypLE Select (Thermo Fisher Scientific, USA). The dissociated cells were plated on poly-l-ornithine (PO) and recombinant mouse Laminin (Thermo Fisher Scientific, USA), or growth-factor-reduced Matrigel (33 × dilution, thin coated; Corning)-coated dishes at a density of 5 × 104–1 × 105 cells/cm2 and cultured in motor neuron medium (MNM) consisting of media hormone mix (MHM) or KBM Neural Stem Cell medium (Kohjin Bio, Japan) [58] supplemented with 2% B27 supplement (Thermo Fisher Scientific, USA), 1% NEAA, 50 nM RA, 500 nM purmorphamine, 10 μM cyclic AMP (cAMP) (Sigma-Aldrich, USA), 10 ng/mL recombinant BDNF (R&D systems, USA), 10 ng/mL recombinant GDNF (R&D systems, USA), 10 ng/mL recombinant human IGF-1 (R&D systems, USA), and 200 ng/mL L-ascorbic acid (Sigma-Aldrich, USA) for up to 4 weeks in 5% O2 atmosphere. 10 nM DHT (Sigma-Aldrich, USA) was added to MN culture at the day of lentiviral infection (4 days after monolayer differentiation). Half of the medium was changed every 2–3 days.
Immunocytochemistry
Cells were fixed in 4% paraformaldehyde for 15–25 min at room temperature. After blocking in blocking buffer (PBS containing 10% FBS and 0.3% Triton X-100), the cells were incubated with primary antibodies overnight at 4 °C. The cells were then rinsed with PBS three times and incubated with Alexa 488-, Alexa 555-, or Alexa 647- conjugated secondary antibodies (Thermo Fisher Scientific) for 1 h at room temperature. Nuclei were stained with 10 μg/ml Hoechst 33258 (Sigma-Aldrich, USA). The cells were then rinsed with PBS three times, mounted on slides, and examined using IX83 (Olympus, Japan). The primary antibodies used in these analyses were listed in Additional file 3: Table S9.
Teratoma formation assay
Each iPSC clone was harvested in dissociation solution, collected into tubes, and centrifuged, and the resulting pellets were suspended in hESC medium with 10 μΜ Y-27632 (Wako, Japan), which is a Rho-associated coiled-coil forming kinase (ROCK) inhibitor. Then, 1 × 105–5 × 105 cells were injected into the testes of NOD/ SCID mice (Charles River, USA). At 8–10 weeks after injection, the tumors were dissected and fixed with PBS containing 4% PFA. Paraffin-embedded tissue was sliced and stained with hematoxylin and eosin. Images were obtained using a BZ-9000 microscope (Keyence, Japan).
CAG repeat sizing
DNA was extracted using DNeasy Blood & Tissue kits (Qiagen, Germany). PCR amplification of the CAG repeat in the AR gene was performed using a fluorescent-labeled forward primer (5′-TCCAGAATCTGTTCCAGAGCGTGC-3′) and an unlabeled reverse primer (5′-GCTGTGAAGGTTGCTGTTCCTCAT-3′). Detailed PCR conditions were described previously [58]. For determining the CAG repeat numbers of each PCR product, capillary electrophoresis and direct sequencing were performed using the 3730xl DNA Analyzer (Thermo Fisher Scientific, USA). Fragment analysis was performed using Peak Scanner™ Software v1.0. Sanger sequencing was performed from both 5′ and 3′ sides using a forward sequence primer (5′- TGCGCGAAGTGATCCAGAAC-3′) and a reverse sequence primer (5′- TTGGGGAGAACCATCCTCAC-3′).
RNA isolation and quantitative RT-PCR analysis
RNA was isolated using a RNeasy mini kit (Qiagen, Germany) and then converted into cDNA using SuperScript III reverse transcriptase (Thermo Fisher Scientific, USA) and Oligo dT primers as described previously [58, 59]. Real-time quantitative RT-PCR were performed as previously described using SYBR Premix ExTaq II and the StepOnePlus or the QuantStudio 7 Real-Time PCR system. The amount of cDNA was normalized to that of human- specific β-ACTIN mRNA. The primer sequences and PCR cycling conditions are listed in Additional file 3: Table S10.
Generation of the HB9e438::Venus lentivirus
Lenti-XTM 293T cells cultured in DMEM supplemented with 10% FBS in 150 mm dishes were transfected with 16 μg of pSIN2-HB9e438-βglo-Venus (a variant of yellow fluorescent protein (YFP) with fast and efficient maturation [60]) or pSIN2-βglo-Venus and 10 μg of each of two packaging vectors (pCMV-VSV-G-RSV-Rev and pCAG-HIVgp, kindly provided by Dr. Hiroyuki Miyoshi) with 200 μL of polyethylenimine (Polysciences, Inc., USA), and the medium was changed to the Freestyle 293 expression medium (Thermo Fisher Scientific, USA) the next day. Three days after the medium change, the culture supernatant was harvested and centrifuged at 25,000 rpm for 90 min at 4 °C in an Optima LE-80 K ultracentrifuge (Beckman Coulter, USA). After discarding the supernatant, 80 μL of PBS/150 mm dish was added to the pellet, which was resuspended by repeated pipetting to obtain the HB9e438::Venus reporter lentivirus. Lentiviral infection was performed on the day 4 of monolayer MN differentiation. For lentiviral infection, HB9e438::Venus in Opti-MEM (Thermo Fisher Scientific, USA) was added to a MN culture, followed by incubation for 2 h, after which the total medium was changed to MNM.
Flow cytometry
For flow cytometric analysis and cell sorting, iPSC-derived MNs were dissociated 4 weeks after infection with the HB9e438::Venus lentivirus using Tissue Dissociation Kits (Miltenyi Biotec) according to the manufacturer’s instructions. The dissociated cells (5 × 104–1 × 105 cells) were suspended in 50–100 μl of Hanks’ balanced salt solution (HBSS) (Thermo Fisher Scientific, USA) containing 2% FBS, 10 mM HEPES, and 1 μg/ml 7-AAD. The cells were then analyzed and sorted based on the expression of the HB9e438::Venus reporter using a FACSAria III cell sorter (BD Biosciences, USA).
RNA sequencing and data analysis
The yield and quality control of total RNA were measured using Nano Drop 2000c (Thermo Fisher scientific, USA) and Agilent RNA6000 Nano Kit (Agilent technologies, USA), respectively. The 2100 Bioanalyzer system (Agilent technologies, USA) was used to qualify RNA integrity number (RIN). RNA samples with sufficient RIN values (more than 9.2) were subjected to the generation of mRNA libraries using Illumina TruSeq protocols for poly-A selection, fragmentation, and adaptor ligation, according to the manufacturer’s instructions (TruSeq RNA Sample Prep Kit v2). Quantification of libraries was performed using Qubit3.0 (Thermo Fisher), Agilent 2200 TapeStaition System (Agilent), and qPCR analysis by Kapa Library Quantification Kit (TakaRa). The multiplexed libraries were sequenced as 75 nt pair end runs on an Illumina NextSeq500 system (San Diego, CA). Sequence reads were mapped to the reference human genome (GRCh38/hg38) using STAR (2-pass mode, version2.7.1a). We then excluded reads mapped to rRNA and tRNA regions. Annotation of the rRNA and tRNA regions were obtained from the UCSC Table Browser. Read counts of transcripts (feature counts [61]) were calculated using FeatureCounts of the Rsubread package. Ensembl gene annotation (Homo_sapiens.GRCh38.96.chr.gtf) was used for the transcript counts. DEGs based on the Wald test were analyzed using DESeq2 [62]. The expression data were grouped using a hierarchical clustering algorithm in Cluster 3.0 software (http://bonsai.hgc.jp/~mdehoon/software/cluster/software.htm) [63] by average linkage with the Euclidean distance, and visualized by Java TreeView software (http://jtreeview.sourceforge.net/) [64].
Gene set enrichment analysis (GSEA)
GSEA is a computational method that determines if a priori defined set of genes show statistically significant, concordant differences between two biological states [23], and is accessible at http://www.broadinstitute.org/gsea/index.jsp. The expression profile data were analyzed using GSEA 4.0.1. The gene sets were downloaded from the Molecular Signatures Database (MSigDB) and C2 (curated gene sets: chemical and genetic perturbation (CGP) and Canonical pathway (CP)) and C5 (Gene ontology gene sets) were used for the GSEA.
Statistical analysis
For the statistical analysis in the quantitative RT-PCR, either the Student’s t-test or the Welch’s t-test was used. For the differential analysis of mRNAs between control and SBMA MNs using DESeq2, adjusted p-value for a false discovery rate (FDR) correction was performed by the Benjamini–Hochberg (B-H) method. The gene set enrichment analysis was performed using the Fisher’s exact test and corrected with the B-H FDR.
Supplementary information
{kind=link}
Additional file 1: Figure S1. Evaluation of established iPSCs. Related to Fig. 1. (A) Immunocytochemical analysis of the established iPSC clones for pluripotent markers OCT4 and NANOG. Scale bar, 100 μm. (B) Hematoxylin and eosin staining of teratomas derived from the established iPSC clones. Scale bar, 200 μm.
{kind=link}
Additional file 2: Figure S2. Karyotype analysis of the established iPSC clones via G-banding analysis. Related to Fig. 2. All clones showed normal karyotypes, 46, XY.
Additional file 3: Table S1. DEGs in heatmap (NT). Related to Fig. 5C. Table S2. DEGs in heatmap (HPF). Related to Fig. 5D. Table S3. Profiles of GSEA. Table S4. Gene ontology terms (down in NT). List of GO terms negatively enriched in SBMA-MNs of NT (FDR < 0.2). Table S5. Gene ontology terms (down in HPF). List of GO terms negatively enriched in SBMA-MNs of HPF (FDR < 0.01). Table S6. Pathway terms (down in NT). List of pathway terms negatively enriched in SBMA-MNs of NT (FDR < 0.01). Table S7. Pathway terms (down in HPF). List of pathway terms negatively enriched in SBMA-MNs of HPF (FDR < 0.01). Table S8. The extracted upregulated genes in SBMA-MNs in GSEA. List of 96 genes that were significantly upregulated in enriched GO and pathway terms with p < 0.05 and significantly higher (> 2-fold) expression levels in SBMA-MNs than in control-MNs. Table S9. Primer sequences and cycling conditions for quantitative RT-PCR. Table S10. Antibodies for immunocytochemistry.
Acknowledgments
We are grateful to Dr. Hirofumi Suemori for KhES1 hESCs, Dr. Shinya Yamanaka for the plasmids (pCXLE-hOCT3/4-shp53, pCXLE-hSK, and pCXLE-hUL), Dr. Steven Goldman for the HB9e438 enhancer element, and Dr. Hiroyuki Miyoshi for the lentiviral vectors. We are also grateful to all members of the Okada laboratory for their encouragement and kind support.
Abbreviations
- AChR
Acetylcholine receptor
- ALS
Amyotrophic lateral sclerosis
- AR
Androgen receptor
- CALCA
Calcitonin-related polypeptide α
- CXCL14
CXC motif chemokine ligand 14
- DHT
Dihydrotestosterone
- DM1
Myotonic dystrophy
- EB
Embryoid body
- FAM135B
Family with sequence similarity 135 member B
- FDR
False discovery rate
- GO
Gene ontology
- GPCR
G protein-coupled receptor
- GRID2
Glutamate ionotropic receptor delta type subunit 2
- GRIK1
Glutamate ionotropic receptor kainate type subunit 1
- GRM2
Glutamate metabotropic receptor 2
- GSEA
Gene set enrichment analysis
- HD
Huntington disease
- hESC
Human embryonic stem cell
- hiPSC
Human induced pluripotent stem cell
- HPF
High positive fraction, purified motor neurons
- IGF-1
Insulin-like growth factor-1
- iPSC
Induced pluripotent stem cell
- Lgr5
Leucine-rich repeat-containing G-protein coupled receptor 5
- MFRP
Membrane-type frizzled-related protein
- MN
Motor neuron
- NES
Normalized enrichment score
- NF
Negative fraction
- NMJ
Neuromuscular junction
- NT
No treatment, unpurified motor neurons
- NTF3, NT-3
Neurotrophin-3
- RGS4
Regulator of G protein signaling 4
- RGS5
Regulator of G protein signaling 5
- RSPO2
R-spondin 2
- SBMA
Spinal bulbar muscular atrophy
- SYT6
Synaptotagmin6
- SYT9
Synaptotagmin9
- WNT2B
Wnt family member 2B
- WNT3A
Wnt family member 3A
Authors’ contributions
KO and YO conceived and designed the study. KO, DS, YI, NK and YO performed the experiments, and analyzed the data. MYano performed RNA sequencing, and MYano, MF, TT analyzed RNA sequence data. MYoshida supported histological analysis. HB, MK, and GS recruited SBMA patients. MO conducted skin biopsy and prepared fibroblasts from the patients. TI, RO, and BAH provided technical assistance. KO, BAH, and YO wrote the manuscript. MK, MD, GS, and HO provided scientific discussion and critical reading. All authors read and approved the final manuscript.
Funding
This work was supported by grants from the Practical Research Project for Intractable Diseases of the Ministry of Health, Labor and Welfare (MHLW), the Japan Agency for Medical Research and Development (AMED) (15ek0109025h0002, 16ek0109025h0003, 17ek0109243h0001, 18ek0109243h0002, and 19ek0109243h0003 to YO, 18 dm0107105 and 19 dm0107105 to MYoshida, 19bm0804003h0003 to HO), a Grant-in-Aid for Scientific Research (B) (19H03576), a Grant-in-Aid for Challenging Research (Exploratory) (17K19465) from the Japan Society for the Promotion of Science (JSPS), a Grant-in-Aid for Scientific Research in Innovative Areas (Brain Protein Aging and Dementia Control) (15H01568, 17H05707) from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) to YO, Program for the Advancement of Research in Core Projects on Longevity of the Keio University Global Research Institute from Keio University to HO, and a Research fund from Japan SBMA Association to KO.
Availability of data and materials
The datasets supporting the conclusions of this article are available in the following repositories; RNA sequence expression data in GEO.
[http://www.ncbi.nlm.nih.gov/geo/].
(accession number: GSE142612)
Ethics approval and consent to participate
All the experimental procedures for human iPSC production and the use of human iPSCs were approved by the ethics committee of the Keio University School of Medicine (approval number 20080016) and the ethics committee of the Aichi Medical University School of Medicine (approval number 14–004). All the experimental procedures for human ESC cultures were approved by the ethics committee of the Keio University School of Medicine (approval number 2002-001).
Consent for publication
Not applicable.
Competing interests
H.O. is a paid member of the Scientific Advisory Board of SanBio Co., Ltd. Y.O. is a scientific advisor of Kohjin Bio Co., Ltd. Other authors declare no conflict of interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary information accompanies this paper at 10.1186/s13041-020-0561-1.
References
- 1.Kennedy WR, Alter M, Sung JH. Progressive proximal spinal and bulbar muscular atrophy of late onset. A sex-linked recessive trait. Neurology. 1968;18:671–680. doi: 10.1212/WNL.18.7.671. [DOI] [PubMed] [Google Scholar]
- 2.Sobue G, Hashizume Y, Mukai E, Hirayama M, Mitsuma T, Takahashi A. X-linked recessive bulbospinal neuronopathy. A clinicopathological study. Brain. 1989;112(Pt 1):209–232. doi: 10.1093/brain/112.1.209. [DOI] [PubMed] [Google Scholar]
- 3.Katsuno M, Tanaka F, Adachi H, Banno H, Suzuki K, Watanabe H, et al. Pathogenesis and therapy of spinal and bulbar muscular atrophy (SBMA) Prog Neurobiol. 2012;99:246–256. doi: 10.1016/j.pneurobio.2012.05.007. [DOI] [PubMed] [Google Scholar]
- 4.Katsuno M, Adachi H, Minamiyama M, Waza M, Tokui K, Banno H, et al. Reversible disruption of dynactin 1-mediated retrograde axonal transport in polyglutamine-induced motor neuron degeneration. J Neurosci. 2006;26:12106–12117. doi: 10.1523/JNEUROSCI.3032-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Katsuno M, Adachi H, Minamiyama M, Waza M, Doi H, Kondo N, et al. Disrupted transforming growth factor-beta signaling in spinal and bulbar muscular atrophy. J Neurosci. 2010;30:5702–5712. doi: 10.1523/JNEUROSCI.0388-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.La Spada AR, Wilson EM, Lubahn DB, Harding AE, Fischbeck KH. Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nature. 1991;352:77–79. doi: 10.1038/352077a0. [DOI] [PubMed] [Google Scholar]
- 7.La Spada AR, Roling DB, Harding AE, Warner CL, Spiegel R, Hausmanowa-Petrusewicz I, et al. Meiotic stability and genotype-phenotype correlation of the trinucleotide repeat in X-linked spinal and bulbar muscular atrophy. Nat Genet. 1992;2:301–304. doi: 10.1038/ng1292-301. [DOI] [PubMed] [Google Scholar]
- 8.Katsuno M, Adachi H, Kume A, Li M, Nakagomi Y, Niwa H, et al. Testosterone reduction prevents phenotypic expression in a transgenic mouse model of spinal and bulbar muscular atrophy. Neuron. 2002;35:843–854. doi: 10.1016/S0896-6273(02)00834-6. [DOI] [PubMed] [Google Scholar]
- 9.Katsuno M, Adachi H, Doyu M, Minamiyama M, Sang C, Kobayashi Y, et al. Leuprorelin rescues polyglutamine-dependent phenotypes in a transgenic mouse model of spinal and bulbar muscular atrophy. Nat Med. 2003;9:768–773. doi: 10.1038/nm878. [DOI] [PubMed] [Google Scholar]
- 10.Katsuno M, Banno H, Suzuki K, Takeuchi Y, Kawashima M, Yabe I, et al. Efficacy and safety of leuprorelin in patients with spinal and bulbar muscular atrophy (JASMITT study): a multicentre, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2010;9:875–884. doi: 10.1016/S1474-4422(10)70182-4. [DOI] [PubMed] [Google Scholar]
- 11.Hashizume A, Katsuno M, Suzuki K, Hirakawa A, Hijikata Y, Yamada S, et al. Long-term treatment with leuprorelin for spinal and bulbar muscular atrophy: natural history-controlled study. J Neurol Neurosurg Psychiatry. 2017;88:1026–1032. doi: 10.1136/jnnp-2017-316015. [DOI] [PubMed] [Google Scholar]
- 12.Cortes CJ, Ling SC, Guo LT, Hung G, Tsunemi T, Ly L, et al. Muscle expression of mutant androgen receptor accounts for systemic and motor neuron disease phenotypes in spinal and bulbar muscular atrophy. Neuron. 2014;82:295–307. doi: 10.1016/j.neuron.2014.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lieberman AP, Yu Z, Murray S, Peralta R, Low A, Guo S, et al. Peripheral androgen receptor gene suppression rescues disease in mouse models of spinal and bulbar muscular atrophy. Cell Rep. 2014;7:774–784. doi: 10.1016/j.celrep.2014.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xu Y, Halievski K, Henley C, Atchison WD, Katsuno M, Adachi H, et al. Defects in neuromuscular transmission may underlie motor dysfunction in spinal and bulbar muscular atrophy. J Neurosci. 2016;36:5094–5106. doi: 10.1523/JNEUROSCI.3485-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 16.Okano H, Yamanaka S. iPS cell technologies: significance and applications to CNS regeneration and disease. Mol Brain. 2014;7:22. doi: 10.1186/1756-6606-7-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Engle SJ, Blaha L, Kleiman RJ. Best practices for translational disease modeling using human iPSC-derived neurons. Neuron. 2018;100:783–797. doi: 10.1016/j.neuron.2018.10.033. [DOI] [PubMed] [Google Scholar]
- 18.Imaizumi Y, Okada Y, Akamatsu W, Koike M, Kuzumaki N, Hayakawa H, et al. Mitochondrial dysfunction associated with increased oxidative stress and alpha-synuclein accumulation in PARK2 iPSC-derived neurons and postmortem brain tissue. Mol Brain. 2012;5:35. doi: 10.1186/1756-6606-5-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fujimori K, Ishikawa M, Otomo A, Atsuta N, Nakamura R, Akiyama T, et al. Modeling sporadic ALS in iPSC-derived motor neurons identifies a potential therapeutic agent. Nat Med. 2018;24:1579–1589. doi: 10.1038/s41591-018-0140-5. [DOI] [PubMed] [Google Scholar]
- 20.Shimojo D, Onodera K, Doi-Torii Y, Ishihara Y, Hattori C, Miwa Y, et al. Rapid, efficient, and simple motor neuron differentiation from human pluripotent stem cells. Mol Brain. 2015;8:79. doi: 10.1186/s13041-015-0172-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Matsumoto T, Fujimori K, Andoh-Noda T, Ando T, Kuzumaki N, Toyoshima M, et al. Functional neurons generated from T cell-derived induced pluripotent stem cells for neurological disease modeling. Stem Cell Reports. 2016;6:422–435. doi: 10.1016/j.stemcr.2016.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Suemori H, Yasuchika K, Hasegawa K, Fujioka T, Tsuneyoshi N, Nakatsuji N. Efficient establishment of human embryonic stem cell lines and long-term maintenance with stable karyotype by enzymatic bulk passage. Biochem Biophys Res Commun. 2006;345:926–932. doi: 10.1016/j.bbrc.2006.04.135. [DOI] [PubMed] [Google Scholar]
- 23.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kondo N, Tohnai G, Sahashi K, Iida M, Kataoka M, Nakatsuji H, et al. DNA methylation inhibitor attenuates polyglutamine-induced neurodegeneration by regulating Hes5. EMBO Mol Med. 2019;11:e8547. doi: 10.15252/emmm.201708547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Minamiyama M, Katsuno M, Adachi H, Doi H, Kondo N, Iida M, et al. Naratriptan mitigates CGRP1-associated motor neuron degeneration caused by an expanded polyglutamine repeat tract. Nat Med. 2012;18:1531–1538. doi: 10.1038/nm.2932. [DOI] [PubMed] [Google Scholar]
- 26.Watanabe M, Abe K, Aoki M, Yasuo K, Itoyama Y, Shoji M, et al. Mitotic and meiotic stability of the CAG repeat in the X-linked spinal and bulbar muscular atrophy gene. Clin Genet. 1996;50:133–137. doi: 10.1111/j.1399-0004.1996.tb02367.x. [DOI] [PubMed] [Google Scholar]
- 27.Nihei Y, Ito D, Okada Y, Akamatsu W, Yagi T, Yoshizaki T, et al. Enhanced aggregation of androgen receptor in induced pluripotent stem cell-derived neurons from spinal and bulbar muscular atrophy. J Biol Chem. 2013;288:8043–8052. doi: 10.1074/jbc.M112.408211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Narayanan G, Sheila M, Chai J, Stanton LW. Generation of sibling-matched induced pluripotent stem cell lines from spinal and bulbar muscular atrophy patients. Stem Cell Res. 2017;20:30–33. doi: 10.1016/j.scr.2017.02.005. [DOI] [PubMed] [Google Scholar]
- 29.Grunseich C, Zukosky K, Kats IR, Ghosh L, Harmison GG, Bott LC, et al. Stem cell-derived motor neurons from spinal and bulbar muscular atrophy patients. Neurobiol Dis. 2014;70:12–20. doi: 10.1016/j.nbd.2014.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Seriola Anna, Spits Claudia, Simard Jodie P., Hilven Pierre, Haentjens Patrick, Pearson Christopher E., Sermon Karen. Huntington's and myotonic dystrophy hESCs: down-regulated trinucleotide repeat instability and mismatch repair machinery expression upon differentiation. Human Molecular Genetics. 2010;20(1):176–185. doi: 10.1093/hmg/ddq456. [DOI] [PubMed] [Google Scholar]
- 31.Camnasio S, Delli Carri A, Lombardo A, Grad I, Mariotti C, Castucci A, et al. The first reported generation of several induced pluripotent stem cell lines from homozygous and heterozygous Huntington's disease patients demonstrates mutation related enhanced lysosomal activity. Neurobiol Dis. 2012;46:41–51. doi: 10.1016/j.nbd.2011.12.042. [DOI] [PubMed] [Google Scholar]
- 32.Liu L, Huang JS, Han C, Zhang GX, Xu XY, Shen Y, et al. Induced pluripotent stem cells in Huntington's disease: disease modeling and the potential for cell-based therapy. Mol Neurobiol. 2016;53:6698–6708. doi: 10.1007/s12035-015-9601-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Du J, Campau E, Soragni E, Jespersen C, Gottesfeld JM. Length-dependent CTG.CAG triplet-repeat expansion in myotonic dystrophy patient-derived induced pluripotent stem cells. Hum Mol Genet. 2013;22:5276–5287. doi: 10.1093/hmg/ddt386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sheila M, Narayanan G, Ma S, Tam WL, Chai J, Stanton LW. Phenotypic and molecular features underlying neurodegeneration of motor neurons derived from spinal and bulbar muscular atrophy patients. Neurobiol Dis. 2019;124:1–13. doi: 10.1016/j.nbd.2018.10.019. [DOI] [PubMed] [Google Scholar]
- 35.Cho K-I, Yoon D, Qiu S, Danziger Z, Grill WM, Wetsel WC, et al. Loss of Ranbp2 in motoneurons causes disruption of nucleocytoplasmic and chemokine signaling, proteostasis of hnRNPH3 and Mmp28, and development of amyotrophic lateral sclerosis-like syndromes. Dis Model Mech. 2017;10:559–579. doi: 10.1242/dmm.027730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Palazzolo Isabella, Burnett Barrington G., Young Jessica E., Brenne Phebe L., La Spada Albert R., Fischbeck Kenneth H., Howell Brian W., Pennuto Maria. Akt blocks ligand binding and protects against expanded polyglutamine androgen receptor toxicity. Human Molecular Genetics. 2007;16(13):1593–1603. doi: 10.1093/hmg/ddm109. [DOI] [PubMed] [Google Scholar]
- 37.Jimenez Garduno AM, Juarez-Hernandez LJ, Polanco MJ, Tosatto L, Michelatti D, Arosio D, et al. Altered ionic currents and amelioration by IGF-1 and PACAP in motoneuron-derived cells modelling SBMA. Biophys Chem. 2017;229:68–76. doi: 10.1016/j.bpc.2017.05.003. [DOI] [PubMed] [Google Scholar]
- 38.Palazzolo I, Stack C, Kong L, Musaro A, Adachi H, Katsuno M, et al. Overexpression of IGF-1 in muscle attenuates disease in a mouse model of spinal and bulbar muscular atrophy. Neuron. 2009;63:316–328. doi: 10.1016/j.neuron.2009.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tessarollo L, Vogel KS, Palko ME, Reid SW, Parada LF. Targeted mutation in the neurotrophin-3 gene results in loss of muscle sensory neurons. Proc Natl Acad Sci U S A. 1994;91:11844–11848. doi: 10.1073/pnas.91.25.11844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Klein R, Silos-Santiago I, Smeyne RJ, Lira SA, Brambilla R, Bryant S, et al. Disruption of the neurotrophin-3 receptor gene trkC eliminates la muscle afferents and results in abnormal movements. Nature. 1994;368:249–251. doi: 10.1038/368249a0. [DOI] [PubMed] [Google Scholar]
- 41.Henderson CE, Camu W, Mettling C, Gouin A, Poulsen K, Karihaloo M, et al. Neurotrophins promote motor neuron survival and are present in embryonic limb bud. Nature. 1993;363:266–270. doi: 10.1038/363266a0. [DOI] [PubMed] [Google Scholar]
- 42.Braun S, Croizat B, Lagrange MC, Warter JM, Poindron P. Neurotrophins increase motoneurons' ability to innervate skeletal muscle fibers in rat spinal cord--human muscle cocultures. J Neurol Sci. 1996;136:17–23. doi: 10.1016/0022-510X(95)00315-S. [DOI] [PubMed] [Google Scholar]
- 43.Garcia N, Santafe MM, Tomas M, Lanuza MA, Besalduch N, Tomas J. Involvement of neurotrophin-3 (NT-3) in the functional elimination of synaptic contacts during neuromuscular development. Neurosci Lett. 2010;473:141–145. doi: 10.1016/j.neulet.2010.02.040. [DOI] [PubMed] [Google Scholar]
- 44.Xie K, Wang T, Olafsson P, Mizuno K, Lu B. Activity-dependent expression of NT-3 in muscle cells in culture: implications in the development of neuromuscular junctions. J Neurosci. 1997;17:2947–2958. doi: 10.1523/JNEUROSCI.17-09-02947.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bennett SA, Tanaz R, Cobos SN, Torrente MP. Epigenetics in amyotrophic lateral sclerosis: a role for histone post-translational modifications in neurodegenerative disease. Transl Res. 2019;204:19–30. doi: 10.1016/j.trsl.2018.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yu Z, Wang AM, Adachi H, Katsuno M, Sobue G, Yue Z, et al. Macroautophagy is regulated by the UPR-mediator CHOP and accentuates the phenotype of SBMA mice. PLoS Genet. 2011;7:e1002321. doi: 10.1371/journal.pgen.1002321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Montague K, Malik B, Gray AL, La Spada AR, Hanna MG, Szabadkai G, et al. Endoplasmic reticulum stress in spinal and bulbar muscular atrophy: a potential target for therapy. Brain. 2014;137:1894–1906. doi: 10.1093/brain/awu114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Poort JE, Rheuben MB, Breedlove SM, Jordan CL. Neuromuscular junctions are pathological but not denervated in two mouse models of spinal bulbar muscular atrophy. Hum Mol Genet. 2016;25:3768–3783. doi: 10.1093/hmg/ddw222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gerber KJ, Squires KE, Hepler JR. Roles for regulator of G protein signaling proteins in synaptic signaling and plasticity. Mol Pharmacol. 2016;89:273–286. doi: 10.1124/mol.115.102210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jiang Y, Choi WH, Lee JH, Han DH, Kim JH, Chung YS, et al. A neurostimulant Para-chloroamphetamine inhibits the arginylation branch of the N-end rule pathway. Sci Rep. 2014;4:6344. doi: 10.1038/srep06344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Meltz Steinberg K, Nicholas TJ, Koboldt DC, Yu B, Mardis E, Pamphlett R. Whole genome analyses reveal no pathogenetic single nucleotide or structural differences between monozygotic twins discordant for amyotrophic lateral sclerosis. Amyotrophic Lateral Sclerosis and Frontotemporal Degeneration. 2015;16:385–392. doi: 10.3109/21678421.2015.1040029. [DOI] [PubMed] [Google Scholar]
- 52.Nakashima H, Ohkawara B, Ishigaki S, Fukudome T, Ito K, Tsushima M, et al. R-spondin 2 promotes acetylcholine receptor clustering at the neuromuscular junction via Lgr5. Sci Rep. 2016;6:28512. doi: 10.1038/srep28512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li J, Ito M, Ohkawara B, Masuda A, Ohno K. Differential effects of spinal motor neuron-derived and skeletal muscle-derived Rspo2 on acetylcholine receptor clustering at the neuromuscular junction. Sci Rep. 2018;8:13577. [DOI] [PMC free article] [PubMed]
- 54.Zhang B, Liang C, Bates R, Yin Y, Xiong WC, Mei L. Wnt proteins regulate acetylcholine receptor clustering in muscle cells. Mol Brain. 2012;5:7. doi: 10.1186/1756-6606-5-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Messeant J, Ezan J, Delers P, Glebov K, Marchiol C, Lager F, et al. Wnt proteins contribute to neuromuscular junction formation through distinct signaling pathways. Development. 2017;144:1712–1724. doi: 10.1242/dev.146167. [DOI] [PubMed] [Google Scholar]
- 56.Shen C, Li L, Zhao K, Bai L, Wang A, Shu X, et al. Motoneuron Wnts regulate neuromuscular junction development. Elife. 2018;7:e34625. [DOI] [PMC free article] [PubMed]
- 57.Wang J, Ruan NJ, Qian L, Lei WL, Chen F, Luo ZG. Wnt/beta-catenin signaling suppresses Rapsyn expression and inhibits acetylcholine receptor clustering at the neuromuscular junction. J Biol Chem. 2008;283:21668–21675. doi: 10.1074/jbc.M709939200. [DOI] [PubMed] [Google Scholar]
- 58.Okada Y, Matsumoto A, Shimazaki T, Enoki R, Koizumi A, Ishii S, et al. Spatiotemporal recapitulation of central nervous system development by murine embryonic stem cell-derived neural stem/progenitor cells. Stem Cells. 2008;26:3086–3098. doi: 10.1634/stemcells.2008-0293. [DOI] [PubMed] [Google Scholar]
- 59.Okada Y, Shimazaki T, Sobue G, Okano H. Retinoic-acid-concentration-dependent acquisition of neural cell identity during in vitro differentiation of mouse embryonic stem cells. Dev Biol. 2004;275:124–142. doi: 10.1016/j.ydbio.2004.07.038. [DOI] [PubMed] [Google Scholar]
- 60.Nagai T, Ibata K, Park ES, Kubota M, Mikoshiba K, Miyawaki A. A variant of yellow fluorescent protein with fast and efficient maturation for cell-biological applications. Nat Biotechnol. 2002;20:87–90. doi: 10.1038/nbt0102-87. [DOI] [PubMed] [Google Scholar]
- 61.Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- 62.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci U S A. 1998;95:14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Saldanha AJ. Java Treeview—extensible visualization of microarray data. Bioinformatics. 2004;20:3246–3248. doi: 10.1093/bioinformatics/bth349. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Figure S1. Evaluation of established iPSCs. Related to Fig. 1. (A) Immunocytochemical analysis of the established iPSC clones for pluripotent markers OCT4 and NANOG. Scale bar, 100 μm. (B) Hematoxylin and eosin staining of teratomas derived from the established iPSC clones. Scale bar, 200 μm.
Additional file 2: Figure S2. Karyotype analysis of the established iPSC clones via G-banding analysis. Related to Fig. 2. All clones showed normal karyotypes, 46, XY.
Additional file 3: Table S1. DEGs in heatmap (NT). Related to Fig. 5C. Table S2. DEGs in heatmap (HPF). Related to Fig. 5D. Table S3. Profiles of GSEA. Table S4. Gene ontology terms (down in NT). List of GO terms negatively enriched in SBMA-MNs of NT (FDR < 0.2). Table S5. Gene ontology terms (down in HPF). List of GO terms negatively enriched in SBMA-MNs of HPF (FDR < 0.01). Table S6. Pathway terms (down in NT). List of pathway terms negatively enriched in SBMA-MNs of NT (FDR < 0.01). Table S7. Pathway terms (down in HPF). List of pathway terms negatively enriched in SBMA-MNs of HPF (FDR < 0.01). Table S8. The extracted upregulated genes in SBMA-MNs in GSEA. List of 96 genes that were significantly upregulated in enriched GO and pathway terms with p < 0.05 and significantly higher (> 2-fold) expression levels in SBMA-MNs than in control-MNs. Table S9. Primer sequences and cycling conditions for quantitative RT-PCR. Table S10. Antibodies for immunocytochemistry.
Data Availability Statement
The datasets supporting the conclusions of this article are available in the following repositories; RNA sequence expression data in GEO.
[http://www.ncbi.nlm.nih.gov/geo/].
(accession number: GSE142612)