

Abstract
The protein-protein interaction between menin and Mixed Lineage Leukemia 1 (MLL1) plays an important role in development of acute leukemia with translocations of the MLL1 gene and in solid tumors. Here, we report the development of a new generation of menin-MLL1 inhibitors identified by structure-based optimization of the thienopyrimidine class of compounds. This work resulted in compound 28 (MI-1481), which showed very potent inhibition of the menin-MLL1 interaction (IC50 = 3.6 nM), representing the most potent reversible menin-MLL1 inhibitor reported to date. The crystal structure of the menin-28 complex revealed a hydrogen bond with Glu366 and hydrophobic interactions, which contributed to strong inhibitory activity of 28. Compound 28 also demonstrates pronounced activity in MLL leukemia cells and in vivo in MLL leukemia models. Thus, 28 is a valuable menin-MLL1 inhibitor that can be used for potential therapeutic applications and in further studies regarding the role of menin in cancer.
Graphical Abstract
Introduction
Menin is a scaffold protein that is involved in a network of protein-protein interactions, among which the interaction with the Mixed Lineage Leukemia 1 (MLL1; hereafter referred as MLL) protein or MLL fusion proteins have been recognized as highly relevant to human diseases, specifically to cancer.1 The MLL1 gene is translocated in 5–10% of acute leukemia cases, and this translocation leads to the expression of MLL fusion proteins, which are critical drivers of leukemogenesis.2–3 The MLL leukemia patients have an approximately 35% five-year survival rate,4–5 which reflects poor response to currently available treatments and supports the need for the development of new therapies. The interaction of menin with MLL fusion proteins plays a key role in leukemogenesis, which was demonstrated before using genetic approaches.2, 6–7 The importance of this interaction in MLL leukemia has been further validated by small molecule inhibitors of the menin-MLL interaction that we have reported in earlier studies.8–12 Furthermore, the interaction of menin with the wild type MLL plays an important role in solid tumors, including castration resistant prostate cancer,13 Ewing sarcoma,14 hepatocellular carcinoma (HCC)15–16 and pediatric gliomas.17 Therefore, the development of small molecule inhibitors of the menin-MLL interaction with optimized drug-like properties may lead to new therapeutics for acute leukemia and a subset of solid tumors.
The development of potent inhibitors with favorable drug-like properties that target protein-protein interactions (PPI) is still a challenge.18–19 The specific architecture of protein-protein interfaces (e.g. the lack of well-defined pockets and large contact areas) frequently implies the need for increasing the molecular weight of PPI inhibitors, which makes it difficult to achieve strong potency and favorable drug-like properties.20 Nevertheless, successful examples of PPI inhibitors exist,21–24 including compounds advanced to clinical trials (e.g. Bcl-2 family inhibitors, p53 activators25–26), thus supporting efforts in this direction. Furthermore, the recent FDA approval of the BCL-2 inhibitor Venetoclax demonstrates that PPI inhibitors can be valuable therapeutics, despite challenges it obtaining favorable drug-like properties for such compounds as a result of inherently higher than average molecular weight (Mw > 800 Da for Venetoclax).
The protein-protein interaction between menin and MLL or MLL fusion proteins involves the N-terminal fragment of MLL (MLL4–43)2, 6, which binds to menin with a strong binding affinity (Kd = 6.8 nM) by utilizing the bivalent binding mode, with MBM1 (menin-binding motif 1, MLL4–15) and MBM2 (menin-binding motif 2, MLL23–43) motifs involved in the interactions with menin.27 Structural studies have revealed that MLL binds to a large central cavity on menin,9, 28 thus supporting the existence of pockets to bind small molecules. Indeed, our previous studies have validated that the menin-MLL interaction can be effectively blocked with small molecules that bind to the MLL binding site on menin.8–12 However, due to the extended menin-MLL interface resulting from the bivalent binding mode of MLL to menin, optimization of the potency and drug-like properties of menin-MLL inhibitors remains challenging.12
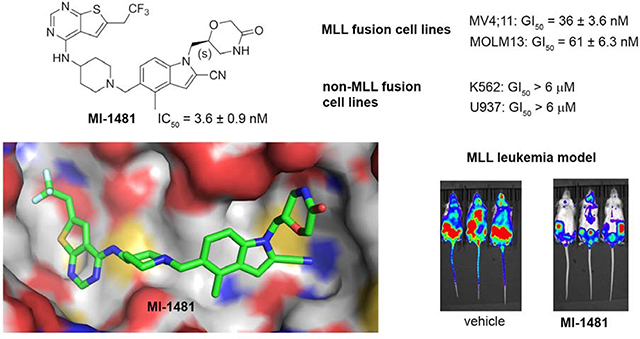
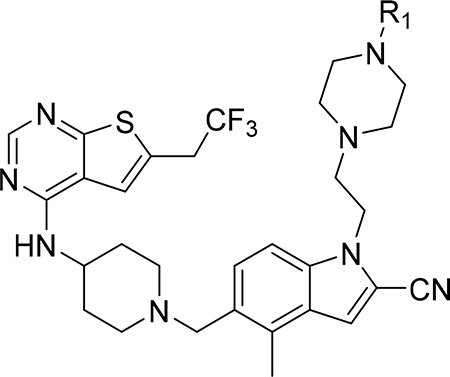
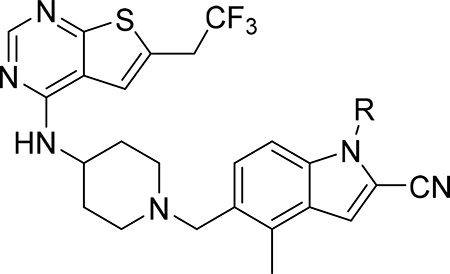
Our previous efforts resulted in the development of the thienopyrimidine class of menin-MLL inhibitors, with MI-463, MI-503 and MI-538 demonstrating the most potent inhibitory activity (Figure 1a).10, 12 These compounds selectively block proliferation of human MLL leukemia cells (GI50 = 200–500 nM), demonstrate specific mechanism of action and delay leukemia progression in mice models of MLL leukemia.10, 12 MI-503 also demonstrated activity in prostate cancer,13 Ewing sarcoma14 and hepatocellular carcinoma16 models. However, menin-MLL inhibitors with improved therapeutic potential over MI-503, namely with increased potency (e.g. IC50 < 10 nM to make them comparable or more potent than MLL4–43, GI50 < 50 nM and >100-fold selectivity to MLL leukemia cells, strong reduction in leukemia burden and >50% decrease in target gene expression in vivo) are still required. In this study we report structure-based optimization of the MI-463 and MI-503 menin-MLL inhibitors by exploring a collection of saturated six-membered rings introduced at the indole nitrogen of these compounds to further improve their activity and drug-like properties. These efforts resulted in the identification of compound 28 (MI-1481), which demonstrated low nanomolar activity (IC50 = 3.6 nM) for the inhibition of the interaction between menin and the bivalent MLL4–43 encompassing the entire menin binding motif on MLL. Thus, 28 demonstrates >10-fold improved inhibitory activity over MI-503, representing the most potent reversible small molecule menin-MLL inhibitor reported to date. Compound 28 also demonstrated substantially increased activity in MLL leukemia cells (~10-fold) over the previous generation of reversible menin-MLL inhibitors,10, 12 high selectivity towards MLL leukemia cells and pronounced in vivo activity in a mice model of MLL leukemia. We also report the co-crystal structure of the menin-28 complex, which revealed that both polar and hydrophobic interactions likely contribute to the strong inhibitory activity of this compound. This compound could serve as a valuable chemical probe to further study the role of menin in pathological and physiological states and could lead to the development of a new therapeutic agent for tumors that rely on menin-MLL interaction.
Figure 1. Structures and binding mode of the previous generation of menin-MLL inhibitors.

a) Chemical structures of the thienopyrimidine menin-MLL inhibitors MI-463, MI-503 and MI-538 together with the inhibitory activity of these compounds measured in the fluorescence polarization (FP) competition assays with menin and the fluorescein labeled MLL4–15 or MLL4–43 peptides. b) Crystal structure of menin in complex with MI-503 (4X5Y in PDB). Protein is shown in ribbon representation and key menin residues involved in interactions with MI-503 are shown as sticks. MI-503 is shown in stick representation with colors corresponding to the atom type (green: carbons, dark blue: nitrogens, yellow: sulfur, light blue: fluorine). Dashed lines represent hydrogen bonds between MI-503 and menin.
Results and Discussion
Biochemical assay to assess potent menin-MLL inhibitors
We have recently reported the thienopyrimidine class of menin-MLL interaction, with the most potent MI-463 (compound 1) and MI-503 (compound 2), Figure 1a, Table 1.10, 12 These compounds bind to menin with Kd ~ 10 nM and block the interaction of menin with a short MLL-derived peptide MBM1 (MLL4–15) with nanomolar inhibitory activities (IC50 ~15 nM for both compounds) as measured in the fluorescence polarization (FP) assay, Figure 1a. However, in the FP competition assay the low end of inhibitor Ki values that can be accurately measured is the Kd value of the fluorescent ligand, and for inhibitors more potent than the fluorescent probe the IC50 values are independent of inhibitor potency but linearly proportional to the Kd value of the fluorescence ligand.29 Based on the modest binding affinity of the fluorescein labeled (FLSN) MBM1 to menin (Kd = 49 nM)27 we concluded that menin-MLL inhibitors with low nanomolar binding affinity or better would reach the detection limit of the competition FP assay that uses FLSN_MBM1. To avoid this issue, we utilized the bivalent fragment of MLL (MLL4–43) in the competition FP assay for testing of the new generation of menin-MLL inhibitors. The MLL4–43 includes the entire menin binding motif (MBM1 and MBM2)6, 27 and binds to menin with Kd = 6.8 nM as determined by the Isothermal Titration Calorimetry (ITC). First, we determined the binding affinity of the fluorescein labeled MLL4–43 peptide (FLSN-MLL4–43) to menin in this new FP assay, resulting in Kd = 1.0 nM Supplementary Figure 1, which remains in a good agreement with the results from the ITC experiment.27 We then applied the FP competition assay with FLSN-MLL4–43 to test the inhibitory activity of MI-463 and MI-503 menin-ML inhibitors, resulting in IC50 values of 32 and 33 nM, respectively, Figure 1a, Table 1. Because MLL4–43 encompasses the entire menin binding motif, these data support that the thienopyrimidine compounds (e.g. MI-463 and MI-503) can effectively displace the bivalent MLL fragment from menin, which is an important feature in the context of functional studies. Overall, based on these results we concluded that the FP assay utilizing the bi-valent MLL4–43 peptide represents an appropriate approach to provide accurate activity measurements for low nanomolar inhibitors of the menin-MLL interactions. Thus, we used this assay to assess the in vitro activity of all compounds reported in this study.
Table 1.
Structure-Activity Relationship (SAR) and properties of MI-463 derivatives with various rings introduced at indole nitrogen.
 | |||||
|---|---|---|---|---|---|
| Compound | R | IC50 MLL4–43 (nM)a | GI50 (μM)b MLL-AF9 | T1/2 (min)c | cLogPd |
| 1 (MI-463) | H | 32 ± 9.9 | 0.23 | 14 | 4.7 |
| 2 (MI-503) |  |
33 ± 8.5 | 0.22 | 21 | 4.4 |
| 3 |  |
15 ± 4.9 | 0.52 | >60 | 4.3 |
| 4 | 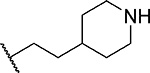 |
14 ± 1.4 | 0.81 | >60 | 5.5 |
| 5 | 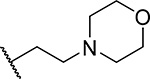 |
22 ± 5.7 | 0.24 | <3 | 4.7 |
| 6 (RS) | 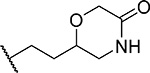 |
28 ± 4.9 | 0.15 | 31 | 4.1 |
| 7 (RS) (MI-568) | 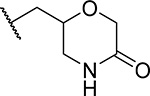 |
7.5 ± 2.1 | 0.05 | >60 | 3.7 |
IC50 values were measured by fluorescence polarization assay using fluorescein labeled MLL4–43, average values from 2–3 independent measurements ± SD are provided.
Growth inhibition (GI50 values) measured in the MTT cell viability assay after 7 days of treatment of murine bone marrow cells transformed with MLL-AF9.
Half-life of compounds in mouse liver microsomes.
Calculated with ChemBioDraw Ultra 14.0.
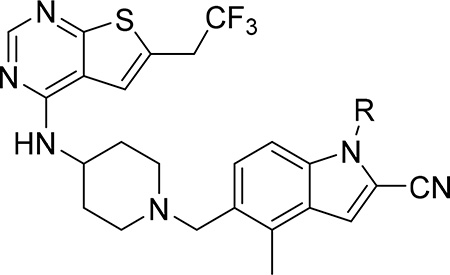
Development of menin-MLL inhibitors with saturated rings substituting indole nitrogen
In our previous studies, we developed MI-463 and its analogues with different substitutions at the indole ring, including aliphatic and aromatic substituents on indole nitrogen (e.g. MI-503), Figure 1a. However, these modifications did not result in increased inhibitory activity as compared to MI-463.12 To further optimize these compounds we performed detailed analysis of the co-crystal structure of menin in complex with MI-503, which revealed three hydrogen bonds (with Tyr276, Trp341 and Glu366) and hydrophobic interactions with menin,10 Figure 1b. Furthermore, we found that the methyl-pyrazole ring substituting indole nitrogen in MI-503 is partly solvent exposed, with two negatively charged residues on menin, Glu363 and Glu366, approaching this ring, Figure 1b. Based on this analysis we designed various saturated rings harboring positively charged and/or uncharged polar groups to be introduced at the indole nitrogen of MI-463 to form electrostatic interactions or hydrogen bonds with this area of the binding site on menin.
First, we explored several saturated rings with short aliphatic linkers substituting indole nitrogen of MI-463, Table 1. Introduction of the piperazine or piperidine rings, which resulted in compounds 3 and 4, Table 1, led to a ~2-fold improved inhibitory activity over MI-463 (IC50 = 15 and 14 nM for 3 and 4, respectively). On the other hand, incorporation of the morpholine (compound 5) or morpholinone (compound 6) rings connected to indole through an ethyl linker resulted in less pronounced activity improvement (<1.5-fold) over MI-463, Table 1. Interestingly, the introduction of the methyl-morpholinone substituent at indole nitrogen (compound 7, MI-568) resulted in an over 4-fold improvement in the IC50 value (IC50 = 7.5 nM for the racemic mixture of 7) as compared to MI-463, Table 1. Thus, although 7 represents the racemic mixture of two enantiomers, this compound demonstrates low nanomolar inhibition of the menin-MLL interaction, outperforming other compounds with various saturated rings introduced to the scaffold, Table 1.
We then assessed the cellular activity in MLL leukemia cells and the microsomal stability of compounds 3-7, Table 1. Notably, compound 7 demonstrated the most pronounced effect on proliferation of the MLL-AF9 transformed leukemia cells (GI50 = 50 nM), representing ~5-fold stronger cellular activity than MI-463, Table 1. The cellular activities of compounds 3-6 are less pronounced and range from 0.15–0.81 μM, Table 1. Interestingly, despite improved inhibitory activity, compounds 3 and 4 showed weaker cell growth inhibition as compared to MI-463, which may have resulted from the positive charge in the piperazine or piperidine rings of these compounds, possibly affecting their cellular permeability. In addition, compounds 3, 4 and 7 showed the best microsomal stability measured in mouse liver microsomes (T1/2 > 60 min), representing substantial improvement over MI-463 or MI-503, Table 1. Furthermore, compound 7 has also the lowest clogP value (Table 1), thus supporting favorable polarity and solubility. Overall, these results support that the analogues of MI-463 with saturated rings introduced at indole nitrogen represent valuable candidates for further optimization.
Optimization of analogues with piperazine ring
Since compound 3 showed 2-fold improvement in inhibitory activity and substantially increased metabolic stability over MI-463 (Table 1), we pursued further optimization of 3 by introducing substituents to the piperazine ring. First, we introduced aliphatic groups, such as methyl (compound 8) and n-propyl (compound 9), but they did not improve substantially the in vitro inhibitory activity or cell growth inhibition in MLL-AF9 cells over 3, Table 2. Analysis of the crystal structure of the menin-MI-503 complex revealed the presence of Arg330,10 which could potentially be involved in hydrogen bond/salt bridge with ligands that harbor the appropriate substituent at the indole nitrogen of MI-463. Therefore, we introduced the formamide group to the piperazine ring (compound 10), but this modification did not improve inhibitory activity (IC50 = 13 nM for 10), although activity in the MLL-AF9 transformed cells demonstrated a 2-fold improvement for 10 over 3, Table 2. This was likely due to the neutralization of the positive charge in 3, which could have positively affected the cellular permeability. Further substitution of the amide with methyl (compound 11) did not significantly change the inhibitory activity, while ethyl analog (compound 12) showed an over 6-fold reduction in inhibitory activity (IC50 = 83 nM) as compared to 10. Interestingly, the analogue with the monofluoromethyl group (compound 13, MI-853) demonstrated slightly improved inhibitory activity (IC50 = 10 nM) over 10, while the di- and trifluoromethyl analogues (14 and 15) were 2–5-fold less potent than 10, Table 2. Improved in vitro activity of 13 resulted in more potent cell growth inhibition of the MLL-AF9 transformed cells observed for this compound (GI50 = 75 nM), Table 2. Furthermore, introduction of a larger hydrophobic group substituting amide in 10, which resulted in compounds 16 and 17, led to a 3–4-fold reduced inhibitory activity, Table 2. Finally, we introduced the 2-propanamide group (compound 18), but did not observe increased inhibitory activity for the racemic mixture of this compound over 10, Table 2.
Table 2.
SAR and properties of N-substituted analogues of compound 3.
 | |||||
|---|---|---|---|---|---|
| Compound | R1 | IC50 MLL4–43 (nM)a | GI50 (μM)b MLL-AF9 | T1/2 (min)c | cLogPd |
| 3 | H | 15 ± 4.9 | 0.40 | >60 | 4.3 |
| 8 | 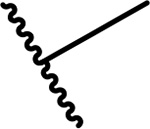 |
13 ± 0.7 | 0.23 | 9.2 | 3.8 |
| 9 | 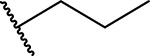 |
39 ± 2.1 | 1.0 | 12 | 4.9 |
| 10 | 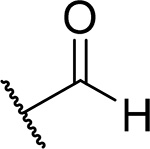 |
13 ± 4.2 | 0.15 | 21 | 4.1 |
| 11 | 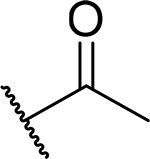 |
18 ± 3.5 | 0.15 | 13 | 4.2 |
| 12 | 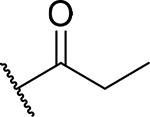 |
83 ± 2.8 | 0.25 | 13 | 4.8 |
| 13 (MI-853) | 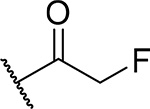 |
10 ± 0.7 | 0.075 | 5.7 | 4.5 |
| 14 |  |
44 ± 17 | 0.18 | 4.9 | 4.8 |
| 15 | 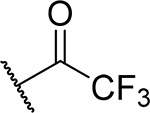 |
75 ± 22 | 0.23 | 14 | 5.4 |
| 16 | 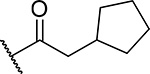 |
57 ± 8.5 | 1.2 | 8.6 | 6.4 |
| 17 | 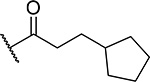 |
42 ± 2.8 | 0.75 | 10 | 6.9 |
| 18 (RS) | 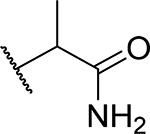 |
21 ± 4.2 | 0.25 | 11 | 3.2 |
| 19 | 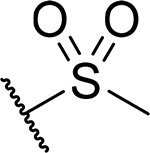 |
13 ± 2.8 | 0.15 | 26 | 4.7 |
| 20 | 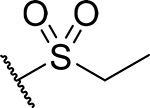 |
55 ± 15 | 0.25 | 19 | 5.2 |
| 21 | 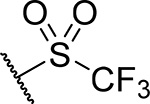 |
71 ± 7.1 | 0.45 | 15 | 5.8 |
| 22 | 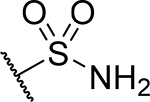 |
26 ± 2.1 | 0.22 | 53 | 4.7 |
| 23 | 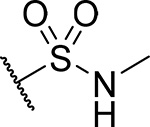 |
32 ± 1.4 | 0.15 | 24 | 4.4 |
IC50 values were measured by fluorescence polarization assay using fluorescein labeled MLL 4–43, average values from 2–3 independent measurements ± SD are provided.
Growth inhibition (GI50 values) measured in the MTT cell viability assay after 7 days of treatment of murine bone marrow cells transformed with MLL-AF9.
Half-life of compounds in mouse liver microsomes.
Calculated with ChemBioDraw Ultra 14.0.
To further explore substitutions at the piperazine ring of 3, we substituted this ring with sulfonyl or sulfonamide groups (compounds 19-23), Table 2. Among these compounds the analogue with the methyl sulfonamide (compound 19), demonstrated the most pronounced inhibitory activity for blocking the menin-MLL interaction (IC50 = 13 nM), Table 2. The structure-activity relationship for the sulfonyl analogues (compounds 19-23) is similar to the corresponding amide derivatives, Table 2, with reduced inhibitory activity upon increasing the size of the aliphatic substituent (e.g. compounds 20 and 21, Table 2). The sulfamide derivatives 22 and 23 showed about 2-fold reduction in inhibitory activity as compared to the methylsulfone analogue 19, Table 2.
Despite comparable or weaker inhibitory activity in in vitro biochemical assay, the majority of compounds with amides, sulfones or sulfonamides substituting the piperazine ring in 3 demonstrated stronger cell growth inhibition of the MLL-AF9 transformed cells (GI50 values below 200 nM) as compared to compound 3, Table 2. This may have resulted from the reduced basicity of these compounds over 3, which could have affected their cellular permeability. Importantly, the piperazine analogues demonstrated good correlation of in vitro inhibitory activity with cell growth inhibition, with compound 13 showing the strongest inhibitory activity (IC50 = 10 nM) for blocking the menin-MLL interaction and inhibition of cell proliferation in MLL-AF9 leukemia cells (GI50 = 75 nM), Table 2. On the other hand, the microsomal stability of the amide and sulfonamide analogues is reduced as compared to compound 3 (Table 2), although compounds 19 and 22 still retain relatively good microsomal stability (T 1/2 > 25 min).
Development of analogues with substituted piperidine ring
Because unsubstituted piperazine (3) and piperidine (4) derivatives have similar inhibitory activity in blocking the menin-MLL interaction, we also explored several analogues derived from 4, Table 3. In contrast to the corresponding piperazine analogues, the introduction of the amide group (compound 24) and its further substitution with methyl (compound 25) resulted in a 5–10 fold reduction in inhibitory activity as compared to 4, Table 3. Similarly, the incorporation of the sulfonamide (compound 26) or sulfamide (compound 27) groups to this scaffold resulted in >2-fold reduced inhibitory activity over 4, Table 3. Based on these results we concluded that the reduced basicity of the piperidine series (Table 3) makes these compound less potent menin-MLL inhibitors than the corresponding piperazine derivatives (Table 2) possibly due to the absence of long-range electrostatic interactions with Glu363 and/or Glu366 side chains, Figure 1b. Interestingly, despite weaker in vitro inhibitory activity, all piperidine analogues demonstrated comparable or increased activity in MLL leukemia cells over 4 (e.g. compound 26 has over 3-fold improved GI50 as compared to 4, Table 3). Furthermore, compounds 24-27 demonstrate good microsomal stability, with the sulfonyl (26) or sulfonamide (27) analogues exhibiting the longest half-life in mouse liver microsomes (T1/2 > 60 min).
Table 3.
SAR and properties of N-substituted analogues of compound 4.
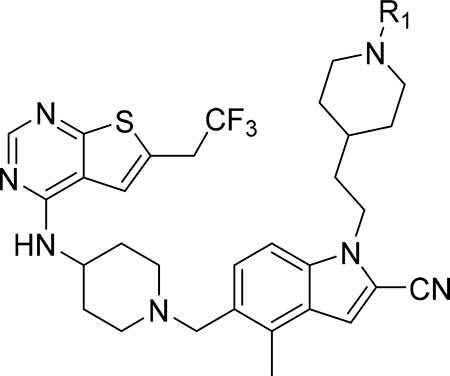 | |||||
|---|---|---|---|---|---|
| Compound | R | IC50 MLL4–43 (nM)a | GI50 (μM)b MLL-AF9 | T1/2 (min)c | cLogPd |
| 4 | H | 14 ± 1.4 | 0.81 | >60 | 5.5 |
| 24 | 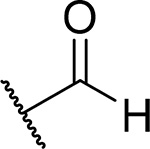 |
63 ± 2.1 | 0.57 | 25 | 4.6 |
| 25 | 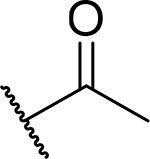 |
130 ± 32 | 0.97 | 26 | 4.6 |
| 26 | 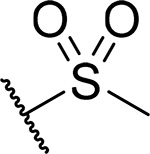 |
40 ± 9.2 | 0.25 | >60 | 5.2 |
| 27 | 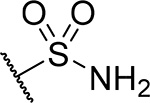 |
38 ± 10 | 0.35 | >60 | 5.6 |
IC50 values were measured by fluorescence polarization assay using fluorescein labeled MLL4–43, average values from 2–3 independent measurements ± SD are provided.
Growth inhibition (GI50 values) measured in the MTT cell viability assay after 7 days of treatment of murine bone marrow cells transformed with MLL-AF9.
Half-life of compounds in mouse liver microsomes.
Calculated with ChemBioDraw Ultra 14.0.
Structural studies reveal the binding mode of piperazine derivatives to menin
To understand the molecular basis of the interactions of the piperazine analogues with menin, we co-crystallized menin in complex with 13, which represents the most potent compound from this series (Table 2) and solved a high-resolution crystal structure of the complex, Figure 2a, Supplementary Table 1. The crystal structure revealed that the binding mode of 13 to menin is similar as that observed for MI-503, with two hydrogen bonds (with Tyr276 and Trp341) and hydrophobic interactions retained in 13 within the core structure preserved in both compounds (Figure 1b, 2a). The differences in the binding mode of 13 versus MI-503 were only observed for binding of the substituent introduced at indole nitrogen, which varies between these two compounds, Table 1, 2. The ethyl linker connecting indole with the piperazine ring in 13 is involved in hydrophobic interactions with the side chains of Glu363, Met322, Val367 and Glu366 on menin, Figure 2a. One of the nitrogen atoms in the piperazine ring of 13 points towards the carboxyl group of Glu366 (distance ~4.5Å), supporting the long-range electrostatic interactions, Figure 2a. Furthermore, one side of the piperidine ring in 13 is involved in the hydrophobic contacts with Val367 and Val371 on menin, while the other side of this ring in exposed to the solvent, Figure 2a. In addition, the oxygen atom from the fluoroacetamide group of 13 forms a hydrogen bond with the side chain of Arg330 (3.3 Å), while the fluoromethyl group is involved in the hydrophobic interactions with the side chains of Cys329 and Arg330 with the fluorine atom approaching Cys329 and Gly326, Figure 2a. Overall, favorable interactions of the fluoroacetamide group in 13 with menin result in the most pronounced activity of this compound among all amide or sulfonamide derivatives, Table 2, 3. The crystal structure also demonstrates limited space in the binding site to accommodate hydrophobic groups larger than acetamide, reflected by the reduced activity of such analogs (e.g. compounds 12 and 14-17, Table 2). Overall, the crystal structure of menin in complex with 13 supports that both hydrophobic contacts and hydrogen bond with Arg330 on menin contribute to the pronounced inhibitory activity of this compound.
Figure 2. Structure-based optimization results in potent menin-MLL inhibitors.

a) Crystal structure of the menin-compound 13 complex (PDB code 6BXH). Residues on menin involved in the interactions with the piperazine ring and the terminal amide of 13 are shown in stick representation. Hydrogen bond with Arg330 is marked. b) Crystal structure of the menin-compound 28 complex (PDB code 6BXY). Residues on menin involved in the interactions with the morpholinone ring in 28 are shown in stick representation. Hydrogen bond with Glu366 is marked. c) Superposition of the crystal structures of menin in complex with MI-503, 28 and MLL peptide encompassing MBM1 and MBM2 motifs (3U88 in PDB). d) Comparison of the binding mode of 28 (left) and 29 (right, PDB code 6BY8) to menin.
Development of compound 28 as a highly potent menin-MLL inhibitor
Exploring different saturated rings introduced at indole nitrogen led to the identification of 7, which demonstrated the most potent inhibitory activity for blocking the menin-MLL interaction (IC50 = 7.5 nM for the racemic mixture), Table 1. This compound harbors a morpholinone ring introduced to the indole nitrogen through a methyl linker, Table 1. In addition to exhibiting the most potent in vitro activity, compound 7 also demonstrated the most pronounced cell growth inhibition in the MLL-AF9 leukemia cells (GI50 = 50 nM), Table 1. To assess the activity of individual enantiomers of this compound we synthesized both of them, resulting in 28 (S enantiomer, MI-1481) and 29 (R enantiomer, MI-1482), Table 4. The S enantiomers (28) demonstrated a ~2-fold improvement in inhibitory activity for blocking the menin-MLL interaction (IC50 = 3.6 nM) over the racemic mixture (compound 7), Figure 3a, Table 4. Thus, 28 demonstrates about 10-fold increase in inhibitory activity over MI-463 and MI-503 (Table 1), representing the most potent reversible menin-MLL inhibitor reported to date. In contrast, the R enantiomer (compound 29) demonstrated a ~35-fold reduction in inhibitory activity in blocking the menin-MLL interaction (IC50 = 123 nM for 29) as compared to 28, Figure 3a, Table 4. Thus, 28 and 29 provide a valuable pair of enantiomers with distinct inhibitory activity to study menin-MLL inhibition in biological systems.
Table 4.
SAR and properties of enantiomers of compound 7.
 | ||||||
|---|---|---|---|---|---|---|
| Compound | R | IC50 MLL4–43 (nM)a | GI50 (μM)b MLL-AF9 | SI | T1/2 (min)c | cLogPd |
| 7 (RS) (MI-568) |  |
7.5 ± 2.1 | 0.05 | 180 | >60 | 3.7 |
| 28 (S) (MI-1481) | 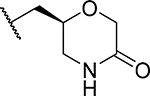 |
3.6 ± 0.9 | 0.034 | >180 | 59 | 3.7 |
| 29 (R)(MI-1482) | 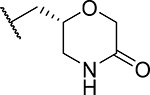 |
122 ± 22 | 0.7 | ND | >60 | 3.7 |
IC50 values were measured by fluorescence polarization assay using fluorescein labeled MLL 4–43, average values from 2–3 independent measurements ± SD are provided.
Growth inhibition (GI50 values) measured in the MTT cell viability assay after 7 days of treatment of murine bone marrow cells transformed with MLL-AF9.
Half-life of compounds in mouse liver microsomes.
Calculated with ChemBioDraw Ultra 14.0. SI, selectivity index calculated as a ratio of GI50 values measured in HM-2 cells (control cell line) and MLL-AF9 transformed murine bone marrow cells. ND – not determined.
Figure 3. Inhibition of the menin-MLL interaction and cellular activity of compound 28.

a) Titration curves from the FP competition assay for inhibition of the menin-MLL4–43 by 28 and 29. Data represent mean values of duplicate samples ± SD. b-d) Titration curves from the MTT cell viability assay performed for 28 or 29 after 7 days of treatment of: MLL-AF9 or Hoxa9/Meis1 (HM-2) transformed murine bone marrow cells treated with 28 (b), human MLL leukemia cell lines (MV4;11 and MOLM-13) and control cell lines (K562 and U937 without MLL translocation) treated with 28, (c); human MLL leukemia cell lines treated with 29 (d). Cell growth inhibition (GI50) values are provided. Data represent mean values of quadruplicates ± SD. The experiments were performed 2 times. Values are normalized with respect to DMSO treated samples. e) qRT-PCR performed in MV4;11 cells after 6 days of treatment with 28. Expression of HOXA9, MEIS1, DLX2 and ITGAM was normalized to 18S and referenced to DMSO-treated cells, mean ± SD, n = 2.
Molecular basis of menin-MLL inhibition by compound 28
To understand the molecular basis of the strong inhibition of the menin-MLL interaction by 28, we co-crystallized menin in complex with this compound, resulting in a high-resolution crystal structure, Supplementary Table 1, Figure 2b. We found a similar interaction pattern of 28 with menin to the one observed in the menin-MI-503 complex for the structural fragment retained in both compounds (e.g. hydrogen bonds with Tyr276 and Trp341 and similar pattern of hydrophobic interactions), Figure 1b, 2b. Again, the differences were found in the binding of substituents introduced at indole nitrogen. Specifically, the methyl group linking indole with the morpholinone ring in 28 is involved in the hydrophobic interactions with Glu363, Met322 and Val 367, Figure 2b. Furthermore, one side of the morpholinone ring in 28 is involved in hydrophobic contacts with the side chains of Glu363, Glu366 and Val367 on menin, while the amino portion of the amide forms a short hydrogen bond (2.6 Å) with the carboxyl group of Glu366 (Figure 2b). This hydrogen bond, which was not observed for the piperazine analogues (e.g. in the menin-13 complex), most likely contributes to the pronounced inhibitory activity of 28. The carbonyl carbon and the adjacent methylene group in 28 are engaged in the hydrophobic contacts with Val371, while the ether oxygen is solvent exposed and does not form major contacts with menin, Figure 2b. In contrast, the pyrazole ring introduced at the indole nitrogen of MI-503 is mostly solvent exposed, with one hydrogen bond formed with Glu366, Figure 1b.
To understand better the improvement in inhibitory activity for 28 over MI-503 we performed the bio-layer interferometry (BLI) studies and compared binding affinities of both compounds to menin. This study demonstrated that the association rates to menin are similar for both compounds, however, the dissociation rate is significantly slower for 28 over MI-503, resulting in the Kd value of ~ 3-fold improved for 28 (Kd = 9 nM) as compared to MI-503 (Kd = 24 nM), Supplementary Figure 2. Furthermore, we have also performed structural analysis of menin interactions with ligands by overlapped the crystal structures of menin in complex with: the bi-valent MLL peptide encompassing both MBM1 and MBM2 motifs, MI-503 and 28, Figure 2c. This analysis have revealed that both compounds occupy the same region of the binding site on menin where MBM1 motif binds, however 28 approaches more closely the region occupied by MBM2, particularly Arg24 from MLL, as compared to MI-503, Figure 2c. Therefore, 28 might be able to more effectively displace the bi-valent MLL peptide from its binding to menin likely due to stronger interference with the MBM2 portion of MLL, as reflected by ~10-fold improved inhibitory activity of 28 over MI-503. Overall, both structural data (e.g. extensive interactions with menin and interference with the MBM2 motif) as well as binding kinetics support 28 as a more effective inhibitor of the menin-MLL interaction over the previous generation of menin-MLL inhibitors (e.g. MI-503 or MI-463), Tables 1, 4. To better understand the molecular basis of the differential inhibitory activity between both enantiomers of 7, we also solved the co-crystal structure of menin in complex with 29 (R enantiomer of 7), Figure 2d, Supplementary Table 1. The core structure of 29 binds to menin in the same way as observed for 28, but the orientation of the morpholinone ring in 29 is different from that of 28, Figure 2d, which results in significantly reduced contacts of this ring with menin. The most striking difference in the binding mode of 29 and 28 is the lack of a hydrogen bond between the amide nitrogen in 29 and the carboxyl group of Glu366 in menin, which is present when 28 binds to menin, Figure 2b,d. Compound 29 also has limited hydrophobic contacts with menin as compared to 28, as 29 engages only a small portion of the morpholinone ring in the interactions with Glu366, Val367 and Val371 on menin, Figure 2d. Overall, we concluded that limited interactions of the morpholinone ring in 29 with menin, in particular the lack of a hydrogen bond with Glu366, result in a 35-fold reduction in inhibitory activity of this compound to menin as compared to 28.
Compound 28 demonstrates potent on target activity in MLL leukemia cells
To assess whether potent inhibition of the menin-MLL interaction by 28 results in strong activity of this compound in functional assays, we tested 28 in several leukemia cell lines. Importantly, 28 markedly reduced cell growth of murine bone marrow cells transformed with MLL-AF9, with a GI50 value of 34 nM (Table 4, Figure 3b). In contrast, limited activity of this compound was observed in murine bone marrow cells transformed with the control oncogenes Hoxa9/Meis1 (HM-2, GI50 > 6 μM), Figure 3b. Thus, 28 demonstrated over 200-fold selectivity towards MLL fusion protein transformed cells, Table 4, demonstrating substantial improvement in selectivity over the previous generation of menin-MLL inhibitors (e.g. selectivity index, SI ~ 27 for MI-503 in MLL leukemia cells).12 When tested in human MLL leukemia cell lines, 28 also showed pronounced cell growth inhibition, with GI50 values of 36 nM in MV4;11 (harboring MLL-AF4 fusion protein) and 61 nM in MOLM13 (harboring MLL-AF9), Figure 3c. Importantly, no substantial cell growth inhibition was observed in the control human leukemia cell lines K562 and U937, which do not harbor MLL translocations (GI50 > 6 μM), Figure 3c. Therefore, 28 shows the most pronounced activity in MLL leukemia cells among all reversible menin-MLL inhibitors reported to date, Tables 1–4,10, 12 demonstrating ~7–10-fold improvement in the activity in MLL leukemia cells over MI-463 or MI-503 (Table 1).10 As pointed out above, the increased cellular activity of 28 is associated with ~10-fold improved inhibitory activity of this compound in blocking the menin-MLL interaction as compared to MI-463 or MI-503, Table 1, 4. In contrast, compound 29, which represents the R enantiomer of 7, showed limited activity in MLL leukemia cells (over 16-fold weaker than 28), Figure 3d, in agreement with the substantially reduced inhibitory activity of 29, Table 4.
To validate the on target activity of 28 in MLL leukemia cells, we assessed the effect of this compound on the expression level of the MLL fusion target genes: HOXA9, MEIS1 and DLX2, all of which are required for the MLL-fusion protein mediated leukemogenesis. Six days of treatment of the MV4;11 human MLL leukemia cells with 28 resulted in a substantial downregulation of the MLL fusion protein target genes, with a strong reduction in the expression level of HOXA9 and MEIS1 at 100 nM of 28, Figure 3e. The effect of 28 on the expression level of DLX2 was even more pronounced, Figure 3e. Furthermore, marked upregulation of the ITGAM differentiation marker was observed upon treatment of MV4;11 cells with 28 (Figure 3e), supporting reprograming of these cells from primitive blasts to more mature blood cells. Overall, 28 demonstrates a pronounced effect on the expression level of MLL fusion target genes, which strongly supports the on target activity of this compound in MLL leukemia cells.
Compound 28 strongly inhibits leukemia progression in vivo
We then performed pharmacokinetic (PK) studies in mice with 28 and found relatively high exposure of this compound in blood plasma using i.v. (intravenous) and i.p. (intraperitoneal) administration (Supplementary Figure 3), however, oral bioavailability of this compound was low (<5%). Based on these results we selected i.p. as a way of administration of 28 for in vivo efficacy studies. To establish a potential therapeutic value of 28, we tested the effect of this compound in a mice model of MLL leukemia. For this purpose, we utilized the xenotransplantation model of the MLL leukemia with MV4;11-luc cells (expressing luciferase) injected into the tail vein of NSG mice. Treatment with 28 (80 mg/kg, i.p., n = 8) or vehicle (n = 8) was initiated 13 days after transplantation of mice with the MV4;11 cells to account for a compound effect in an advanced leukemia model, and was continued for 21 consecutive days. No reduction in the body weight of mice was observed during the entire treatment period with 28, Supplementary Figure 4. To assess treatment efficacy, the bioluminescence level in mice was measured at days 0, 7, 14 and 21 after initiating the treatment. Very pronounced (~75%) and consistent reduction in the luciferase signal was observed over the course of the experiment upon treatment of mice with 28 as compared to the vehicle treated mice, Figure 4a,b. These results demonstrate the pronounced effect 28 on leukemia progression in an advanced model of MLL leukemia.
Figure 4. Compound 28 strongly inhibits leukemia progression in mice.

a, b) Bioluminescence level in MV4;11 leukemia mice measured at days 0, 7, 14 and 21 after initiating treatment with vehicle or 28. n = 8 mice per group. b) Representative images of MV4;11 mice treated with vehicle or 28 at day 21 of treatment. c) Spleen weight and pictures of MV4;11 mice after 6 days of treatment with vehicle or 28 (n = 3 mice / group). d) Level of hCD45+ cells in MV4;11 mice after 6 days of treatment with vehicle or 28 measured by flow cytometry (n = 3 mice / group). e) Expression of HOXA9, MEIS1 and ITGAM in the bone marrow (BM) and spleen samples of the MV4;11 leukemia mice after 6 days of treatment with 28. Expression was normalized to GAPDH and referenced to vehicle treated mice, mean ± SD, n = 3.
To validate the on-target in vivo activity of 28 we performed gene expression studies in bone marrow and spleen samples isolated from mice transplanted with MV4;11 cells and treated with 28 or vehicle. Treatment with 28 was initiated 21 days after transplantation of mice with the leukemic cells to account for the effect of 28 in advanced leukemia model. Compound 28 (or vehicle) was administered for six consecutive days, followed by collection of the bone marrow and spleen samples. Importantly, substantial reduction in the spleen weight and size was observed for mice treated with 28 compared to the vehicle control, Figure 4c. Furthermore, the level of leukemic blasts (human CD45+ cells) was very high (~75%) in the bone marrow cells isolated from vehicle treated mice, which was indicative of advanced leukemia, while the level of leukemic blasts was substantially reduced (to ~35%) in mice treated with 28, Figure 4d. We then performed gene expression studies in bone marrow and spleen samples isolated from mice treated with 28 or vehicle using qRT-PCR and found that treatment with 28 resulted in a strong reduction (2–5 fold) in the expression level of MLL fusion target genes MEIS1 and HOXA9, Figure 4e. Furthermore, an over 4–6-fold increase in the expression of ITGAM differentiation marker was observed in the bone marrow and spleen samples upon treatment with 28, Figure 4e. Overall, the results from gene expression studies demonstrate pronounced, on-target activity of 28 in the advanced mouse model of MLL leukemia, thus supporting the potential therapeutic value of this compound.
Chemistry
Synthesis of compounds 3 and 4 was performed using the synthetic route shown in Scheme 1. Two major intermediates: 5-formyl-4-methyl-1H-indole-2-carbonitrile (30) and N-(piperidin-4-yl)-6-(2,2,2-trifluoroethyl)thieno[2,3-d]pyrimidin-4-amine (33) were obtained according to the procedure described before.10 The alcohols 31a,b were converted into mesylates followed by the substitution with the aldehyde 30 in the presence of cesium carbonate, which afforded aldehydes 32a,b. (Scheme 1). Compounds 32a,b were then used in the reductive amination reaction with the secondary amine 33 utilizing sodium triacetoxyborohydride as the reducing agent to afford compounds 34a,b. Removal of the tert-butoxycarbonyl (Boc) group in the presence of tin (IV) chloride resulted in compounds 3 and 4. Compound 5 bearing the morpholinone ring was obtained using the strategy depicted in Scheme 2. The 4-(2-chloroethyl)morpholine (35) was used for the indole alkylation of aldehyde 30, resulting in compound 36, Scheme 2. The reductive amination of aldehyde 36 with the amine 33 resulted in compound 5, Scheme 2.
Scheme 1. Synthesis of compounds 3 and 4a.

a Reagents and conditions: a) MsCl, TEA, DCM −67°C- rt; b) 30, Cs2CO3, DMF, 90–110°C; c) 33, Na(AcO)3BH; TEA, rt; d) SnCl4, tartaric acid.
Scheme 2: Synthesis of compound 5.a.

a Reagents and conditions: a) 30, Cs2CO3, TBAI, DMF, 90°C; b) 33, Na(AcO)3BH; TEA, rt.
The racemic mixtures of compounds 6 and 7 were prepared according to the synthetic route presented in Scheme 3. The intermediates 39a and 39b were obtained using the methods described before.30–31 Briefly, the commercially available 3-amino-1,2-propanediol (37a) and methyl 4-amino-3-hydroxybutanoate (37b) were subjected to acylation with the chloroacetic acid, resulting in compounds 38a and 38b, respectively. Direct cyclisation of 38a with potassium t-butoxide afforded alcohol 39a. Homologous alcohol 39b was achieved by reduction of the ester group in 38b followed by the analogous cyclisation with potassium t-butoxide as a base (Scheme 3). The alcohols 39a,b were then converted to the corresponding mesylates and used for alkylation of the indol 30 to afford aldehydes 40a and 40b. Reductive amination reactions of the intermediate 33 with aldehydes 40b or 40a led to the target compounds 6 and 7, respectively (Scheme 3).
Scheme 3: Synthesis of compounds 6 and 7a.

a Reagents and conditions: a) chloroacetic acid chloride, MeOH, AcCN, TEA, 0°C; b) t-BuOK, amyl alcohol, rt (for 38a); c) LiBH4, THF, 0°C and d) t-BuOK, amyl alcohol, 0°C (for 38b); e) MsCl, TEA, DCM, −67°C- rt; f) 30, Cs2CO3, DMA, 105°C; g) 33, Na(AcO)3BH; AcOH, 50°C.
The synthetic route for compounds 8 and 9 harboring the piperazine ring in the structure is shown in Scheme 4. Briefly, both compounds were prepared from 3 using either formaline (for 8) or propionaldehyde (for 9) in the reductive amination reaction, Scheme 4. The piperazine and piperidine analogues 10–27 were obtained according to the synthetic strategy depicted in Scheme 5. In general, the amide series (compounds 10–17 and 24–25) was obtained by acylation of the amines 3 or 4. The formamides 10 and 24 were prepared by formylation of 3 or 4, respectively, with the methyl formate. The corresponding acyl chlorides were used to convert the amine 3 into compounds 11, 12 or 17 with DIPEA serving as a base, Scheme 5. The analogous reaction was performed with the amine 4 to afford compound 25. Synthesis of the amides 13,14,16 was performed by utilizing the coupling reaction between the amine 3 and the corresponding carboxylic acids in the presence of 2-(1H-Benzotriazole-1-yl)-1,1,3,3-tetramethylaminium tetrafluoroborate (TBTU) serving as a condensing reagent, Scheme 5. Compound 15 with the trifluoroacetyl moiety was prepared using the triflate anhydride as an acylating agent. Alkylation of the nitrogen in the piperazine ring of 3 by the 2-bromopropionamide led to the amine 18. Scheme 5.
Scheme 4: Synthesis of compounds 8 and 9a.

a Reagents and conditions: a) formaline, Na(AcO)3BH, AcCN, rt (for 8); b) n-propanal, Na(AcO)3BH; TEA, DCM, rt (for 9).
Scheme 5: Synthesis of compounds 10–27a.

a Reagents and conditions: a) HCOOMe, reflux (for 10,24); b) R2COCl, DIPEA, DCM or AcCN, rt (for 11,12,17, 25); c) R2COOH, TBTU, THF (for 13,14,16); d) (CF3CO)2O, DCM, 0°C-rt (for 15); e) rac-BrCH(CH3)CONH2, DMF (for 18); f) R1SO2Cl; DIPEA, DCM, rt (for 19,20,21,23,26); g) CbzNHSO2Cl, DIPEA, DCM and h) Pd/C, MeOH, ammonium formate (for 22,27).
The sulfonamide series (compounds 19–21, 26) was obtained using the sulfonylation reactions with compounds 3 or 4 and the appropriate sulfonyl chlorides, Scheme 5. The two-step synthetic approach was applied to afford compounds 22 and 27. The benzyl (chlorosulfonyl)carbamate was used for sulfonylation of the amines 3 or 4, followed by the cleavage of the Cbz-protected amino group performed with Pd/C and ammonium formate, which led to sulfamides 22 or 27, Scheme 5. Conversion of the amine 3 to compound 23 was achieved using the methylsulfamoyl chloride in the presence of DIPEA.
Synthesis of compounds 28 and 29, which represent single enantiomers of 7, was performed by applying the same methodology as for compound 7, Scheme 3 through utilizing the non-racemic 3-aminopropane-1,2-diols: 37a(S) or 37a(R).
Conclusions
Identification of potent inhibitors targeting protein-protein interactions and optimization of their drug-like properties represents a challenge as such compounds frequently have large molecular weight, which is required for effective blocking of PPI interfaces.18–20 This is particularly true for compounds that target extended protein-protein interfaces, including inhibitors of the BCL-2 family of proteins 26 and others.1 On the other hand, recent FDA approval of the BCL-2 inhibitor Venetoclax demonstrates that PPI inhibitors with large molecular weight (>800 Da) can serve as valuable therapeutics. In the current study, we report optimization of the small molecule inhibitors of the menin-MLL interaction, with the major goal of improving the potency and drug-like properties of these compounds. The MLL binding site on menin is large (over 5,000 Å3 9) as menin binds long, bivalent MLL peptide (MLL4–43). This suggests that compounds with large molecular weight may be required for effective inhibition of the menin-MLL interaction.
Using the crystal structure of menin in complex with MI-503 we designed, synthesized and evaluated a series of new analogues by exploring various saturated rings that substitute indole nitrogen in MI-503, Tables 1–4. The goal of this work was to increase the inhibitory activity and metabolic stability of menin-MLL inhibitors by introducing substituents with different physicochemical properties (e.g. charged versus uncharged substituents, hydrogen bond donor or acceptor groups, etc.). These efforts resulted in compound 28, which demonstrates very potent inhibition of the menin-MLL interaction (IC50 = 3.6 nM with MLL4–43). Therefore, 28 represents the most potent reversible small molecule inhibitor of the menin-MLL interaction reported to date 10, 12. About 10-fold improved inhibitory activity for 28 over previous generation of reversible menin-MLL inhibitors (e.g. MI-503, MI-463) results from stronger binding affinity of this compound to menin, but also likely from more pronounced overlap with MBM2 binding to menin, as supported by the structural data, Figure 2c. Furthermore, 28 demonstrates stronger effect (>8-fold) and better selectivity towards MLL leukemia cells than MI-503, and it has also more pronounced effect on the target gene expression in mouse models of MLL leukemia, Figure 5E.10 On the other hand, increased polarity of 28 (clogP value reduced) and increased molecular weight (by ~30 Da) over MI-503 (Mw = 597 Da for 28) negatively affected oral bioavailability of this compound. These results demonstrate the complexity of blocking bi-valent protein-protein interactions with large molecular interfaces, where achievement of potent inhibition requires effective interference with both motifs occupying different regions of the binding site. The increase in the molecular weight of inhibitors targeting bi-valent PPIs (e.g. > 500 Da) can negatively affect their drug-like properties, making the lead optimization process extensive and complex.
Importantly, strong in vitro inhibition induced by 28 correlates well with pronounced activity of this compound in MLL leukemia cells (GI50 = 30–60 nM), which represents almost 10-fold improvement over MI-503 or any other reversible menin-MLL inhibitor 10, 12. In addition, compound 28 demonstrates high selectivity (>100-fold) and specific mechanism of action in MLL leukemia cells. Furthermore, the pronounced effect of 28 in inhibiting leukemia progression in the disseminated model of MLL leukemia combined with the on-target mechanism of action in vivo make this compound a very attractive candidate compound for further studies. During the preparation of this article, irreversible menin-MLL inhibitors with distinct molecular scaffold were reported,32 but these compounds did not show stronger in vitro inhibitory activity over 28 despite covalent binding to menin. Furthermore, biological activity of these irreversible menin-MLL inhibitors, especially in the in vivo models of MLL leukemia, and their potential off-target effects remain to be assessed.
Overall, compound 28 identified in this study represents a valuable molecule to further investigate the role of menin-MLL interaction in normal and pathological states. This compound may also lead to the development of new therapeutic agents for MLL leukemia and possibly solid tumors, including prostate cancer 13 Ewing sarcoma,14 HCC,16 and other tumors that may rely on the intact menin-MLL interaction. Our work demonstrates that, despite challenges associated with the development of potent PPI inhibitors targeting bi-valent protein-protein interfaces, successful identification of such compounds is possible and can lead to valuable chemical tools for the scientific community and potentially to new therapeutic agents.
Experimental Section
Expression and purification of menin
The expression and purification of menin have been described previously.8–9 In order to generate biotinylated menin, we introduced Avi-tag sequence (AAALEGLNDIFEAQKIEWHE)33 to the C-terminus of menin. Protein was co-expressed in E. Coli BL21 (DE3) cells with the construct encoding BirA enzyme33 and purified using the same protocol as for the intact menin.
Biochemical characterization of menin-MLL inhibitors
Inhibition of the menin-MLL interaction by small molecules was assessed by fluorescence polarization (FP) assay. The fluorescein-labeled MLL4–43 peptide (FLSN-MLL4–43) at 4 nM, menin at 4 nM and varying concentrations of compounds were used for IC50 determination in the FP buffer (50 mM Tris, pH 7.5, 50 mM NaCl, 1 mM TCEP). The complex of menin with the FLSN-MLL 4–43 peptide was incubated for 1h. Compounds (5% final DMSO concentration) were then added to the menin-FLSN_MLL4–43 complex and incubated for 3h before changes in fluorescence polarization (mP values) were measured using the PHERAstar microplate reader (BMG). The IC50 values were calculated in Origin 7.0 (OriginLab) by plotting the mP values measured for each compound as a function of compound concentration.
Bio-Layer Interferometry (BLI) binding assay
BLI assay was performed in 96-well microplates at room temperature with continues shaking at 1000 rpm using the Octet Red 96 system (Fortebio, Menlo Park, CA, USA). Biotinylated menin was bound on Super Streptavidin (SSA) biosensors (ForteBio) by dipping sensors into 200 nM biotinylated menin in the assay buffer composed of 50 mM Tris, pH 7.5, 50 mM NaCl, 1 mM TCEP, 0.05% BSA, 0.01% Tween-20 and 2% DMSO. Menin binding was performed for 600 s followed by equilibration in the assay buffer for 1200 s. Compounds (28 and MI-503) were used at 150 and 200 nM concentrations and binding was performed for 600 s followed by 1200 s dissociation. Data were fit globally and generated automatically by Octet User Software (ForteBio, Inc.). Analysis of binding kinetics was performed in Prism (GraphPad Software, Inc.). Kobs values were extracted using one phase association equation, and Koff values were extracted using one phase decay equation. Subsequently, Kon was derived using Kobs = Kon × [Ligand] + Koff, where [Ligand] is a free ligand concentration in solution. KD was calculated as Koff/Kon.
Crystallization of menin complexes with small molecule inhibitors
For co-crystallization experiments 2.5 mg/mL menin was incubated with small molecule inhibitors at 1:3 molar ratio. Crystals were obtained using the sitting drop technique at 10 °C by applying the procedure described previously. 9 Prior to data collection, crystals were transferred into a cryo-solution containing 20% PEG550 MME and flash-frozen in liquid nitrogen.
Crystallographic data collection and structure determination
Diffraction data for menin and menin complexes were collected at the 21-ID-D and 21-ID-F beamlines at the Life Sciences Collaborative Access Team at the Advanced Photon Source. Data were processed with HKL-2000.34 Structures of the complexes were determined by molecular replacement using MOLREP with the apo-structure of human menin (PDB code: 4GPQ) as a search model in molecular replacement. The model was refined using REFMAC,35 COOT36, CCP4 package 37 and PHENIX 38.Validation of the structures was performed using MOLPROBITY39 and ADIT.40 Details of data processing and refinement are summarized in Supplementary Table 1. Coordinates and structure factors for menin-inhibitor complexes have been deposited in the Protein Data Bank under the following codes: 6BXH (menin-compound 13), 6BXY (menin-compound 28), 6BY8 (menin-compound 29).
Viability assays
The MLL-AF9, HM-2 and E2A-HLF transformed murine bone marrow cells (BMCs) were prepared as described previously.41 MV4;11, MOLM-13, K562 and U937 cells were cultured in RPMI-1640 medium with 10% FBS, 1% penicillin/streptomycin and NEAA (Non-essential amino acid) solution. For viability assay, MOLM-13 (1×105/mL), MV4;11 (1×105/mL), K562 (1×105/mL) and U937 (1×105/mL) human leukemia cells as well as MLL-AF9, HM-2 and E2A-HLF murine bone marrow cells (2.5×104/mL) were plated (1 mL/well), treated with compounds or 0.25% DMSO and cultured at 37 ᵒC for 7 days. Media were changed at day four with viable cell number restored to the original concentration and compounds were re-supplied. 100 μL of cell suspension were transferred to 96 well plates for each sample in quadruplicates. A Vybrant MTT cell proliferation assay kit (Molecular Probes) was employed. Plates were read for absorbance at 570 nm using a PHERAstar BMG microplate reader. The experiments were performed 2–3 times in quadruplicated with calculation of mean and standard deviation for each condition.
Real-Time PCR
Effect of 28 on the expression level of MLL fusion target genes was assessed by Real-time quantitative PCR (qRT-PCR) using the protocol described previously.10
Microsomal stability studies
The metabolic stability was assessed using CD-1 mouse liver microsomes. 1 μM compounds were incubated with 1.3 mg/mL microsomes and 1.7 mM cofactor NADPH in 0.1 M phosphate buffer (pH =7.4) containing 3.3 mM MgCl2 at 37 °C. The DMSO concentration was less than 0.1% in the final incubation system. At 0, 3, 5, 10, 15, 30, 45 and 60 min of incubation, the reactions were stopped by adding 3-fold excess of acetonitrile containing 100 ng/mL of internal standard for quantification. The collected fractions were centrifuged at 3000 g for 20 min to collect the supernatant for LC-MS/MS analysis, from which the amount of compound remaining was determined. The natural log of the amount of compound remaining was plotted against time to determine the disappearance rate and the half-life of tested compounds.
Pharmacokinetic studies in mice
All animal experiments in this study were approved by the University of Michigan Committee on Use and Care of Animals and Unit for Laboratory Animal Medicine (ULAM).
The pharmacokinetics of 28 was determined in female C57BL/6 mice following intravenous (iv) dosing at 15 mg/kg and 30 mg/kg for oral (po) and intraperitoneal (ip) dosing. For iv and po administration, 28 was dissolved in the vehicle containing 25% (v/v) DMSO, 25% (v/v) PEG400 and 50% (v/v) PBS. For ip administration, 28 was dissolved in the vehicle containing 20% HP-β-cyclodextrin and 5% Cremophor. Serial blood samples were collected over 7h, centrifuged at 15,000 rpm for 10 min and saved for analysis. Plasma concentrations of 28 were determined by the LC-MS/MS method developed and validated for this study. The LC-MS/MS method consisted of an Agilent 1200 HPLC system and chromatographic separation of 28 was achieved using an Agilent Zorbax Extend-C18 column (5 cm x 2.1 mm, 3.5 μm; Waters). An AB Sciex QTrap 3200 mass spectrometer equipped with an electrospray ionization source (ABI-Sciex, Toronto, Canada) in the positive-ion multiple reaction monitoring (MRM) mode was used for detection. Pharmacokinetic parameters were calculated by noncompartmental methods using WinNonlin® version 3.2 (Pharsight Corporation, Mountain View, CA, USA).
In vivo efficacy studies
All animal experiments in this study were approved by the University of Michigan Committee on Use and Care of Animals and Unit for Laboratory Animal Medicine (ULAM).
For in vivo efficacy studies, 6 to 8-week-old female NSG mice (Jackson laboratory) were intravenously injected with 1×107 luciferase-expressing MV4;11 cells. At day 13 post-transplantaion, mice were split intro vehicle and the treatment groups of 8 mice per group and were treated for 21 consecutive days with once daily intraperitoneal injections of vehicle (20% HP-β-cyclodextrin and 5% Cremophor) or 80 mg/kg of 28 dissolved in vehicle. Imaging of mice was performed at days 0, 7, 14 and 21 of treatment with 28. For imaging, mice were kept anesthetized with Isoflurane, and luciferase at 50 mg/kg was administered by intraperitoneal injection. Photonic emission was imaged using the in Vivo Imaging System BLI (IVIS, 200) with total imaging time of 10 seconds for each mouse. Survival of mice was monitored to assess treatment efficacy.
For the in vivo gene expression studies, 21 days after transplantation of mice with MV4;11 cells mice were grouped into vehicle or compound 28 treatment groups (n = 3 / group), and the treatment was initiated and continued for 6 consecutive days (80 mg/kg, i.p., once daily). The next day after completing the treatment mice were sacrificed and bone marrow samples were isolated for gene expression studies by qRT-PCR.
Flow cytometric analysis of mice samples
To assess levels of leukemia burden in the mice transplanted with leukemic cells, cells from bone marrow, peripheral blood, and spleen were buffered with PBS and 1% FBS for flow cytometery analysis. Red blood cells were lysed with ACK (Lonza). All flow cytometry experiments were performed on a LSR II FACSCanto or FACSAria (BD Biosciences), and all data were analyzed with FlowJo software (Tree Star, Inc.).
General Chemistry
All commercially available solvents and reagents were used without further preparation unless otherwise indicated. The 1H and 13C NMR data were taken on Bruker Avance III 600 MHz or Varian MR400. Chemical shifts are reported in ppm relative to tetramethylsilane or residual solvent signal. Some signals that overlapped with solvents were identified using 1H-13C HSQC spectrum (multiplicity not known). The mass measurements were determined on a Micromass LCT time-of-flight mass spectrometer using positive mode and electrospray ionization. The exact mass measurements were determined on Agilent Q-TOF time-of-flight mass spectrometer using positive ion mode and electrospray ionization. Analytical TLC was performed on Merck TLC aluminum plates precoated with F254 silica gel 60 (UV, 254 nm, and iodine). The purity analysis of final compounds was determined on Shimadzu Prominence HPLC system (20 series: binary pump, UV/vis at 254 nm, heated column compartment 28 °C), using Restek Ultra C18 (5 μm) 150 mm × 4.6 mm column. LR-MS was recorder on Shimadzu LC-2020 system (DUIS-ESI). The solvents were programmed to run at gradient starting 20% CH3CN in water to 80% during 8 min run. If not indicated, the purity of all final compounds was >95% as determined by HPLC via integration of UV spectra at 254 nm. Optical rotation was measured using Jasco P2000 polarimeter (path lenght = 100 mm). All final (unless indicated otherwise) compounds 1–29 were tested as hydrochloride salts. The general procedure for HCl salt formation: free base was dissolved in minimum volume of methanol followed by addition of 1 equiv of 1 M HCl in water and thorough drying. During NMR experiment, most of the hydrochloride salts of these compounds exist as a mixture of two forms in solution of either DMSO or MeOH with the approximate ratio of 10:1; NMR is reported for the major rotamer or a free base as indicated.
tert-butyl 4-(2-(2-cyano-5-formyl-4-methyl-1H-indol-1-yl)ethyl)piperazine-1-carboxylate (32b)
To a stirred solution of tert-butyl 4-(2-hydroxyethyl)piperazine-1-carboxylate (31a, 1 g 4.3 mmol) and trimethylamine (1.5 eq, 898 μL, 6.45 mmol) in DCM (14 ml) methanosulfonyl chloride (1.2 eq, 399 μL, 5.16 mmol) was added dropwise at 0°C. Mixture was allowed to warm to the room temperature and stirred for 3 h. The organic phase was diluted with DCM (40 mL) and washed with water, brine and dried over sodium sulfate. Solvent was evaporated (partial decomposition was observed when heated over 35°C during evaporation) and crude product (1.25 g) was used in the next step. The 5-formyl-4-methyl-1H-indole-2-carbonitrile (30, 300 mg, 1.63 mmol) was suspended in DMF (3.2 mL) and cesium carbonate was added (1.63 g, 4.68 mmol, 3 eq). The tert-butyl 4-(2-((methylsulfonyl)oxy)ethyl)piperazine-1-carboxylate, from the previous step (753 mg, 2.45 mmol, 1.5 eq) was then added and resulting suspension was stirred at room temperature for 22 h. Reaction was quenched with water and extracted with DCM (3×40 mL). Organic phase was evaporated with silica gel and separated using flash chromatography (silica gel, hexanes:ethyl acetate 3:1 to 1:1, v:v). 316 mg of compound 32b was isolated (48% yield). MS (m/z) found for [M+H+]: 397 Da.
tert-butyl 4-(2-(2-cyano-4-methyl-5-((4-((6-(2,2,2-trifluoroethyl)thieno[2,3-d]pyrimidin-4-yl)amino)piperidin-1-yl)methyl)-1H-indol-1-yl)ethyl)piperazine-1-carboxylate (34b)
Compound 32b (880 mg, 2.79 mmol, 1 eq) and amine 33 (1.01 g, 3.06 mmol, 1.1 eq) were dissolved in DCM (28 mL). Triethylamine was added (1.6 mL, 4 eq) followed by sodium triacetoxyborohydride (1.2 g, 5.6 mmol, 2 eq). Reaction was stirred overnight at room temperature. Mixture was diluted with DCM (50 mL) and washed with 1M NaOHaq. Organic phase was dried over sodium sulfate and evaporated with silica gel. Purified using flash chromatography (silica gel, dichloromethane:methanol with 5% ammonia, 0% to 5%, v:v). 1.2 g of product 34b was isolated (62% yield). 1H NMR (600 MHz, CDCl4/METHANOL-d4) δ 8.29 (s, 1H), 7.31 – 7.39 (m, 2H), 7.17 – 7.24 (m, 2H), 4.34 (t, J = 6.2 Hz, 2H), 4.10 – 4.19 (m, 1H), 3.65 (q, J = 10.27 Hz, 2H), 3.61 – 3.70 (m, 2H), 3.33 – 3.39 (m, 4H), 2.91 – 3.10 (m, 2H), 2.72 (t, J = 6.42 Hz, 2H), 2.54 (s, 3H), 2.38 – 2.46 (m, 4H), 2.19 – 2.38 (m, 2H), 1.95 – 2.06 (m, 2H), 1.55 – 1.72 (m, 2H), 1.40 (s, 9H); MS (m/z) found for [M+H+]: 697 Da.
4-methyl-1-(2-(piperazin-1-yl)ethyl)-5-((4-((6-(2,2,2-trifluoroethyl)thieno[2,3-d]pyrimidin-4-yl)amino)piperidin-1-yl)methyl)-1H-indole-2-carbonitrile (3)
Compound 34b (180 mg, 0.26mmol) was dissolved in acetonitrile (2.6 mL) and tin (IV) chloride (677 mg, 304 μL, 2.6 mmol, 10 eq) was added at 0°C. The reaction was stirred for 1h at room temperature and then cooled again to 0°C. Ammonia solution (30%aq) was added slowly and DCM was evaporated. Water was added (2 mL) followed by tartaric acid (750 mg). The mixture was sonicated until the solution became clear. The product was extracted with DCM:MeOH (10:1). Purified on preparative TLC (silica gel, dichloromethane:methanol with 5% ammonia, 5%, v:v). 88 mg of compound 3 was isolated (57%). 1H NMR (600 MHz, CHLOROFORM-d) δ 8.47 (s, 1H), 7.35 (d, J = 8.44 Hz, 1H), 7.18 (s, 1H), 7.14 – 7.17 (m, 1H), 7.10 (s, 1H), 5.21 (d, J = 7.70 Hz, 1H), 4.33 (t, J = 6.60 Hz, 2H), 4.14 – 4.29 (m, 1H), 3.57 – 3.68 (m, 4H), 2.87 – 2.97 (m, 6H), 2.66 – 2.74 (m, 2H), 2.54 (s, 3H), 2.46 – 2.53 (m, 4H), 2.22 – 2.30 (m, 2H), 2.04 – 2.12 (m, 2H), 1.53 – 1.65 (m, 2H); 13C NMR (151 MHz, CHLOROFORM-d) δ 166.8, 156.1, 154.3, 136.4, 131.2, 129.0, 128.6, 128.0, 127.3, 124.7 (J = 277 Hz) 118.6, 116.4, 114.0, 111.6, 109.8, 107.2, 60.1, 57.8, 53.9, 52.3, 51.3, 48.0, 45.5, 43.3, 35.7 (J = 32 Hz), 32.4, 15.1; HRMS (ESI): [M+H+] calculated 597.2736; found 597.2731.
4-methyl-1-(2-morpholinoethyl)-5-((4-((6-(2,2,2-trifluoroethyl)thieno[2,3-d]pyrimidin-4-yl)amino)piperidin-1-yl)methyl)-1H-indole-2-carbonitrile (5)
Compound 30 (84 mg, 0.46 mmol, 1 eq) and 4-(2-chloroethyl)morpholine (35) (126 mg, 0.68 mmol, 1.5 eq) were dissolved in DMF (0.92 mL). TBAI (85 mg, 0.23 mmol, 0.5 eq) was added followed by cesium carbonate (905 mg, 1.76 mmol, 6 eq). The suspension was heated at 60°C for 1h. DMF was evaporated with silica gel and product was isolated using flash chromatography (silica gel, hexane:ethyl acetate, 1:1, v:v) to afford 63 mg of compound 36 (47% yield). Compound 36 (22 mg, 0.07 mmol) was then suspended in DCM (0.7 mL) and compound 33 (32 mg, 0.09 mmol, 1.2eq) and triethylamine (20 μL, 0.11 mmol, 1.4 eq) were added. The suspension was stirred for 15 min and then sodium triacetoxyborohydride (23 mg, 0.11 mmol, 1.5 eq) was added. The reaction was stirred overnight. DCM was added to dilute the solution and organic phase was washed with 1M NaOHaq and brine. DCM was evaporated and the product was recrystallized from MeOH affording 30 mg of compound 5 (67% yield). 1H NMR CD3OD (600 MHz): δ 8.35 (s, 1H), 7.56 (m, 3H), 7.45 (s, 1H), 4.54 (m, 2H), 4.49 (m, 3H), 3.87 (q, J = 11 Hz, 2H), 3.65 (m, 6H), 2.79 (m, 2H), 2.70 (s, 3H), 2.53 (m, 4H), 2.35 (m, 2H), 1.99 (m, 2H). 13C NMR CD3OD (150 MHz): δ 167.1, 157.7, 154.9, 138.8, 135.1, 130.6, 130.0, 128.8, 126.6 (q, J = 276 Hz), 121.9, 121.2, 118.3, 114.6, 113.4, 112.6, 110.6, 67.9, 59.0, 58.8, 55.0, 53.1, 47.2, 44.2, 35.9, 35.7 (q, J = 33 Hz), 30.1, 15.9. HRMS (ESI): [M+H+] calculated 598.2570; found 598.2573.
4-methyl-1-(2-(4-propylpiperazin-1-yl)ethyl)-5-((4-((6-(2,2,2-trifluoroethyl)thieno[2,3-d]pyrimidin-4-yl)amino)piperidin-1-yl)methyl)-1H-indole-2-carbonitrile (9)
Compound 3 (20 mg, 0.03 mmol, 1eq) and propionaldehyde (3 μL, 0.034 mmol, 1.2 eq) were dissolved in DCM (0.3 mL). After stirring for 15 min sodium triacetoxyborohydride (13 mg, 0.06 mmol, 2 eq) was added. The mixture was directly evaporated with silica gel and compound 9 (10 mg, 56% yield) was purified using flash chromatography (silica gel, dichloromethane:methanol with 5% ammoniaaq, 5%, v:v). 1H NMR CD3OD (600 MHz): δ 8.36 (s, 1H), 7.63 (m, 1H), 7.56 (m, 2H), 7.56 (m, 2H), 7.45 (s, 1H), 4.55 (m, 2H), 4.49 (m, 2H), 3.86 (q, J = 11 Hz, 2H), 3.62 (m, 2H), 3.49 (m, 2H), 3.06 (m, 5H), 2.84 (m, 2H), 2.70 (s, 3H), 2.55 (m, 2H), 2.34 (m, 2H), 2.04 (m, 2H), 1.76 (m, 2H), 1.00 (t, J = 7.3 Hz, 3H). 13C NMR CD3OD (150 MHz): δ 176.4, 166.6, 157.7, 154.7, 154.7, 138.7, 135.1, 130.8, 130.1, 128.7, 126.8 (q, J = 276 Hz), 121.8, 121.1, 118.2, 114.8, 113.3, 112.6, 110.4, 59.4, 58.8, 57.7, 53.3, 52.9, 51.3, 47.1, 44.4, 35.6 (q, J = 33 Hz), 29.9, 22.1, 18.5, 15.8, 11.1. HRMS (ESI): [M+H+] calculated 639.3200; found 639.3198.
1-(2-(4-formylpiperazin-1-yl)ethyl)-4-methyl-5-((4-((6-(2,2,2-trifluoroethyl)thieno[2,3-d]pyrimidin-4-yl)amino)piperidin-1-yl)methyl)-1H-indole-2-carbonitrile (10)
Compound 3 (750 mg, 1.25 mmol) was dissolved in methyl formate (10 mL) and stirred overnight. Solvent was evaporated with silica gel and compound 10 (700 mg, 89% yield) was isolated using flash chromatography (silica gel, dichloromethane:methanol with 5% ammoniaaq, 5%, v:v). 1H NMR CD3OD (600 MHz): δ 8.34 (s, 1H), 7.98 (m, 1H), 7.61 (m, 1H), 7.55 (m, 2H), 7.45 (s, 1H), 4.55 (m, 2H), 4.49 (m, 3H), 3.86 (q, J = 11 Hz, 2H), 3.64 (m, 2H), 3.48 (m, 2H), 3.43 (m, 2H), 3.35 (m, 2H), 2.85 (m, 2H), 2.70 (s, 3H), 2.58 (m, 2H), 2.50 (m, 2H), 2.34 (m, 2H), 2.03 (m, 2H). 13C NMR CD3OD (150 MHz): δ 167.0, 163.2, 157.8, 154.9, 138.9, 135.2, 130.8, 130.1, 128.9, 126.8 (q, J = 276 Hz), 121.9, 121.2, 118.3, 114.7, 113.4, 112.6, 110.6, 59.0, 58.2, 54.9, 53.8, 53.0, 47.2, 46.7, 44.4, 40.9, 35.7 (q, J = 33 Hz), 30.1, 16.0. HRMS (ESI): [M+H+] calculated 625.2679; found 625.2682.
General procedure for synthesis of compounds (11, 12, 17)
Compound 3 (20 mg, 0.03 mmol) was dissolved in DCM or AcCN (0.2 mL) and DIPEA (12 μL, 0.05 mmol) was added. Mixture was cooled to 0°C and corresponding acid chloride was added. Mixture was allowed to warm to the room temperature and was stirred until no starting material was detected by TLC (30 min).
1-(2-(4-acetylpiperazin-1-yl)ethyl)-4-methyl-5-((4-((6-(2,2,2-trifluoroethyl)thieno[2,3-d]pyrimidin-4-yl)amino)piperidin-1-yl)methyl)-1H-indole-2-carbonitrile (11)
Reaction carried out in AcCN with acetyl chloride. Isolated 16 mg (86% yield). 1H NMR CD3OD (600 MHz): δ 8.38 (s, 1H), 7.69 (m, 2H), 7.58 (s, 1H), 7.53 (s, 1H), 4.56 (m, 2H), 4.49 (m, 1H), 3.87 (q, J = 11 Hz, 2H), 3.80 (m, 2H), 3.63 (m, 2H), 3.35 (m, 2H), 3.19 (m, 4H), 2.71 (s, 3H), 2.34 (m, 2H), 2.13 (s, 3H), 2.04 (m, 2H). 13C NMR CD3OD (150 MHz): 176.5, 171.9, 165.9, 157.7, 154.4, 139.0, 135.5, 131.3, 130.5, 128.9, 126.8 (q, J = 276 Hz), 122.0, 121.7, 118.3, 114.6, 114.2, 111.9, 110.6, 58.9, 56.5, 53.8, 53.6, 53.0, 47.4, 45.5, 42.0, 40.7, 35.7 (q, J = 33 Hz), 30.0, 27.4, 22.2, 21.1, 16.0. HRMS (ESI): [M+H+] calculated 639.2836; found 639.2841.
4-methyl-1-(2-(4-propionylpiperazin-1-yl)ethyl)-5-((4-((6-(2,2,2-trifluoroethyl)thieno[2,3-d]pyrimidin-4-yl)amino)piperidin-1-yl)methyl)-1H-indole-2-carbonitrile (12)
The reaction was carried out in DCM with propionic acid chloride. Isolated 19 mg (99% yield). 1H NMR CD3OD (600 MHz): δ 8.35 (s, 1H), 7.61 (m, 2H), 7.54 (s, 1H), 7.48 (s, 1H), 4.61 (m, 2H), 4.55 (m, 2H), 4.47 (m, 1H), 3.86 (q, J = 11 Hz, 2H), 3.62 (m, 2H), 3.60 (m, 2H), 3.35 (m, 2H), 2.70 (s, 3H), 2.34 (m, 2H), 2.40 (q, J = 7.6 Hz, 2H), 2.35 (m, 2H), 2.00 (m, 2H), 1.10 (t, J = 7.6 Hz, 3H). 13C NMR CD3OD (150 MHz): 174.8, 166.8, 157.7, 154.7, 138.8, 135.2, 130.8, 130.0, 128.8, 126.8 (q, J = 276 Hz), 121.7, 121.2, 118.2, 114.4, 110.5, 58.8, 54.3, 54.0, 52.9, 47.1, 35.7 (q, J = 33 Hz), 30.0, 27.1, 15.8, 9.8. HRMS (ESI): [M+H+] calculated 653.2992; found 653.2989.
1-(2-(4-(3-cyclopentylpropanoyl)piperazin-1-yl)ethyl)-4-methyl-5-((4-((6-(2,2,2-trifluoroethyl)thieno[2,3-d]pyrimidin-4-yl)amino)piperidin-1-yl)methyl)-1H-indole-2-carbonitrile (17)
The reaction was carried out in DCM with 3-cyclopentylpropanoyl chloride. Reaction time: 30 min. Isolated 13 mg, 52% yield. 1H NMR (600 MHz, METHANOL-d4) δ 8.36 (s, 1H), 7.50 – 7.64 (m, 3H), 7.44 (s, 1H), 4.48 (t, J = 5.9 Hz, 2H), 4.33 – 4.47 (m, 3H), 3.88 (q, J = 10.6 Hz, 2H), 3.48 – 3.60 (m, 6H), 3.10 – 3.24 (m, 2H), 2.78 (t, J = 5.9Hz,. 2H), 2.70 (s, 3H), 2.48 – 2.54 (m, 2H), 2.41 – 2.45 (m, 2H), 2.36 – 2.40 (m, 2H), 2.26 – 2.33 (m, 2H), 2.21 – 2.26 (m, 1H), 1.93 – 2.12 (m, 2H), 1.77 – 1.86 (m, 2H), 1.62 – 1.69 (m, 2H), 1.51 – 1.61 (m, 4H), 1.08 – 1.19 (m, 2H); 13C NMR (151 MHz, METHANOL-d4) δ 179.5, 174.3, 167.1, 157.7, 154.9, 138.7, 134.6, 130.5, 129.9, 128.8, 126.6 (J = 276.20Hz) 121.8, 118.2, 114.7, 113.1, 112.4, 110.2, 58.3, 54.7, 54.2, 47.0, 44.6, 42.8, 41.1, 35.7 (J = 31.9 Hz), 35.9, 33.5, 33.5, 33.4, 33.3, 32.9, 26.1, 15.7; HRMS (ESI): [M+H+] calculated 721.3618; found 721.3619.
General procedure for synthesis of compounds (13, 14, 16)
Compound 3 (30 mg, 0.05 mmol) was dissolved in THF (0.5 mL). Fluoroacetic acid (3.1 μL, 0.06 mmol, 1.2 eq), difluoroacetic acid (3.8 μl, 0.06 mmol, 1.2 eq) or 2-cyclopentylacetic acid (70 mg, 0.5 mmol, 1.1 eq) were added followed by TBTU (22.8 mg, 0.07 mmol, 1.3 eq for 13 and 14) or EDCI (11 mg, 0.06 mmol, 1.1 eq for 16). Reaction was carried out at room temperature until the starting material was not detected by LC-MS. (0.5–24h). Crude mixture was loaded on preparative TLC plate and purified using DCM:methanol (20:1, v:v).
1-(2-(4-(2-fluoroacetyl)piperazin-1-yl)ethyl)-4-methyl-5-((4-((6-(2,2,2-trifluoroethyl)thieno[2,3-d]pyrimidin-4-yl)amino)piperidin-1-yl)methyl)-1H-indole-2-carbonitrile (13)
Reaction time: 3h. Isolated 24 mg, 73 % yield. 1H NMR CD3OD (600 MHz): δ 8.34 (s, 1H), 7.59 (m, 1H), 7.54 (m, 2H), 7.44 (s, 1H), 5.07 (d, J = 40 Hz, 2H), 4.54 (m, 2H), 4.47 (m, 3H), 3.86 (q, J = 11 Hz, 2H), 3.63 (m, 2H), 3.53 (m, 2H), 3.35 (m, 2H), 2.77 (m, 2H), 2.70 (s, 3H), 2.50 (m, 2H), 2.45 (m, 2H), 2.34 (m, 2H), 1.99 (m, 2H). 13C NMR CD3OD (150 MHz): δ 168.2 (d, J = 40 Hz), 167.3, 158.0, 155.1, 139.0, 135.3, 130.8, 130.2, 129.0, 126.9 (q, J = 276 Hz), 122.0, 121.3, 118.4, 114.9, 113.4, 112.9, 110.7, 80.5 (d, J = 40 Hz), 59.1, 58.5, 54.5, 54.2, 53.2, 47.4, 45.5, 44.8, 43.1, 35.7 (q, J = 33 Hz), 30.2, 16.1. HRMS (ESI): [M+H+] calculated 657.2742; found 657.2745.
1-(2-(4-(2,2-difluoroacetyl)piperazin-1-yl)ethyl)-4-methyl-5-((4-((6-(2,2,2-trifluoroethyl)thieno[2,3-d]pyrimidin-4-yl)amino)piperidin-1-yl)methyl)-1H-indole-2-carbonitrile (14)
Reaction time: 24h. Purified two times on the prep-TLC plate isolated 15 mg (32%) 1H NMR CD3OD (600 MHz): δ 8.34 (s, 1H), 7.56 (m, 3H), 7.43 (s, 1H), 6.43 (t, J = 53 Hz, 1H), 4.47 (m, 5H), 3.86 (q, J = 11 Hz, 2H), 3.56 (m, 2H), 3.23 (m, 2H), 2.77 (m, 2H), 2.69 (s, 3H), 2.52 (m, 2H), 2.47 (m, 2H), 2.30 (m, 2H), 2.00 (m, 2H). 13C NMR CD3OD (150 MHz): δ 167.2, 162.6 (d, J = 25 Hz), 162.5, 157.9, 155.0, 138.8, 135.0, 130.6, 130.1, 130.1, 128.9, 126.9 (q, J = 276 Hz), 121.9, 118.3, 114.8, 113.2, 112.7, 111.4, 110.4, 109.8, 108.2, 58.3, 54.5, 54.0, 46.2, 44.7, 43.7, 39.0, 35.7 (q, J = 33 Hz), 15.9. HRMS (ESI): [M+H+] calculated 675.2647; found 675.2643.
1-(2-(4-(2-cyclopentylacetyl)piperazin-1-yl)ethyl)-4-methyl-5-((4-((6-(2,2,2-trifluoroethyl)thieno[2,3-d]pyrimidin-4-yl)amino)piperidin-1-yl)methyl)-1H-indole-2-carbonitrile (16)
Reaction time: 30 min. Isolated 19 mg, 54% yield. 1H NMR (600 MHz, METHANOL-d4) δ 8.36 (s, 1H), 7.55 – 7.62 (m, 3H), 7.45 (s, 1H), 4.50 – 4.60 (m, 2H), 4.45 – 4.53 (m, 3H), 3.88 (q, J = 10.6 Hz, 2H), 3.63 (d, J = 12.8 Hz, 2H), 3.49 – 3.57 (m, 4H), 3.30 – 3.43 (m, 2H, overlapped with the residual methanol signal), 2.78 (t, J = 5.5Hz,. 2H), 2.72 (s, 3H), 2.50 (t, J = 5.14 Hz, 2H), 2.42 (t, J = 5.14 Hz, 2H), 2.39 (d, J = 7.34 Hz, 2H), 2.27 – 2.37 (m, 2H), 2.13 – 2.24 (m, 1H), 1.94 – 2.10 (m, 2H), 1.77 – 1.87 (m, 2H), 1.62 – 1.69 (m, 2H), 1.52 – 1.61 (m, 2H), 1.14 – 1.25 (m, 2H); 13C NMR (151 MHz, METHANOL-d4) δ 173.8, 167.1, 157.7, 154.9, 138.8, 135.0, 130.5, 130.0, 128.8, 126.6 (J = 277 Hz), 121.9, 121.2, 118.2, 114.6, 113.2, 112.6, 110.5, 58.9, 58.3, 54.7, 54.3, 52.9, 47.0, 44.6, 42.8, 39.9, 38.2, 35.7 (J = 32 Hz), 33.5, 30.0, 25.8, 15.8; HRMS (ESI): [M+H+] calculated 707.3462; found 707.3466.
4-methyl-1-(2-(4-(2,2,2-trifluoroacetyl)piperazin-1-yl)ethyl)-5-((4-((6-(2,2,2-trifluoroethyl)thieno[2,3-d]pyrimidin-4-yl)amino)piperidin-1-yl)methyl)-1H-indole-2-carbonitrile (15)
Compound 3 (30 mg, 0.05 mmol) was dissolved in DCM (0.25 ml) and cooled to 0°C. The triflate anhydride (7 μL, 0.05 mmol) in DCM (0.25 mL) was added at 0°C. The mixture was warmed to room temperature and solvents were evaporated with silica gel. Compound 15 (21 mg, 60% yield) was purified using flash chromatography (silica gel, DCM:methanol) 1H NMR CD3OD (600 MHz): δ 8.35 (s, 1H), 7.55(m, 3H), 7.43 (s, 1H), 4.50 (m, 5H), 3.85 (q, J = 11 Hz, 2H), 3.61 (m, 4H), 2.83 (m, 2H), 2.70 (s, 3H), 2.57 (m, 4H), 2.33 (m, 2H), 2.00 (m, 2H). 13C NMR CD3OD (150 MHz): δ 166.6, 157.7, 154.7, 138.8, 135.2, 130.6, 130.1, 128.8, 121.8, 121.1, 118.2, 114.6, 113.3, 112.6, 110.5, 58.9, 58.0, 54.2, 53.7, 52.9, 47.1, 46.8, 44.3, 35.7 (m), 30.0, 15.8; HRMS (ESI): [M+H+] calculated 693.2553; found 693.2558.
2-(4-(2-(2-cyano-4-methyl-5-((4-((6-(2,2,2-trifluoroethyl)thieno[2,3-d]pyrimidin-4-yl)amino)piperidin-1-yl)methyl)-1H-indol-1-yl)ethyl)piperazin-1-yl)propanamide (18)
Compound 3 (57 mg, 0.1 mmol) was dissolved in DCM (0.5 mL) and 2-bromopropionamide (7 mg, 0.5eq) was added. After 24h the reaction was diluted with water (despite the presence of starting bromide) and extracted with DCM:MeOH (10:1, v:v). Compound 18 was purified using preparative TLC (silica gel, DCM:MeOH:NH4OH, 10:1:0.05, v:v:v). 12 mg (18% yield) of the product was isolated. 1H NMR (600 MHz, METHANOL-d4) δ 8.33 (s, 1H), 7.55 (s, 1H), 7.46 – 7.54 (m, 2H), 7.40 (s, 1H), 4.45 (t, J = 6.2 Hz, 2H), 4.37 – 4.43 (m, 1H), 4.29 – 4.36 (m, 2H), 3.85 (q, J = 10.6 Hz, 2H), 3.47 (d, J = 12.8 Hz, 2H), 2.97 – 3.15 (m, 3H), 2.72 – 2.80 (m, 2H), 2.66 (s, 3H) 2.61 – 2.69 (m, 2H), 2.49 – 2.61 (m, 6H), 2.21 – 2.30 (m, 2H), 1.87 – 2.03 (m, 2H), 1.24 (d, J = 6.97 Hz, 3H); 13C NMR (151 MHz, METHANOL-d4) δ 178.4, 167.0, 157.7, 154.9, 138.6, 134.4, 130.5, 129.8, 128.7, 126.4 (J = 276.2 Hz), 123.20, 121.8, 118.2, 114.6, 113.1, 112.1, 110.1; 65.2, 59.2, 58.3, 54.2, 52.8, 51.0, 47.5, 44.4, 35.8 (J = 31.8 Hz), 30.8, 30.3, 15.7, 14.3; HRMS (ESI): [M+H+] calculated 668.3107; found 668.3098
General procedure for synthesis of compounds (19, 20, 21, 23, 26)
Compound 3 (1 eq), DIPEA (1.5 eq) were dissolved in DCM (6 ml/mmol). At 0°C the corresponding sulfonyl or sulfamoyl chloride (1 eq) was added and reaction was left to warm to the room temperature. The crude reaction was loaded on the silica gel column and purified using DCM:methanol.
4-methyl-1-(2-(4-(methylsulfonyl)piperazin-1-yl)ethyl)-5-((4-((6-(2,2,2-trifluoroethyl)thieno[2,3-d]pyrimidin-4-yl)amino)piperidin-1-yl)methyl)-1H-indole-2-carbonitrile (19)
The reaction was performed with the methanesulfonyl chloride. Isolated 13 mg, 58% yield. 1H NMR CD3OD (600 MHz): δ 8.36 (s, 1H), 7.59 (m, 2H), 7.54 (s, 1H), 7.46 (s, 1H), 4.55 (m, 4H), 4.47 (m, 1H), 3.86 (q, J = 11 Hz, 2H), 3.63 (m, 2H), 3.34 (m, 2H), 3.25 (m, 4H), 2.94 (m, 2H), 2.84 (s, 3H), 2.73 (m, 4H), 2.70 (s, 3H), 2.4 (m, 2H), 2.00 (m, 2H). 13C NMR CD3OD (150 MHz): δ 166.8, 157.8, 154.8, 138.9, 135.3, 130.9, 130.2, 128.9, 126.8 (q, J = 276 Hz), 121.9, 121.3, 118.3, 114.6, 113.7, 112.5, 110.6, 59.0, 57.7, 53.9, 53.0, 47.3, 46.6, 44.0, 35.7 (q, J = 33 Hz), 34.9, 30.1, 15.9. HRMS (ESI): [M+H+] calculated 675.2506; found 675.2503.
4-methyl-1-(2-(4-(ethylsulfonyl)piperazin-1-yl)ethyl)-5-((4-((6-(2,2,2-trifluoroethyl)thieno[2,3-d]pyrimidin-4-yl)amino)piperidin-1-yl)methyl)-1H-indole-2-carbonitrile (20)
The reaction was performed with the ethanesulfonyl chloride. Isolated 20 mg, 58% yield. 1H NMR CD3OD (600 MHz): δ 8.34 (s, 1H), 7.60 (m, 1H), 7.54 (m, 2H), 7.44 (s, 1H), 4.54 (m, 2H), 4.49 (m, 3H), 3.86 (q, J = 11 Hz, 2H), 3.63 (m, 2H), 3.34 (m, 2H), 3.26 (m, 4H), 3.02 (q, J = 7.6 Hz, 2H), 2.85 (m, 2H), 2.70 (s, 3H), 2.60 (m, 4H), 2.34 (m, 2H), 2.01 (m, 2H). 13C NMR CD3OD (150 MHz): δ 167.2, 157.9, 155.1, 139.0, 135.4, 130.9, 130.3, 129.0, 126.8 (q, J = 276 Hz), 122.0, 121.3, 118.4, 114.9, 113.6, 112.8, 110.7, 59.1, 58.2, 54.4, 53.2, 47.4, 46.8, 44.9, 44.5, 35.9 (q, J = 33 Hz), 30.2, 16.1, 8.3. HRMS (ESI): [M+H+] calculated 689.2662; found 689.2658.
4-methyl-5-((4-((6-(2,2,2-trifluoroethyl)thieno[2,3-d]pyrimidin-4-yl)amino)piperidin-1-yl)methyl)-1-(2-(4-((trifluoromethyl)sulfonyl)piperazin-1-yl)ethyl)-1H-indole-2-carbonitrile (21)
Trifluoromethanosulfonyl chloride was added to the reaction mixture at −78°C. Isolated 9 mg, 25% yield. 1H NMR CD3OD (600 MHz): δ 8.34 (s, 1H), 7.54 (m, 3H), 7.43 (s, 1H), 4.46 (m, 2H), 4.42 (m, 3H), 3.86 (q, J = 11 Hz, 2H), 3.53 (m, 2H), 3.45 (m, 2H), 3.19 (m, 4H), 2.80 (m, 2H), 2.68 (s, 3H), 2.55 (m, 4H), 2.28 (m, 2H), 1.97 (m, 2H). 13C NMR CD3OD (150 MHz): δ 167.0, 157.7, 154.9, 138.6, 134.7, 130.5, 129.9, 128.8, 126.8 (q, J = 276 Hz), 122.6, 121.8, 120.5, 118.2, 114.8, 113.1, 112.5, 110.2, 58.1, 54.0, 44.5, 35.6 (q, J = 33 Hz), 15.7. HRMS (ESI): [M+H+] calculated 729.2223; found 729.2218.
4-(2-(2-cyano-4-methyl-5-((4-((6-(2,2,2-trifluoroethyl)thieno[2,3-d]pyrimidin-4-yl)amino)piperidin-1-yl)methyl)-1H-indol-1-yl)ethyl)-N-methylpiperazine-1-sulfonamide (23, tested as a free base)
Reaction performed with the methylsulfamoyl chloride added to the reaction mixture at the room temperature. Isolated 11 mg, 23% yield. 1H NMR CD3OD (600 MHz): δ 8.30 (s, 1H), 7.52 (s, 1H), 7.39 (m, 3H), 4.41 (m, 2H), 4.19 (m, 1H), 3.83 (q, J = 11 Hz, 2H), 3.76 (m, 2H), 3.12 (m, 4H), 3.07 (m, 2H), 2.75 (m, 2H), 2.61 (s, 3H), 2.59 (s, 3H), 2.51 (m, 4H), 2.39 (m, 2H), 2.03 (m, 2H). 13C NMR CD3OD (150 MHz): δ 166.9, 157.9, 155.1, 138.2, 132.9, 130.5, 129.7, 129.6, 128.9, 126.8 (q, J = 276 Hz), 122.0, 118.3, 115.2, 112.8, 111.5, 109.2, 60.7, 58.3, 54.0, 53.8, 47.2, 44.5, 35.5 (q, J = 33 Hz), 32.3, 30.9, 29.7, 15.5, 15.5. HRMS (ESI): [M+H+] calculated 690.2615; found 690.2611.
4-(2-(2-cyano-4-methyl-5-((4-((6-(2,2,2-trifluoroethyl)thieno[2,3-d]pyrimidin-4-yl)amino)piperidin-1-yl)methyl)-1H-indol-1-yl)ethyl)piperazine-1-sulfonamide (22)
Compound 3 (59 mg, 0.1 mmol) and DIPEA (25 μL, 0.15 mmol, 1.5 eq) was dissolved in DCM (1 mL). Benzyl (chlorosulfonyl)carbamate, CbzNHSO2Cl) (26 mg, 0.11 mmol, 1.1 eq), prepared as described before 42 was then added and reaction was carried out at room temperature for 30 min. Crude mixture was loaded on preparative TLC plate and Cbz-protected compound 22 was purified (silica gel, DCM:MeOH:NH3OH, 10:1:0.5 v:v:v). The product was then dissolved in MeOH (2.25mL) and Pd/C (6 mg) and ammonium formate (9 mg, 0.13 mmol, 4 eq) were added. The mixture was heated at 60°C for 12 h. Mixture was filtered through Celite and evaporated. Purified using preparative TLC (silica gel, DCM:MeOH:NH3OH, 10:1:0.5 v:v:v). 5.6 mg (8% yield) of compound 22 was isolated. 1H NMR CD3OD (600 MHz): δ 8.51 (s, 1H), 7.76 (m, 1H), 7.67 (m, 2H), 7.58 (s, 1H), 4.56 (m, 4H), 4.42 (m, 3H), 3.92 (q, J = 11 Hz, 2H), 3.63 (m, 4H), 3.58 (m, 2H), 3.51 (m, 4H), 3.35 (m, 2H), 2.71 (s, 3H), 2.34 (m, 2H), 2.08 (m, 2H). 13C NMR CD3OD (150 MHz): δ 157.7, 153.7, 152.7, 139.1, 135.6, 131.6, 129.9, 129.7, 129.5, 129.0, 126.6 (q, J = 276 Hz), 122.3, 121.8, 118.5, 115.1, 114.0, 111.6, 110.6, 69.2, 63.3, 58.9, 55.6, 53.1, 52.9, 47.8, 45.1, 41.1, 35.5 (q, J = 33 Hz), 33.2, 30.9, 29.9, 26.2, 23.9, 16.0. HRMS (ESI): [M+H+] calculated 676.2458; found 676.2463.
(S)-4methyl-1-((5-oxomorpholin-2-yl)methyl)-5-((4-((6-(2,2,2-trifluoroethyl)thieno[2,3-d]pyrimidin-4-yl)amino)piperidin-1-yl)methyl)-1H-indole-2-carbonitrile (28)
(S)-3-aminopropane-1,2-diol (37a, 5 g, 55 mmol, 1 eq) was dissolved in methanol (20 ml) and the solution was diluted by AcCN (60 ml) and trimethylamine was added, stirred and cooled at −20 °C. Chloroacetyl chloride was added slowly into the reaction mixture over 1 h and the temperature was controlled to –15°C. The reaction mixture was slowly warmed to –10°C and stirred for 1 h, then warmed to the room temperature and stirred overnight. Solvents were removed and the residue was washed by ethyl acetate (800 mL), filtered and concentrated to give the desired compound (S)-38a. (S)-2-chloro-N-(2,3-dihydroxypropyl)acetamide, (S)-38a, from the previous step (2.0 g, 12 mmol) in t-amyl alcohol (32 ml) was added dropwise to the solution of t-BuOK (4.7 g, 42 mmol, 3.5 eq) in t-amyl alcohol (45 mL) at the room temperature for 2.5 h. The mixture was stirred for an additional 1 h. 1M HClaq (15 ml) was added to the mixture (to adjust pH to ~2) and the mixture was stirred for additional 30 min. Solvents were removed and the (S)-6-(hydroxymethyl)morpholin-3-one (S)-39a was purified using column chromatography (silica gel, hexane:ethyl acetate then DCM:MeOH, 0–10% v:v). 1H NMR (600 MHz, DMSO-d6) δ 7.90 (br. s., 1H), 4.82 (t, J = 5.69 Hz, 1H), 3.92 – 4.10 (m, 2H), 3.60 – 3.71 (m, 1H), 3.45 – 3.53 (m, 1H), 3.36 – 3.45 (m, 1H), 3.18 (td, J = 3.85, 12.10 Hz, 1H), 3.02 – 3.13 (m, 1H). Compound (S)-39a (1 g, 7.6 mmol) was dissolved in DCM (12 mL) and TEA (1.4 mL, 9.9 mmol, 1.3 eq) was added. Mixture was cooled to 0°C and methanesulfonyl chloride (823 μL, 9.9 mmol, 1.3 eq) was added dropwise. The mixture was warmed to the room temperature and stirred overnight. Solvent was evaporated and the residue was purified using column chromatography (silica gel, DCM:MeOH, 0–10% v:v) providing 1.2 g of the(S)-(5-oxomorpholin-2-yl)methyl methanesulfonate, which was directly used in the next step. The mesylate from the previous step (1.2 g) was dissolved in DMA (22 mL) and cesium carbonate (7.5 g, 22.8 mmol, 4 eq) and compound 30 (1.1 g, 5.7 mmol, 1 eq) were added. Mixture was stirred at 105°C for 4 h. After cooling to the room temperature, ethyl acetate (150 ml) was added along with water (3×15 mL). Organic phase was then separated and concentrated. The residue was purified using column chromatography (silica gel, DCM:MeOH, 0–10% v:v) resulting in 420 mg of compound (S)-40a, (S)-5-formyl-4-methyl-1-((5-oxomorpholin-2-yl)methyl)-1H-indole-2-carbonitrile, MS (m/z) found for [M+AcCN+H+]: 339 Da. Compound (S)-40a (400 mg, 1.4 mmol) was dissolved in THF (48 mL) followed by addition of compound 33 (638 mg, 2.0 mmol, 1.5 eq) and acetic acid (116 μL). Resulting mixture was stirred at 50°C for 30min, then cooled to the room temperature. Sodium triacetoxyborohydride (572 mg, 2.69 mmol, 2 eq) was then added and the mixture was stirred for 5 h at 50°C. Cooled mixture was then partitioned between EtOAc (60 mL) and 1M NaOHaq (25 mL). Organic phase was concentrated and product was purified by column chromatography (silica gel, DCM:MeOH, 0–10% v:v). 520 mg (65% yield) of compound 28 was obtained. 1H NMR (600 MHz, DMSO-d6) δ 10.68 (br. s., 1H), 8.38 (s, 1H), 8.20 – 8.29 (m, 1H), 8.00 – 8.03 (m, 1H), 7.76 – 7.81 (m, 1H), 7.75 (bs. 1H), 7.62 – 7.66 (m, 2H), 4.57 – 4.62 (m, 1H), 4.31 – 4.45 (m, 4H), 4.01 – 4.10 (m, 3H), 4.00 (d, J = 16.4 Hz, 1H), 3.89 (d, J = 16.5 Hz, 1H), 3.35 – 3.47 (m, 3H, overlapped with water), 3.21 – 3.32 (m, 2H), 3.18 (t, J = 11.37 Hz, 1H), 2.64 (s, 3H), 2.01 – 2.14 (m, 4H); 13C NMR (151 MHz, DMSO-d6) δ 166.7, 155.7, 153.4, 137.3, 133.3, 130.0, 127.2, 126.5, 125.4 (J = 277 Hz) 121.5, 120.4, 116.2, 113.5, 113.2, 110.2, 109.3, 71.5, 66.9, 56.1, 50.6, 50.5, 46.8, 45.7, 42.7, 33.8 (J = 31 Hz), 28.2, 15.5; 1H NMR (600 MHz, METHANOL-d4) δ 8.41 (s, 1H), 7.57 – 7.67 (m, 3H), 7.49 (s, 1H), 4.61 (dd, J = 2.75, 15.22 Hz, 1H), 4.56 (s, 2H), 4.47 – 4.54 (m, 2H), 4.14 – 4.21 (m, 1H), 4.15 (AB, d, J = 16.51 Hz, 1H), 4.00 (AB, d, J = 16.51 Hz, 1H), 3.83 – 3.93 (m, 2H), 3.65 (d, J = 11.74 Hz, 2H), 3.48 (dd, J = 2.38, 12.29 Hz, 1H), 3.33 – 3.41 (m, 3H, partially overlapped with MeOH), 2.71 (s, 3H), 2.35 (bd, J = 13.20 Hz, 2H), 2.06 (q, J = 12.47 Hz, 2H); 13C NMR (151 MHz, METHANOL-d4) δ 170.6, 165.1, 157.6, 153.9, 139.6, 135.0, 130.8, 130.5, 128.6, 126.6 (J = 276 Hz), 125.6, 121.9, 121.2, 118.3, 114.2, 114.0, 112.5, 73.3, 68.0, 58.8, 52.9, 52.9, 48.2, 47.3, 44.4, 35.6 (J = 32 Hz), 29.9, 15.8; [α]24D −17.5 (c 1.0, MeOH); HR-MS (ESI): [M+H+] calculated 598.2212, found 598.2208.
Synthesis of compounds 4, 6, 7, 8, 24, 25, 26, 27, 29 was outsourced to the contract research organization (CRO). These compounds were prepared using the general methodology presented in Schemes 1, 3, 4, 5. Final compounds were characterized in-house and the purity was confirmed by HPLC before their biological activity was evaluated.
4-methyl-1-(2-(piperidin-4-yl)ethyl)-5-((4-((6-(2,2,2-trifluoroethyl)thieno[2,3-d]pyrimidin-4-yl)amino)piperidin-1-yl)methyl)-1H-indole-2-carbonitrile (4)
1H NMR (600 MHz, DMSO-d6) δ 11.02 (br. s., 1H), 9.08 (br. s., 1H), 8.90 (br. s., 1H), 8.79 (br. s., 1H), 8.49 (s, 1H), 7.74 – 7.91 (m, 2H), 7.65 (s, 1H), 7.57 (d, J = 8.80 Hz, 1H), 4.31 – 4.47 (m, 5H), 4.08 (q, J = 10.88 Hz, 2H), 3.41 (d, J = 11.37 Hz, 2H), 3.18 – 3.30 (m, 4H), 2.80 (q, J = 11.74 Hz, 2H), 2.65 (s, 3H), 2.04 – 2.22 (m, 4H), 1.88 (d, J = 12.84 Hz, 2H), 1.70 (q, J = 6.72 Hz, 2H), 1.55 – 1.65 (m, 1H), 1.36 – 1.48 (m, 2H) 13C NMR (151 MHz, DMSO-d6) δ 155.2, 151.6, 136.8, 133.5, 130.2, 129.9, 128.3, 126.6, 125.4 (J = 276 Hz), 121.7, 120.3, 116.5, 113.5, 113.1, 108.8, 108.8, 56.0, 50.4, 46.2, 42.8, 42.7, 35.9, 33.7 (J = 31 Hz) 30.7, 28.0, 28.0, 15.6; HRMS (ESI): [M+H+] calculated 596.2783; found 596.2776.
4-methyl-1-(2-(5-oxomorpholin-2-yl)ethyl)-5-((4-((6-(2,2,2-trifluoroethyl)thieno[2,3-d]pyrimidin-4-yl)amino)piperidin-1-yl)methyl)-1H-indole-2-carbonitrile (6)
1H NMR (600 MHz, DMSO-d6) δ 10.91 (br. s., 1H), 8.72 – 8.88 (m, 1H), 8.43 – 8.59 (m, 1H), 7.91 – 7.98 (m, 1H), 7.86 (s, 1H), 7.82 (d, J = 8.80 Hz, 1H), 7.65 (s, 1H), 7.53 (d, J = 8.80 Hz, 1H), 4.33 – 4.57 (m, 5H), 4.09 (q, J = 11.0 Hz, 2H), 4.05 (d, J = 16.1 Hz, 1H), 3.92 (d, J = 16.1 Hz, 1H), 3.49 – 3.58 (m, 1H), 3.36 – 3.46 (m, 2H), 3.19 – 3.30 (m, 2H), 3.11 – 3.18 (m, 1H), 3.02 – 3.10 (m, 1H), 2.65 (s, 3H), 2.06 – 2.26 (m, 4H), 1.99 – 2.07 (m, 1H), 1.85 – 1.95 (m, 1H); 13C NMR (151 MHz, DMSO-d6) δ 167.0, 155.3, 151.6, 136.9, 133.5, 130.1, 128.3, 126.6, 125.4 (J = 277 Hz), 121.7, 120.3, 116.5, 113.4, 113.1, 109.1, 108.8, 70.0, 66.8, 56.0, 50.5, 50.4, 46.2, 44.9, 41.6, 33.7 (J = 31 Hz), 32.3, 28.0, 15.6 HRMS (ESI): [M+H+] calculated 612.2369; found 612.2369.
4-methyl-1-((5-oxomorpholin-2-yl)methyl)-5-((4-((6-(2,2,2-trifluoroethyl)thieno[2,3-d]pyrimidin-4-yl)amino)piperidin-1-yl)methyl)-1H-indole-2-carbonitrile (7)
1H NMR (600 MHz, DMSO-d6) δ 10.67 (br. s., 1H), 8.52 – 8.63 (m, 1H), 8.45 (s, 1H), 8.01 (d, J = 3.67 Hz, 1H), 7.74 – 7.83 (m, 2H), 7.59 – 7.67 (m, 2H), 4.59 (dd, J = 2.75, 15.22 Hz, 1H), 4.33 – 4.47 (m, 4H), 4.03 – 4.14 (m, 3H), 3.99 (d, J = 16.1 Hz, 1H), 3.89 (d, J = 16.5 Hz, 1H), 3.36 – 3.47 (m, 3H), 3.21 – 3.33 (m, 2H), 3.18 (t, J = 11.37 Hz, 1H), 2.64 (s, 3H), 2.03 – 2.24 (m, 4H); 13C NMR (151 MHz, DMSO-d6) δ 166.7, 155.5, 152.4, 137.4, 133.3, 130.0, 127.9, 126.6, 125.4 (J = 277Hz) 121.6, 120.4, 116.4, 113.5, 113.2, 110.2, 109.3, 71.5, 66.9, 56.1, 50.6, 50.5, 46.8, 46.0, 42.7, 33.7 (J = 31 Hz), 28.1, 15.5; HRMS (ESI): [M+H+] calculated 598.2212; found 598.2205.
4-methyl-1-(2-(4-propylpiperazin-1-yl)methyl)-5-((4-((6-(2,2,2-trifluoroethyl)thieno[2,3-d]pyrimidin-4-yl)amino)piperidin-1-yl)methyl)-1H-indole-2-carbonitrile (8)
1H NMR (600 MHz, DMSO-d6) δ 10.66 (bs,1H), 8.38 (s, 1H), 8.25 (d, J = 6.60 Hz, 1H), 7.78 (d, J = 8.80 Hz, 1H), 7.74 (s, 1H), 7.58 – 7.70 (m, 2H), 4.50 (m, 2H), 4.40 (d, J = 5.14 Hz, 2H), 4.31 – 4.39 (m, 1H), 4.05 (q, J = 11.13 Hz, 2H), signals overlapped with water: 3.39 (HSQC), 3.36 (HSQC), 3.20 – 3.29 (m, 2H), 3.02 – 3.15 (m, 2H), 2.93 – 3.02 (m, 2H), 2.80 – 2.90 (m, 2H), 2.75 (s, 3H), 2.64 (s, 3H), 2.01 – 2.22 (m, 4H); 13C NMR (151 MHz, DMSO-d6) δ 155.6, 136.8, 133.4, 130.0, 126.6, 121.4, 116.2, 109.0, 69.1, 56.7 (HSQC) 56.1, 52.0, 50.6, 49.3, 45.8, 42.7 (HSQC), 41.9, 33.6, 28.2, 15.5; HRMS (ESI): [M+H+] calculated 611.2892; found 611.2873.
1-(2-(1-formylpiperidin-4-yl)ethyl)-4-methyl-5-((4-((6-(2,2,2-trifluoroethyl)thieno[2,3-d]pyrimidin-4-yl)amino)piperidin-1-yl)methyl)-1H-indole-2-carbonitrile (24)
1H NMR (600 MHz, DMSO-d6) δ 10.42 (br. s., 1H), 8.36 – 8.43 (m, 1H), 8.22 (d, J = 7.34 Hz, 1H), 7.96 (s, 1H), 7.71–7.77 (m, 2H), 7.65 (s, 1H), 7.58 (d, J = 8.80 Hz, 1H), 4.37 – 4.45 (m, 4H), 4.29 – 4.36 (m, 1H), 4.13 (d, J = 13.20 Hz, 1H), 4.05 (q, J = 11.00 Hz, 2H), 3.66 (d, J = 13.20 Hz, 1H), 3.42 (m, 2H, HSQC, overlapped with water) 3.20 – 3.29 (m, 2H), 2.94 – 3.03 (m, 1H), 2.64 (s, 3H), 2.54 – 2.61 (m, 1H), 2.08 – 2.20 (m, 2H), 1.96 – 2.07 (m, 2H), 1.73 – 1.84 (m, 2H), 1.71 (q, J = 6.97 Hz, 2H), 1.52 – 1.62 (m, 1H), 0.96 – 1.16 (m, 2H); 13C NMR (151 MHz, DMSO-d6) δ 160.6, 155.7, 153.3, 136.8, 133.5, 130.0, 127.3, 126.6, 125.5 (J = 277 Hz), 121.4, 120.3, 116.2, 113.5, 113.1, 108.9, 108.9, 56.2, 50.6, 45.7, 44.8, 42.8, 38.8, 36.2, 33.8 (J = 30 Hz), 33.2, 32.1, 30.8, 28.2, 15.5; HRMS (ESI): [M+Na+] calculated 646.2552; found 646.2544.
1-(2-(1-acetylpiperidin-4-yl)ethyl)-4-methyl-5-((4-((6-(2,2,2-trifluoroethyl)thieno[2,3-d]pyrimidin-4-yl)amino)piperidin-1-yl)methyl)-1H-indole-2-carbonitrile (25)
1H NMR (600 MHz, DMSO-d6) δ 10.22 (bs, 1H), 8.36 (s, 1H), 8.11 (d, J = 6.97 Hz, 1H), 7.69 – 7.74 (m, 2H), 7.65 (s, 1H), 7.58 (d, J = 8.80 Hz, 1H), 4.36 – 4.46 (m, 4H), 4.30 – 4.35 (m, 2H), 4.05 (q, J = 10.9 Hz, 2H), 3.78 (d, J = 13.2 Hz, 1H), 2.97 (t, J = 13.2 Hz, 1H), 2.63 (s, 3H), 2.44 – 2.49 (m, 1H), 2.10–2.15 (m, 2H), 1.97 (s, 3H), 1.92 – 2.02 (m, 2H), 1.76 (d, J = 13.2 Hz, 1H), 1.66 – 1.74 (m, 3H), 1.48 – 1.57 (m, 1H), 1.15 (m, 1H), 0.96 – 1.07 (m, 1H); HSQC (1H-13C): signals overlapped with water: 3.43 (CH2), 3.25 (CH2); 13C NMR (151 MHz, DMSO-d6) δ 167.8, 165.6, 155.7, 153.7, 136.8, 133.5, 130.0, 127.2, 126.6, 125.50 (J = 277Hz), 121.4, 120.2, 116.9, 116.2, 113.5, 113.0, 108.9 56.3, 50.7, 45.7, 45.6, 42.9, 40.8, 36.2, 33.8 ( J = 32 Hz), 32.9, 32.0, 31.3, 28.3, 21.2, 15.5 HRMS (ESI): [M+Na+] calculated 660.2703; found 660.2699.
4-methyl-1-(2-(1-(methylsulfonyl)piperidin-4-yl)ethyl)-5-((4-((6-(2,2,2-trifluoroethyl)thieno[2,3-d]pyrimidin-4-yl)-amino)piperidin-1-yl)methyl)-1H-indole-2-carbonitrile (26)
1H NMR (600 MHz, DMSO-d6) δ 10.75 (bs, 1H), 8.60 (bs, 1H), 8.46 (s, 1H), 7.81 (s, 1H), 7.79 (d, J = 8.80 Hz, 1H), 7.65 (s, 1H), 7.58 (d, J = 8.80 Hz, 1H), 4.32 – 4.45 (m, 5H), 4.08 (q, J = 11.00 Hz, 2H), 3.54 (d, J = 12.1 Hz, 2H), 3.42 (d, J = 12.1 Hz, 2H), 3.20 – 3.31 (m, 2H), 2.83 (s, 3H), 2.65 – 2.69 (m, 2H), 2.64 (s, 3H), 2.04 – 2.24 (m, 4H), 1.79 – 1.87 (m, 2H), 1.69 – 1.78 (m, 2H), 1.40 (m, 1H), 1.22 – 1.30 (m, 2H) 13C NMR (151 MHz, DMSO-d6) δ 155.4, 152.2, 136.8, 133.5, 130.1, 126.6, 126.4, 121.6, 120.3, 116.4, 113.5, 113.1, 108.9, 108.9, 108.9, 56.1, 50.5, 46.1, 45.3, 42.8, 35.9, 34.1, 33.7 (J = 30 Hz), 32.1, 30.9, 28.1, 15.5; HRMS (ESI): [M+H+] calculated 674.2559; found 674.2548.
4-(2-(2-cyano-4-methyl-5-((4-((6-(2,2,2-trifluoroethyl)thieno[2,3-d]pyrimidin-4-yl)amino-) piperidin-1-yl)methyl)-1H-indol-1-yl)ethyl)piperidine-1-sulfonamide (27)
1H NMR (600 MHz, DMSO-d6) δ 10.58 (bs, 1H), 8.42 (s, 1H), 8.34 – 8.41 (m, 1H), 7.73 – 7.83 (m, 2H), 7.65 (s, 1H), 7.58 (d, J = 8.80 Hz, 1H), 6.65 (br. s., 2H), 4.32 – 4.46 (m, 5H), 4.06 (q, J = 11.13 Hz, 2H), 3.38 – 3.47 (m, 4H), 3.17 – 3.35 (m, 2H), 2.64 (s, 3H), 2.44 – 2.48 (m, 2H), 2.02 – 2.21 (m, 4H), 1.77 – 1.88 (m, 2H), 1.66 – 1.75 (m, 2H), 1.30 – 1.40 (m, 1H), 1.19 – 1.30 (m, 2H); 13C NMR (151 MHz, DMSO-d6) δ 155.6, 152.8, 136.8, 133.5, 130.0, 127.6, 126.6, 126.4, 124.5, 121.5, 120.3, 116.3, 113.5, 113.1, 108.9, 56.1, 50.6, 45.9, 42.9, 40.1, 36.0, 33.75 (J = 31Hz), 32.3, 30.6, 28.2, 15.5; HRMS (ESI): [M+H+] calculated 675.2511; found 675.2505.
(R)-4-methyl-1-((5-oxomorpholin-2-yl)methyl)-5-((4-((6-(2,2,2-trifluoroethyl)thieno[2,3-d]pyrimidin-4-yl)amino)piperidin-1-yl)methyl)-1H-indole-2-carbonitrile (29)
1H NMR (600 MHz, DMSO-d6) δ 10.68 (br. s., 1H), 8.48 – 8.56 (m, 1H), 8.44 (s, 1H), 8.01 (d, J = 3.67 Hz, 1H), 7.75 – 7.81 (m, 2H), 7.61 – 7.66 (m, 2H), 4.59 (dd, J = 2.75, 15.22 Hz, 1H), 4.33 – 4.45 (m, 4H), 4.03 – 4.11 (m, 3H), 4.00 (d, J = 16.5 Hz, 1H), 3.89 (d, J = 16.5 Hz, 1H), 3.37 – 3.47 (m, 3H), 3.21 – 3.32 (m, 2H), 3.18 (t, J = 11.37 Hz, 1H), 2.64 (s, 3H), 2.06 – 2.15 (m, 4H); 13C NMR (151 MHz, DMSO-d6) δ 166.7, 155.5, 152.5, 137.4, 133.3, 130.0, 127.8, 126.4, 125.4 (J = 276Hz) 121.6, 120.4, 116.4, 113.5, 113.2, 110.2, 109.3, 71.5, 66.9, 56.1, 50.6, 50.5, 46.8, 46.0, 42.7, 33.7 (J = 32 Hz), 28.1, 15.5; [α]24D 18.1 (c 1.0, MeOH); HR-MS (ESI): [M+H+] calculated 598.2212, found 598.2207.
Supplementary Material
Acknowledgements
This work was funded by the National Institute of Health (NIH) grants R01 (1R01CA160467) to J.G. and R01 (1R01CA200660) to J.G., a Leukemia and Lymphoma Society LLS Scholar (1215-14) to J.G., LLS TAP (Therapy Acceleration Program) to J.G., American Cancer Society Research Scholar grant RSG-13-130-01-CDD to J.G., and the Department of Pathology, University of Michigan. Use of the Advanced Photon Source was supported by the US Department of Energy, Office of Science, Office of Basic Energy Sciences under contract number DE-AC02-06CH11357. Use of the LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor for the support of this research program (grant 085P1000817). The mouse work was performed under oversight of UCUCA at the University of Michigan.
Conflict of interest. The authors declare the following competing financial interest(s): Drs. Grembecka and Cierpicki receive research support from Kura Oncology, Inc. They have also an equity ownership in the company. Other coauthors declare no potential conflict of interest.
Abbreviations used
- FP
Fluorescence Polarization
- MLL
Mixed Lineage Leukemia
- PPI
Protein-Protein Interaction
- NEAA
Non-essential amino acids
- FBS
fetal bovine serum
- AcCN
acetonitrile
- AcOH
acetic acid
- DIPEA
N,N-Diisopropylethylamine
- EDCI
N-(3-Dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride
- TEA
trimethylamine
- TBTU
O-(Benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium tetrafluoroborate
- TBAI
Tetrabutylammonium iodide
Footnotes
Supporting Information
Supplementary Table 1 with crystallography data, Supplementary Figure 1 with the Kd results from the FP assay for MLL4–43 binding to menin, Supplementary Figure 2 with BLI studies for menin-MLL inhibitors, Supplementary Figure 3 with PK results for 28 and Supplementary Figure 4 with mice body weight from in vivo efficacy studies with 28. Molecular Formula Strings are available. This material is available free of charge via the Internet at http://pubs.acs.org.
Accession Codes
The atomic coordinate files have been deposited in PDB with the PDB codes: 6BXH (menin-compound 13), 6BXY (menin-compound 28), 6BY8 (menin-compound 29).
References
- 1.Cierpicki T; Grembecka J Challenges and opportunities in targeting the menin-MLL interaction. Future Med. Chem 2014, 6 (4), 447–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yokoyama A; Somervaille TC; Smith KS; Rozenblatt-Rosen O; Meyerson M; Cleary ML The menin tumor suppressor protein is an essential oncogenic cofactor for MLL-associated leukemogenesis. Cell. 2005, 123 (2), 207–218. [DOI] [PubMed] [Google Scholar]
- 3.Marschalek R Mechanisms of leukemogenesis by MLL fusion proteins. Br. J. Haematol 2011, 152 (2), 141–154. [DOI] [PubMed] [Google Scholar]
- 4.Dimartino JF; Cleary ML Mll rearrangements in haematological malignancies: lessons from clinical and biological studies. Br. J. Haematol 1999, 106 (3), 614–626. [DOI] [PubMed] [Google Scholar]
- 5.Popovic R; Zeleznik-Le NJ MLL: how complex does it get? J. Cell Biochem 2005, 95 (2), 234–242. [DOI] [PubMed] [Google Scholar]
- 6.Caslini C; Yang Z; El-Osta M; Milne TA; Slany RK; Hess JL Interaction of MLL amino terminal sequences with menin is required for transformation. Cancer Res. 2007, 67 (15), 7275–7283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yokoyama A; Cleary ML Menin critically links MLL proteins with LEDGF on cancer-associated target genes. Cancer Cell. 2008, 14 (1), 36–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Grembecka J; He S; Shi A; Purohit T; Muntean AG; Sorenson RJ; Showalter HD; Murai MJ; Belcher AM; Hartley T; Hess JL; Cierpicki T Menin-MLL inhibitors reverse oncogenic activity of MLL fusion proteins in leukemia. Nat. Chem. Biol 2012, 8 (3), 277–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shi A; Murai MJ; He S; Lund G; Hartley T; Purohit T; Reddy G; Chruszcz M; Grembecka J; Cierpicki T Structural insights into inhibition of the bivalent menin-MLL interaction by small molecules in leukemia. Blood 2012, 120 (23), 4461–4469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Borkin D; He S; Miao H; Kempinska K; Pollock J; Chase J; Purohit T; Malik B; Zhao T; Wang J; Wen B; Zong H; Jones M; Danet-Desnoyers G; Guzman ML; Talpaz M; Bixby DL; Sun D; Hess JL; Muntean AG; Maillard I; Cierpicki T; Grembecka J Pharmacologic inhibition of the Menin-MLL interaction blocks progression of MLL leukemia in vivo. Cancer Cell 2015, 27 (4), 589–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.He S; Senter TJ; Pollock J; Han C; Upadhyay SK; Purohit T; Gogliotti RD; Lindsley CW; Cierpicki T; Stauffer SR; Grembecka J High-affinity small-molecule inhibitors of the menin-mixed lineage leukemia (MLL) interaction closely mimic a natural protein-protein interaction. J. Med. Chem 2014, 57 (4), 1543–1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Borkin D; Pollock J; Kempinska K; Purohit T; Li X; Wen B; Zhao T; Miao H; Shukla S; He M; Sun D; Cierpicki T; Grembecka J Property focused structure-based optimization of small molecule inhibitors of the protein-protein interaction between menin and Mixed Lineage Leukemia (MLL). J. Med. Chem 2016, 59 (3), 892–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Malik R; Khan AP; Asangani IA; Cieslik M; Prensner JR; Wang X; Iyer MK; Jiang X; Borkin D; Escara-Wilke J; Stender R; Wu YM; Niknafs YS; Jing X; Qiao Y; Palanisamy N; Kunju LP; Krishnamurthy PM; Yocum AK; Mellacheruvu D; Nesvizhskii AI; Cao X; Dhanasekaran SM; Feng FY; Grembecka J; Cierpicki T; Chinnaiyan AM Targeting the MLL complex in castration-resistant prostate cancer. Nat. Med 2015, 21 (4), 344–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Svoboda LK; Bailey N; Van Noord RA; Krook MA; Harris A; Cramer C; Jasman B; Patel RM; Thomas D; Borkin D; Cierpicki T; Grembecka J; Lawlor ER Tumorigenicity of Ewing sarcoma is critically dependent on the trithorax proteins MLL1 and menin. Oncotarget 2017, 8 (1), 458–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xu B; Li SH; Zheng R; Gao SB; Ding LH; Yin ZY; Lin X; Feng ZJ; Zhang S; Wang XM; Jin GH Menin promotes hepatocellular carcinogenesis and epigenetically up-regulates Yap1 transcription. Proc. Natl. Acad. Sci. U S A 2013, 110 (43), 17480–17485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kempinska K; Malik B; Borkin D; Klossowski S; Shukla S; Miao H; Wang J; Cierpicki T; Grembecka J Pharmacologic inhibition of the menin-MLL interaction leads to transcriptional repression of PEG10 and blocks hepatocellular carcinoma. Mol. Cancer Ther 2018, 17 (1), 26–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Funato K; Major T; Lewis PW; Allis CD; Tabar V Use of human embryonic stem cells to model pediatric gliomas with H3.3K27M histone mutation. Science 2014, 346 (6216), 1529–1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fry DC Protein-protein interactions as targets for small molecule drug discovery. Biopolymers 2006, 84 (6), 535–552. [DOI] [PubMed] [Google Scholar]
- 19.Buchwald P Small-molecule protein-protein interaction inhibitors: therapeutic potential in light of molecular size, chemical space, and ligand binding efficiency considerations. IUBMB Life 2010, 62 (10), 724–731. [DOI] [PubMed] [Google Scholar]
- 20.Azzarito V; Long K; Murphy NS; Wilson AJ Inhibition of alpha-helix-mediated protein-protein interactions using designed molecules. Nat. Chem 2013, 5 (3), 161–173. [DOI] [PubMed] [Google Scholar]
- 21.Wells JA; McClendon CL Reaching for high-hanging fruit in drug discovery at protein-protein interfaces. Nature 2007, 450 (7172), 1001–1009. [DOI] [PubMed] [Google Scholar]
- 22.Arkin MR; Whitty A The road less traveled: modulating signal transduction enzymes by inhibiting their protein-protein interactions. Curr. Opin. Chem. Biol 2009, 13 (3), 284–290. [DOI] [PubMed] [Google Scholar]
- 23.Smith MC; Gestwicki JE Features of protein-protein interactions that translate into potent inhibitors: topology, surface area and affinity. Expert Rev. Mol. Med 2012, 14, e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cierpicki T; Grembecka J Targeting protein-protein interactions in hematologic malignancies: still a challenge or a great opportunity for future therapies? Immunol. Rev 2015, 263 (1), 279–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bai L; Wang S Targeting apoptosis pathways for new cancer therapeutics. Annu. Rev. Med 2014, 65, 139–155. [DOI] [PubMed] [Google Scholar]
- 26.Nero TL; Morton CJ; Holien JK; Wielens J; Parker MW Oncogenic protein interfaces: small molecules, big challenges. Nat. Rev. Cancer 2014, 14 (4), 248–262. [DOI] [PubMed] [Google Scholar]
- 27.Grembecka J; Belcher AM; Hartley T; Cierpicki T Molecular basis of the mixed lineage leukemia-menin interaction: implications for targeting mixed lineage leukemias. J. Biol. Chem 2010, 285 (52), 40690–40698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Murai MJ; Chruszcz M; Reddy G; Grembecka J; Cierpicki T Crystal structure of menin reveals binding site for mixed lineage leukemia (MLL) protein. J. Biol. Chem 2011, 286 (36), 31742–31748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huang X Fluorescence polarization competition assay: the range of resolvable inhibitor potency is limited by the affinity of the fluorescent ligand. J. Biomol. Screen 2003, 8 (1), 34–38. [DOI] [PubMed] [Google Scholar]
- 30.Brenner E; Baldwin RM; Tamagnan G Asymmetric synthesis of (+)-(S,S)-reboxetine via a new (S)-2-(hydroxymethyl)morpholine preparation. Org. Lett 2005, 7 (5), 937–939. [DOI] [PubMed] [Google Scholar]
- 31.Ghosh M; Seekamp CK; Zhang X; Tarrant JG; Lee C-W; Kaiser B; Zablocki MM; Chenard BL; Doller D; Yamaguchi Y; Wustrow DJ; Li G Aminopiperidines and Realted Compounds. U.S. Patent, WO2008016811A2, 2007.
- 32.Xu S; Aguilar A; Xu T; Zheng K; Huang L; Stuckey J; Chinnaswamy K; Bernard D; Fernandez-Salas E; Liu L; Wang M; McEachern D; Przybranowski S; Foster C; Wang S Design of the first-in-class, highly potent irreversible inhibitor targeting the menin-MLL protein-protein interaction. Angew. Chem. Int. Ed. Engl 2018, 57 (6), 1601–1605. [DOI] [PubMed] [Google Scholar]
- 33.Fairhead M; Howarth M Site-specific biotinylation of purified proteins using BirA. Methods Mol. Biol 2015, 1266, 171–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Otwinowski Z; Minor W [20] Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997, 276, 307–326. [DOI] [PubMed] [Google Scholar]
- 35.Murshudov GN; Vagin AA; Dodson EJ Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr 1997, 53 (Pt 3), 240–255. [DOI] [PubMed] [Google Scholar]
- 36.Emsley P; Cowtan K Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr 2004, 60 (Pt 12 Pt 1), 2126–2132. [DOI] [PubMed] [Google Scholar]
- 37.Collaborative Computational Project, N. The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr 1994, 50 (Pt 5), 760–763. [DOI] [PubMed] [Google Scholar]
- 38.Adams PD; Afonine PV; Bunkoczi G; Chen VB; Davis IW; Echols N; Headd JJ; Hung LW; Kapral GJ; Grosse-Kunstleve RW; McCoy AJ; Moriarty NW; Oeffner R; Read RJ; Richardson DC; Richardson JS; Terwilliger TC; Zwart PH PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr 2010, 66 (Pt 2), 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Davis IW; Leaver-Fay A; Chen VB; Block JN; Kapral GJ; Wang X; Murray LW; Arendall WB 3rd; Snoeyink J; Richardson JS; Richardson DC MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007, 35 (Web Server issue), W375–W383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yang H; Guranovic V; Dutta S; Feng Z; Berman HM; Westbrook JD Automated and accurate deposition of structures solved by X-ray diffraction to the Protein Data Bank. Acta Crystallogr. D Biol. Crystallogr 2004, 60 (Pt 10), 1833–1839. [DOI] [PubMed] [Google Scholar]
- 41.Muntean AG; Tan J; Sitwala K; Huang Y; Bronstein J; Connelly JA; Basrur V; Elenitoba-Johnson KS; Hess JL The PAF complex synergizes with MLL fusion proteins at HOX loci to promote leukemogenesis. Cancer Cell 2010, 17 (6), 609–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Krueger AC; Madigan DL; Jiang WW; Kati WM; Liu D; Liu Y; Maring CJ; Masse S; McDaniel KF; Middleton T; Mo H; Molla A; Montgomery D; Pratt JK; Rockway TW; Zhang R; Kempf DJ Inhibitors of HCV NS5B polymerase: synthesis and structure-activity relationships of N-alkyl-4-hydroxyquinolon-3-yl-benzothiadiazine sulfamides. Bioorg. Med. Chem. Lett 2006, 16 (13), 3367–3370. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.