

Abstract
Methyl-CpG-binding protein 2 (MeCP2) mutations are the primary cause of Rett syndrome, a severe neurodevelopmental disorder. Cortical parvalbumin GABAergic interneurons (PV) make exuberant somatic connections onto pyramidal cells in the visual cortex of Mecp2-deficient mice, which contributes to silencing neuronal cortical circuits. This phenotype can be rescued independently of Mecp2 by environmental, pharmacological, and genetic manipulation. It remains unknown how Mecp2 mutation can result in abnormal inhibitory circuit refinement. In the present manuscript, we examined the development of GABAergic circuits in the primary visual cortex of Mecp2-deficient mice. We identified that PV circuits were the only GABAergic interneurons to be upregulated, while other interneurons were downregulated. Acceleration of PV cell maturation was accompanied by increased PV cells engulfment by perineuronal nets (PNNs) and by an increase of PV cellular and PNN structural complexity. Interestingly, selective deletion of Mecp2 from PV cells was sufficient to drive increased structure complexity of PNN. Moreover, the accelerated PV and PNN maturation was recapitulated in organotypic cultures. Our results identify a specific timeline of disruption of GABAergic circuits in the absence of Mecp2, indicating a possible cell-autonomous role of MeCP2 in the formation of PV cellular arbors and PNN structures in the visual cortex.
Keywords: development, GABA, perineuronal nets, Rett syndrome, visual cortex
Introduction
Rett syndrome (RTT, OMIM ID 312750) is a severe progressive neurodevelopmental disorder that mainly affects girls. It is characterized by early neurological regression followed by loss of acquired cognitive, social, and motor skills, together with development of autistic behavior and seizures (Hagberg 2002). Typical RTT is due to de novo mutations in an X-linked gene that encodes for methyl-CpG-binding protein 2 (MECP2) (Amir et al. 1999). MeCP2 is a multifunctional chromatin protein that regulates long gene expression (Sugino et al. 2014; Gabel et al. 2015) by either repressing or activating transcription or by functioning at a post-transcriptional level (Chahrour et al. 2008; Lyst and Bird 2015). Recently, it has also been demonstrated that MeCP2 promotes cell reprogramming and stimulates cell proliferation through regulating cell cycle protein expression (Zhang et al. 2018).
Co-morbidity of epilepsy with RTT is approximately 80% and suggests an inhibitory signaling deficit contributing to the progression of the disease (Jian et al. 2006). Indeed, density of γ-amino butyric acid (GABA) receptors is increased in young patients (Blue et al. 1999), while benzodiazepine binding to GABAA receptor is decreased in adulthood (Yamashita et al. 1998). Furthermore, Mecp2-deficient (KO) mice show reduced expression of the cation-chloride cotransporter KCC2 (K+/Cl- exporter), responsible for establishing the chloride ion gradient in neurons (Banerjee et al. 2016). In particular, patch clamp recordings demonstrated that the reversal potential for GABA is more depolarized in mutant mice. KCC2 expression has also been shown to be significantly decreased in cerebrospinal fluid of RTT patients (Duarte et al. 2013). Experiments in Mecp2 KO mice have reported contrasting GABAergic abnormalities in the thalamus (Zhang et al. 2010), medulla (Medrihan et al. 2008), and cortices (Durand et al. 2012; Tomassy et al. 2014; Krishnan et al. 2015) but all of them agree that such alterations precede the onset of RTT symptoms.
Neocortical GABAergic interneurons account for almost 30% of neurons in the mammalian cerebral cortex. The majority of interneurons arise from the medial and caudal ganglionic eminences (MGE and CGE) of the basal ganglia. As MGE cells mature, they express somatostatin (SST) and parvalbumin (PV) (Wonders and Anderson 2006), whereas CGE-derived cells mainly express calretinin (CR) and calbindin (CB) (Xu et al. 2010). GABAergic interneurons contain significantly higher levels of MeCP2 than to pyramidal cells (Akbarian et al. 2001; Chao et al. 2010; Mierau et al. 2015) and selective deletion of Mecp2 from them is sufficient to elicit RTT phenotypes and induce significant deficits in GABA immunoreactivity (Chao et al. 2010; Durand et al. 2012). This highlights the critical role of MeCP2 in GABA-releasing neurons. Selective Mecp2 deletion from PV-expressing cells is sufficient to alter experience-dependent plasticity (Durand et al. 2012; He et al. 2014) and results in the onset of a wide range of RTT-related features (motor, sensory, social, and cognitive deficits), while the deletion of Mecp2 from SST cells only causes a subset of RTT-related features (seizures and stereotypic behaviors) (Ito-Ishida et al. 2015). It was, therefore, suggested that the stronger neurological phenotype induced by PV cells reflects their main role in maintaining excitatory/inhibitory balance (E/I) in the cortex, a role that SST neurons do not share (Xue et al. 2014). Intriguingly, restoring MeCP2 function in PV-expressing interneurons ameliorates RTT-like behavioral phenotypes and is sufficient to partially restore proper neural network activity (Goffin et al. 2014). These data support the hypothesis that PV circuits may critically contribute to the onset of RTT disorder (Durand et al. 2012; Krishnan et al. 2015).
In the present study, we first characterized the impact of the loss of Mecp2 on the expression of multiple inhibitory-related genes throughout development and then evaluated the level of maturation of different sub-classes of GABAergic interneurons in the visual cortex. Interestingly, MGE-derived interneurons showed divergent phenotypes. PV-expressing neurons were upregulated as early as eye-opening, whereas SST-expressing neurons were downregulated, starting at postnatal day 30 (P30). CGE cells were also downregulated from P30 onward. The anatomical and functional maturation of PV cells is accompanied by the progressive appearance of the engulfing perineuronal net (PNN), a specialized extracellular milieu enriched in chondroitin sulfate proteoglycans (Dityatev and Schachner 2003). We, therefore, analyzed the PNN development in Mecp2 KO mice and found an increase in the percentage of PV cells enwrapped by PNNs. In addition, both PV interneurons and PNN structures displayed a more complex arborization in Mecp2 KO mice. We also revealed that the selective loss of Mecp2 from PV cells was enough to induce an increase in PV cell and PNN structure complexity. All of these phenotypes were recapitulated in organotypic culture, suggesting that the accelerated maturation of PV cells and the hyper-formation of PNN structures are driven by the loss of MeCP2 rather than only by sensory experience.
Materials and Methods
Animals
All procedures were approved by Institutional Animal Care and Use Committees (IACUC) at Boston Children’s Hospital and CHU Ste-Justine Research Center and Université de Montréal. Mecp2-deficient mouse line (B6.129P2(C)-Mecp2tm1.1Bird/J) crossed with C57BL6 (Guy et al. 2001) animals were used. PV-Cre (B6;129P2-Pvalbtm1(cre)Arbr/J) (Hippenmeyer et al. 2005) were mated with female heterozygous Mecp2flox/x (Guy et al. 2001).
Light-rearing (LR) and dark-rearing (DR) mice were socially housed in groups of 2 to 5 animals. LR mice were house in ventilated Optimice cages, whereas DR mice were housed in static cages. No environmental enrichment toys were added to the cages, only an extra kimwipe tissue. LR mice were maintained on a 12 h light/dark cycles, whereas DR litters were kept in complete darkness from birth until adulthood (P60). All mice were provided with food and water ad libitum.
All experiments were performed blind to the genotype of the animals and were conducted using male mice. All control animals were wild-type (WT) age-matched littermates of the mutant mice. Animals across multiple litters were randomly assigned to an experimental group.
Reverse transcription quantitative polymerase chain reaction (qRT-PCR)
Total RNA from visual cortical tissue was extracted from P30 and P60 Mecp2-deficient mice and control littermates (WT) using the Lipid Tissue Mini Kit (Qiagen). Total RNA was reverse transcribed to complimentary DNA (cDNA) and amplified using the High Capacity cDNA Reverse Transcription Kit (Thermo Fisher). Messenger RNA (mRNA) transcript levels were quantified using TaqMan fluorescent probes and the StepOnePlus real-time PCR system (Thermo Fisher).
Fluorescence data from qRT-PCR experiments was analyzed using LinRegPCR software from the Heart Failure Research Center (Amsterdam, the Netherlands). Raw fluorescence data was collected from the StepOnePlus qRT-PCR system and entered into LinRegPCR. For each experiment, LinRegPCR was used to determine a baseline fluorescence signal, from which all fluorescence signals were then subtracted. After taking the logarithm of the resulting fluorescence data, linear regression was used to determine the period during which amplification of the fluorescence signal was linear. The slope of the best-fit line during this period, also called the “Window-of-Linearity” (Ramakers et al. 2003), was used as the amplification efficiency in subsequent calculations. The traditional method of analyzing comparative qRT-PCR data, the delta-delta-Ct method, incorrectly assumes perfect amplification efficiency in every reaction. Using LinRegPCR to correct for this assumption, we then calculated the expression of the target gene relative to the expression of a normalizing gene (glyceraldehyde-phosphate-dehydrogenase, GAPDH). Finally, relative expression data was statistically analyzed using GraphPAD PRISM. The following TaqMan assays were used: GAD67 (Gad2 Mm00484623_m1), GAD65 (Gad1 Mm00725661_s1), Gephyrin (Gphn Mm00556895_m1), Gabra1 (Mm00439046_m1), Gabra2 (Mm00433435_m1), Kv3.1 (KCNC Mm00657708_m1), VGAT (Slc32a1 Mm00494138_m1), parvalbumin (Pvalb Mm00443100_m1), calbindin (Calb1 Mm00486647_m1), calretinin (Calb2 Mm00801461_m1), and somatostatin (Sst Mm00436671_m1).
Immunohistochemistry and Confocal Microscopy in Ex-Vivo Slices
Immunohistochemistry and confocal microscopy were performed as described in Durand et al. (2012). Mice were transcardially perfused with saline, followed by 45 ml of phosphate-buffered 4% paraformaldehyde solution. Brains were extracted, post-fixed in the same fixative, and cryoprotected in ascending sucrose solution (10%, 20%, and 30%) and sectioned with a cryostat. A 30 μm cryosections of primary visual cortex (V1) were first incubated for 30 min at room temperature in blocking solution (StartingBlock Blocking Buffers, Thermo Scientific) in phosphate-buffered saline (PBS), then transferred for overnight 4°C incubation into primary antibody solution (StartingBlock Blocking Buffers, Thermo Scientific) with rabbit anti-parvalbumin (Swant, 1:2500 dilution), guinea pig anti-parvalbumin (Frontier Institute Co., Ltd, 1:10000), mouse-calbindin (Swant, 1:2500), rabbit-calretinin (Swant, 1:1000), rat anti-somatostatin (Millipore, 1:500), rabbit anti-somatostatin (Peninsula Laboratories, llc, 1:500), and Lectin from Wisteria floribunda Biotin conjugate antibody, (WFA, Sigma, 1:500) (Hartig et al. 1992). A 40 μm cryosections of primary somatosensory cortex (S1) were first incubated for 30 min at room temperature with glycine 0.3 M in PBS followed by 1 h blocking at room temperature (10% Normal Goal serum [NGS], 0.3% Triton 10X in PBS), then transferred for overnight 4°C incubation into primary antibody solution (5% NGS, 0.1% Triton 10X in PBS) with guinea pig anti-calretinin (Synaptic Systems, 1:500), mouse anti-EF1α (elongation factor 1A, Millipore Sigma, 1:500), and rabbit anti-parvalbumin (Swant, 1:2500). The sections were washed in PBS and then incubated for 1 h at room temperature in secondary antibody solution with a combination of the following secondary antibodies, Alexa 488 (Invitrogen, 1:1000), Alexa 594 (Invitrogen, 1:500), Alexa 546 (Invitrogen, 1:800), and Alexa 647 (Invitrogen, 1:800) in PBS. Sections were again washed and mounted on glass slides with DAPI Fluoromount-G mounting medium (Southern Biotech).
Quantitative analyses were performed on a minimum of 3 mice per genotypes. All quantifications were performed blind to genotype, age, region of analysis, and repeated by 2 independent investigators.
V1 sections were acquired with a laser scanning confocal microscope (Olympus FluoView, FV1000), whereas S1 sections were acquired with another laser scanning confocal microscope (Zeiss LSM710). The multi-channel acquisition mode was used for both areas to avoid fluorescence cross-talk (pinhole: 1.0 airy unit for each channel). A minimum of 5 anatomically matched sections per animal were acquired and quantified per experiment.
For mean intensity and interneuron cell density quantification, 1024×1024 fields of V1 were imaged using a 20× objective (0.75 numerical aperture). Mean pixel intensity for each class of interneurons was measured across V1 images (from pial surface to white matter) using MacBiophotonics ImageJ software (http://www.macbiophotonics.ca/imagej/) using the analyze measure function that allows to quantify the mean gray values. We then estimated the numerical density of interneuron-positive cells using ImageJ software. After a manual threshold to the single channel, we automatically counted total number of cells per each channel. We counted all interneuron- and DAPI-positive cells contained within an individual area but not touching the exclusion sides (top and left side of each optical section). Finally, the number of cells was divided by the volume of the neuropil examined, resulting in cell density.
The parvalbumin cell volume was analyzed on images acquired with a ×60 oil-immersion objective (1.42 numerical aperture) and the pinhole set at 1 Airy unit using image-analysis program Volocity (PerkinElmer). On the same images, we also analyzed the PNNs structure. The volume containing the individual PNNs was extracted using maximum projection function using MacBiophotonics ImageJ software. Total branch length and total branch number were then manually measured. Between 100 and 150 cells were analyzed per genotype at each age.
For interneuron subtype experiment in S1, 2048×2048 fields, with a line average of 4, were imaged using a 10× objective (0.45 numerical aperture; 850.19 μm × 850.19 μm). Elongation factor 1A (EF1α) served as a cell marker and was manually counted across the S1 images (from pial surface to the white matter) using the cell counter on ImageJ. We then manually counted CR- and PV-positive cell density in S1 as described in Tomassy et al. (2014). Briefly, 10 equally sized superimposed digital boxes (850.19 μm in width) serving as bins were placed across the region of interest from the pial surface to the white matter. In comparison to DAPI staining, these bins were assigned a cortical layer (layer I: bin 1; layer II/III: bins 2–3; layer IV: bins 4–5; layer V: bins 6–7, and layer VI: 8–10). CR- and PV-positive cells were manually counted in each bin, and relative distribution was measured as the percentage of interneurons in each bin/layer relative to the total number of interneurons. Cell density was measure as total number of cells divided by either the area of the cortex analyzed (cells/mm2) or divided by the number of EF1α-positive cells (IN/EF1α).
Cortical Organotypic Culture and Biolistic Transfection
Slice culture preparation and gene gun transfection were performed as described in Chattopadhyaya et al. (2004). Briefly, P4 Mecp2 KO and control littermates mice were decapitated, brains were rapidly removed, and immersed in culture medium (containing DMEM, 20% horse serum, 1 mM glutamine, 13 mM glucose, 1 mM CaCl2, 2 mM MgSO4, 0.5 μM/mL insulin, 30 mM HEPES, 5 mM NaHCO3, and 0.001% ascorbic acid). Coronal brain slices, 400 μm thick, were cut with a manual chopper (Stoelting). Slices were then placed on transparent Millicell membrane inserts (Millipore), usually 2–3 slices per insert, in 30 mm petri dishes containing 750 μL of culture medium. Finally, they were incubated in a humidified incubator at 34°C with a 5% CO2-enriched atmosphere. Medium was changed every alternate day. All procedures were performed under sterile conditions. Constructs to be transfected were incorporated into “bullets” made using 1.6 μm gold particles coated with 25 μg of the G67-GFP (green fluorescent protein) plasmid. The PG67 promoter is expressed in large majority by GABAergic basket cells in cortical organotypic cultures (Chattopadhyaya et al. 2004, 2007). These bullets were used to biolistically transfect slices by gene gun (Bio-Rad, USA) at high pressure (180ψ) 6 days after slice culture preparation. The transfected slices were incubated until they reached Equivalent Postnatal Day (EP) 18 (P4 + 14 days in vitro), under the same conditions as described above.
Immunohistochemistry and Confocal Microscopy in Organotypic Cortical Slices
At EP18, slices were fixed and immunostained as described previously (Chattopadhyaya et al. 2013). Neuronal somata were visualized using anti-NeuN (monoclonal antibody, 1:400; Millipore), followed by incubation with Alexa Fluor 633-conjugated goat anti-mouse IgG (1:400; Invitrogen). Three non-overlapping fields of axonal innervation for each well-isolated basket cells were acquired with a 63× glycerol-immersion objective (NA 1.3; Leica) using a confocal microscope (LEICA TCS SPE). Z-stacks were acquired with 1 μm steps, exported as TIFF files, and analyzed with Neurolucida software (MicroBrightField). Green and blue channels were separated, and GFP-positive axons and boutons were traced in 3 dimensions in the green channel; tracings were then superimposed on the blue channel showing pyramidal cell somata (identified by NeuN immunofluorescence). Care was taken to analyze only those pyramidal cell somata that were complete and well isolated. The following parameters were analyzed for each basket cell: 1) perisomatic bouton density and 2) axonal terminal branching around contacted somata, using a 3D Sholl-based analysis using Sholl spheres with a 1 μm increment from the center of each pyramidal somata. Axon branch complexity around a single pyramidal cell soma was quantified as the average number of intersection between basket cell axons and each Sholl sphere within the first 9 μm from the center of the pyramidal cell soma. We chose 9 μm as the limiting radius for Sholl spheres because it approximates the average radius of NeuN-positive somata. Bouton density around each NeuN-positive cell soma was measured as the number of boutons present in a 9 μm Sholl-sphere, then averaged among all the analyzed targeted pyramidal neurons for each basket cell. Only pyramidal cell somata with Sholl spheres that intersect basket cell axons in the first 9 μm from the center of their soma were taken for analysis. Between 15–20 pyramidal neurons located not more than 200 μm around the basket cell soma were analyzed for each basket neuron. For each genotype, analyzed basket cells were selected from at least 3 different animals. By using this method, we obtained an unbiased estimate of the number of presumptive boutons within a given distance of a labeled pyramidal soma (NeuN profiles), rather than manually choosing the boutons that potentially target the neuronal soma, which could potentially bias the measure.
To analyze the dendritic complexity of Mecp2 KO and WT littermate basket neurons, complete dendrite fields around individual basket cell soma was acquired. The 17 EP18 parvalbumin cells (7 from controls and 10 from mutants) were put together as a mosaic and dendrites were then traced using Neurolucida software (MicroBrightField). Total branch length, total branch number, branch order, and somata volume were calculated and 3D Sholl analysis was performed. For the Sholl analysis, a series of concentric 3D spheres were drawn centered at the soma with radius increasing at 10 μm intervals and the number of dendritic intersections with each sphere plotted.
PNNs were visualized around neuronal somata (labeled with anti-NeuN monoclonal antibody, 1:400; Millipore), as described above, in EP18 organotypic slices, using WFA antibody (1:400, Sigma) followed by Streptavidin-568 Alexa Fluor conjugate secondary antibody (1:400, Invitrogen). In organotypic cultures, images of WFA staining were acquired from at least 3 cortical slices per animal with a 63× glycerol-immersion objective (NA 1.3; Leica) using a confocal microscope (LEICA TCS SPE). Z-stacks were acquired every 1 μm, and exported as TIFF stacks. Since there was an obvious observable dramatic increase in PNN complexity in Mecp2 KO slices versus WT littermates, laser power and photomultiplier gain had to be optimized for the 2 different genotypes with the goal of visualizing PNN branching in their entirety. The volume containing the individual PNNs was extracted using maximum projection function using MacBiophotonics ImageJ software. Total branch length and total branch number were then manually measured. The 150 cells were analyzed per genotype.
Statistical Analysis
All data are presented as mean ± standard error. All data were tested for D’Agostino and Pearson omnibus normality test. Statistical analysis was then done by either Mann–Whitney, one-way or two-way analysis of variance (ANOVA), as specifically indicated in the text of each figure legend. P < 0.05 was used to define statistical significance. All statistics were performed using GraphPad Prism (version 5.0) software.
Results
The Parvalbumin Circuit Is the First GABAergic System to be Disrupted in the Visual Cortex of Mecp2-Deficient Mice
Neuronal cortical activity is severely reduced in the absence of Mecp2 with a shift of E/I in favor of inhibition (Dani et al. 2005; Durand et al. 2012; Kron et al. 2012). To assess the mechanism driving such imbalance, we investigated how GABAergic interneurons mature in Mecp2-deficient (KO) mice. Quantitative qRT-PCR of visual cortical homogenates confirmed a general downregulation of major inhibitory markers at P60 (Table 1, Supplementary Fig. 1) (Durand et al. 2012). In particular, mRNA levels of Gad2 and Gad1, the two isoforms of glutamate decarboxylase responsible for GABA synthesis were decreased. Likewise, mRNA levels of Kcnc1, which encodes for a subunit of the Kv3 voltage-gated potassium ion channel responsible for regulating high-frequency neuronal firing, and levels of Slc32a1, which encodes for a vesicular inhibitory amino acid transporter, were significantly downregulated. Calbindin (Calb1) and calretinin (Calb2) and somatostatin (Sst) mRNA levels were also significantly downregulated in Mecp2 KO homogenates, whereas parvalbumin (Pvalb) levels were upregulated, confirming previous reports (Chao et al. 2010; Durand et al. 2012; Krishnan et al. 2015). We then used immunofluorescence confocal microscopy to semi-quantify the protein levels of the different classes of GABAergic interneurons. Immunofluorescence analysis confirmed an upregulation of PV marker and a downregulation of all of the other interneuron markers (Fig. 1). Though mRNA levels of all genes tested did not differ between WT and KO at P30 (Table 1, Supplementary Fig. 1), PV immunofluorescence intensity was already significantly upregulated at P15, immediately after eye opening. On the other side, CR, SST, and calbindin (data not shown) intensity were significantly downregulated starting from P30 onward. Both the upregulation of PV immunofluorescence and the downregulation of the other interneuron markers were possibly caused by an increase (PV cells) and decrease (CR, SST) of axonal and dendritic neurite complexity (Fig. 1A,B), rather than alteration of total cell density (Fig. 1C).
Table 1.
Adult dysregulation of inhibitory system in visual cortex of Mecp2 KO mice
| P30 | P60 | |||
|---|---|---|---|---|
| Gene | Mecp2 WT | Mecp2 KO | Mecp2 WT | Mecp2 KO |
| Gad2 | 1.1 ± 0.04 | 1.1 ± 0.03 | 0.9 ± 0.02## | 0.8 ± 0.02$$$** |
| Gad1 | 1.1 ± 0.1 | 1.1 ± 0.03 | 1.0 ± 0.04 | 0.9 ± 0.03$$$ |
| Kcnc1 | 1.1 ± 0.1 | 1.2 ± 0.1 | 1.0 ± 0.02 | 0.9 ± 0.03$ |
| Slc32a1 | 1.0 ± 0.1 | 1.2 ± 0.03 | 1.1 ± 0.03 | 1.0 ± 0.01$$ |
| Gphn | 0.9 ± 0.1 | 1.3 ± 0.1 | 1.0 ± 0.04 | 1.0 ± 0.04 |
| Gabra1 | 1.0 ± 0.1 | 0.1 ± 0.03 | 0.9 ± 0.04 | 0.9 ± 0.01 |
| Gabra2 | 1.8 ± 0.1 | 1.0 ± 0.1 | 1.0 ± 0.02 | 0.9 ± 0.03 |
| Pvalb | 1.0 ± 0.05 | 1.1 ± 0.03 | 0.9 ± 0.03 | 1.1 ± 0.03** |
| Calb1 | 1.5 ± 0.2 | 1.4 ± 0.1 | 0.9 ± 0.1### | 0.7 ± 0.03$$$ |
| Calb2 | 1.5 ± 0.3 | 1.5 ± 0.1 | 0.9 ± 0.03## | 0.7 ± 0.03$$$ |
| Sst | 1.2 ± 0.1 | 1.2 ± 0.1 | 1.2 ± 0.1 | 0.9 ± 0.1* |
Quantitative mRNA expression level of visual cortical homogenates demonstrated no differences between Mecp2 WT and Mecp2 KO at P30. At P60, a general downregulation of some of the inhibitory markers. Only parvalbumin (Pvalb) mRNA levels were upregulated. P30, WT, KO n = 3; P60, WT, KO n = 4–6 mice. One-way ANOVA, *P < 0.05; **P < 0.01, Bonferroni post-test. # indicates significance between P30 and P60 WT, $ indicates significance between P30 and P60 KO and * indicates significance between P60 WT and P60 KO. Mean ± s.e.m.
Figure 1.
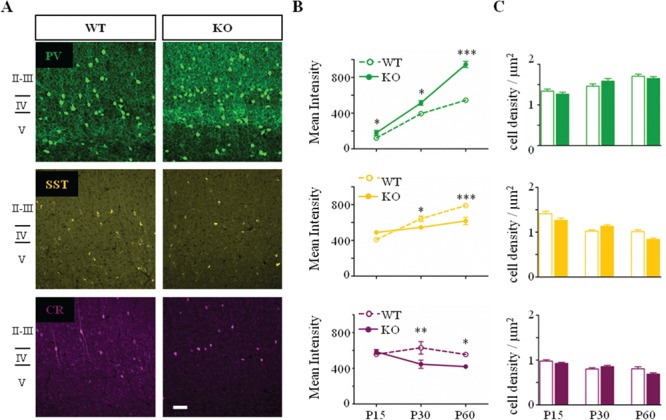
Selective developmental alteration of interneuron circuits in Mecp2 KO mice. (A) Representative confocal images of PV (green), SST (yellow), and CR (purple)-positive interneurons in mature visual cortex (P60) of Mecp2 WT and KO mice. No visual difference in interneuron organization. Scale bar: 50 μm. (B) PV immunostaining intensity is significantly upregulated from P15 onward. SST and CR immunostaining intensity are instead downregulated from P30 onward. (C) No differences in the PV, SST, and CR-positive interneurons cell density. n = 5–6 animals per age and genotype. Two-way ANOVA, *P < 0.05; **P < 0.01; ***P < 0.001, Bonferroni’s post-test. Mean ± s.e.m.
As previously reported, Mecp2 KO mice exhibit a decrease in the thickness of the visual cortex at P60 (WT, 745 ± 1.8; P60 KO, 653 ± 2 μm; P = 0.05, Mann–Whitney, n = 3 mice per age and genotype) (Chen et al. 2001; Durand et al. 2012; Tomassy et al. 2014). Despite the cortical shrinkage, we did not detect any change in PV-, CR-, and SST-positive cell density at the time points analyzed (Fig. 1C).
Tomassy et al. (2014) previously reported that the organization and density of CR-positive cells, in the somatosensory cortex (S1), is altered in mutant animals at early postnatal developmental stages. In particular, the density of CR-positive cells is significantly increased as early as 2 weeks of age in mutant mice, whereas the density of PV-positive cells increases 2 weeks later, around P30 (Tomassy et al. 2014). Therefore, we quantified the density of both bipolar and non-bipolar CR-positive cells in V1 (Caputi et al. 2009) but we did not observe changes at any of the ages we examined (Table 2). These data indicated that the loss of Mecp2 mainly affects PV expressing cell development in V1 and raise the possibility that different cortices may show different phenotypes. To test this idea, we analyzed the density and the distribution of CR- and PV-positive cells at P15 in S1 (Supplementary Fig. 2A). We confirmed that the thickness of the cortex did not differ between Mecp2 KO and Mecp2 WT (Supplementary Fig. 2B). We, then, quantified the total cell density by manually counting elongation factor 1 alpha (eEF1α)-positive cells throughout S1. We did not detect any differences between control and mutant mice (Supplementary Fig. 2A,C). Also, the total density of both CR- and PV-positive interneurons was not different between the 2 genotypes (Supplementary Fig. 2D–G). Finally, we analyzed the distribution of CR- and PV-positive cells, dividing the entire S1 cortex in 10 sampling areas (bins) that spanned from pial surface to the white matter (Tomassy et al. 2014). We, again, failed to detect any differences in the interneuron distribution throughout the cortical area (Supplementary Fig. 2F). Our results both in V1 and S1 indicated that interneuron cell density and distribution were not impacted by the loss of Mecp2.
Table 2.
No differences in the CR-positive cells populations
| CR bipolar cells/μm2 | CR non-bipolar cells/μm2 | % SST-CR | ||||
|---|---|---|---|---|---|---|
| Age | Mecp2 WT | Mecp2 KO | Mecp2 WT | Mecp2 KO | Mecp2 WT | Mecp2 KO |
| P15 | 0.23 ± 0.02 | 0.28 ± 0.01 | 0.75 ± 0.04 | 0.64 ± 0.03 | 31 ± 2 | 32 ± 2 |
| P30 | 0.11 ± 0.01 | 0.13 ± 0.01 | 0.69 ± 0.03 | 0.72 ± 0.03 | 35 ± 2 | 35 ± 1 |
| P60 | 0.12 ± 0.01 | 0.09 ± 0.01 | 0.68 ± 0.04 | 0.60 ± 0.03 | 38 ± 2 | 43 ± 2 |
The cell density of different CR interneuron subtypes did not underline differences between the Mecp2 WT and KO at all age examined. P15, P30, P60, WT, KO n = 5–6 mice. Mann–Whitney, P ≥ 0.05. Mean ± s.e.m.
We previously reported that sensory deprivation from birth until adulthood (dark-rearing, DR) or partial genetic deletion of the NMDA receptor 2A (Grin2A) subunits are sufficient to prevent the upregulation of PV expression and its hyper-connectivity in Mecp2 KO mice (Durand et al. 2012). Here, we tested whether these 2 paradigms were also sufficient to restore PV phenotype in Mecp2 KO mice. Surprisingly, we found that not only PV, but all inhibitory markers were unaltered, both adult DR Mecp2 KO and adult Mecp2 KO/Grin2A Het mice, as compared to their control littermates (Supplementary Fig. 3). Altogether, these data suggest that early upregulation of PV circuits may lead to homeostatic compensatory mechanisms resulting in the downregulation of other inhibitory sub-circuits. Interestingly, activity-dependent re-normalization of PV circuits is sufficient to prevent dampening of other GABAergic circuits in Mecp2 KO mouse visual cortex.
The Parvalbumin Circuit Maturates Faster in the Visual Cortex of Mecp2-Deficient Mice
It is well known that visual cortical structural and functional development and plasticity correlates with PV cell maturation (Hensch 2005). The degree of PV neuron maturation is accompanied by their progressive engulfment by a specialized extracellular milieu, called PNN (Dityatev and Schachner 2003). During the postnatal development of cortical circuits, the number of PNNs enwrapping PV cells and their complexity increases in an experience-dependent manner contributing to the consolidation of mature PV circuits and closing of critical period plasticity (Pizzorusso et al. 2002; Sugiyama et al. 2008; Nowicka et al. 2009). PNNs mainly surround the soma and proximal dendrites of PV interneurons in mature visual cortex (Figs 2A and 3A) (Dityatev and Schachner 2003).
Figure 2.
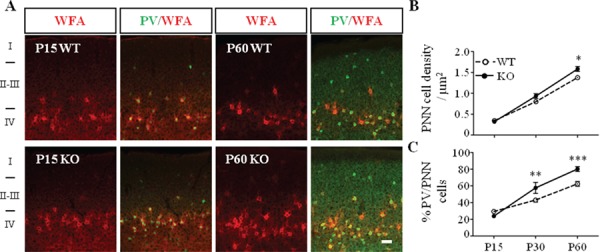
Precocious maturation of PNNs enwrapping PV cells in Mecp2 KO mice. (A) Double immunostaining of PNNs with WFA (red) and PV (green) in immature (P15) and mature visual cortex (P60) of Mecp2 WT and KO mice. Scale bar: 50 μm. (B) PNN cell density, measured as WFA-positive structures, is increased in Mecp2 KO mice. (C) Percentage of PV cells enwrapped by WFA is also increased in Mecp2 KO mice. n = 5–6 animals per age and genotype. Two-way ANOVA, *P < 0.05; **P < 0.01; ***P < 0.001, Bonferroni’s post-test. Mean ± s.e.m.
Figure 3.
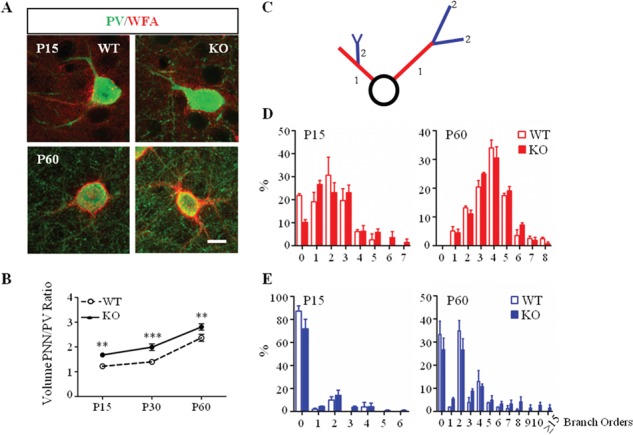
Increased complexity of PNNs enwrapping PV cells in Mecp2 KO mice. (A) Double immunostaining of PNNs with WFA (red) and PV cells (green) in immature (P15) and mature (P60) visual cortex of Mecp2 WT and KO mice. Scale bar: 10 μm. (B) Somata volume of PNN around PV-positive cell body increased in Mecp2 KO throughout animal life. (C) Schematic diagram showing classification of WFA-positive primary and secondary branches. (D) Percentage of primary PV-positive branches surrounded by WFA is increased in Mecp2 KO compared to littermates WT at P15. No differences at P60. (E) Percentage of secondary PV-positive branches is upregulated in Mecp2 KO both at P15 and P60. n = 5–6 animals per age and genotype. Two-way ANOVA, *P < 0.05; **P < 0.01; ***P < 0.001, Bonferroni’s post-test. Mean ± s.e.m.
To study the time course of PNNs maturation in the visual cortex of Mecp2 KO mice, we labeled PNNs with WFA, an established and highly sensitive PNN marker (see Materials and Methods). We found that the density of WFA-labeled PNNs increased with age in the visual cortex of both Mecp2 WT and KO mice. Interestingly at P60, the density of WFA-labeled PNNs was significantly higher in mutant animals (Fig. 2B). Shortly after eye opening (P15), PNNs were highly immature in both genotypes, in fact WFA staining was weak and diffuse, and only 20% of PV-positive cells were surrounded by PNN (Fig. 2A,C). Starting from P30, the percentage of PV-positive cells surrounded by PNN was significantly higher in Mecp2 KO animals (Fig. 2C). Interestingly, WFA-positive cell density and the percentage of PV-positive cells enwrapped by PNNs were renormalized in both adult DR Mecp2 KO and in adult Mecp2 KO/Grin2A Het mice (Supplementary Fig. 4). Taken together, these results demonstrate a precocious maturation of PV-positive interneurons that was accompanied by aberrant processing controlling PNN structures organization. This phenotype can be prevented by early restoration of overall cortical activity.
As previously demonstrated, pyramidal and GABAergic interneuronal soma size is decreased across all cortical layers in Mecp2 KO mice (Chen et al. 2001; Durand et al. 2012; Tomassy et al. 2014). Despite the developmental shrinkage of the PV-positive soma, the volume of PNN enwrapped around PV-positive soma increased throughout the animal life (Fig. 3A and Supplementary Fig. 5A) in Mecp2 KO mice. We, then, performed morphometric analysis of PNN structure along PV-positive cell dendrites. We acquired PV-positive cells enwrapped by WFA in the visual cortex of adult Mecp2 WT and KO animals and manually traced the WFA-positive structures (see Methods). The overall average dendritic branch area and length were higher in Mecp2 KO samples as compared to controls (Supplementary Fig. 5B,C). To better characterize how PNNs were distributed along single PV-positive cells, we defined primary branches as all the ramifications directly in contact with the PV-cell soma (red, Fig. 1C), whereas the higher order branches were all grouped together (blue, Fig. 1C). We discovered a significant increase in the number of primary branches enwrapped by PNN at eye opening (P15, Fig. 3D, left). Interestingly, this phenotype was no longer present at P30 and P60 stages (Fig. 3D, right and Supplementary Fig. 5D). On the contrary, the percentage of secondary PV-positive branches enwrapped by PNNs was higher already at P15 and this phenotype persist until adulthood in Mecp2 KO compared to WT littermates (Fig. 3E and Supplementary Fig. 5E). Altogether, these data showed that, PV cells exhibit a rapid maturation after eye opening (Krishnan et al. 2015) as reflected by more complex enwrapping PNN despite a significant reduction in soma size as regression takes place.
To evaluate whether the early maturation of PV circuits drives the abnormal development of PNNs, we selectively removed Mecp2 from PV cells, crossing Mecp2lox/x females (Guy et al. 2001) with Pvalb-Cre males (PV-Cre), (Hippenmeyer et al. 2005). Using this PV-Cre line, we previously demonstrated that Mecp2 is gradually yet selectively deleted from PV cells, reaching full removal only by P90 (Durand et al. 2012). We, therefore, decided to analyze the effect of Mecp2 deletion at P200 (Fig. 4A). Triple immunolabeling for MeCP2, PV, and WFA confirmed total deletion of MeCP2 from PV cells (Fig. 4B) and increased PV mean intensity (Fig. 4C) (Durand et al. 2012) without affecting PV cell density (c-WT, 1.5 ± 0.08; c-KO, 1.5 ± 0.05 μm2, n = 3 mice per genotype). Remarkably, the loss of MeCP2 was sufficient to induce a significantly upregulation of WFA mean intensity (Fig. 4D) and an upregulation of the percentage of PV-positive cells surrounded by WFA-positive structures (Fig. 4E), without changing WFA-positive cell density (c-WT, 1.1 ± 0.08; c-KO, 1.2 ± 0.05 μm2, n = 3 mice per genotype). Our results suggest a cell-autonomous role of MeCP2 in the formation of PNNs in the visual cortex.
Figure 4.
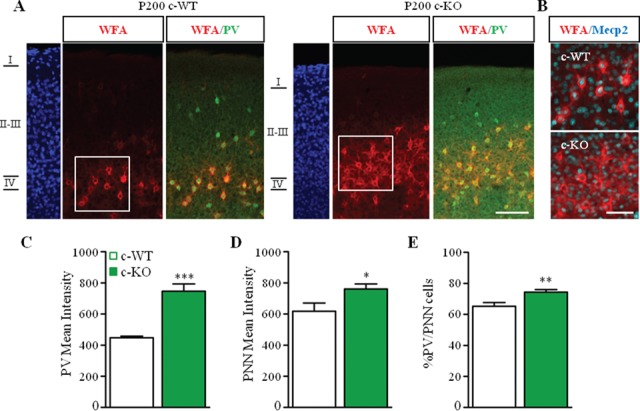
Mecp2 deletion in PV cells promotes the formation of PNNs around PV cells. (A) Triple staining for WFA (red), PV (green), and MeCP2 (cyan) in the visual cortex of Mecp2+/y/PV-Cre Het (c-WT) and Mecp2lox/y/PV-Cre Het (c-KO) at P200. (B) Selective deletion of Mecp2 in WFA-positive cells. (C, D) PV (C) and WFA (D) immunofluorescent mean intensity are upregulated in c-KO compared to c-WT littermates. (D) Percentage of PV-positive cells enwrapped by WFA is increased in c-KO compared to c-WT. Scale bar: 100 μm (A); 50 μm (B). n = 3 animals per genotype. Mann–Whitney test, *P < 0.05; **P < 0.01; ***P < 0.001. Mean ± s.e.m.
Parvalbumin Cells Are Morphologically More Complex in Mecp2 KO Mice
We have previously shown an increased number of PV-positive perisomatic puncta in Mecp2 KO mouse cortex, as compared to WT controls, at eye opening (Durand et al. 2012). However, it is unknown whether this upregulation is concomitant with increased PV cellular complexity. To study dendritic and axonal maturation in basket cells, we took advantage of a well-established organotypic slice preparation (Chattopadhyaya et al. 2004, 2007, 2013). We biolistically transfected cortical organotypic slices prepared from Mecp2 KO mice and control littermates with a construct that drives GFP expression under the promoter PG67 (PG67-GFP) and enables GFP-labeling of individual basket neurons at high resolution (Chattopadhyaya et al. 2004, 2007, 2013). Slices at equivalent postnatal day (EP) 10 (culture prepared at P4 + 6 DIV) were biolistically transfected and GFP-labeled basket cell were acquired at EP18.
Remarkably, we noticed that basket-positive cells in Mecp2 KO mice covered a denser area of the cortical territory as compared to their WT littermates (Fig. 5A). We then analyzed the extent of their perisomatic innervation of pyramidal neurons by quantifying their terminal axonal branching and bouton density around NeuN-labeled postsynaptic pyramidal cell somata. We have previously shown, by localization of pre- and post-synaptic markers and electron microscopy (Chattopadhyaya et al. 2004), that the vast majority of GFP-labeled boutons in this experimental condition likely represent presynaptic terminals. We observed that, at EP18 in Mecp2 KO-derived cultures, GFP-labeled basket neurons showed more intricate axonal branching around neuronal somata than did basket neurons from WT animals, with multiple axonal terminals bearing more numerously clustered boutons (Fig. 5A,B). Both density of perisomatic boutons (Fig. 5B) and axonal complexity (Fig. 5C) in GFP-positive cells were significantly increased in Mecp2 KO mice. Moreover, these properties of EP18 Mecp2 KO-derived PV cells were comparable to values observed at older ages in Mecp2 WT basket cells (P28) (Chattopadhyaya et al. 2007, 2013).
Figure 5.
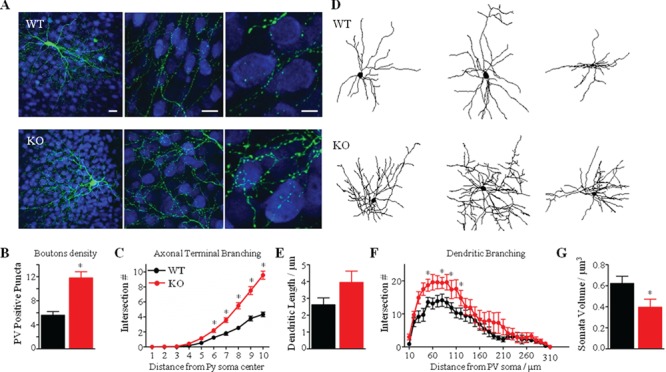
Axonal and dendritic hyper-ramification of PV cells in Mecp2 KO mice. (A) Mecp2 WT and KO cortical organotypic cultures were transfected via biolistics with a Pg67-GFP construct. A KO basket neuron (lower panel, green) among other NeuN immunostained neurons (blue) shows elaborate axonal branching and numerous boutons on the postsynaptic somata, as compared to a WT basket neuron (higher panel). Scale bar: 20 μm (left), 20 μm (middle), 5 μm (right). (B, C) Axonal bouton density (B) and complexity of terminal branching (C) are significantly increase in Mecp2 KO basket neurons (Mann–Whitney test, *P < 0.01). (D) Representative Neurolucida-traced basket cells in Mecp2 WT and KO animals. (E, F) Sholl-analysis reveals a significant increase in the total dendritic length (E) and in the complexity of dendritic branching (F) in Mecp2 KO. (G) PV-positive somata volume of Mecp2 KO basket cells is significantly reduced, as compared to WT littermates. n = 7 WT basket cells; n = 10 KO basket neurons. Mann–Whitney test, *P < 0.01. Mean ± s.e.m.
We further asked whether the loss of Mecp2 alters basket cell dendritic arborization. We acquired the dendrites of 17 EP18 basket cells and reconstructed them in Neurolucida (Fig. 5D). Reconstructed dendrites demonstrated that the total number of branches per basket cell was significantly increased in Mecp2 KO-derived cells, as compared to WT control cells (Supplementary Fig. 6A). Total dendrite length was also increased in mutant animals (Fig. 5E), suggesting that in the absence of Mecp2 basket cells develop longer dendritic arborization.
Sholl analysis revealed that, starting at ~ 40–50 μm from the soma, arbor complexity (number of Sholl intersections per concentric sphere) is increased in Mecp2 KO mice (Fig. 5F). These results suggest that, farther than 50 μm from the soma, PV cells are more likely to bifurcate and extend laterally leading to a denser dendritic arbor in Mecp2 KO cortex. We then analyzed the branching organization of Mecp2 KO basket cells and found that they have more branches of higher order (>1), even beginning with branches of the first order (Supplementary Fig. 6C). In fact, control basket cells do not usually have branches beyond orders 6–8, whereas the vast majority of Mecp2 KO basket cells had branches of order ≥7 (Supplementary Fig. 6C). Average dendritic branch length per order was generally higher in Mecp2 KO-derived basket cells (Supplementary Fig. 6B). Despite this increased dendritic complexity, organotypically cultured Mecp2 KO basket cells still exhibited reduced soma volume than WT basket cells under the same conditions (Fig. 5G). Taken together, these results indicate that loss of Mecp2 significantly alters PV cell development by reducing its cell body size, while dramatically increasing axonal bouton density, as well as axonal and dendritic arborization and complexity.
PNNs in Mecp2-Deficient Cortex Demonstrate Hypercomplex Phenotype in Organotypic Culture
We then asked if the observed increased complexity of axonal and dendritic arbors in Mecp2 KO basket interneurons was accompanied by an increase in PNN envelopment of the same cell. As previously shown, WFA-positive PNN structure is well preserved in organotypic cortical slices (Fig. 6) (Bruckner and Grosche 2001). PNN structures were present in both Mecp2 WT and KO animals (Fig. 6A). At EP18, PNNs in control slices were highly immature. In fact, WFA-positive signal was weak and diffuse, and only a few NeuN-positive neurons were enwrapped by PNNs. On the contrary, PNNs were quite distinct and well formed in mutant slices (Fig. 6A). The density of WFA-labeled PNNs surrounding NeuN-positive cells was also increased in EP18 Mecp2 KO-derived cultures (Fig. 6B), with no change in the distribution of NeuN-cells enwrapped by PNNs (data not shown). Total dendrite length was also increased in Mecp2 KO-derived basket cells (Fig. 6C). Finally, we analyzed PNN complexity by manually tracing WFA-positive structure in culture (see Material and Method). We noticed that overall WFA-labeled PNNs were less developed and complex in the organotypic cultures compared to in vivo structures at comparable ages (Fig. 3D vs. Fig. 6D). However, Mecp2 KO in organotypic cultures were still characterized by a higher complexity compared to their control littermates. In particular, we found a significant increase in the WFA-labeled PNNs in Mecp2 KO mice (Fig. 6A,B) and their length was longer in mutant compared to control littermates (Fig. 6C). The percentage of both primary and secondary PNN structures was also increased in Mecp2 KO mice (Fig. 6D,E). Taken together, our results support a direct relationship between higher PV cellular complexity and increased PNN structure complexity in the absence of MeCP2 even in the absence of sensory experience.
Figure 6.
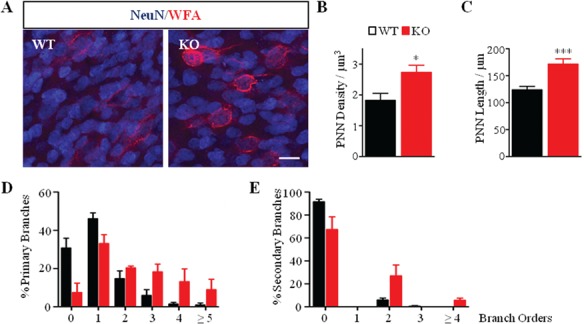
Complexity of PNNs is increased in Mecp2 KO organotypic cultures. (A) Double immunostaining of PNNs (red) and NeuN (blue) in cortical organotypic cultures of Mecp2 WT and KO mice. Scale bar: 20 μm. (B, C) Quantification of the number of WFA-positive cells reveals an increase in the density of PNN-positive enwrapped neurons (B) and in the WFA-positive dendritic length (C) in Mecp2 KO culture. Mann–Whitney test, *P ˂ 0.05; ***P ˂ 0.001. (D, E) Percentage of both WFA-positive primary (D) and secondary neurites (E) is increased in Mecp2 KO cultures as compared to WT cultures. n = 5 animals per genotype. Two-way ANOVA, ***P < 0.001, Bonferroni’s post-test. Mean ± s.e.m.
Discussion
Our results showed that precocious maturation of cortical PV-basket interneurons was followed by downregulation of SST, CR, and calbindin-positive interneurons. Interestingly, PV axonal and dendritic arborization complexity was increased in Mecp2 KO mice and this phenotype was accompanied by an increase in PNN structure complexity. Late selective deletion of MeCP2 from PV cells was sufficient to promote PNN hyper-formation. Both environmental (DR) and genetic (GluN2A) manipulations prevented such changes. Interestingly, increased complexity of PV and PNN structures were replicated in organotypic culture, suggesting that they may mainly depend from the loss of Mecp2 and independently from the external sensory experience. Altogether, our data support the hypothesis that MeCP2 is differentially required for the correct development of each subset of GABAergic interneurons.
The Loss of Mecp2 Selectively Affects Subclasses of Interneurons
Using 2 complementary techniques, we identified that the overall maturation of the GABAergic system is disrupted in the visual cortex of Mecp2 KO mice. In particular, our mRNA analysis showed significant differences mainly at P60, whereas the immunofluorescence analysis detected changes as early as P15. The correlation between expression levels of mRNA and protein can be poor (Vogel and Marcotte 2012) due to measurement noise, particularly when performed from homogenate of tissue containing mix of cell types as glia, excitatory, and inhibitory cells, as in our case (Maier et al. 2009). Single cell mRNA sequencing may provide the necessary resolution to detect finer changes in GABAergic neurons over development and disease (Renthal et al. 2018).
Longitudinal analysis of major interneuron markers revealed that the loss of MeCP2 does not affect equally the postnatal development of each interneuron subpopulation. While we found no differences in the density or distribution of all interneurons at any ages examined, our data showed that the PV-positive cells were the first to be disrupted and upregulated, followed by downregulation of the SST, CR, and calbindin circuits. This could be either a direct effect of the loss of MeCP2 or driven by homeostatic mechanisms to dampen excessive inhibition in the cortical circuits (Dani et al. 2005; Ramocki and Zoghbi 2008; Kron et al. 2012).
Though MeCP2 directly binds to the promoter regions of Pvalb and Sst genes (Chahrour et al. 2008; Durand et al. 2012) and preferentially influences the maturation of MGE-derived interneurons, it does not exclude a possible indirect role in adult function of other major classes of cortical interneurons. Our results are in apparent contrast with previous work showing that somatosensory cortex is characterized by precocious maturation of CR circuit, followed by disruption of PV but not SST circuits (Tomassy et al. 2014). One possible explanation for this discrepancy is the use of 2 different Mecp2 mutant lines. Here we studied Mecp2 mutant mice generated by the Bird laboratory (Guy et al. 2001), whereas Tomassy et al. (2014) analyzed Mecp2 mutant mice generated by the Jaenisch laboratory (Chen et al. 2001). One of the major differences between these 2 lines is that the Bird strain has no signs of mRNA and MeCP2 protein (Santos et al. 2007), whereas the Jaenisch one expresses some mRNA Mecp2 at 8 weeks of age (Jordan et al. 2007). Studies have reported distinct neurological phenotypes (Monteggia and Powell, 2012) and brain region-specific alterations in these 2 types of Mecp2 mutant mice (Belichenko et al. 2008, 2009). These results suggest a possible MeCP2-dose dependent effect on GABAergic circuit maturation and raise the question of how different truncation and missense mutations, found in RTT patients, may differentially impact inhibitory neurons and their overall network function. Indeed, missense mutations like T158 M and R306C induce a dramatically milder phenotype and degree of symptom presentation than truncating mutations like R168X and R255X (Li and Pozzo-Miller 2012). Future studies should be devoted to fully characterize GABAergic circuits in mice with common MeCP2 mutations in order to design target therapeutic intervention.
Intrinsic and Extrinsic Factors in Structural Maturation
Although it is well established that loss of MeCP2 induces a dramatic reduction in dendritic branching and spine formation/stabilization on pyramidal cortical neurons (Belichenko et al. 1994; Armstrong et al. 1995; Kishi and Macklis 2004; Fukuda et al. 2005), little is known about the impact of MeCP2 on interneuron cell ramifications.
Here, we began exploring the branching complexity of PV neurons using organotypic slice cultures and discovered that PV basket cells have increased both axonal and dendritic arborization complexity in the V1 of Mecp2 KO mice, despite the shrinkage of their cell bodies and independently from sensory experience. This enrichment in complexity persisted into adulthood despite a decrease in GABA content (Chao et al. 2010; Durand et al. 2012) and other essential GABA markers, like gad1, gad2, Kcnc1, and Slc32a1 (Table 1) (Durand et al. 2012).
Such significant impact of loss of MeCP2 on structural complexity raises the question whether cell-specific signal pathways and/or cell extrinsic factors are at play in determining the size and shape of a neuron’s dendritic arbors (Miller and Kaplan 2003). For example, brain-derived neurotrophic factor (BDNF) and homeoprotein Otx2 (orthodenticle homeobox 2) contribute to the structural and functional maturation of PV cells (Huang et al. 1999; Sugiyama et al. 2008; Riffault et al. 2014). While BDNF has been extensively implicated in structural deficits pyramidal neurons and over-expression or modulation of its signal pathway is sufficient to rescue several excitatory circuit and behavioral deficits in Mecp2 mutant mice (Li and Pozzo-Miller 2014), no specific analysis has been performed to quantify the impact on inhibitory neurons’ dendritic and axonal structure in health and diseases, like RTT.
Another major regulator of PV circuit’s maturation is the homeoprotein Otx2. It has been demonstrated that Otx2 is sufficient to drive the development of PV cells and their PNN and determine the expression of critical period plasticity (Sugiyama et al. 2009). Otx2 is produced in the retina and choroid plexus and it is transported to the cerebral cortex, where it gets preferentially internalized and accumulated in PV positive cells surrounded by PNN. No data are available about Otx2 expression and localization in Mecp2 mutant mice. Future work should be conducted as it could open new avenues of intervention for RTT circuit dysfunction.
Finally, microglia may contribute to the hypercomplexity of PV cells, as it actively participate in dendritic, axonal, and synaptic remodeling and pruning during normal and pathological conditions (Gomez-Nicola and Perry 2015). For example, a mis-regulated microglia could fail to prune PV cells, while over-pruning other neurons. However, this explanation seems unlikely as Mecp2 KO mice do not show any gross alteration of the microglia cells throughout the development and their activation only happens in late stages of the phenotypic regression (Cronk et al. 2015; Schafer et al. 2016). Thus, microglia may not actively drive circuit defects in Mecp2 KO mice but rather contribute to later stages of the disorder (Cronk et al. 2015; Schafer et al. 2016).
Impact of Early Maturation and Increased of PV and PNN Complexity on Circuit Activity
After eye opening, cortical PV-basket cells undergo substantial maturation in morphology, connectivity, and intrinsic and synaptic properties through an activity-dependent process that is necessary for the onset and expression of critical period plasticity and overall proper network processing in health and diseases (Hensch 2005). The accelerated growth of PV interneurons’ is accompanied by mature-like PNN, larger spontaneous inhibitory currents, diminished evoked inhibitory current amplitude, and synaptic depression during trains of action potentials in V1 (Krishnan et al. 2015). The misregulation of excitatory synaptic inputs onto PV cells, including increased intracortical excitatory marker VGlut1 (Morello et al. 2018), enhanced thalamocortical excitatory marker VGlut2 (Sigel, Bae, Bogart, Hensch, and Zhuang, personal communication) and precocious maturation of NMDA receptor composition along PV-positive soma and proximal dendrites (Mierau et al. 2015) favors PV cell activation and further contributes to shifting the E/I network activity. As a result, there is a more coherent inhibitory network, premature expression of critical period for ocular dominance plasticity (Krishnan et al. 2015), and gradual dampening of global cortical network activity (Durand et al. 2012).
In conclusion, we described progressive changes in selective inhibitory networks that could result in E/I unbalance silencing cortical circuits and driving the regression and late onset of epilepsy, previously described in mice and in RTT patients. An early detection of specific cellular and molecular abnormalities in RTT may lead to the identification of new therapeutic targets and/or biomarkers that may be translated to the clinical setting.
Authors Contributions
A.P. and M.F. conceived and designed the project; A.P. performed most of the in vivo studies; P.N.A. performed some of the immunofluorescence experiments; C.L. performed most of the qRT-PCR studies; B.C. performed most of the organotypic cultures studies; A.P., C.L., P.N.A., and B.C. analyzed the data; A.P., B.C., P.N.A., C.L., G.D.C., and M.F. wrote the manuscript.
Funding
RettSyndrome.org (to A.P. and to M.F.); Nancy Lurie Marks Foundation (to A.P.); NIH NINDS (National Institute of Neurological Disorders and Stroke) ROS1NS095959 (to M.F.); Rett Syndrome Research Trust (to M.F.); Simon Foundation (to M.F.); Ste. Justine Foundation (to B.C), and Scottish Rite Charitable Foundation (to G.D.C.).
Notes
We thank E. Centofante for helping in the neurolucida cell reconstruction, D. Surin for helping with the immunohistochemistry analysis, A.D. Hill, C.V. Innocent and Boston Children’s Hospital IDDRC imaging core (NIH-P30-HD-18655) for imaging software and Dr Hensch and members of the Fagiolini and Hensch labs for helpful discussion. Conflict of Interest: The authors declare no conflict of interest.
Supplementary Material
References
- Akbarian S, Chen RZ, Gribnau J, Rasmussen TP, Fong H, Jaenisch R, Jones EG. 2001. Expression pattern of the Rett syndrome gene MeCP2 in primate prefrontal cortex. Neurobiol Dis. 8:784–791. [DOI] [PubMed] [Google Scholar]
- Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. 1999. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 23:185–188. [DOI] [PubMed] [Google Scholar]
- Armstrong D, Dunn JK, Antalffy B, Trivedi R. 1995. Selective dendritic alterations in the cortex of Rett syndrome. J Neuropathol Exp Neurol. 54:195–201. [DOI] [PubMed] [Google Scholar]
- Banerjee A, Rikhye RV, Breton-Provencher V, Tang X, Li C, Li K, Runyan CA, Fu Z, Jaenisch R, Sur M. 2016. Jointly reduced inhibition and excitation underlies circuit-wide changes in cortical processing in Rett syndrome. Proc Natl Acad Sci U S A. 113:E7287–E7296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belichenko NP, Belichenko PV, Li HH, Mobley WC, Francke U. 2008. Comparative study of brain morphology in Mecp2 mutant mouse models of Rett syndrome. J Comp Neurol. 508:184–195. [DOI] [PubMed] [Google Scholar]
- Belichenko PV, Oldfors A, Hagberg B, Dahlstrom A. 1994. Rett syndrome: 3-D confocal microscopy of cortical pyramidal dendrites and afferents. Neuroreport. 5:1509–1513. [PubMed] [Google Scholar]
- Belichenko PV, Wright EE, Belichenko NP, Masliah E, Li HH, Mobley WC, Francke U. 2009. Widespread changes in dendritic and axonal morphology in Mecp2-mutant mouse models of Rett syndrome: evidence for disruption of neuronal networks. J Comp Neurol. 514:240–258. [DOI] [PubMed] [Google Scholar]
- Blue ME, Naidu S, Johnston MV. 1999. Altered development of glutamate and GABA receptors in the basal ganglia of girls with Rett syndrome. Exp Neurol. 156:345–352. [DOI] [PubMed] [Google Scholar]
- Bruckner G, Grosche J. 2001. Perineuronal nets show intrinsic patterns of extracellular matrix differentiation in organotypic slice cultures. Exp Brain Res. 137:83–93. [DOI] [PubMed] [Google Scholar]
- Caputi A, Rozov A, Blatow M, Monyer H. 2009. Two calretinin-positive GABAergic cell types in layer 2/3 of the mouse neocortex provide different forms of inhibition. Cereb Cortex. 19:1345–1359. [DOI] [PubMed] [Google Scholar]
- Chahrour M, Jung SY, Shaw C, Zhou X, Wong ST, Qin J, Zoghbi HY. 2008. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science. 320:1224–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao HT, Chen H, Samaco RC, Xue M, Chahrour M, Yoo J, Neul JL, Gong S, Lu HC, Heintz N et al. 2010. Dysfunction in GABA signalling mediates autism-like stereotypies and Rett syndrome phenotypes. Nature. 468:263–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chattopadhyaya B, Baho E, Huang ZJ, Schachner M, Di Cristo G. 2013. Neural cell adhesion molecule-mediated Fyn activation promotes GABAergic synapse maturation in postnatal mouse cortex. J Neurosci. 33:5957–5968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chattopadhyaya B, Di Cristo G, Higashiyama H, Knott GW, Kuhlman SJ, Welker E, Huang ZJ. 2004. Experience and activity-dependent maturation of perisomatic GABAergic innervation in primary visual cortex during a postnatal critical period. J Neurosci. 24:9598–9611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chattopadhyaya B, Di Cristo G, Wu CZ, Knott G, Kuhlman S, Fu Y, Palmiter RD, Huang ZJ. 2007. GAD67-mediated GABA synthesis and signaling regulate inhibitory synaptic innervation in the visual cortex. Neuron. 54:889–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen RZ, Akbarian S, Tudor M, Jaenisch R. 2001. Deficiency of methyl-CpG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nat Genet. 27:327–331. [DOI] [PubMed] [Google Scholar]
- Cronk JC, Derecki NC, Ji E, Xu Y, Lampano AE, Smirnov I, Baker W, Norris GT, Marin I, Coddington N et al. 2015. Methyl-CpG binding protein 2 regulates microglia and macrophage gene expression in response to inflammatory stimuli. Immunity. 42:679–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dani VS, Chang Q, Maffei A, Turrigiano GG, Jaenisch R, Nelson SB. 2005. Reduced cortical activity due to a shift in the balance between excitation and inhibition in a mouse model of Rett syndrome. Proc Natl Acad Sci U S A. 102:12560–12565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dityatev A, Schachner M. 2003. Extracellular matrix molecules and synaptic plasticity. Nat Rev Neurosci. 4:456–468. [DOI] [PubMed] [Google Scholar]
- Duarte ST, Armstrong J, Roche A, Ortez C, Perez A, O'Callaghan Mdel M, Pereira A, Sanmarti F, Ormazabal A, Artuch R et al. 2013. Abnormal expression of cerebrospinal fluid cation chloride cotransporters in patients with Rett syndrome. PLoS One. 8:e68851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand S, Patrizi A, Quast KB, Hachigian L, Pavlyuk R, Saxena A, Carninci P, Hensch TK, Fagiolini M. 2012. NMDA receptor regulation prevents regression of visual cortical function in the absence of Mecp2. Neuron. 76:1078–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda T, Itoh M, Ichikawa T, Washiyama K, Goto Y. 2005. Delayed maturation of neuronal architecture and synaptogenesis in cerebral cortex of Mecp2-deficient mice. J Neuropathol Exp Neurol. 64:537–544. [DOI] [PubMed] [Google Scholar]
- Gabel HW, Kinde B, Stroud H, Gilbert CS, Harmin DA, Kastan NR, Hemberg M, Ebert DH, Greenberg ME. 2015. Disruption of DNA-methylation-dependent long gene repression in Rett syndrome. Nature. 522:89–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goffin D, Brodkin ES, Blendy JA, Siegel SJ, Zhou Z. 2014. Cellular origins of auditory event-related potential deficits in Rett syndrome. Nat Neurosci. 17:804–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Nicola D, Perry VH. 2015. Microglial dynamics and role in the healthy and diseased brain: a paradigm of functional plasticity. Neuroscientist. 21:169–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guy J, Hendrich B, Holmes M, Martin JE, Bird A. 2001. A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat Genet. 27:322–326. [DOI] [PubMed] [Google Scholar]
- Hagberg B. 2002. Clinical manifestations and stages of Rett syndrome. Ment Retard Dev Disabil Res Rev. 8:61–65. [DOI] [PubMed] [Google Scholar]
- Hartig W, Brauer K, Bruckner G. 1992. Wisteria floribunda agglutinin-labelled nets surround parvalbumin-containing neurons. Neuroreport. 3:869–872. [DOI] [PubMed] [Google Scholar]
- He LJ, Liu N, Cheng TL, Chen XJ, Li YD, Shu YS, Qiu ZL, Zhang XH. 2014. Conditional deletion of Mecp2 in parvalbumin-expressing GABAergic cells results in the absence of critical period plasticity. Nat Commun. 5:5036. [DOI] [PubMed] [Google Scholar]
- Hensch TK. 2005. Critical period mechanisms in developing visual cortex. Curr Top Dev Biol. 69:215–237. [DOI] [PubMed] [Google Scholar]
- Hippenmeyer S, Vrieseling E, Sigrist M, Portmann T, Laengle C, Ladle DR, Arber S. 2005. A developmental switch in the response of DRG neurons to ETS transcription factor signaling. PLoS Biol. 3:e159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang ZJ, Kirkwood A, Pizzorusso T, Porciatti V, Morales B, Bear MF, Maffei L, Tonegawa S. 1999. BDNF regulates the maturation of inhibition and the critical period of plasticity in mouse visual cortex. Cell. 98:739–755. [DOI] [PubMed] [Google Scholar]
- Ito-Ishida A, Ure K, Chen H, Swann JW, Zoghbi HY. 2015. Loss of MeCP2 in parvalbumin-and somatostatin-expressing neurons in mice leads to distinct Rett syndrome-like phenotypes. Neuron. 88:651–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jian L, Nagarajan L, de Klerk N, Ravine D, Bower C, Anderson A, Williamson S, Christodoulou J, Leonard H. 2006. Predictors of seizure onset in Rett syndrome. J Pediatr. 149:542–547. [DOI] [PubMed] [Google Scholar]
- Jordan C, Li HH, Kwan HC, Francke U. 2007. Cerebellar gene expression profiles of mouse models for Rett syndrome reveal novel MeCP2 targets. BMC Med Genet. 8:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishi N, Macklis JD. 2004. MECP2 is progressively expressed in post-migratory neurons and is involved in neuronal maturation rather than cell fate decisions. Mol Cell Neurosci. 27:306–321. [DOI] [PubMed] [Google Scholar]
- Krishnan K, Wang BS, Lu J, Wang L, Maffei A, Cang J, Huang ZJ. 2015. MeCP2 regulates the timing of critical period plasticity that shapes functional connectivity in primary visual cortex. Proc Natl Acad Sci U S A. 112:E4782–E4791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kron M, Howell CJ, Adams IT, Ransbottom M, Christian D, Ogier M, Katz DM. 2012. Brain activity mapping in Mecp2 mutant mice reveals functional deficits in forebrain circuits, including key nodes in the default mode network, that are reversed with ketamine treatment. J Neurosci. 32:13860–13872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Pozzo-Miller L. 2012. Beyond widespread Mecp2 deletions to model Rett syndrome: conditional spatio-temporal knockout, single-point mutations and transgenic rescue mice. Autism Open Access. 2012:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Pozzo-Miller L. 2014. BDNF deregulation in Rett syndrome. Neuropharmacology. 76(Pt C):737–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyst MJ, Bird A. 2015. Rett syndrome: a complex disorder with simple roots. Nat Rev Genet. 16:261–275. [DOI] [PubMed] [Google Scholar]
- Maier T, Guell M, Serrano L. 2009. Correlation of mRNA and protein in complex biological samples. FEBS Lett. 583:3966–3973. [DOI] [PubMed] [Google Scholar]
- Medrihan L, Tantalaki E, Aramuni G, Sargsyan V, Dudanova I, Missler M, Zhang W. 2008. Early defects of GABAergic synapses in the brain stem of a MeCP2 mouse model of Rett syndrome. J Neurophysiol. 99:112–121. [DOI] [PubMed] [Google Scholar]
- Mierau SB, Patrizi A, Hensch TK, Fagiolini M. 2015. Cell-specific regulation of N-methyl-D-aspartate receptor maturation by Mecp2 in cortical circuits. Biol Psychiatry 79:746–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller FD, Kaplan DR. 2003. Signaling mechanisms underlying dendrite formation. Curr Opin Neurobiol. 13:391–398. [DOI] [PubMed] [Google Scholar]
- Monteggia LM, Powell CM. 2012. The autisms: molecules to model systems. New York City (NY): Oxford University Press [Google Scholar]
- Morello N, Schina R, Pilotto F, Phillips M, Melani R, Plicato O, Pizzorusso T, Pozzo-Miller L, Giustetto M. 2018. Loss of Mecp2 causes atypical synaptic and molecular plasticity of parvalbumin-expressing interneurons reflecting Rett syndrome-like sensorimotor defects. eNeuro. Sep 24;5(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowicka D, Soulsby S, Skangiel-Kramska J, Glazewski S. 2009. Parvalbumin-containing neurons, perineuronal nets and experience-dependent plasticity in murine barrel cortex. Eur J Neurosci. 30:2053–2063. [DOI] [PubMed] [Google Scholar]
- Pizzorusso T, Medini P, Berardi N, Chierzi S, Fawcett JW, Maffei L. 2002. Reactivation of ocular dominance plasticity in the adult visual cortex. Science. 298:1248–1251. [DOI] [PubMed] [Google Scholar]
- Ramakers C, Ruijter JM, Deprez RH, Moorman AF. 2003. Assumption-free analysis of quantitative real-time polymerase chain reaction (PCR) data. Neurosci Lett. 339:62–66. [DOI] [PubMed] [Google Scholar]
- Ramocki MB, Zoghbi HY. 2008. Failure of neuronal homeostasis results in common neuropsychiatric phenotypes. Nature. 455:912–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renthal W, Boxer LD, Hrvatin S, Li E, Silberfeld A, Nagy MA, Griffith EC, Vierbuchen T, Greenberg ME. 2018. Characterization of human mosaic Rett syndrome brain tissue by single-nucleus RNA sequencing. Nat Neurosci. 21:1670–1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riffault B, Medina I, Dumon C, Thalman C, Ferrand N, Friedel P, Gaiarsa JL, Porcher C. 2014. Pro-brain-derived neurotrophic factor inhibits GABAergic neurotransmission by activating endocytosis and repression of GABAA receptors. J Neurosci. 34:13516–13534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos M, Silva-Fernandes A, Oliveira P, Sousa N, Maciel P. 2007. Evidence for abnormal early development in a mouse model of Rett syndrome. Genes Brain Behav. 6:277–286. [DOI] [PubMed] [Google Scholar]
- Schafer DP, Heller CT, Gunner G, Heller M, Gordon C, Hammond T, Wolf Y, Jung S, Stevens B. 2016. Microglia contribute to circuit defects in Mecp2 null mice independent of microglia-specific loss of Mecp2 expression. Elife. Jul 26;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugino K, Hempel CM, Okaty BW, Arnson HA, Kato S, Dani VS, Nelson SB. 2014. Cell-type-specific repression by methyl-CpG-binding protein 2 is biased toward long genes. J Neurosci. 34:12877–12883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiyama S, Di Nardo AA, Aizawa S, Matsuo I, Volovitch M, Prochiantz A, Hensch TK. 2008. Experience-dependent transfer of Otx2 homeoprotein into the visual cortex activates postnatal plasticity. Cell. 134:508–520. [DOI] [PubMed] [Google Scholar]
- Sugiyama S, Prochiantz A, Hensch TK. 2009. From brain formation to plasticity: insights on Otx2 homeoprotein. Dev Growth Differ. 51:369–377. [DOI] [PubMed] [Google Scholar]
- Tomassy GS, Morello N, Calcagno E, Giustetto M. 2014. Developmental abnormalities of cortical interneurons precede symptoms onset in a mouse model of Rett syndrome. J Neurochem. 131:115–127. [DOI] [PubMed] [Google Scholar]
- Vogel C, Marcotte EM. 2012. Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat Rev Genet. 13:227–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wonders CP, Anderson SA. 2006. The origin and specification of cortical interneurons. Nat Rev Neurosci. 7:687–696. [DOI] [PubMed] [Google Scholar]
- Xu X, Roby KD, Callaway EM. 2010. Immunochemical characterization of inhibitory mouse cortical neurons: three chemically distinct classes of inhibitory cells. J Comp Neurol. 518:389–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue M, Atallah BV, Scanziani M. 2014. Equalizing excitation-inhibition ratios across visual cortical neurons. Nature. 511:596–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita Y, Matsuishi T, Ishibashi M, Kimura A, Onishi Y, Yonekura Y, Kato H. 1998. Decrease in benzodiazepine receptor binding in the brains of adult patients with Rett syndrome. J Neurol Sci. 154:146–150. [DOI] [PubMed] [Google Scholar]
- Zhang W, Feng G, Wang L, Teng F, Wang L, Li W, Zhang Y, Zhou Q. 2018. MeCP2 deficiency promotes cell reprogramming by stimulating IGF1/AKT/mTOR signaling and activating ribosomal protein-mediated cell cycle gene translation. J Mol Cell Biol 10:515–526. [DOI] [PubMed] [Google Scholar]
- Zhang ZW, Zak JD, Liu H. 2010. MeCP2 is required for normal development of GABAergic circuits in the thalamus. J Neurophysiol. 103:2470–2481. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.