

Abstract
Selective access to a targeted isomer is often critical in the synthesis of biologically active molecules. Whereas small-molecule reagents and catalysts often act with anticipated site- and stereoselectivity, this predictability does not extend to enzymes. Further, the lack of access to catalysts that provide complementary selectivity creates a challenge in the application of biocatalysis in synthesis. Here, we report an approach for accessing biocatalysts with complementary selectivity that is orthogonal to protein engineering. Through the use of a sequence similarity network (SSN), a number of sequences were selected and the corresponding biocatalysts were evaluated for reactivity and selectivity. With a number of biocatalysts identified that operate with complementary site- and stereoselectivity, these catalysts were employed in the stereodivergent, chemoenzymatic synthesis of azaphilone natural products. Specifically, the first syntheses of trichoflectin, deflectin-1a, and lunatoic acid A were achieved. In addition, chemoenzymatic syntheses of these azaphilones supplied enantioenriched material for reassignment of the absolute configuration of trichoflectin and deflectin-1a based on optical rotation, CD spectra, and X-ray crystallography.
Keywords: biocatalysis, azaphilones, chemoenzymatic synthesis, total synthesis, flavin-dependent monooxygenases, sequence similarity networks
Graphical Abstract
INTRODUCTION
Biocatalytic approaches can provide numerous advantages in synthesis. These include high levels of selectivity, mild reaction conditions, and opportunities for cascade reactions that enable the rapid construction of complex molecules.1 These attractive qualities have drawn the attention of the synthetic community, sparking growth in the application of biocatalysis in the synthesis of bioactive compounds.2–7 When a particular biological activity motivates the synthesis of a target molecule, the ability to access individual isomers of the final compound is critical.8 Traditional chemical strategies for selective synthesis can provide tools to arrive at a predicted selectivity outcome. For example, the desired enantiomer can be accessed by employing chiral ligands that can be obtained from commercial sources or synthesized as either enantiomer.9, 10 This strategy is not currently applicable to biocatalytic approaches, as methods for generation of mirror-image proteins are in the early stages of development.11, 12 Protein engineering is commonly employed to address this problem (Figure 1A); however, there is no unified approach for enabling access to diverse selectivity outcomes biocatalytically. Here, we seek to address this drawback by employing an alternative approach to identifying panels of catalysts which deliver complementary selectivity outcomes in a synthetically valuable transformation.
Figure 1.
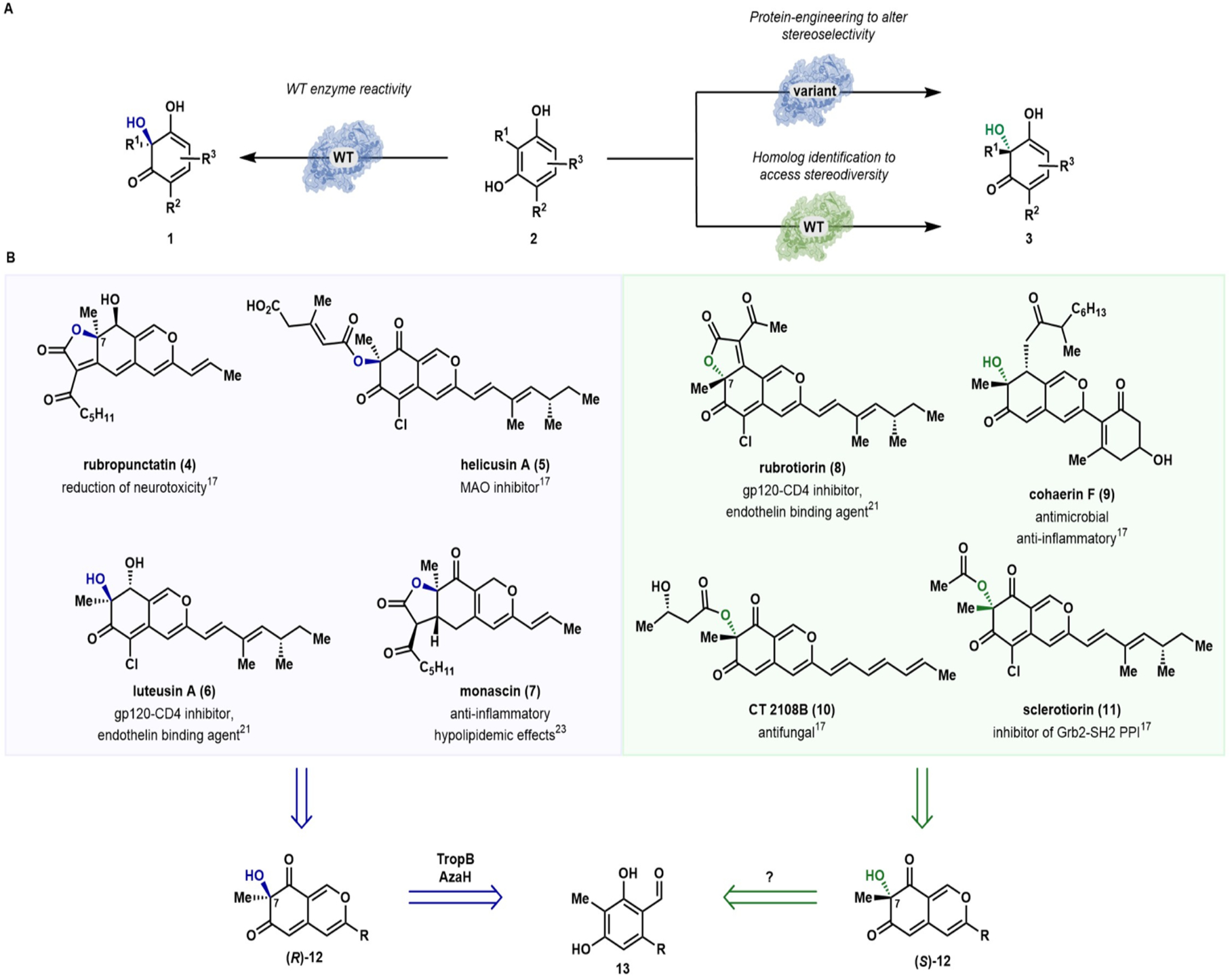
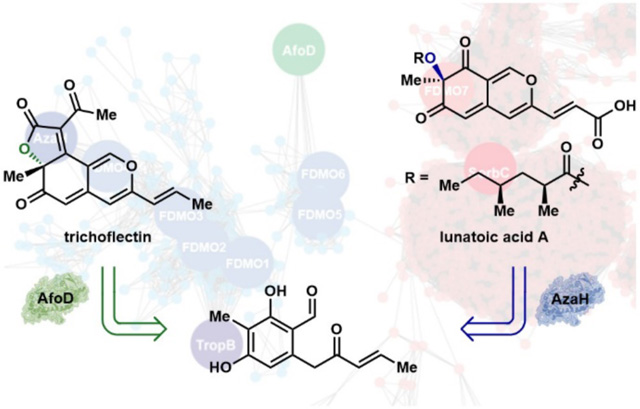
A) Approaches to enabling biocatalytic stereodiversity. B) Biologically active azaphilone natural products feature both the R- and S-configuration at the C7 stereocenter.17,21,23 Biocatalytic oxidative dearomatization gives access to either enantiomer via AzaH or AfoD mediated oxidation.
Previously, we demonstrated the utility of select biocatalysts for site- and stereoselective oxidative dearomatization of resorcinol compounds.13 The flavin-dependent enzymes used in this study provide distinct advantages over small-molecule reagents and catalysts for asymmetric dearomatization, as these biocatalysts are perfectly site-selective and avoid overoxidation that leads to undesired byproducts.13–15 However, the set of biocatalysts reported previously hydroxylate to afford products with only the R-configuration at C3 (see (R)-12, Figure 1B) limiting the potential applications of this method. To access the enantiomeric set of products, we considered two approaches: protein engineering to achieve a switch in stereoselectivity or use of a natural enzyme with complementary selectivity.
Over the last decade, there has been exponential growth in the number of annotated protein sequences available, due in part to the continuing decrease in the price of sequencing.16 This sequencing revolution coupled with improved bioinformatic tools for predicting enzyme class and function provides an alternative to protein engineering campaigns, making it increasingly possible to identify a natural enzyme that can perform a given biocatalytic reaction with the desired selectivity (Figure 1A). Based on the number of natural products that could biosynthetically arise through enantioselective hydroxylative dearomatization, we anticipated it would be possible to identify enzymes capable of providing the complementary stereoisomer to the flavin-dependent monooxygenases TropB and AzaH employed in our previous study (Figure 1B).13 Once stereocomplementary catalysts have been identified, we envision that this suite of biocatalysts will enable the stereodivergent, chemoenzymatic synthesis of high-value molecules.
To showcase this strategy, we targeted the azaphilone family of natural products. These compounds are characterized by an oxygenated pyranoquinone bicyclic core bearing a single tetrasubstituted carbon (Figure 1B).17 Isolated from fungal sources, this large family of natural products is known to contain a diverse array of structural features that impart a wide range of biological properties including anticancer,18–20 antiviral,21 and anti-inflammatory activities.20, 22 For example, luteusin A (6) and rubrorotiorin (8) were found to inhibit the binding of the HIV surface glycoprotein gp120 to the human CD4 protein, making these natural products potential starting points for the development of therapeutic agents against the virus.21 In addition, monascin (7), a tricyclic azaphilone natural product, has been shown to down-regulate steatohepatitis in a mouse model, indicating that these secondary metabolites have potential as therapeutics for non-alcoholic fatty liver disease.23 As illustrated in Figure 1B, azaphilone natural products can contain either the R- or S-configuration at the C7-position. Several cases of epimeric azaphilones are known, in which each C7-epimer is produced by a distinct fungal source.24, 25 The pharmaceutical potential of these molecules has been demonstrated through initial in vitro and cell-based assays of isolated natural products;26–28 however, to gain a more comprehensive understanding of the therapeutic potential of these molecules, the challenge of constructing the densely functionalized core and C7-configuration for exploration of this stereocenter’s impact on biological activity must be addressed.24, 25, 29
Existing approaches toward azaphilones rely either on the cyclization of linear precursors30 or, more commonly, the oxidative dearomatization of highly substituted resorcinol intermediates.31–33 This biomimetic, dearomative strategy has proven valuable as it allows for the installation of structural complexity through well-established arene functionalization methods prior to the generation of the bicyclic core. Established approaches for oxidative dearomatization of azaphilone precursors have employed oxidants such as hypervalent iodide reagents33–35 or Pb(OAc)4.31 Racemic syntheses of azaphilones have been reported using these reagents; however, achieving high levels of enantioselectivity can be a challenge. Enantioenriched azaphilones have been accessed using Porco and coworkers’ copperoxo (−)-sparteine dearomatization strategy,36 achieving the total synthesis of (−)-mitorubrin with 97% ee,37 as well as that of (+)-sclerotiorin and (+)-8-O-methylsclerotiorinamine from a common precursor obtained in 98% ee.29 This method requires superstoichiometric quantities of both the oxidant and chiral ligand. To date, there has yet to be a report of a highly enantioselective, catalytic oxidative dearomatization using a small- molecule catalyst. Furthermore, the application of asymmetric oxidative dearomatization to the synthesis of tricyclic members of the azaphilone family, such as rubropunctatin (4) and rubrotiorin (8), has yet to be reported. As such, the absolute configuration of many of these natural products, which are typically assigned by analogy, has yet to be confirmed.34, 38
RESULTS AND DISCUSSION
To survey the selectivity of flavin-dependent monooxygenases related by sequence, we constructed a sequence similarity network (SSN) of flavin-dependent monooxygenases (Figure 2A).39–41 SSNs are visual representations of the relatedness of protein sequences, which cluster based on similarity thresholds. This approach can enable the rapid and logical investigation of proteins likely to demonstrate similar reactivity and/or selectivity, dramatically reducing the time and search space required during screening, considerations critical to improving the compatibility of biocatalytic approaches and traditional synthetic strategies. The protein family (Pfam01494) of flavin-dependent monooxygenases includes over 45,000 enzymes, which are included in the full SSN (Supporting Information Figure S4). This data set was truncated by limiting the search to edges possessing an alignment score greater than 110, which returned 1,211 sequences (Figure 2A). We noted that the enzymes we had previously investigated, AzaH, TropB, and SorbC, were each located in distinct groups or “clusters” within the network, and that a fourth tight clustering of sequences formed between the TropB and SorbC clusters containing an enzyme associated with asperfuranone biosynthesis, AfoD.42–44 Previous in vivo studies44 and bioinformatic analysis43 indicated that AfoD is responsible for oxidative dearomatization of an asperfuranone precursor with the same site-selectivity as TropB and AzaH;43 however, the absolute configuration of asperfuranone suggests that AfoD carries out this transformation with the opposite facial selectivity.42
Figure 2.
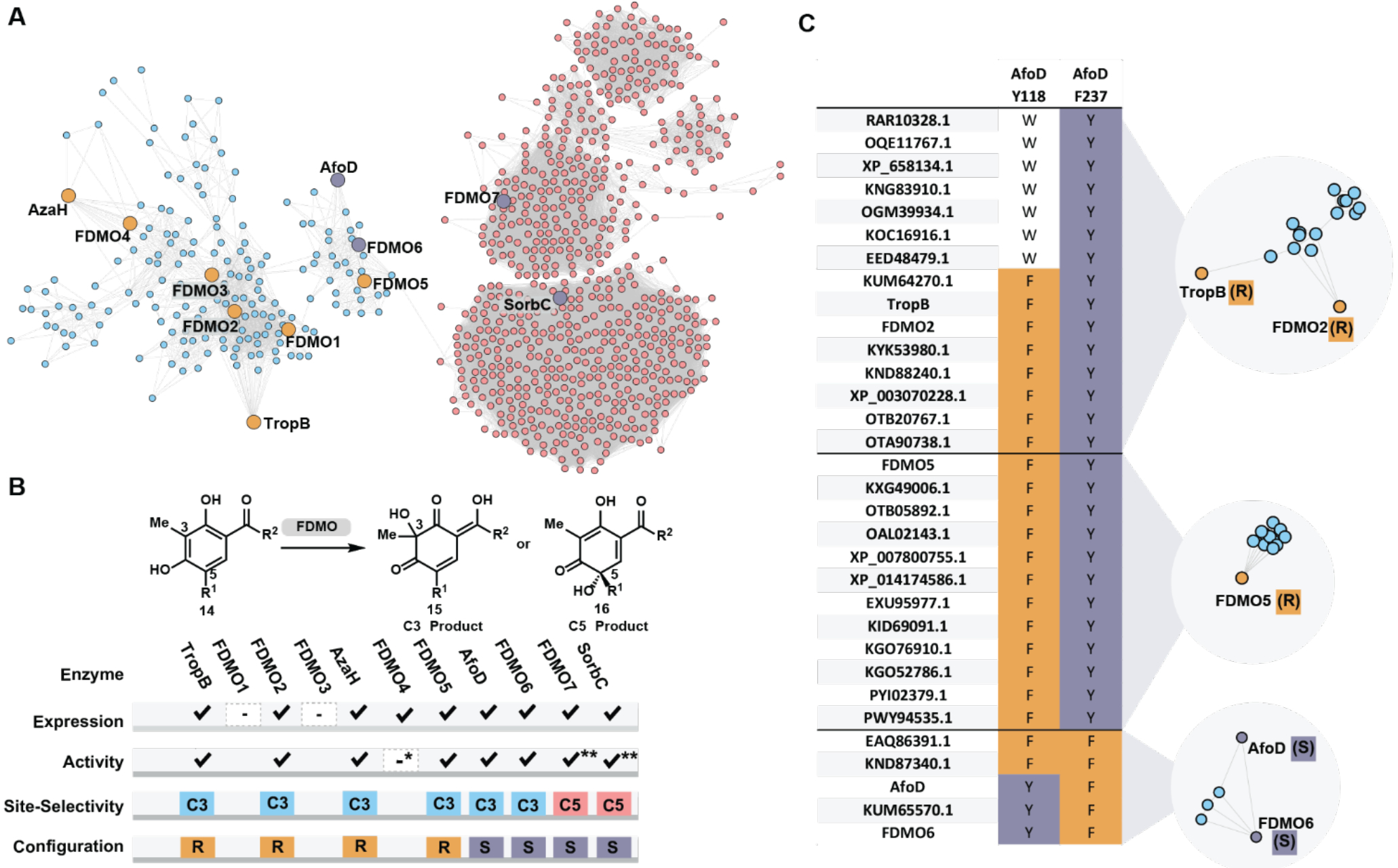
A) Sequence similarity network (SSN) of FAD-dependent monooxygenases (Pfam01494) using a sequence alignment score of 110. B) Results of expression, activity with model substrate S9 or S14**, site- and stereoselectivity of enzymes chosen from the SSN in panel A. *FDMO4 demonstrated <10% conversion by UPLC with substrate S9. C) Selected clusters from a more stringent SSN generated with an alignment score of 150 and corresponding analysis of the multisequence alignment of each cluster.
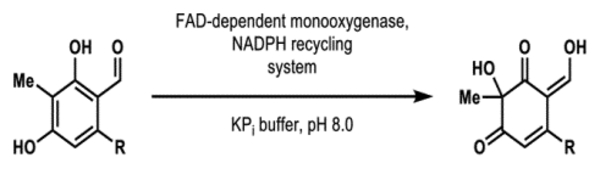
We hypothesized that sequences with high similarity to tropB, azaH, afoD and sorbC would encode for enzymes that possess the ability to carry out oxidative dearomatization reactions, and further questioned if trends in site- and stereoselectivity could be predicted based on sequence. To test this hypothesis, seven sequences were selected based on proximity to either TropB, AzaH, AfoD or SorbC in the SSN (labeled FDMO1–7 in Figure 2A). Synthetic genes corresponding to the four known enzymes and FDMO1–7 were transformed into E. coli BL21(DE3) cells. Under standard expression conditions, nine proteins were successfully obtained, whereas FDMO1 and FDMO3 proved insoluble under these conditions (Figure 2B). The reactivity of the nine soluble enzymes was evaluated with two substrates, S9 and S14. Gratifyingly, all nine enzymes showed activity with one of the two model substrates, eight of which displayed sufficiently high activity for product isolation from preparative-scale reactions (Figure 2B). A single product was obtained from each biocatalytic reaction and a strong trend in site-selectivity was clear from this data: FDMO7, the enzyme most similar to SorbC, afforded C5-hydroxylated product (see 16), whereas the remaining enzymes more similar to TropB, AzaH and AfoD delivered C3-hydroxylated products (see 15). However, the trend between location on the SSN (Figure 2A) and stereoselectivity was not apparent. Increasing the alignment score from 110 to 150 produced an SSN that provided greater insight to the relationships between sequences in this family (see Supporting Information Figure S5 for full SSN). Notably, previously clustered sequences associated with divergent selectivity, such as AfoD and FDMO5, now clearly separated into distinct clusters (see Figure 2C). Analysis of sequence alignments between each of these clusters revealed conserved residues at positons 118 and 237 (AfoD numbering). In general, catalysts that generate products with the R-configuration possess a tyrosine at position 237 and an aromatic residue such as phenyalanine or tryptophan at position 118. In contrast, these conserved residues switch positions in biocatalysts that afford products with the S-configuration. We previously demonstrated that a two point coordination of the phenolate substrate by Tyr237 and an arginine residue is critical for positioning the substrate within the active site in the R-selective enzyme, TropB.45 To probe the role of Tyr118 in the stereoselectivity provided by AfoD, the AfoD Y118F variant was generated. AfoD Y118F was reacted with resorcinol S9 to afford dearomatized product in 53:47 (S:R) e.r. (Supporting Information Figure S16). This data provides additional evidence for the importance of this tyrosine residue in controlling stereoselectivity in these biocatalytic reactions and supports the consideration of this sequence feature in choosing biocatalysts for a specific synthetic application. Obtaining additional structure and function data on these proteins will aid in demystifying the elements that contribute to the precise stereocontrol exhibited by these catalysts. Beyond the importance of Tyr position in the active site, it is clear that other mechanisms for stereocontrol have evolved in this class of enzymes. For example, catalysts AzaH and SorbC break from this Tyr control mechanism, and in the case of SorbC, we have proposed an alternative mechanism for control of substrate position in the active site.47
Ultimately, this survey of sequence space surrounding known enzymes provides a greater understanding of the sequence features that can predict site- and stereoselectivity and has increased the number of biocatalysts vetted for this transformation. With catalysts capable of delivering enantiomeric products in hand, we chose to pursue an enantiodivergent synthetic strategy with AzaH and AfoD based on the robust expression and reactivity of these enzymes, in addition to the excellent stereoselectivity of each catalyst. Under reaction conditions to dearomatize model substrate S9 using AzaH and AfoD, enantiomeric bicycles (R)- and (S)-S10 were accessed in >99% ee and 98% ee, respectively (Supporting Information Figure S16).
With access to either enantiomer of azaphilone S10, we sought to complete the stereodivergent syntheses of two biologically active azaphilone natural products. To identify a common intermediate that would provide versatility in accessing an array of azaphilone natural products (Figure 1B), a number of aromatic substrates were synthesized and subjected to oxidative dearomatization with stereo-complementary biocatalysts. We hypothesized that the orcinaldehyde motif and homobenzylic ketone group would be critical for substrate recognition. In addition, we desired a functional handle to enable late-stage functionalization of the eastern portion of the azaphilone scaffold. Interestingly, the AzaH native substrate was not converted by AfoD (Table 1, entry 1). We hypothesize that the β-hydroxy group prevents the substrate from engaging in productive binding interactions and leads to the lack of activity of AfoD as the native substrate of AfoD features a hydrophobic group at this position.43 This is supported by the activity of the saturated ketone substrate with AfoD (entry 2), which does not contain this polar functional group.
Table 1.
Substrates screened for conversion by AfoD (0.8 mol %) and AzaH (0.8 mol %). *NADPH recycling system: G6P (2 equiv), NADP(+) (0.4 equiv), G6PDH (1 U/mL).
 | |||
|---|---|---|---|
| Entry | R | AfoD % conv. | AzaH % conv. |
| 1 | 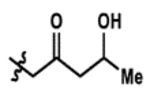 |
0% | 73% |
| 2 | 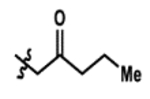 |
24% | 64% |
| 3 | 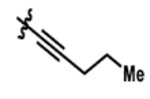 |
0% | 48% |
| 4 | 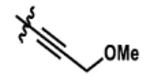 |
0% | >99% |
| 5 | 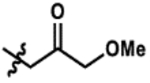 |
19% | >99% |
| 6 | 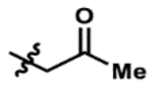 |
19% | 94% |
| 7 | 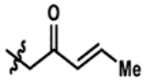 |
83% | 95% |
Promisingly, two alkynyl substrates that are intermediates in the synthesis of the ketone substrates are accepted by AzaH (entries 3 and 4); however, neither alkyne substrate was converted by AfoD. The α-methoxy ketone substrate (entry 5) was initially pursued as a promising intermediate as the dicarbonyl was fully converted by AzaH, but gave low conversions with AfoD. A simple methyl ketone derivative (entry 6) demonstrated activity with both enzymes as well; however, its low activity with AfoD ultimately disqualified it as the common substrate. The enone substrate depicted in entry 7 was efficiently dearomatized by both AfoD and AzaH in 83% and 95% conversion, respectively. The high conversions observed for this compound with both enzymes and the synthetic handle afforded by the double bond made it an ideal candidate for the common intermediate in our envisioned divergent synthesis of azaphilone natural products.
Angular azaphilone, trichoflectin (17), was the first natural product target chosen as a model for route development that would enable the synthesis of more complex tricyclic azaphilones. First isolated by Sterner in 1998, trichoflectin exhibits moderate antimicrobial activity as well as the ability to inhibit DHN-melanin biosynthesis in certain fungal species.27 Despite interest in the biological activity of this compound, no total synthesis of trichoflectin has been reported to date. Our proposed retrosynthesis is described in Figure 3A. We envisioned constructing the butenolide ring of trichoflectin through an intramolecular Knoevenagel condensation following acylation of bicycle 18.34, 38 To obtain the reported (−)-trichoflectin, the C7-stereocenter would be set by stereoselective oxidative dearomatization of enone 19 with AzaH.
Figure 3.
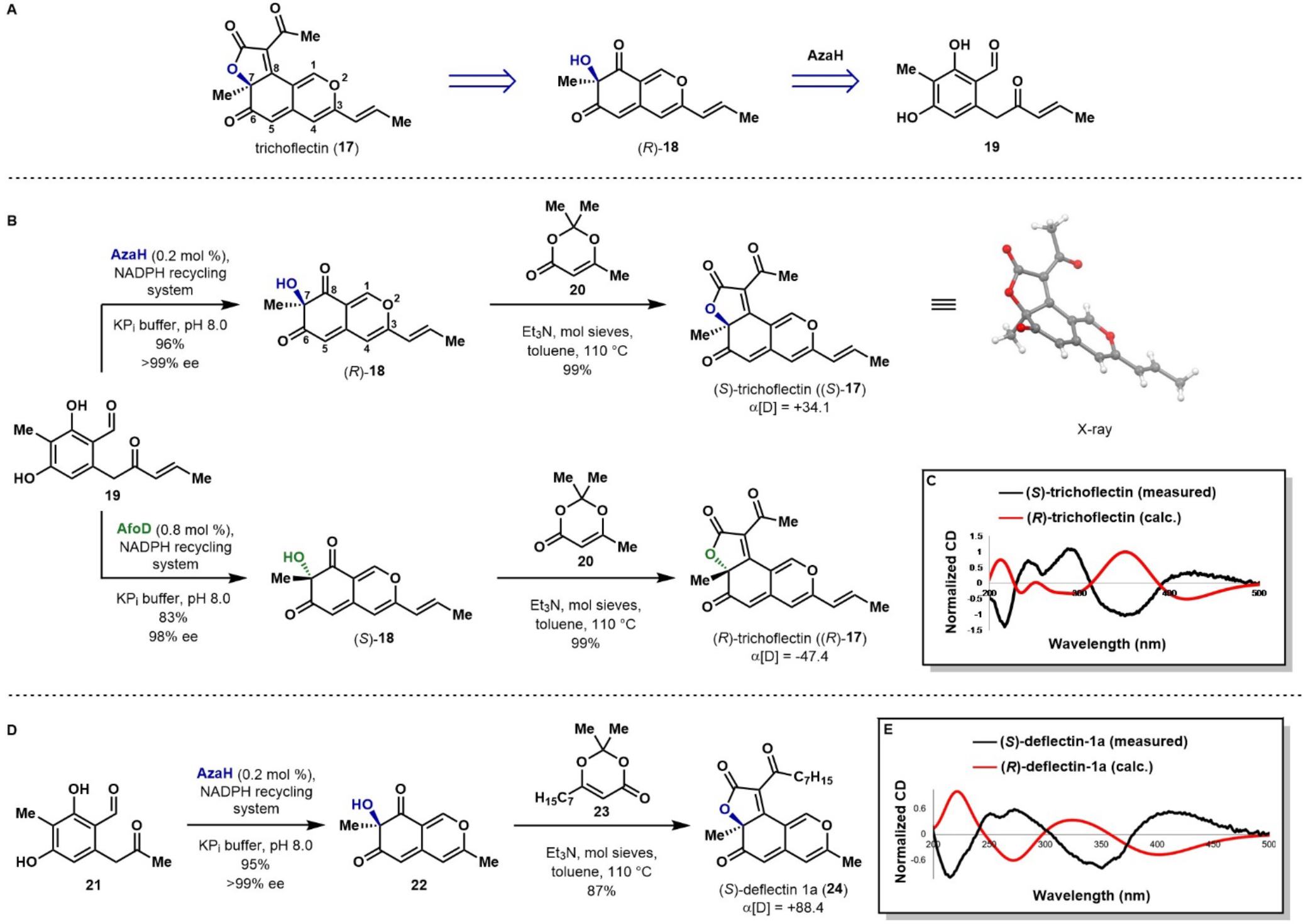
A) Retrosynthetic analysis of the natural product trichoflectin (17). B) Total synthesis of (S)- and (R)-trichoflectin. C) Calculated and measured CD data for (S)- and (R)-trichoflectin. D) Total synthesis of (S)-deflectin-1a (24). E) Calculated and measured CD data for (S)- and (R)-deflectin-1a. *NADPH recycling system: G6P (2 equiv), NADP(+) (0.4 equiv), G6PDH (1 U/mL).
The synthesis of trichoflectin was initiated through a five-step route to enone 19 (see Supporting Information). Dearomatization of 19 with 0.2 mol % AzaH afforded azaphilone scaffold (R)-18 in 96% yield and >99% ee (Figure 3B). Next, acylation of (R)-18 with the acylketene generated in situ from precursor 20 followed by Knoevenagel condensation provided (S)-trichoflectin ((S)-17) in 99% yield. This level of selectivity was not expected as Franck and coworkers reported the isolation of both angular and linear tricycles when constructing the butenolide ring of a similar azaphilone scaffold, proposing the distribution of these products is controlled by the steric properties of the acyl group.38 However, in our system, the desired angular tricycle was produced exclusively. We attribute this to a difference in electronic properties of the extended π-system and have observed through attempts to reduce the C8-ketone that this position is more electrophilic than the C6-ketone.
Unexpectedly, the sign of the optical rotation of our synthetic (S)-17 was opposite to that which is reported for the natural product (measured +34.1, reported −121).27 To rule out the possibility of stereocenter inversion during the construction of the butenolide ring or that the enzyme performed the dearomatization on this particular substrate with unanticipated facial selectivity, a CD spectrum was calculated and compared to the measured spectrum of synthetic (S)-17. As depicted in Figure 3C, the calculated CD spectrum for (R)-trichoflectin is equal and opposite to the measured spectrum from (S)-trichoflectin, initially suggesting that the C7-stereocenter had been installed as anticipated. Ultimately, an X-ray crystal structure was obtained of the AzaH-produced tricycle, unambiguously confirming the C7-configuration of (+)-trichoflectin as S (Figure 3B). Based on this data, we suspected that the natural product was misassigned. A 1976 report, which has been the basis for assignment of absolute configuration in the case of trichoflectin and many other azaphilone natural products, suggests that the optical rotation of these compounds is controlled solely by the C7-stereocenter.48 However, we have found that the electronic properties of the azaphilone core, as well as the presence of other stereocenters in the compound, can have a significant impact on the optical rotation based on computational modeling (see Supporting Information Part X). To confirm the configuration of the natural product as R rather than S as assigned upon isolation, the enantiomeric tricycle was synthesized from the AfoD generated product (S)-18 (Figure 3B). The optical rotation of (R)-17 was measured as −47.4, agreeing in sign with the characterization of the isolated natural product. Therefore, it is proposed that the structure of the natural product be revised to the R-configuration.
This revision prompted a careful inspection of the absolute configuration of structurally related angular tricycles. An azaphilone series called the deflectins, which are reported to possess inhibitory activity of bacteria and erythrocytes as well as cytotoxicity towards carcinoma cells, share the same tricyclic core as trichoflectin (Figure 3D).49 Upon isolation, the absolute C7-configuration was assigned solely by optical rotation, as was done in the case of 17.27 Comparison of the calculated CD spectrum of deflectin-1a to that of trichoflectin showed a good correlation (see Supporting Information Figures S14 and S18), indicating that these compounds should exhibit similar optical properties. The deflectins are reported to possess the R-configuration at C7 and a corresponding optical rotation in the positive direction. This assignment disagrees with the data collected for the trichoflectin enantiomers. Thus, it was deemed likely that this series was also assigned incorrectly.
To answer the question surrounding the absolute configuration of these molecules, we sought to complete the total synthesis of (S)-deflectin-1a (24, Figure 3D). As this compound contains a methyl group in place of the 1,2-disubstituted double bond found in trichoflectin, methyl ketone 21 was dearomatized with AzaH to produce bicycle 22 in 95% yield and >99% ee. Although AfoD also accepts 21 as a substrate (Table 1, entry 6), AzaH was used due to higher turnover and isolated yield in this reaction. Acylation and subsequent Knoevenagel condensation with the acylketene derived from precursor 23 furnished the desired butenolide to deliver (S)-deflectin-1a in 87% yield. The spectral data for 24 matched all available reported values. A measured optical rotation of +88.4 for this S-enantiomer confirms the need for structural revision of the natural product from the R-configuration to S at C7.
With both trichoflectin and deflectin-1a in hand, we set out to construct the natural product lunatoic acid A (25), which was reported to possess the opposite configuration at C7 to the newly established configuration of natural trichoflectin and deflectin-1a. Isolated by Marumo, this natural product exhibited interesting antibacterial properties, as well as the ability to act as an antifungal agent by inducing chlamydospore-like cells in certain fungi.26, 50 As depicted by the retrosynthesis in Figure 4A, we envisioned accessing the carboxylic acid moiety of 25 through a cross metathesis with the C9,10 double bond of intermediate 26.37 We anticipated that the chiral aliphatic ester of 26 could be constructed through an acylation of the C7-hydroxyl group of (R)-18, which would be accessed through dearomatization of common intermediate 19 with AzaH.
Figure 4.
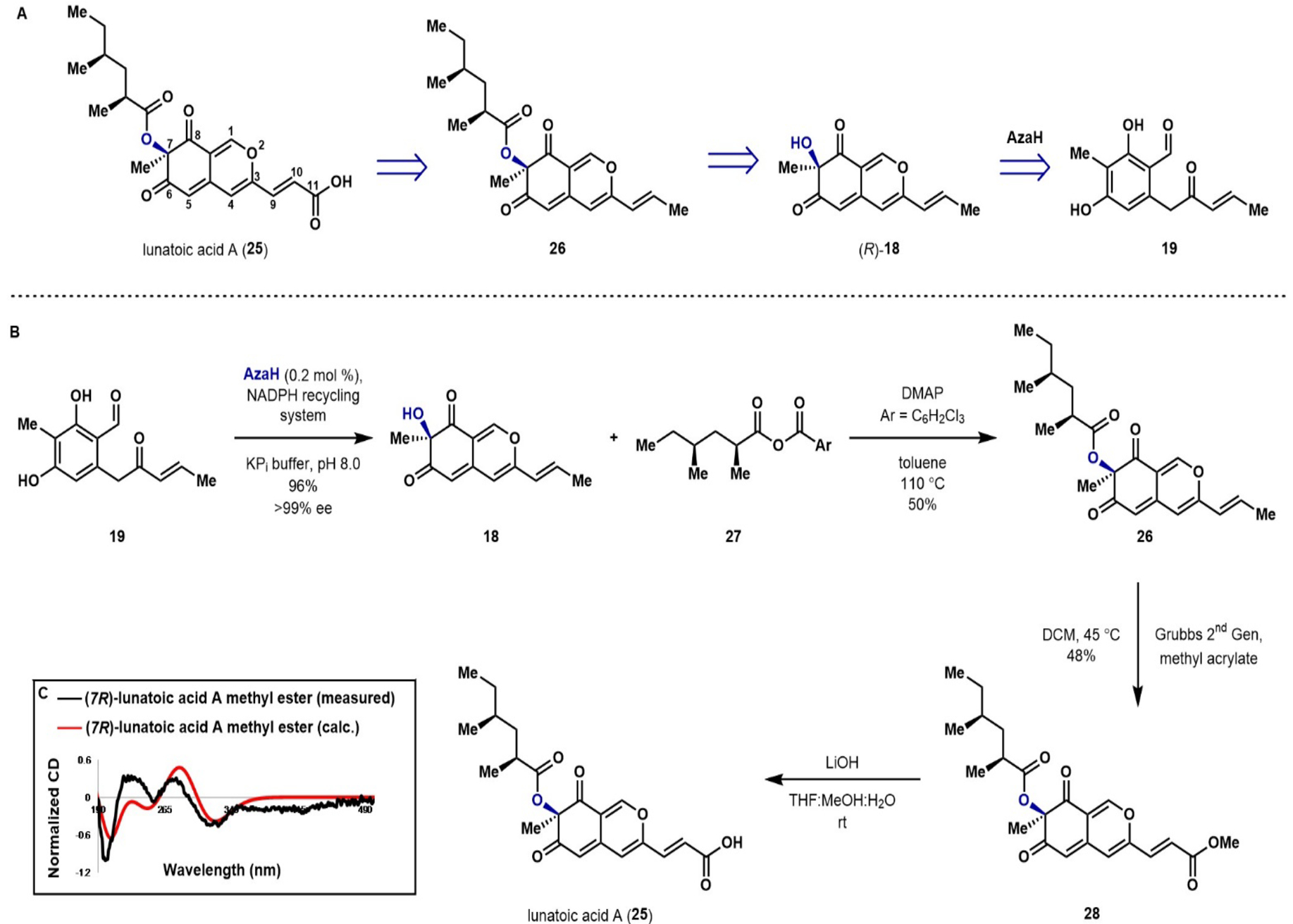
A) Retrosynthetic analysis of azaphilone natural product lunatoic acid A (25). B) Total synthesis of lunatoic acid A (25). C) Calculated and measured CD data for lunatoic acid A methyl ester* (28). *NADPH recycling system: G6P (2 equiv), NADP(+) (0.4 equiv), G6PDH (1 U/mL)
Having established a robust method for biocatalytic dearomatization of enone 19, the first challenge we encountered en route to lunatoic acid A was the acylation of tertiary alcohol 18. While the acylation had proceeded smoothly with the ketene en route to trichoflectin and deflectin-1a, our attempts at acylation of 18 using acyl chlorides or symmetric anhydrides consistently failed to produce chiral ester 26, affording only starting material or under more forcing conditions, complete decomposition of 18. Ultimately, we discovered that Yamaguchi esterification with 27 (Figure 4B) did afford the desired product, although after optimization of reaction conditions, a maximum 50% yield was obtained.51 Mixed anhydride 27 was generated in situ from the corresponding carboxylic acid, prepared as previously reported.52 The carboxylic acid moiety present in lunatoic acid A was appended as the masked methyl ester through olefin metathesis with methyl acrylate and Grubbs 2nd generation catalyst in 48% yield.53 Our attempts to perform the metathesis prior to esterification resulted in decomposition of the starting material, indicating that protection of the C7-hydroxyl group increases the stability of the compound to the reaction conditions. The expected mass of lunatoic acid A was observed by TOF-MS upon saponification of methyl ester 28 using LiOH; however, sufficient quantities of the pure compound for NMR studies could not be obtained. This observed instability parallels the nature of the free acid from culture extracts. These extracts were methylated to obtain lunatoic acid A methyl ester (28), which was fully characterized in lieu of the free acid.26 All spectral data from the fully synthetic 28 match with reported literature values, including an agreement in the sign of the optical rotation and Cotton effects from the reported CD, indicating in this case that the original assignment of R at C7 is accurate.26
CONCLUSIONS
A set of complementary biocatalysts for oxidative dearomatization have been identified and applied to the enantiodivergent, chemoenzymatic synthesis of azaphilone natural products. Comparison of sequences related to characterized enzymes involved in natural product biosynthesis, has provided an expanded set of biocatalysts for this transformation and insight on the sequence features that govern site- and stereoselectivity. A focused substrate library was created to identify a suitable common intermediate for dearomatization by both AzaH and AfoD. These enzymes were then used to construct both enantiomers of scaffold 18 to access the desired natural products containing either the R-or S-configuration at C7. The AzaH-produced scaffold (R)-18 was initially used to access the natural product trichoflectin ((S)-17); however, it was discovered that this tricycle contained the opposite stereocenter from the natural product based on its optical rotation, CD spectral data, and X-ray crystal structure. Synthesis of the opposite enantiomer of trichoflectin ((R)-18) was achieved using the dearomatized scaffold accessed through AfoD, with an optical rotation that led to the reassignment of the C7-stereocenter of trichoflectin to S from the original literature report of R. This prompted the construction of related tricycle deflectin-1a (24) using analogous substrates, which allowed for the reassignment of its C7-stereocenter. Lunatoic acid A (25) was then constructed from (R)-18 and its structure confirmed through CD spectra and optical rotation. We anticipate these methods will enable the rapid construction of libraries of natural and unnatural azaphilones. By utilizing modern tools in bioinformatics, we have rapidly identified homologs with desired properties, an orthogonal approach to protein engineering.
Supplementary Material
ACKNOWLEDGMENTS
This research was supported by funds from the University of Michigan Life Sciences Institute, the University of Michigan Department of Chemistry and the National Institutes of Health R35 GM124880 (A.R.H.N.) and R01 DK042303 (J.L.S.), and the Margaret J. Hunter Professorship (J.L.S.). A.R.B. and S.A.B.D. were supported by the National Institutes of Health Chemistry Biology Interface Training Grant (T32 GM008597). In addition, A. R. B. was supported by a Graduate Assistance of Areas in National Need Training Grant (GAANN P200A150164) and a Rackham Merit Fellowship. The authors thank Professor Yi Tang from the University of California Los Angeles for providing a plasmid containing azaH, Dr. Vikram Shende for his assistance in the synthesis of the enantioenriched carboxylic acid, and Dr. Meagan Hinze for her assistance in performing CD measurements.
Footnotes
Supporting Information. Experimental details, NMR spectra, full compound characterization, UPLC and LCMS traces, details of protein expression and purification, SFC traces, computational and experimental CD data, and crystallographic information. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
REFERENCES
- 1.Devine PN; Howard RM; Kumar R; Thompson MP; Truppo MD; Turner NJ, Extending the application of biocatalysis to meet the challenges of drug development. Nat. Rev. Chem 2018, 2, 409–421. [Google Scholar]
- 2.France SP; Aleku GA; Sharma M; Mangas-Sanchez J; Howard RM; Steflik J; Kumar R; Adams RW; Slabu I; Crook R; Grogan G; Wallace TW; Turner NJ, Biocatalytic Routes to Enantiomerically Enriched Dibenz[c,e]azepines. Angew. Chem. Int. Ed 2017, 56, 15589–15593. [DOI] [PubMed] [Google Scholar]
- 3.Savile CK; Janey JM; Mundorff EC; Moore JC; Tam S; Jarvis WR; Colbeck JC; Krebber A; Fleitz FJ; Brands J; Devine PN; Huisman GW; Hughes GJ, Biocatalytic Asymmetric Synthesis of Chiral Amines from Ketones Applied to Sitagliptin Manufacture. Science 2010, 329, 305–309. [DOI] [PubMed] [Google Scholar]
- 4.Bajaj P; Sreenilayam G; Tyagi V; Fasan R, Gram-Scale Synthesis of Chiral Cyclopropane-Containing Drugs and Drug Precursors with Engineered Myoglobin Catalysts Featuring Complementary Stereoselectivity. Angew. Chem. Int. Ed 2016, 55, 16110–16114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Loskot SA; Romney DK; Arnold FH; Stoltz BM, Enantioselective Total Synthesis of Nigelladine A via Late-Stage C–H Oxidation Enabled by an Engineered P450 Enzyme. J. Am. Chem. Soc 2017, 139, 10196–10199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Truppo MD, Biocatalysis in the Pharmaceutical Industry: The Need for Speed. ACS Med. Chem. Lett 2017, 8, 476–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hernandez KE; Renata H; Lewis RD; Kan SBJ; Zhang C; Forte J; Rozzell D; McIntosh JA; Arnold FH, Highly Stereoselective Biocatalytic Synthesis of Key Cyclopropane Intermediate to Ticagrelor. ACS Catal. 2016, 6, 7810–7813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.FDA’S policy statement for the development of new stereoisomeric drugs. Chirality 1992, 4, 338–340. [DOI] [PubMed] [Google Scholar]
- 9.Beletskaya IP; Nájera C; Yus M, Stereodivergent Catalysis. Chem. Rev 2018, 118, 5080–5200. [DOI] [PubMed] [Google Scholar]
- 10.Liu C; Lin ZW; Zhou ZH; Chen HB, Stereodivergent synthesis of all the four stereoisomers of antidepressant reboxetine. Org. Biomol. Chem 2017, 15, 5395–5401. [DOI] [PubMed] [Google Scholar]
- 11.Weinstock MT; Jacobsen MT; Kay MS, Synthesis and folding of a mirror-image enzyme reveals ambidextrous chaperone activity. Proc. Natl. Acad. Sci. U.S.A 2014, 111, 11679–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vinogradov AA; Evans ED; Pentelute BL, Total synthesis and biochemical characterization of mirror image barnase. Chem. Sci 2015, 6, 2997–3002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Baker Dockrey SA; Lukowski AL; Becker MR; Narayan ARH, Biocatalytic site- and enantioselective oxidative dearomatization of phenols. Nat. Chem 2018, 10, 119–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nicolaou KC; Vassilikogiannakis G; Simonsen KB; Baran PS; Zhong Y-L; Vidali VP; Pitsinos EN; Couladouros EA, Biomimetic Total Synthesis of Bisorbicillinol, Bisorbibutenolide, Trichodimerol, and Designed Analogues of the Bisorbicillinoids. J. Am. Chem. Soc 2000, 122, 3071–3079. [Google Scholar]
- 15.Bosset C; Coffinier R; Peixoto PA; El Assal M; Miqueu K; Sotiropoulos J-M; Pouységu L; Quideau S, Asymmetric Hydroxylative Phenol Dearomatization Promoted by Chiral Binaphthylic and Biphenylic Iodanes. Angew. Chem. Int. Ed 2014, 53, 9860–9864. [DOI] [PubMed] [Google Scholar]
- 16.Gerlt JA, Tools and strategies for discovering novel enzymes and metabolic pathways. Perspect. Sci 2016, 9, 24–32. [Google Scholar]
- 17.a) Stierle AA; Stierle DB; Bugni T Sequoiatones A and B: Novel Antitumor Metabolites Isolated from a Redwood Endophyte. J. Org. Chem 1999, 64, 5479. [DOI] [PubMed] [Google Scholar]; b) Yoshida E; Fujimoto H: Baba M; Yamazaki M Four New Chlorinated Azaphilones, Helicusins A-D, Closely Related to 7-epi-Sclerotiorin, from an Ascomycetous Fungus, Talaromyces helicus. Chem. Pharm. Bull 1995, 43, 1307. [Google Scholar]; c) Quang DN; Stadler M; Fournier J; Tomita A; Hashimoto T Cohaerins C–F, four azaphilones from the xylariaceous fungus Annulohypoxylon cohaerens. Tetrahedron 2006, 62, 6349. [Google Scholar]; d) Laakso JA; Raulli R; McElhaney-Feser GE; Actor P; Underiner T; Hotovec BJ; Mocek U; Cihlar RL; Broedel SE Jr. CT2108A and B: New Fatty Acid Synthase Inhibitors as Antifungal Agents. J. Nat. Prod 2003, 66, 1041. [DOI] [PubMed] [Google Scholar]; e) Nam JY; Son KH; Kim HK; Han MY; Kim SU; Choi JD; Kwon BM Fungal pyranose oxidases: occurrence, properties and biotechnical applications in carbohydrate chemistry. J. Microbiol. Biotechnol 2000, 10, 544. [DOI] [PubMed] [Google Scholar]
- 18.Kaur K; Wu X; Fields JK; Johnson DK; Lan L; Pratt M; Somoza AD; Wang CCC; Karanicolas J; Oakley BR; Xu L; De Guzman RN, The fungal natural product azaphilone-9 binds to HuR and inhibits HuR-RNA interaction in vitro. PLOS ONE 2017, 12, e0175471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang W; Liao Y; Chen R; Hou Y; Ke W; Zhang B; Gao M; Shao Z; Chen J; Li F, Chlorinated Azaphilone Pigments with Antimicrobial and Cytotoxic Activities Isolated from the Deep Sea Derived Fungus Chaetomium sp. NA-S01-R1. Mar. Drugs 2018, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yasukawa K; Takahashi M; Natori S; Kawai K. i.; Yamazaki M; Takeuchi M; Takido M, Azaphilones Inhibit Tumor Promotion by 12-O-Tetradecanoylphorbol-13-Acetate in Two-Stage Carcinogenesis in Mice. Oncology 1994, 51, 108–112. [DOI] [PubMed] [Google Scholar]
- 21.Matsuzaki KTH, Inokoshi J, Tanaka H, Masuma R,; O. S, New brominated and halogen-less derivatives and structure-activity relationship of azaphilones inhibiting gp120-CD4 binding. J. Antibiot 1998, 51, 1004–1011. [DOI] [PubMed] [Google Scholar]
- 22.Tang J-L; Zhou Z-Y; Yang T; Yao C; Wu L-W; Li G-Y, Azaphilone Alkaloids with Anti-inflammatory Activity from Fungus Penicillium sclerotiorum cib-411. J. Agric. Food Chem 2019, 67, 2175–2182. [DOI] [PubMed] [Google Scholar]
- 23.Hsu W-H; Chen T-H; Lee B-H; Hsu Y-W; Pan T-M Monascin and ankaflavin act as natural AMPK activators with PPARα agonist activity to down-regulate nonalcoholic steatohepatitis in high-fat diet-fed C57BL/6 mice. Food Chem. Toxicol 2014, 64, 94–103. [DOI] [PubMed] [Google Scholar]
- 24.Udagawa S, (−)-Sclerotiorin, A Major Metabolite of Penicillium hirayamae. Chem. Pharm. Bull 1963, 11, 366–367. [Google Scholar]
- 25.Whalley WB; Ferguson G; Marsh WC; Restivo RJ, The chemistry of fungi. Part LXVIII. The absolute configuration of (+)-sclerotiorin and of the azaphilones. J. Chem. Soc., Perkin Trans. 1 1976, 13, 1366–1369. [Google Scholar]
- 26.Nukina M; Marumo S, Lunatoic acid A and B, aversion factor and its related metabolite of cochliobolus lunata. Tetrahedron Lett 1977, 18, 2603–2606. [Google Scholar]
- 27.Thines E; Anke H; Sterner O, Trichoflectin, a Bioactive Azaphilone from the Ascomycete Trichopezizella nidulus. J. Nat. Prod 1998, 61, 306–308. [DOI] [PubMed] [Google Scholar]
- 28.Park J-H; Choi GJ; Jang KS; Lim HK; Kim HT; Cho KY; Kim J-C, Antifungal activity against plant pathogenic fungi of chaetoviridins isolated from Chaetomium globosum. FEMS Microbiol. Lett 2005, 252, 309–313. [DOI] [PubMed] [Google Scholar]
- 29.Germain AR; Bruggemeyer DM; Zhu J; Genet C; O’Brien P; Porco JA, Synthesis of the azaphilones (+)-sclerotiorin and (+)-8-O-methylsclerotiorinamine utilizing (+)-sparteine surrogates in copper-mediated oxidative dearomatization. J. Org. Chem 2011, 76, 2577–2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stark LM; Pekari K; Sorensen EJ, A nucleophile-catalyzed cycloisomerization permits a concise synthesis of (+)-harziphilone. Proc. Natl. Acad. Sci. U.S.A 2004, 101, 12064–12066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chong R; Gray RW; King RR; Whalley WB, The synthesis of (±) mitorubrin. J. Chem. Soc. D 1970, 2, 101a–101a. [DOI] [PubMed] [Google Scholar]
- 32.Marsini MA; Gowin KM; Pettus TRR, Total Synthesis of (±)-Mitorubrinic Acid. Org. Lett 2006, 8, 3481–3483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhu J; Germain AR; Porco JA Jr., Synthesis of Azaphilones and Related Molecules by Employing Cycloisomerization of o-Alkynylbenzaldehydes. Angew. Chem. Int. Ed 2004, 43, 1239–1243. [DOI] [PubMed] [Google Scholar]
- 34.Makrerougras M; Coffinier R; Oger S; Chevalier A; Sabot C; Franck X, Total Synthesis and Structural Revision of Chaetoviridins A. Org. Lett 2017, 19, 4146–4149. [DOI] [PubMed] [Google Scholar]
- 35.Kang H; Torruellas C; Liu J; Kozlowski MC, Total Synthesis of Chaetoglobin A via Catalytic, Atroposelective Oxidative Phenol Coupling. Org. Lett 2018, 20, 5554–5558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhu J; Grigoriadis NP; Lee JP; Porco JA, Synthesis of the Azaphilones Using Copper-Mediated Enantioselective Oxidative Dearomatization. J. Am. Chem. Soc 2005, 127, 9342–9343. [DOI] [PubMed] [Google Scholar]
- 37.Zhu J; Porco JA, Asymmetric Syntheses of (−)-Mitorubrin and Related Azaphilone Natural Products. Org. Lett 2006, 8, 5169–5171. [DOI] [PubMed] [Google Scholar]
- 38.Peixoto PA; Boulangé A; Ball M; Naudin B; Alle T; Cosette P; Karuso P; Franck X, Design and Synthesis of Epicocconone Analogues with Improved Fluorescence Properties. J. Am. Chem. Soc 2014, 136, 15248–15256. [DOI] [PubMed] [Google Scholar]
- 39.Zallot R; Oberg NO; Gerlt JA, ‘Democratized’ genomic enzymology web tools for functional assignment. Curr. Opin. Chem. Biol 2018, 47, 77–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gerlt JA, Genomic Enzymology: Web Tools for Leveraging Protein Family Sequence–Function Space and Genome Context to Discover Novel Functions. Biochemistry 2017, 56, 4293–4308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gerlt JA; Bouvier JT; Davidson DB; Imker HJ; Sadkhin B; Slater DR; Whalen KL, Enzyme Function Initiative-Enzyme Similarity Tool (EFI-EST): A web tool for generating protein sequence similarity networks. Biochim. Biophys. Acta. Proteins Proteom 2015, 1854, 1019–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chiang Y-M; Szewczyk E; Davidson AD; Keller N; Oakley BR; Wang CCC, A Gene Cluster Containing Two Fungal Polyketide Synthases Encodes the Biosynthetic Pathway for a Polyketide, Asperfuranone, in Aspergillus nidulans. J. Am. Chem. Soc 2009, 131, 2965–2970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Davison J; al Fahad A; Cai M; Song Z; Yehia SY; Lazarus CM; Bailey AM; Simpson TJ; Cox RJ, Genetic, molecular, and biochemical basis of fungal tropolone biosynthesis. Proc. Natl. Acad. Sci 2012, 109, 7642–7647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Somoza AD; Lee K-H; Chiang Y-M; Oakley BR; Wang CCC, Reengineering an azaphilone biosynthesis pathway in Aspergillus nidulans to create lipoxygenase inhibitors. Org. Lett 2012, 14, 972–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rodríguez Benítez A; Tweedy SE; Baker Dockrey SA; Lukowski AL; Wymore T; Khare D; Brooks CL; Palfey BA; Smith JL; Narayan ARH, Structural Basis for Selectivity in Flavin-Dependent Monooxygenase-Catalyzed Oxidative Dearomatization. ACS Catal. 2019, 9, 3633–3640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kelley LA; Mezulis S; Yates CM; Wass MN; Sternberg MJE, The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc 2015, 10, 845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dockrey SAB; Suh CE; Benítez AR; Wymore T; Brooks CL; Narayan ARH, Positioning-Group-Enabled Biocatalytic Oxidative Dearomatization. ACS Cent. Sci 2019, 5, 1010–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Steyn PS; Vleggaar R, The structure of dihydrodeoxy-8-epi-austdiol and the absolute configuration of the azaphilones. J. Chem. Soc., Perkin Trans. 1 1976, 2, 204–206. [PubMed] [Google Scholar]
- 49.Anke H; Kemmer T; Höfle G, Deflectins, new antimicrobial azaphilones from Aspergillus deflectus. J. Antiobiot 1981, 34, 923–928. [DOI] [PubMed] [Google Scholar]
- 50.Marumo S; Nukina M; Kondo S; Tomiyama K, Lunatoic Acid A, a Morphogenic Substance Inducing Chlamydospore-like Cells in Some Fungi. Agric. Biol. Chem, 1982, 46, 2399–2401. [Google Scholar]
- 51.Kawanami Y; Ito Y; Kitagawa T; Taniguchi Y; Katsuki T; Yamaguchi M, Asymmetric alkylation of carboxyamides by using trans2,5-disubstituted pyrrolidines as chiral auxiliaries. Tetrahedron Lett 1984, 25, 857–860. [Google Scholar]
- 52.Myers AG; Yang BH; Chen H; McKinstry L; Kopecky DJ; Gleason JL, Pseudoephedrine as a Practical Chiral Auxiliary for the Synthesis of Highly Enantiomerically Enriched Carboxylic Acids, Alcohols, Aldehydes, and Ketones. J. Am. Chem. Soc 1997, 119, 6496–6511. [Google Scholar]
- 53.Blackwell HE; O’Leary DJ; Chatterjee AK; Washenfelder RA; Bussmann DA; Grubbs RH, New Approaches to Olefin Cross-Metathesis. J. Am. Chem. Soc 2000, 122, 58–71. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.