

Abstract
Scope:
We previously showed that two hydrogenated xanthohumol (XN) derivatives, α,β-dihydro-XN (DXN) and tetrahydro-XN (TXN), improved parameters of metabolic syndrome (MetS), a critical risk factor of cardiovascular disease (CVD) and type 2 diabetes, in a diet-induced obese murine model. We hypothesized that improvements in obesity and MetS are linked to changes in the composition of the gut microbiota, bile acid metabolism, intestinal barrier function and inflammation.
Methods and results:
To test this hypothesis, we sequenced 16S rRNA genes and measured bile acids in fecal samples from C57BL/6J mice fed a high-fat diet (HFD) or HFD containing XN, DXN or TXN. We measured the expression of genes associated with epithelial barrier function, inflammation, and bile acid metabolism, in the colon, white adipose tissue (WAT), and liver, respectively. Administration of XN derivatives decreased intestinal microbiota diversity and abundance, specifically Bacteroidetes and Tenericutes, altered bile acid metabolism, and reduced inflammation. In WAT, TXN supplementation decreased pro-inflammatory gene expression by suppressing macrophage infiltration. Transkingdom network analysis connected changes in the microbiota to improvements in MetS in the host.
Conclusion:
Changes in the gut microbiota and bile acid metabolism may explain, in part, the improvements in obesity and MetS associated with administration of XN and its derivatives.
Keywords: bile acid, gut microbiota, metabolic syndrome, xanthohumol
Graphic Abstract
Xanthohumol (XN) is a prenylated flavonoid isolated from hops. Prior studies show XN derivatives reduce diet-induced weight gain, improve glucose tolerance and inhibit accumulation of triglycerides and inflammation in the liver in mouse models of obesity. The present findings demonstrate that XN and its derivatives improve obesity and metabolic syndrome, in part, by changing gut microbiota and bile acid metabolism.
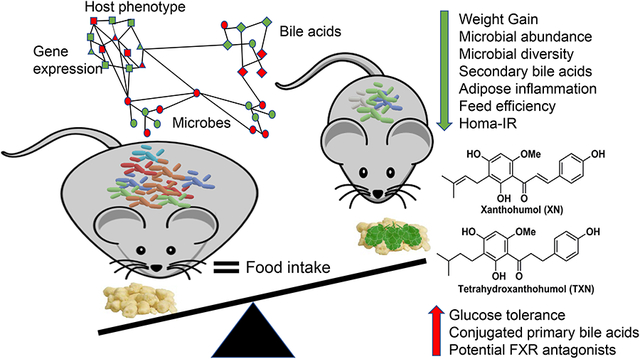
1. Introduction
Administration of xanthohumol (XN), a prenylated flavonoid found in hops (Humulus lupulus L.), improves several parameters of obesity and metabolic syndrome (MetS) in animal models[1–5]. Obesity alone affects more than 107.7 million children and 603.7 million adults worldwide[6]. It costs society about $2 trillion every year[7]. There are concerns with treating individuals with high doses of XN, because gut microbes can metabolize XN into a potent phytoestrogen, 8-prenylnaringenin (8-PN)[8]. To address these concerns, we developed two hydrogenated XN derivatives, α, β-dihydro-XN (DXN), a minor constituent of hops and gut microbe metabolite[9–10] and tetrahydro-XN (TXN), a synthetic, natural product derivative[11]. These two derivatives cannot be metabolically converted into 8-PN due to lack of an α, β-unsaturated keto moiety required for intramolecular cyclization, and that have weak affinity for estrogen receptors[1]. We showed in a prior study that administering XN, DXN or TXN at 30 mg/kg body weight (BW)/day for 13 weeks in the diet decreased weight gain and improved glucose and lipid homeostasis in a preclinical MetS and diet-induced obesity (DIO) mouse model[1].
Numerous studies indicate that gut microbiota contribute to obesity, MetS, and CVD[15–16]. One hypothesis is that intestinal microbiota promote DIO and its associated complications through altering bile acid composition and the farnesoid-X-receptor (FXR) pathway[14]. In addition to their classical role as detergents in the digestion of lipids and fat, both primary and secondary bile acids act as natural endogenous ligands for various host nuclear (FXR, VDR, PXR) and G protein-coupled receptors (TGR-5, S1PR2)[15–16]. Others and we previously showed that XN could function as a ligand for FXR, a nuclear receptor that regulates gluconeogenesis and de novo lipogenesis[17–18].
Based on these findings, we hypothesized that DXN and TXN supplementation improves DIO through changes in the intestinal microbiota, bile acid metabolism, intestinal barrier function and inflammation. To test our hypothesis, we sequenced the16S rRNA genes and determined bile acid profiles of mouse fecal samples from our prior study[1]. We also measured mRNA expression of genes associated with pro-inflammatory cytokine production, bile acid metabolism, and intestinal barrier function in the liver, white adipose tissue (WAT), and colon. To link MetS criteria to the fecal microbiota and bile acid composition, we integrated microbiota, host phenotypic features, and gene expression data using transkingdom network analysis[19]. Our results demonstrate that XN and its derivatives affect microbiota composition, bile acid metabolism, and highlights a potential mechanism for their role in improving in obesity and MetS.
2. Experimental Section
2.1. Animals, diets and experimental design
C57BL/6J male mice were fed a high-fat diet (HFD, control, 60% kcal from fat) or a HFD containing XN or one of its two non-estrogenic derivatives, DXN and TXN for 13 weeks at a dose of 30 mg/kg body weight (BW)/day as described previously[1]. In brief, all animal experiments were performed in accordance with the relevant guidelines and regulations of NIH and were approved by Institutional Animal Care and Use Committee (IACUC) at Oregon State University (protocol # 4501). Male C57BL/6J mice were purchased from The Jackson Laboratory (Bar Harbor, ME, USA) and at nine weeks of age, they were distributed into four groups of 12 animals each as described previously[1]. All animals were housed individually in plastic cages under a 12–12-hr light-dark cycle. Group 1 (control) was fed a high-fat diet (HFD, 60 % kcal from fat, 20% kcal from carbohydrate and 20% kcal from protein) whereas the test groups were fed a HFD containing XN, DXN or TXN as described above. The test compounds were first dissolved in OPT (oleic acid:propylene glycol:Tween 80, 0.9:1:1 by weight) before mixing with the diet. The diets were prepared in pellet form by Dyets, Inc. (Bethlehem, PA, USA). Weekly food intake and body weights were recorded for 13 weeks. Heparinized blood was collected by cardiac puncture under anesthesia. The mice were euthanized by cervical dislocation followed by collection of liver, skeletal muscle and other tissues for analyses.
2.2. Tissue RNA preparation and gene expression analysis
Tissue RNAs were isolated using Direct-zol RNA kits according to manufacturer’s protocol (Zymo Research, Irvine, CA). Tissues were homogenized using nuclease-free 1.6-mm stainless steel beads in a Precellys24 homogenizer (Bertin Corp., MD, USA). 0.25 μg RNA was converted into cDNA using iScript reverse transcriptase and random hexamer primers (Bio-Rad Laboratories, CA, USA). Gene expression was determined by qRT-PCR using SsoAdvanced Universal Probes Supermix (Bio-Rad Laboratories). The primers and probes used in this study were purchased from IDT (Integrated DNA Technologies, IA, USA) and are listed in Supporting Information Table S1. All the threshold cycle (Ct) numbers were normalized to the reference gene Ywhaz.
2.3. Fecal DNA isolation and 16S amplicon sequencing
Fresh fecal pellets were collected from each mouse, frozen in liquid nitrogen and stored at −80°C at the end of the feeding study. DNA was extracted from 2–3 fecal pellets using the PowerFecal DNA Isolation Kit (MoBio, Carlsbad, CA, USA) per the manufacturer’s protocol. Using the same amount of fecal genomic DNA, amplification of 16S rRNA, library preparation and sequencing were performed according to established methods[21].
2.4. Processing of 16S rRNA sequence data
For identification of the presence and abundance of gut microbial taxa, we analyzed microbial communities with the DADA2 1.2 pipeline[22]. Taxonomy was assigned using the Ribosomal Database Project’s Training Set 16 and the 11.5 release of the RDP database, after building the ASV table and removing chimeras[23].
2.5. Data visualization and statistical analyses of 16S rRNA sequence data
Phyloseq (v1.24.2)[24] and ggplot2 (v3.0.0) were used to visualize community composition at both phylum and family levels[25], with taxa above 0.1 and 0.3% for phylum and family filtered respectively, for better visualization. ASV that were not assigned taxonomy to the corresponding levels were also removed for plotting purposes. Changes in the community composition at the taxa level were investigated with DESeq2 (v1.20.0)[26].
2.6. Beta diversity
R package phyloseq was used to calculate ordinations and conduct Principal Coordinate Analysis (PCoA)[27]. To test the effect of supplementation with different xanthohumol compounds in a HFD on group differences based on Bray-Curtis distance, a permutation analysis of variance (PERMANOVA) and the adonis function were used in the vegan package[28].
2.7. Fecal metabolite extraction and analysis
Lyophilized feces were placed in a 2 mL reinforced screw-top tube with ten 2 mm silica beads. Deuterated chenodeoxycholic acid (CDCA-d4) was added as an internal standard at a final concentration of 10 μM with 110 μL of cold ethanol: methanol (1:1, v/v). Samples were homogenized at 6.5K rpm, 2 × 45 seconds, then centrifuged 10 minutes at 10K rpm at 4°C. The supernatant was used to repeat the extraction with 80 μL of cold ethanol: methanol (1:1, v/v). A quality control (QC) sample was prepared by pooling 5 μL of each fecal sample to monitor suitability, repeatability and stability of the LC-MS system. Bile acid standards were weighed and dissolved in cold ethanol: methanol (1:1, v/v) at a final concentration of 5 μM.
Samples were analyzed using a Waters ACQUITY UPLC I class system coupled to a Waters Synapt G2 HDMS mass spectrometer. Chromatographic separation was performed on an ACQUITY UPLC HSS T3 (C18) column (2.1 × 150 mm, 100Å 1.8 μm, Waters Corporation, Milford, MA, USA), the column temperature was held at 45°C. The total run time was 20 minutes at a flow rate of 0.45 mL/min. The solvent system consisted of a mobile phase A (0.1% HCOOH in water), and phase B (0.1% HCOOH in CH3CN). The run started at 1% B, increased to 36% B at 3 min; then linearly increased to 85% at 12 min; from 12 min to 15 min, B was increased to 99% and held at 99% until 16 min; then rapidly decreased from 99% to 1% in one minute and held at 1% B for the last three minutes.
Acquisition of MS and MS/MS (MSE) data were recorded in negative ionization mode. Voltages of the electro spray capillary, sampling cone, and extraction cone were 2800 V, 35 V, and 4.5 V, respectively. The source and desolvation temperatures were 120°C and 600°C, respectively. The desolvation and cone gas flows were 550 L/hr and 45 L/hr. MSE mode was used with a low energy acquisition set at 4 eV and a high energy acquisition ramping from 20 V to 50 V to induce collision induced dissociation. A scan range of m/z 50–1200 was used. A QC sample was injected every five injections to evaluate chromatographic reproducibility and platform stability over time.
Data was processed using XCMS (https://xcmsonline.scripps.edu/). Chromatograms were aligned using the obiwarp method and the features were extracted using the centWave method at a signal/noise ratio of 3. Selected features were then normalized by the feces dry weight and internal standard. Bile acids were identified by matching their retention time, isotopic pattern, accurate mass of the [M-H]– ion and fragmentation pattern with those of authentic commercial standards. The area of the base peak extracted ion chromatogram was used for relative quantitation.
2.8. Transkingdom network analysis
Microbial abundance data was cumulative sum scaling transformed and quantile normalized[29]. An element (microbe, bile acid, gene expression, and phenotype) was considered differentially abundant due to treatment (DXN, TXN) compared to control (HFD) if it had the same direction of fold change in both comparisons (DXN vs HFD, TXN vs HFD) and t-test FDR < 15% in at least one comparison. Meta-analysis of Spearman’s correlations in DXN and TXN was used to identify connections between differentially abundant elements[19]. For within data type pairs, an edge was considered significant if it passed the principles of causality[30], had the same sign of correlation with a p-value < 30% in DXN and TXN, Fisher p-value < 5%, and FDR < 15%. The microbial network showed four distinct sub-networks when visualized in Cytoscape. For edges between different data types, FDR was calculated separately per bile acid type and microbial subnetwork, and a per-treatment correlation p-value cutoff was not applied. Bipartite betweenness centrality analysis was used to identify key microbes connecting a microbial network to bile acids, genes and phenotypes[31].
2.9. Statistical Analysis
Analysis of variance procedures for continuous data and Fisher’s exact test for binary data were used for statistical comparisons. P-values of orthogonal a priori comparisons of the HFD control group vs. each of the supplement groups are shown in the corresponding tables and figures. Additional details of statistical analyses are described in the corresponding figure legends.
3. Results
3.1. DXN and TXN decrease fecal microbial counts and diversity compared to mice on a high-fat diet
To determine the effect of treating mice with XN and its derivatives on composition of the fecal microbiota, we characterized microbial abundance and diversity of all mice that completed the study (47 of 48 mice) by sequencing the V3–V4 region of the 16S rRNA gene (Supporting Information Figure S1). We obtained 3,013,231 forward reads with an average read length of 250 base pairs. The median sequencing depth per sample was 48,446 reads. We excluded the reverse reads due to their low quality and analyzed the forward reads. From a total of 1619 amplicon sequence variant (ASV) with ≥2 reads (Control [HFD]: 1080, XN: 1145, DXN: 1064, TXN: 942), 1364 (Control: 945, XN: 996, DXN: 918, TXN: 748) were annotated as bacterial. Of the 1364 reads, 339 (Control: 325, XN: 325, DXN: 290, TXN: 273) comprised ≥ 0.5% of the counts, and 33 ASV were present in all mice. Supplementary File 2 shows the annotated ASV.
In Table 2, we report fecal microbial counts to show the effect of supplementation on the microbiota. The counts used in our analyses reflect the rate at which specific taxa were sampled during the DNA sequencing process. While 16S rRNA gene sequencing affords powerful insight into the composition of the gut microbiome, it cannot offer direct insight into the absolute abundance of microbiota. Rather, the DNA sequencing process samples a finite set of molecules from the total population of 16S rRNA amplicons and sequences this set. The counts produced, provide insight into the number of times a 16S rRNA molecule associated with a particular taxon was sampled. As a result, these counts afford insight into the frequency at which this taxon was detected in our data, and consequently into the abundance of the taxon relative to other taxa in the community (as opposed to the taxon’s absolute abundance). We also confirmed that correcting for read depth, non-normal relative abundance distributions, and false discovery rate generally did not affect these results (data not shown). The effect of supplementation on microbial diversity (ASV number) is shown in Table 3. The effect of supplementation on relative abundance of surviving microbial ASV (counts/ASV number), a.k.a. microbial abundance, is shown in Table 4. Phyla and families within phyla are organized in the tables in the order of microbial number.
Table 2.
Supplementation with 30 mg/kg BW/day XN or its derivatives DXN and TXN decreases the relative fecal abundancea
| CON | XN | DXN | TXN | SEM | XN vs CON | DXN vs CON | TXN vs CON | |
|---|---|---|---|---|---|---|---|---|
| Microbial Number (Counts/DNA) | P-values (Comparisons) | |||||||
| Total | 48,896 | 46,062 | 39,634 | 33,259 | 2,683 | 0.42 | 0.01 | <0.001 |
| Firmicutes | 27,397 | 26,676 | 28,295 | 25,034 | 2,555 | 0.84 | 0.80 | 0.51 |
| Lachnospiraceae | 17,737 | 18,185 | 19,467 | 17,125 | 1871 | 0.86 | 0.50 | 0.81 |
| Ruminococcaceae | 5,696 | 7,237 | 6,797 | 6,715 | 761 | 0.14 | 0.29 | 0.34 |
| Erysipelotrichaceae | 406 | 500 | 1,172 | 1,064 | 161 | 0.67 | 0.001 | 0.005 |
| Eubacteriaceae | 3,087 | 339 | 62 | 54 | 180 | <0.001 | <0.001 | <0.001 |
| Peptostreptococcaceae | 276 | 272 | 217 | 44 | 78 | 0.97 | 0.58 | 0.04 |
| Clostridiaceae | 19.9 | 0.8 | 0.3 | 0.1 | 3.7 | <0.001 | <0.001 | <0.001 |
| Halobacteroidacea | 102.4 | 73.0 | 0.3 | 0 | 8.4 | 0.01 | <0.001 | <0.001 |
| Lactobacillaceae | 15 | 4 | 1 | 1 | 3 | 0.01 | 0.002 | 0.002 |
| Paenibacillaceae | 0.1 | 0.1 | 1.5 | 1.2 | 0.3 | 0.05 | <0.001 | 0.007 |
| Natranaerovirga | 0.1 | 0 | 545.1 | 1.2 | 287.1 | 1 | 0.17 | 1 |
| Enterococcaceae | 5.0 | 1.3 | 0.4 | 2.7 | 1.4 | 0.06 | 0.02 | 0.24 |
| Staphylococcaceae | 2.3 | 1.8 | .3 | 0.4 | 0.6 | 0.49 | 0.24 | 0.03 |
| Bacillaceae | 0.7 | 1.3 | 1.8 | 0.9 | 0.6 | 0.41 | 0.15 | 0.77 |
| Bacteroidetes | 17,887 | 16,192 | 2,375 | 590 | 1,330 | 0.35 | <0.001 | <0.001 |
| Porphyromonadaceae | 17,838 | 16,154 | 2,367 | 584 | 1,328 | 0.35 | <0.001 | <0.001 |
| Flavobacteriaceae | 2.6 | 3.1 | 4.9 | 3.5 | 1.2 | 0.76 | 0.16 | 0.60 |
| Chitinophagaceae | 1.4 | 1.3 | 2.4 | 1.6 | 0.5 | 0.81 | 0.15 | 0.76 |
| Rikenellaceae | 2.6 | 1.9 | 0.5 | 0.3 | 0.6 | 0.42 | 0.01 | 0.008 |
| Verrucomicrobia | 3,534 | 3,061 | 8,824 | 7,576 | 2,833 | 0.90 | 0.18 | 0.31 |
| Verrucomicrobiaceae | 3,534 | 3,061 | 8,824 | 7,576 | 2,833 | 0.90 | 0.18 | 0.31 |
| Tenericutes | 50 | 0 | 0 | 0 | 0 | <0.001 | <0.001 | <0.001 |
| Anaeroplasmataceae | 50 | 0 | 0 | 0 | 0 | <0.001 | <0.001 | <0.001 |
| Actinobacteria | 111 | 109 | 81 | 23 | 24 | 0.96 | 0.37 | 0.01 |
| Coriobacteriaceae | 111 | 109 | 81 | 23 | 24 | 0.96 | 0.37 | 0.01 |
| Proteobacteria | 18 | 24 | 60 | 36 | 11 | 0.67 | 0.008 | 0.25 |
| Enterobacteriaceae | 4 | 6 | 31 | 10 | 8 | 0.87 | 0.02 | 0.62 |
| Comamonadaceae | 3.3 | 3.4 | 6.6 | 5.7 | 1.5 | 0.94 | 0.11 | 0.25 |
| Oxalobacteraceae | 2.4 | 3.8 | 5.1 | 4.1 | 1.0 | 0.33 | 0.06 | 0.23 |
| Sphingomonadaceae | 2.3 | 3.1 | 3.8 | 3.9 | 0.7 | 0.46 | 0.14 | 0.13 |
| Methylobacteriaceae | 0.1 | 0.1 | 0.8 | 1.0 | 0.4 | 1 | 0.21 | 0.14 |
| Pseudomonas | 0.3 | 0.3 | 0.5 | 0.3 | 0.2 | 0.77 | 0.57 | 0.84 |
Phyla and families within phyla are organized in the order of microbial number.
Table 3.
Supplementation with 30 mg/kg BW/day XN or its derivatives DXN and TXN decreases the fecal microbial diversityb
| CON | XN | DXN | TXN | SEM | XN vs CON | DXN vs CON | TXN vs CON | |
|---|---|---|---|---|---|---|---|---|
| Microbial Diversity ASV Number (Mice n with counts) | P-values (Comparisons) | |||||||
| Total | 399 | 362 | 323 | 318 | 9 | 0.004 | <0.001 | <0.001 |
| Firmicutes | 292 | 258 | 218 | 213 | 9 | 0.01 | <0.001 | <0.001 |
| Lachnospiraceae | 161 | 142 | 121 | 119 | 4 | 0.003 | <0.001 | <0.001 |
| Ruminococcaceae | 66 | 60 | 37 | 36 | 2.0 | 0.04 | <0.001 | <0.001 |
| Erysipelotrichaceae | 39 | 37 | 46 | 46 | 2.9 | 0.60 | 0.10 | 0.11 |
| Eubacteriaceae | 4.3 (12) | 2.8 (12) | 1.4 (12) | 1.6 (10) | 0.2 | <0.001 | <0.001 | <0.001 |
| Peptostreptococcaceae | 3.7 (12) | 2.8 (11) | 2.1 (10) | 1.0 (6) | 0.4 | 0.17 | 0.01 | <0.001 |
| Clostridiaceae | 1.2 (12) | 0.3 (4) | 0.25 (3) | 0.09 (1) | 0.1 | <0.001 | <0.001 | <0.001 |
| Halobacteroidacea | 1.0 (12) | 1.0 (12) | 0.08 (1) | 0 (0) | 0.04 | 1 | <0.001 | <0.001 |
| Lactobacillaceae | 1.0 (11) | 0.7 (8) | 0.4 (5) | 0.7 (8) | 0.1 | 0.09 | 0.005 | 0.18 |
| Paenibacillaceae | 0.1 (1) | 0.5 (6) | 0.8 (9) | 0.8 (9) | 0.1 | 0.02 | <0.001 | <0.001 |
| Natranaerovirga | 0.1 (1) | 0 (0) | 1.6 (11) | 0.4 (2) | 0.2 | 0.80 | <0.001 | 0.40 |
| Enterococcaceae | 0.8 (9) | 0.7 (8) | 0.2 (2) | 0.7 (8) | 0.1 | 0.65 | 0.003 | 0.90 |
| Staphylococcaceae | 0.7 (8) | 0.6 (7) | 0.6 (7) | 0.4 (4) | 0.1 | 0.69 | 0.69 | 0.16 |
| Bacillaceae | 0.6 (6) | 0.8 (7) | 0.8 (7) | 0.6 (6) | 0.2 | 0.61 | 0.45 | 0.87 |
| Bacteroidetes | 67.4 | 54.6 | 18.9 | 12.4 | 2.6 | <0.001 | <0.001 | <0.001 |
| Porphyromonadaceae | 64.0 | 51.1 | 15.3 | 9.3 | 2.4 | <0.001 | <0.001 | <0.001 |
| Flavobacteriaceae | 1.7 (10) | 1.8 (11) | 2.0 (12) | 1.6 (10) | 0.3 | 0.85 | 0.44 | 0.95 |
| Chitinophagaceae | 0.8 (9) | 0.9 (9) | 1.3 (10) | 1.0 (6) | 0.2 | 0.80 | 0.22 | 0.63 |
| Rikenellaceae | 0.8 (10) | 0.8 (9) | 0.4 (5) | 0.3 (3) | 0.1 | 0.66 | 0.03 | 0.005 |
| Verrucomicrobia | 7.1 | 4.8 | 7.6 | 7.6 | 2.0 | 0.40 | 0.86 | 0.84 |
| Verrucomicrobiaceae | 7.1 | 4.8 | 7.6 | 7.6 | 2.0 | 0.40 | 0.86 | 0.84 |
| Tenericutes | 1 (12) | 0 (0) | 0 (0) | 0 (0) | 0 | <0.001 | <0.001 | <0.001 |
| Anaeroplasmataceae | 1 (12) | 0 (0) | 0 (0) | 0 (0) | 0 | <0.001 | <0.001 | <0.001 |
| Actinobacteria | 3.1 | 2.8 | 2.7 | 2.2 | 0.2 | 0.41 | 0.17 | 0.005 |
| Coriobacteriaceae | 3.1 | 2.8 | 2.6 | 2.1 | 0.2 | 0.25 | 0.09 | 0.002 |
| Proteobacteria | 9.1 | 10.1 | 12.2 | 12.2 | 1.2 | 0.53 | 0.06 | 0.06 |
| Enterobacteriaceae | 2.3 (12) | 2.5 (11) | 3.8 (11) | 3.0 (10) | 0.4 | 0.76 | 0.01 | 0.24 |
| Comamonadaceae | 1.3 (11) | 1.2 (12) | 1.3 (12) | 1.2 (10) | 0.2 | 0.45 | 0.70 | 0.50 |
| Oxalobacteraceae | 2.0 (12) | 2.1 (11) | 2.3 (11) | 2.3 (10) | 0.4 | 0.87 | 0.51 | 0.59 |
| Sphingomonadaceae | 1.4 (10) | 1.8 (11) | 2.0 (10) | 2.5 (10) | 0.3 | 0.33 | 0.17 | 0.02 |
| Methylobacteriaceae | 0.1 (1) | 0.1 (1) | 0.4 (5) | 0.2 (2) | 0.1 | 1 | 0.04 | 0.54 |
| Pseudomonas | 0.3 (4) | 0.3 (3) | 0.3 (3) | 0.3 (3) | 0.1 | 0.66 | 0.66 | 0.76 |
phyla and families within phyla are organized in the order of microbial number; mice with counts are not shown for phyla and families, who have counts for all mice.
Table 4.
Supplementation with 30 mg/kg BW/day XN or its derivatives DXN and TXN alters the relative fecal microbial abundancec
| CON | XN | DXN | TXN | SEM | XN vs CON | DXN vs CON | TXN vs CON | |
|---|---|---|---|---|---|---|---|---|
| Microbial Abundance (counts/detected ASV) | P-values (Comparisons) | |||||||
| Total | 122 | 126 | 124 | 105 | 7 | 0.65 | 0.84 | 0.07 |
| Firmicutes | 93 | 102 | 128 | 115 | 9 | 0.44 | 0.005 | 0.07 |
| Lachnospiraceae | 109 | 127 | 159 | 142 | 12 | 0.30 | 0.005 | 0.06 |
| Ruminococcaceae | 15 | 6 | 3 | 2 | 5 | 0.10 | 0.06 | 0.02 |
| Erysipelotrichaceae | 10 | 12 | 23 | 22 | 2 | 0.49 | <0.001 | 0.001 |
| Eubacteriaceae | 744 | 119 | 45 | 30 | 62 | <0.001 | <0.001 | <0.001 |
| Peptostreptococcaceae | 67 | 94 | 74 | 25 | 28 | 0.56 | 0.82 | 0.22 |
| Clostridiaceae | 18 | 3 | 1 | 1 | 19 | 0.19 | 0.21 | 0.42 |
| Halobacteroidacea | 102 | 73 | 3 | 0 | 39 | 0.08 | 0.02 | <0.001 |
| Lactobacillaceae | 86 | 121 | 182 | 184 | 14 | 0.08 | <0.001 | <0.001 |
| Paenibacillaceae | 1.0 | 1.7 | 2.0 | 1.4 | 0.9 | 0.48 | 0.28 | 0.63 |
| Natranaerovirga | 1 | 0 | 120 | 3 | 375 | <0.001 | 0.44 | 0.99 |
| Enterococcaceae | 6.7 | 2.0 | 2.5 | 3.8 | 4.1 | 0.11 | 0.36 | 0.31 |
| Staphylococcaceae | 3.5 | 3.0 | 2.3 | 1.0 | 1.0 | 0.67 | 0.30 | 0.08 |
| Bacillaceae | 1.1 | 1.7 | 1.8 | 1.3 | 0.3 | 0.11 | 0.07 | 0.54 |
| Bacteroidetes | 262 | 293 | 87 | 33 | 24 | 0.37 | <0.001 | <0.001 |
| Porphyromonadaceae | 276 | 314 | 101 | 41 | 28 | 0.32 | <0.001 | <0.001 |
| Flavobacteriaceae | 1.5 | 1.9 | 2.1 | 2.1 | 0.4 | 0.44 | 0.25 | 0.25 |
| Chitinophagaceae | 1.7 | 1.4 | 1.9 | 1.6 | 0.3 | 0.41 | 0.71 | 0.85 |
| Rikenellaceae | 3.1 | 2.6 | 1.2 | 1.0 | 1.4 | 0.63 | 0.16 | 0.20 |
| Verrucomicrobia | 254 | 220 | 517 | 451 | 158 | 0.88 | 0.25 | 0.39 |
| Verrucomicrobiaceae | 254 | 220 | 517 | 451 | 166 | 0.88 | 0.25 | 0.39 |
| Tenericutes | 50 | 0 | 0 | 0 | 15 | <0.001 | <0.001 | <0.001 |
| Anaeroplasmataceae | 50 | 0 | 0 | 0 | 0 | <0.001 | <0.001 | <0.001 |
| Actinobacteria | 34 | 40 | 30 | 11 | 8 | 0.65 | 0.75 | 0.06 |
| Coriobacteriaceae | 34 | 40 | 30 | 11 | 8 | 0.64 | 0.78 | 0.06 |
| Proteobacteria | 1.9 | 2.3 | 4.5 | 2.9 | 0.8 | 0.72 | 0.02 | 0.37 |
| Enterobacteriaceae | 1.8 | 2.1 | 6.7 | 2.8 | 1.7 | 0.86 | 0.03 | 0.65 |
| Comamonadaceae | 2.5 | 3.0 | 4.6 | 5.1 | 1.0 | 0.71 | 0.12 | 0.07 |
| Oxalobacteraceae | 1.2 | 1.8 | 2.2 | 1.6 | 0.3 | 0.07 | 0.004 | 0.20 |
| Sphingomonadaceae | 1.6 | 1.6 | 1.8 | 1.6 | 0.2 | 0.99 | 0.58 | 0.98 |
| Methylobacteriaceae | 1.0 | 1.0 | 2.0 | 5.5 | 2.4 | 1.00 | 0.72 | 0.18 |
| Pseudomonas | 1.0 | 1.0 | 2.0 | 1.0 | 0.5 | 1 | 0.14 | 1 |
Phyla and families within phyla are organized in the order of microbial number.
We visualized differences in fecal microbiota counts and diversity using principal coordinate analysis (PCoA; Adonis; R2 = 0.396; adj-P = 0.001; permutations = 999) with three different distance matrixes: Bray-Curtis (Figure 1A), unweighted UniFrac (Figure 1B), and weighted UniFrac (Figure 1C). Consistent with the significant changes in body weight and metabolic parameters[1], we observed decreased fecal microbial counts (DXN: −19%; P = 0.01; TXN: −32%; P < 0.001; Table 2) that coincided with decreasing microbial diversity (DXN: −19%; P < 0.001; TXN: −27%; P < 0.001; Table 3) in the mice treated with DXN and TXN. Compared with the XN derivatives[1], mice treated with XN showed fewer significant changes in metabolic parameters and this coincided with a smaller effect on microbial diversity (XN: −9%; P = 0.04). The treatment-induced decrease in microbial diversity is visualized in Figure 1D using the alpha-diversity index.
Figure 1. Principal coordinates analyses (PCoA) of gut microbiota based upon different distance matrices for the HFD-CON and HFD-XN, HFD-DXN and HFD-TXN supplementation.
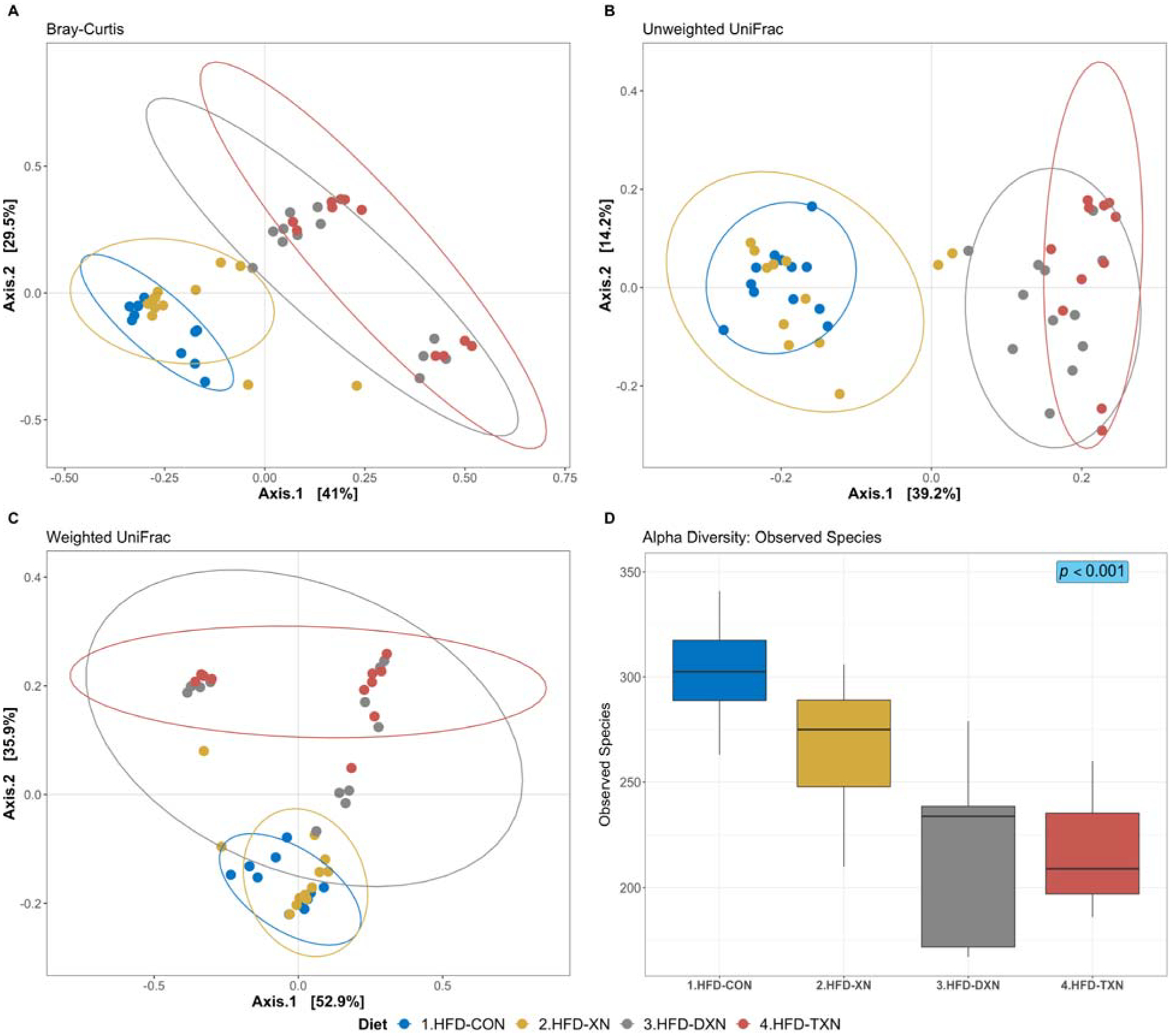
Each point represents a mouse fecal sample, plotted by a principal component on the X-axis and another principal component on the Y-axis. The percentage on each axis indicates the contribution value to discrepancy among samples. (A) Bray-Curtis dissimilarity. (B) Unweighted UniFrac distance. (C) Weighted UniFrac distance. Ellipses are drawn at 0.95 C.I., t-distribution. Significant dissimilarity by dietary treatments across samples is observed. (ADONIS; adj-p = 0.001, R2 = 0.396; permutation = 999). (D) Alpha diversity index (observed species) was calculated on the rarefied ASV count data (chi-squared = 26.0, df = 3, p-value = 9.4 × 10–6; Kruskal-Wallis rank sum test). Metrics are plotted against HFD control and different xanthohumol treatments, i.e., XN, DXN, and TXN; with median (line), and hinges as first and third quartiles (25th and 75th percentiles).
XN derivatives did not affect all phyla equally. Between the two most abundant phyla, Bacteroidetes counts were most affected (DXN: −87%; P < 0.001; TXN: −97%; P < 0.001; Table 2); specifically, the families Porphyromonadaceae and Rikenellaceae, were generally sensitive to the XN derivatives. Treatment with the XN derivatives dramatically decreased microbial diversity (DXN: −72%; P < 0.001; TXN: −82%; P < 0.001; Table 3) and relative abundance in Bacteroidetes (counts/detected ASV; DXN: −67%; P < 0.001; TXN: −87%; P < 0.001; Table 4). The XN derivatives also dramatically decreased the microbial diversity and relative abundance of Porphyromonadaceae and resulted in fewer mice with detectable Rikenellaceae (DXN: −50%; P = 0.09; TXN: −63%; P = 0.01; Fisher’s exact test) (Tables 3–4).
The effects of XN derivative treatment on Firmicutes were more complex; as DXN and TXN decreased microbial diversity (DXN: −25%; P < 0.001; TXN: −27%; P < 0.001; Table 3) but increased microbial counts of detected species (DXN: +38%; P = 0.005; TXN: +24%; P = 0.07; Table 4). A smaller effect on microbial diversity was observed with XN treatment (XN: −12%; P = 0.01; Table 3). The effect of XN derivative treatments differed among Firmicutes families and genera (Table 2–4). Most, but not all of the prominent Firmicutes families were sensitive to DXN and TXN and lost a significant number of ASV (Lachnospiraceae: DXN: −35%, TXN: −36%; Ruminococcaceae: DXN: −44%, TXN: −45%; Eubacteriaceae: DXN: −67%, TXN: −63%; Table 3). Compared with control mice, fewer mice receiving XN derivatives carried Halobacteroidacea (DXN: 1 vs. 12; P < 0.001; TXN: 0 vs. 12; P < 0.001; Fisher’s exact test) and Clostridiaceae (DXN: 3 vs. 12; P < 0.001; TXN: 1 vs. 12; P < 0.001) and more carried Paenibacillaceae (DXN: 9 vs. 1; P = 0.003; TXN: 9 vs. 1; P = 0.003; Table 3). Peptostreptococcaceae (TXN: 6 vs. 12; P = 0.01) and Natranaerovirga (DXN: 11 vs. 1; P < 0.001) only differed for DXN compared with the control (Table 3). Relative microbial abundance differed among families and genera, as over 2-fold higher counts/detected ASV were measured in Lactobacillaceae and Erysipelotrichaceae and over 2-fold lower counts/detected ASV were measured in Ruminococcaceae for DXN and TXN compared with the control (Table 4). The most intriguing ASV, present in all mice, were from the Lachnospiraceae family: ASV 22, 31, and 65 were each at least 10-fold higher and ASV62 was at least 10-fold lower in TXN vs. control mice. ASV133, ASV212, and ASV346 (the first two Ruminococcaceae and the third Lachnospiraceae) were present in all HFD-fed mice but not in DXN/TXN supplemented mice. None of the ASV were unique to supplemented mice.
Among the less abundant phyla, growth of Proteobacteria (+233%; P = 0.008), especially Enterobacteriaceae (+675%; P = 0.02), was only promoted by DXN treatment (Table 2) primarily by increasing relative microbial abundance (Proteobacteria: +137%; P = 0.01; Enterobacteriaceae: +272%; P = 0.03; Table 4). Actinobacteria were only sensitive to TXN treatment, which decreased counts 79% (P = 0.01; Table 2) by decreasing microbial diversity (−32%; P = 0.002; Table 3) and abundance (−68%; P = 0.06; Table 4). Tenericutes were extremely sensitive to treatments with XN and its derivatives as we only detected it in control mice (Table 3). Large variations in Verrucomicrobiaceae within DXN and TXN-supplemented mice did not allow us to detect significant group differences (Tables 2–4). This also resulted in non-significant group differences using the Shannon index (Supporting Information Figure S2).
3.2. XN derivatives modulate fecal bile acid metabolism
We measured fecal bile acid composition to determine the potential impact of treatment with XN and its derivatives on changes in fecal microbiota and microbiome-host metabolic interactions. Administration of XN derivatives altered fecal bile acid metabolism, which is visualized using PCA plots (Supporting Information Figure S3). Total fecal bile acid levels were higher in mice treated with DXN (+53%; P = 0.02) and TXN (+54%; P = 0.02) than in control mice (Table 5). The higher bile acid levels reflected significant increases in taurine-conjugated primary bile acids TCA, TαMCA and TβMCA (Table 5), suggesting a decrease in deconjugation by the microbiota. We also observed a decrease in microbial conversion of secondary bile acids DCA, UDCA, and HDCA in mice treated with the XN derivatives. Fecal levels of conjugated, dehydroxylated bile acids TωMCA, TDCA and TUDCA did not differ among treatment groups. DCA is the 7-α-dehydroxylation product of CA.
Table 5.
Supplementation with 30 mg/kg BW/day XN or its derivatives DXN and TXN alters the fecal bile acid profile
| Bile Acid Group | CON | XN | DXN | TXN | SEM | XN vs CON | DXN vs CON | TXN vs CON |
|---|---|---|---|---|---|---|---|---|
| Bile Acid | Peak Intensity (LS mean) | P-values (Comparisons) | ||||||
| Total | 18,866 | 15,352 | 29,309 | 29,618 | 3,200 | 0.40 | 0.02 | 0.02 |
| Unconjugated Primary | 1,555 | 1,384 | 997 | 864 | 198 | 0.51 | 0.04 | 0.01 |
| CA | 450 | 383 | 414 | 257 | 109 | 0.64 | 0.80 | 0.20 |
| αMCA | 107 | 92 | 43 | 35 | 9 | 0.24 | <0.001 | <0.001 |
| βMCA | 998 | 909 | 539 | 572 | 118 | 0.56 | 0.004 | 0.01 |
| Conjugated Primary | 13,920 | 10,660 | 25,868 | 26,350 | 2,828 | 0.36 | 0.008 | 0.01 |
| TCA | 1,378 | 1,069 | 5,851 | 6,202 | 908 | 0.79 | <0.001 | <0.001 |
| TCDCA | 358 | 857 | 399 | 205 | 150 | 0.01 | 0.83 | 0.46 |
| TαMCA | 748 | 621 | 1,009 | 1,471 | 154 | 0.53 | 0.20 | 0.001 |
| TβMCA | 11,435 | 8,113 | 18,608 | 18,471 | 1,988 | 0.20 | 0.08 | 0.01 |
| Unconj. Dehydroxylated | 1,420 | 1,289 | 427 | 314 | 147 | 0.50 | <0.001 | <0.001 |
| DCA | 1,160 | 1,047 | 385 | 282 | 126 | 0.49 | <0.001 | <0.001 |
| UDCA | 22 | 12 | 4 | 3 | 3 | 0.009 | <0.001 | <0.001 |
| HDCA | 238 | 229 | 38 | 29 | 23 | 0.77 | <0.001 | <0.001 |
| Conjug. Dehydroxylated | 1,972 | 2,019 | 2,017 | 2,090 | 408 | 0.93 | 0.93 | 0.83 |
| TDCA | 179 | 224 | 264 | 325 | 66 | 0.60 | 0.33 | 0.11 |
| TUDCA | 939 | 678 | 742 | 507 | 288 | 0.49 | 0.60 | 0.27 |
| TωMCA | 854 | 1,116 | 1,011 | 1,258 | 228 | 0.38 | 0.60 | 0.20 |
| Ratios | ||||||||
| CA/DCA | 0.36 | 0.40 | 1.78 | 2.83 | 0.50 | 0.95 | 0.05 | 0.002 |
| CA/TCA | 0.46 | 0.47 | 0.12 | 0.09 | 0.08 | 0.87 | 0.002 | 0.001 |
| αMCA/TαMCA | 0.208 | 0.180 | 0.048 | 0.023 | 0.036 | 0.56 | 0.001 | 0.0005 |
| βMCA/TβMCA | 0.102 | 0.133 | 0.032 | 0.032 | 0.013 | 0.07 | 0.0001 | 0.0002 |
| Conjugated/Unconj. | 5.02 | 5.06 | 28.2 | 48.2 | 7.1 | 0.99 | 0.02 | <0.0001 |
We observed an increase in the ratio of CA/DCA in DXN- and TXN-treated mice compared with control and XN-treated animals (Table 5) suggesting that XN derivatives decreased 7-α-dehydroxylation activity in the gut. We also observed an increase in conjugated/unconjugated fecal bile acid ratios in XN derivative treated mice compared with control mice (Table 5) suggesting a decrease in bile salt hydrolase (BSH) enzyme activity. In summary, supplementation with XN derivatives altered microbial conversion of bile acids in the intestine, coincident with reduced microbial abundance and diversity.
3.3. XN derivatives alter expression of host genes involved in bile acid metabolism
We did not assess gene expression in our prior study[1]. To determine the effect of treatment on the expression of host genes involved in bile acid metabolism, we measured expression of Cyp7a1, Cyp27b1, Cyp8b1, Fxr, Shp and Bsep mRNAs in the liver. Mice fed XN derivatives vs. control diet had an approximately 2-fold higher gene expression of Cyp7a1 and 3-fold increase in nuclear hormone receptor Shp (Table 6). An approximately 2-fold lower expression of the hepatic bile salt export pump Bsep was observed (Table 6). No statistically significant changes for Fxr, Cyp27a1 and Cyp8b1 were observed.
Table 6.
Supplementation with 30 mg/kg BW/day XN or its derivatives DXN and TXN alters expression of genes involved in hepatic bile acid metabolismd
| Bile Acid Metabolism | CON | XN | DXN | TXN | SEM | XN vs CON | DXN vs CON | TXN vs CON |
|---|---|---|---|---|---|---|---|---|
| Gene | ΔCT | P-values (Comparisons) | ||||||
| Bile Acid Synthesis | ||||||||
| Cyp7a1 | Ref. | 1.15 | 1.64 | 2.16 | 0.77 | 0.69 | 0.16 | 0.04 |
| Cyp27a1 | Ref. | 1.21 | −1.08 | −1.15 | 0.87 | 0.33 | 0.68 | 0.49 |
| Cyp8b1 | Ref. | 1.29 | 1.16 | 1.22 | 0.86 | 0.21 | 0.47 | 0.34 |
| Bile Acid Secretion | ||||||||
| Fxr | Ref. | −1.07 | 1.28 | 1.27 | 0.86 | 0.76 | 0.22 | 0.25 |
| Shp | Ref. | 1.91 | 3.62 | 2.98 | 0.74 | 0.12 | 0.003 | 0.01 |
| Bsep | Ref. | 1.03 | −1.56 | −1.84 | 0.86 | 0.91 | 0.04 | 0.008 |
All the threshold cycle (Ct) numbers were normalized to a reference gene, tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation protein, Ywhaz, which others and we determined to be relatively stable in most mouse tissues (data not shown)[17]. The fold-change compared to the control (HFD) was calculated using the 2−ΔΔCt method. Ref: HFD control mice were used as the reference.
3.4. XN derivatives reduce inflammation in white adipose tissue (WAT)
To evaluate the impact of treatment with XN and its derivatives on inflammation, we measured expression of Il-1β and Tnfα in the liver, and Ccl2/Mcp1, F4/80, Il-1β, Il-6 and Tnfα in WAT (Table 7). For TXN-supplemented mice, we observed decreased expression of the major pro-inflammatory cytokines Il-6 and Tnfα in WAT as compared to the control HFD mice. This decrease coincided with a statistically significant decrease in the expression of the monocyte chemotactic factor Ccl2/Mcp1 and a concomitant decrease in macrophage infiltration as reflected by the reduced expression of the macrophage-specific marker F4/80 (Adgre1) (Table 7).
Table 7.
Supplementation with 30 mg/kg BW/day XN or its derivatives DXN and TXN alters expression of genes involved in inflammation and colonic barrier function
| Tissue | CON | XN | DXN | TXN | SEM | XN vs CON | DXN vs CON | TXN vs CON |
|---|---|---|---|---|---|---|---|---|
| Gene | ΔCT | P-values (Comparisons) | ||||||
| Colon | ||||||||
| Il-22 | Ref. | −1.67 | −2.56 | −2.77 | 0.84 | 0.04 | <0.001 | <0.001 |
| Ocln | Ref. | 1.47 | 1.60 | 1.72 | 0.85 | 0.09 | 0.04 | 0.02 |
| Liver | ||||||||
| Il−1β | Ref. | 1.30 | −1.21 | −1.70 | 0.80 | 0.40 | 0.53 | 0.10 |
| Tnfα | Ref. | 1.16 | 1.05 | −1.48 | 0.82 | 0.58 | 0.85 | 0.15 |
| WAT | ||||||||
| Il−1β | Ref. | 1.05 | 1.23 | −1.09 | 0.83 | 0.86 | 0.42 | 0.75 |
| Tnfα | Ref. | −1.09 | −1.21 | −1.87 | 0.81 | 0.77 | 0.50 | 0.04 |
| Il-6 | Ref. | −1.19 | −1.27 | −2.39 | 0.79 | 0.59 | 0.46 | 0.01 |
| Ccl2 | Ref. | −1.33 | −1.22 | −2.68 | 0.76 | 0.45 | 0.60 | 0.01 |
| F4/80 | Ref. | −1.14 | −1.19 | −3.79 | 0.73 | 0.76 | 0.68 | 0.004 |
To evaluate the effect of treatment on gene expression in the colon, we measured expression of the genes for the cytokine Il-22 which is upregulated after gastrointestinal infection or damage and the tight junction protein occludin (Table 7). Treatment with XN and both derivatives decreased Il-22 gene expression significantly in the colon as compared to the control. Occludin (Ocln) expression increased with each treatment but was statistically significant for only DXN and TXN.
3.5. Transkingdom network analysis identifies putative key microbial players associated with the beneficial effects of XN derivatives
To visualize the connection between changes in intestinal microbiota, MetS outcomes and bile acid metabolism, we used transkingdom network analysis (Figure 2). To allow statistical convergence, we restricted the analysis to ASV with an abundance of at least 0.5% and reconstructed a bacterial co-abundance network. To infer bacteria that may have driven changes in host outcomes from TXN treatment, we searched for “bottleneck” bacterial nodes (microbes with high betweenness centrality) that link microbes with strong connections to host phenotypes, gene expression and bile acid metabolism using the bipartite betweenness centrality (BiBC) metric[31]. Those ASV with high BiBC values are more likely key regulators of host outcomes than ASV with low values. TXN-treated mice gained significantly less body weight at the same food intake (i.e., lower food conversion or feed efficiency) compared with the other groups, which was linked to an increase of an ASV from the genus Acetatifactor of the family Lachnospiraceae. Feed efficiency was also linked to improvements in the other phenotypic outcomes (e.g. plasma glucose, insulin and leptin, weight gain, and body and liver weight). Srebp1c and Cyp7a1 mRNA expression were also associated with feed efficiency.
Figure 2. Transkingdom microbe-gene-host phenotype regulatory network –
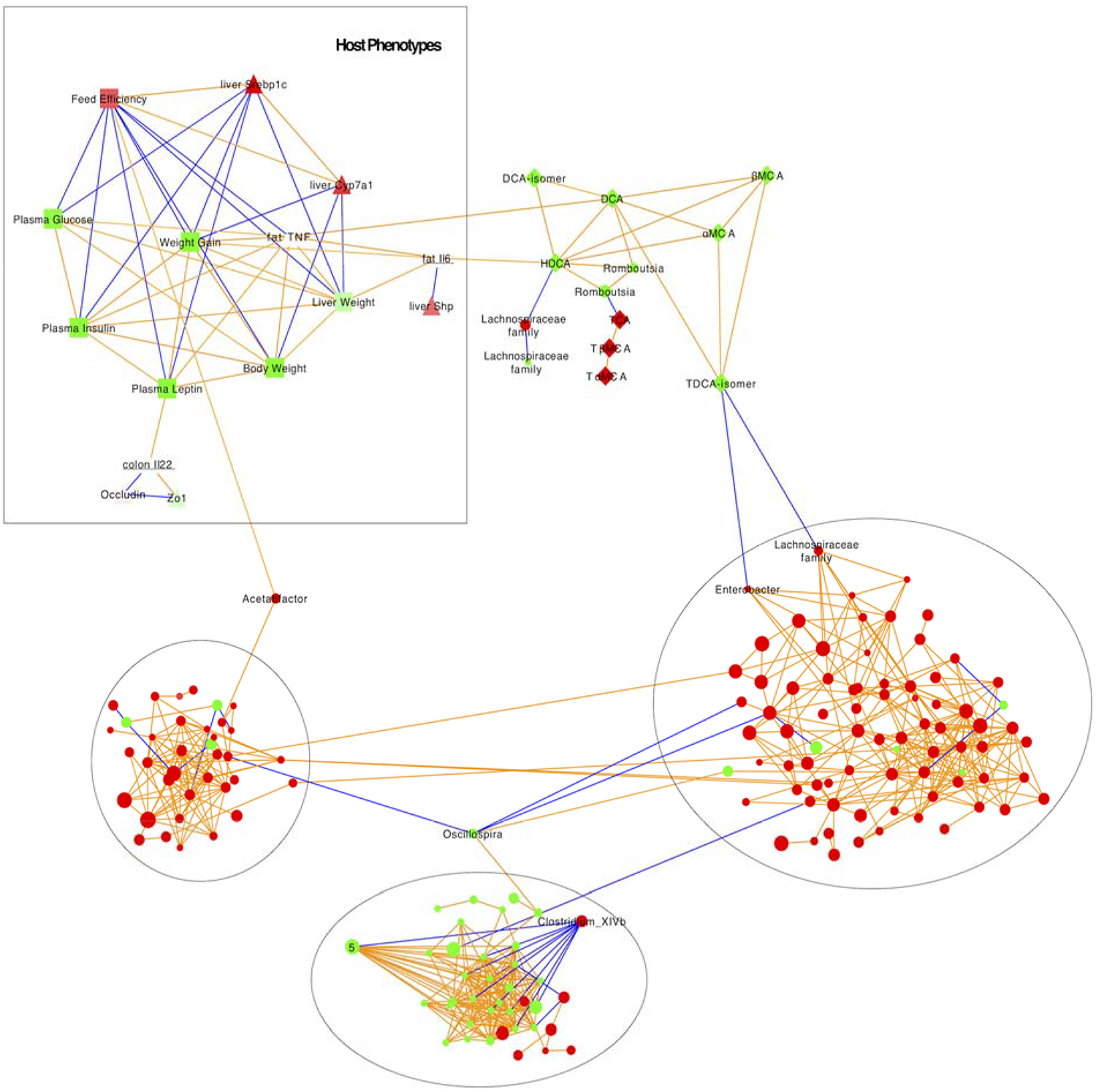
the network reconstructed from microbiota abundances (Tables 2–4), bile acid levels (Table 5), and host gene expression (Tables 6–7) in mice consuming either CON or TXN. Microbes – circles; host genes – triangles; host phenotypes – squares; orange edges denote positive correlations; blue edges denote negative correlations; three major microbial subnetworks defined by black circles; green color indicates a decrease; red color indicates an increase.
The top three ASV associated with fecal bile acid were from the family Lachnospiraceae and the genera Romboutsia and Enterobacter. A decrease in secondary bile acid DCA and HDCA and concomitant increase in the conjugated primary bile acid TCA and secondary bile acid TDCA were linked to a decreased bacterial number in a Romboutsia ASV and an increase in an Enterobacter ASV. A decrease in secondary bile acids DCA and HDCA was associated with decreased Tnfα and Il-6 expression in the WAT, respectively.
4. Discussion
In our prior study, treatment with XN and its derivatives DXN and TXN improved parameters of MetS in a DIO mouse model.[1] At a 30 mg/kg BW dose, we observed a reduction in weight gain associated with a decrease in feed conversion for TXN-treated mice and improved glucose clearance in XN-, DXN- and TXN-treated mice. Only TXN-treated mice showed decreases in liver weight and fasting plasma glucose and insulin. Lower plasma leptin was observed for both DXN- and TXN-treated animals. Our previous study suggested that one mechanism by which TXN mediated its benefits was possibly increasing energy expenditure through mild mitochondrial uncoupling. Prior studies also indicate that XN may improve glucose homeostasis by activating AMPK in mouse liver[1, 41]; however, we did not observe AMPK activation in the liver with DXN or TXN treatment suggesting they may have other effects[1]. At the dose used in the prior study, DXN and TXN appeared more efficacious than XN. The hydrogenation of the α,β-unsaturated keto moiety of XN in DXN and TXN resulted in greater steady-state concentrations in tissues, loss of affinity for the estrogen receptor, and retention or even enhancement of the beneficial effects of XN on HFD-induced dysfunctional glucose metabolism.
Studies implicate intestinal microbiota in the etiology of obesity and MetS[35–40]. Because oral administration of the these compounds would allow them to reach much higher concentrations in the gut than in other tissues, we hypothesized that improvements in obesity and MetS from administering XN and its non-estrogenic derivatives DXN and TXN are linked to changes in the composition of the gut microbiota and bile acid metabolism. In this current study, we report for the first time that improvements in obesity and MetS from administration of XN and its derivatives are linked to significant changes in fecal microbiota composition and bile acid metabolism. Furthermore, gene expression results indicate reduced macrophage infiltration and inflammatory cytokine expression in WAT.
Previously, PCR-denaturing gradient gel electrophoresis (DGGE) fingerprinting analysis of rats consuming 100 mg XN/kg BW in water for four weeks did not detect changes in the fecal microbiome[42]. The improved resolution of 16S rRNA sequencing methodology enabled us to detect decreases in relative microbiota abundance and diversity in mice consuming XN and its derivatives at 30 mg/kg BW for 13 weeks. These findings are consistent with established antifungal, antiviral and antibacterial activities of XN and its derivatives, but the exact mechanism by which these compounds alter the composition of the microbiota remains to be determined.[43–47] Interestingly, the largest decreases in relative microbial abundance were observed with TXN > DXN >> XN. These changes in abundance paralleled the efficacy of these compounds in mitigating obesity and MetS in the mice (Table 1) and suggests that additional hydrogenation of XN (TXN is more hydrogenated than DXN) may affect its bioactivity. The dose of XN and its derivatives used in this study is equivalent to 175 mg/day for a 70 kg adult[48], a realistic dose for a dietary supplement.
Table 1.
Metabolic parameters of mice fed experimental diets in the previous study. Data are expressed in means ± SE of 11–12 animals.
| Parameters | CON | XN | DXN | TXN |
|---|---|---|---|---|
| Body weight gain | 16.6 ± 1.3 | 16.1 ± 1.3 | 15.1 ± 1.3 | 9.70 ± 1.4* |
| food intake | 2.69 ± 0.09 | 2.59 ± 0.09 | 2.66 ± 0.09 | 2.51 ± 0.09 |
| Liver weight | 1.17 ± 0.07 | 1.14 ± 0.07 | 1.11 ± 0.07 | 0.93 ± 0.07* |
| Fasting glucose (mg/dL) | 163 ± 7.4 | 169 ± 8.3 | 150 ± 13.0 | 118 ± 7.2* |
| Fasting insulin (ng/mL) | 2.70 ± 0.76 | 2.61 ± 0.65 | 2.08 ± 0.69 | 0.49 ± 0.08* |
| HOMA-IR | 32.2 ± 1.5 | 31.3 ± 1.7 | 22.0 ± 5.2* | 4.10 ± 0.3* |
| Fasting leptin (ng/mL) | 11.4 ± 1.0 | 10.6 ± 1.0 | 8.48 ± 1.0* | 6.39 ± 1.0* |
p < 0.05 versus CON.
DXN and TXN administration significantly increased levels of taurine conjugated primary bile acids and lowered levels of secondary bile acids in the feces. This finding coincides with a decrease in microbial abundance and diversity and is consistent with observations in germ-free and antibiotic-treated mouse models[32, 49]. While most bile acids are reabsorbed in the distal ileum, some escape into the colon where they can undergo microbial modification including deconjugation by BSH and dehydroxylation attributed to bacteria from Firmicutes (Lachnospiraceae, Clostridiaceae, Erysepelotrichaceae, Ruminococcaceae, Lactobacillus) and Bacteroidetes (Bacteroides)[50]. We observed that XN derivatives decreased the levels and diversity of Bacteroidetes and nearly eliminated Clostridiaceae and Lactobacillaceae, suggesting that part of the effect of XN derivative treatment on intestinal bile acid metabolism may be mediated by changes in the microbiota. Transkingdom network analysis linked a decrease in the genus Romboutsia to elevated levels of TCA, TαMCA and TβMCA suggesting it as a potential candidate genus for the elevated levels of conjugated, fecal primary bile acids following treatment with the XN derivatives. Sequence analysis of the genome of Romboutsia ilealis CRIBT indicates that it possesses a gene that encodes a BSH that deconjugates bile salts[51–52].
Bile acids can act as natural endogenous ligands for various host nuclear (FXR, VDR, PXR) and G protein-coupled receptors (TGR-5, S1PR2)[15–16]. Others and we previously showed that XN could function as a ligand for FXR, the nuclear receptor that serves as a master control for downregulating gluconeogenesis and de novo lipogenesis[17–18]. Our results suggest that XN derivative supplementation may also alter the production of bile acid ligands for FXR by increasing antagonists (TαMCA and TβMCA) and reducing agonists (CA)[32, 49]. We expect that a shift in bile acid composition that favors antagonists would affect FXR target gene expression. As reported previously for XN supplementation[18, 33, 34], TXN supplementation increased expression of hepatic Cyp7a1, the rate-determining enzyme in the classic bile acid biosynthetic pathway[53], linking the currently observed changes in the microbiota with changes in hepatic and intestinal bile acid metabolism. Other bile acid synthesizing enzymes, Cyp8b1 and Cyp27a1 were unchanged. Although, we did not observe a change in Fxr gene expression, XN derivative supplementation decreased hepatic gene expression of Bsep. Shp and Srebp-1c increased in the liver, contrary to prior observations[18]. While the increase in Srebp-1c mRNA expression is not consistent with the improvements in obesity, it was reported that XN improves obesity in mice by suppressing activation of the SREBP-1C protein by blocking its cleavage[33]. Thus, an increase of mRNA expression may not significantly increase SREBP-1C activity that is regulated post-translationally.
One would expect that elevated hepatic Shp expression would suppress Cyp7a1 gene expression; however, one possible explanation is that the elevated FXR antagonists, TαMCA and TβMCA in the intestine may suppress intestinal FXR activity which is required for expression of Fgf15 in the ileum, an endocrine hormone that suppresses Cyp7a1 gene transcription[54]. Although we did not have ileum samples from the prior study for gene expression analysis, our hypothesis, that remains to be tested, is that the shift in bile acid composition favoring antagonists reduces intestinal FGF15 production that would normally down-regulate Cyp7a1 gene expression in the liver via binding to FGFR4[54]. In support of our hypothesis, studies in germ-free and antibiotic-treated mice show significant decreases of intestinal Fgf15 gene expression and significant increases of hepatic Cyp7a1 expression[32].
Most if not all symptoms of MetS are associated with a chronic inflammatory state caused by circulating cytokines and macrophage infiltration into the adipose tissue[55]. Supplementation with TXN decreased chronic inflammation, as indicated by reduced expression of the major pro-inflammatory cytokines Il-6 and Tnfα in WAT and to a smaller extent in liver tissue. To determine the potential role of macrophage infiltration in the suppressed inflammation, we measured Ccl2/Mcp1, a chemotactic factor involved in the recruitment of monocytes, and macrophage marker F4/80 in WAT and observed decreased expression of both suggesting that TXN supplementation may protect WAT from macrophage infiltration.
In summary, we provide support for our hypothesis that improvements in obesity and MetS from administering XN and its non-estrogenic derivatives DXN and in particular, TXN, are linked to changes in the composition of the gut microbiota, metabolism of bile acids, and reduced adipose inflammation. While these findings do not address causation, they lay the foundation for future germ-free transplantation studies to test causation and identify the microbe(s) that are key in mediating the health benefits of these hops compounds.
Supplementary Material
Acknowledgements
We thank Miles V. Rouches for help extracting fecal microbial DNA, and Zachary Foster for helpful suggestions on microbiota data visualization.
Funding
The National Institutes of Health (NIH grants 5R01AT009168 to AFG, CSM and JFS; DK103761 to NS and AM; and 1S10RR027878 to CSM and JFS), the Linus Pauling Institute (LPI), the OSU College of Pharmacy, Hopsteiner, Inc., New York and the OSU Foundation Buhler-Wang Research Fund supported this research. The Marion T. Tsefalas Graduate Fellowship from the LPI, the Charley Helen, Nutrition Science and Margy J. Woodburn Fellowships from the School of Biological and Population Health Sciences at OSU supported YZ.
Abbreviations:
- XN
xanthohumol
- DXN
α,β-dihydro-xanthohumol
- TXN
tetrahydro-xanthohumol
- HFD
high-fat diet
- WAT
white adipose tissue
- MetS
metabolic syndrome
- 8-PN
8-prenylnaringenin
- DIO
diet-induced obesity
Footnotes
Conflict of interest statement
We declare that we have no conflicts of interest.
5. References
- [1].Miranda CL, Johnson LA, de Montgolfier O, Elias VD, Ullrich LS, Hay JJ, Paraiso IL, Choi J, Reed RL, Revel JS, Kioussi C, Bobe G, Iwaniec UT, Turner RT, Katzenellenbogen BS, Katzenellenbogen JA, Blakemore PR, Gombart AF, Maier CS, Raber J, Stevens JF, Sci. Rep, 2018, 8, 613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Miranda CL, Elias VD, Hay JJ, Choi J, Reed RL, Stevens JF, Arch Biochem Biophys, 2016, 599, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Kirkwood JS, L Legette L, Miranda CL, Jiang Y, Stevens JF, J Biol Chem, 2013, 288, 19000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Legette LL, Luna AYM, Reed RL, Miranda CL, Bobe G, Proteau RR, Stevens JF, Phytochemistry, 2013, 91, 236. [DOI] [PubMed] [Google Scholar]
- [5].Wickramasekara SI, Zandkarimi F, Morré J, Kirkwood J, Legette LL, Jiang Y, Gombart AF; Stevens JF, Maier CS, Metabolites, 2013, 3, 701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].The GBD 2015 Obesity Collaborators, N Engl J Med, 2017, 377, 13.28604169 [Google Scholar]
- [7].Dobbs R, Sawers C, Thompson F, Manyika J, Woetzel JR, Child P, McKenna A Spatharou, Overcoming obesity: an initial economic analysis, McKinsey Global Institute; 2014. [Google Scholar]
- [8].Possemiers S, Verstraete W, Environ Microbiol Rep, 2009, 1, 100. [DOI] [PubMed] [Google Scholar]
- [9].L Paraiso I, Plagmann LS, Yang L, Zielke R, Gombart AF, Maier CS, Sikora AE, Blakemore PR, Stevens JF, Mol Nutr Food Res, 2019, 63, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].für Zeitschrift C Naturforschung, 1999, 54, 7–8. [Google Scholar]
- [11].Rodriguez RJ, Miranda CL, Stevens JF, Deinzer ML, Buhler DR, Food Chem Toxicol, 2001, 39, 5. [DOI] [PubMed] [Google Scholar]
- [12].Sanz Y, Santacruz A, Gauffin P, Proc Nutr Soc, 2010, 69, 434. [DOI] [PubMed] [Google Scholar]
- [13].Kelly TN, Bazzano LA, Ajami NJ, He H, Zhao J, Petrosino JF, Adolfo C, Jiang H, Circ Res, 2016, 119, 956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Parséus A, Sommer N, Sommer F, Caesar R, Molinaro A, Ståhlman M, Greiner TU, Perkins R, Bäckhed F, Gut, 2017, 66, 429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Schaap FG, Trauner M, Jansen PLM, Nat Rev Gastroenterol Hepatol, 2014, 11, 55. [DOI] [PubMed] [Google Scholar]
- [16].de Aguiar Vallim TQ, Tarling EJ, Edwards PA, Cell Metab, 2013, 17, 657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Yang L, Broderick D, Campbell Y, Gombart AF, Stevens JF, Jiang Y, Hsu VL, Bisson WH, Maier CS, Biochim Biophys Acta, 2016, 1864, 1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Nozawa H, Biochem Biophys Res Commun, 2005, 336, 754. [DOI] [PubMed] [Google Scholar]
- [19].Rodrigues RR, Shulzhenko N, Morgun A, In Microbiome Analysis: Methods and Protocols, (Eds: Beiko RG, Hsiao W, Parkinson J), Springer, New York: 2018, 227. [Google Scholar]
- [20].Bruce KD, Sihota KK, Byrne CD, Cagampang FR, Liver Int, 2012, 32, 1315. [DOI] [PubMed] [Google Scholar]
- [21].Illumina, 16S Sample Preparation Guide, https://support.illumina.com/documents/documentation/chemistry_documentation/16s/16s-metagenomic-library-prep-guide-15044223-b.pdf
- [22].Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP, Nat Methods, 2016, 13, 581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Cole JR, Wang Q, Cardenas E, Fish J, Chai B, Farris RJ, Kulam-Syed-Mohideen AS, McGarrell DM, Marsh T, Garrity GM, Tiedje JM, Nucleic Acids Res, 2009, 37, D141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].McMurdie PJ, Holmes S, PLoS One, 2013, 8, e61217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Wickham H, ggplot2: Elegant Graphics for Data Analysis, Springer, 2016. [Google Scholar]
- [26].Love M, Anders S, Huber W, Genome Biol, 2014, 15, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Jovel J, Patterson J, Wang W, Hotte N, O’Keefe S, Mitchel T, Perry T, Kao D, Mason AL, Madsen KL, Wong GK-S, Front Microbiol, 2016, 7, 459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Oksanen J, Multivariate analysis of ecological communities in R: vegan tutorial, Univ. of Oulu, Oulu, 2007. [Google Scholar]
- [29].Paulson JN, Stine OC, Bravo HC, Pop M, Nat Methods, 2013, 10, 1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Yambartsev A, Perlin MA, Kovchegov Y, Shulzhenko N, Mine KL, Dong X, Morgun A, Biol Direct, 2016, 11, 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Dong X, Yambartsev A, Ramsey SA, Thomas LD, Shulzhenko N, Morgun A, Bioinform Biol Insights, 2015, 9, 61–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Sayin SI, Wahlström A, Felin J, Jäntti S, Marschall H-U, Bamberg K, Angelin B, Hyötyläinen T, Orešič M, Bäckhed F, Cell Metab, 2013, 17, 225. [DOI] [PubMed] [Google Scholar]
- [33].Miyata S, Inoue J, Shimizu M, Sato R, J Biol Chem, 2015, 290, 20565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Hirata H, Uto-Kondo H, Ogura M, Ayaori M, Shiotani K, Ota A, Tsuchiya Y, Ikewaki K, J Nutr Biochem, 2017, 47, 29. [DOI] [PubMed] [Google Scholar]
- [35].Winer DA, Luck H, Tsai S, Winer S, Cell Metab, 2016, 23, 413. [DOI] [PubMed] [Google Scholar]
- [36].Cani PD, Amar J, Iglesias MA, Poggi M, Knauf C, Bastelica D, Neyrinck AM, Fava F, Touhy KM, Chabo C, Waget A, Delmée E, Cousin B, Sulpice T, Chamontin B, Ferrières J, Tanti J-F, Gibson GR, Casteilla L, Delzenne NM, Alessi MC, Burcelin R, Diabetes, 2007, 56, 1761. [DOI] [PubMed] [Google Scholar]
- [37].Cani PD, Bibiloni R, Knauf C, Waget A, Neyrinck AM, Delzenne NM, Burcelin R, Diabetes, 2008, 57, 1470. [DOI] [PubMed] [Google Scholar]
- [38].van der Heijden RA, Sheedfar F, Morrison MC, Hommelberg PPH, Kor D, Kloosterhuis NJ, Gruben N, Youssef SA, de Bruin A, Hofker MH, Kleemann R, Koonen DPY, Heeringa P, Aging, 2015, 7, 256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Le Chatelier E, Nielsen T, Qin J, Prifti E, Hildebrand F, Falony G, Almeida M, Arumugam M, Batto J-M, Kennedy S, Leonard P, Li J, Burgdorf K, Grarup N, Jorgensen T, Brandslund I, Nielsen HB, Juncker AS, Bertalan M, Levenez F, Pons N, Rasmussen S, Sunagawa S, Tap J, Tims S, Zoetendal EG, Brunak S, Clément K, Doré J, Kleerebezem M, Kristiansen K, Renault P, Sicheritz-Ponten T, de Vos WM, Zucker J-D, Raes J, Hansen T, Consortium M, Bork P Wang J, Ehrlich SD, Pedersen O, Nature, 2013, 500, 541. [DOI] [PubMed] [Google Scholar]
- [40].Federico A, Dallio M, DI R. Sarno, Giorgio V, Miele L, Minerva Gastroenterol Dietol, 2017, 63, 337. [DOI] [PubMed] [Google Scholar]
- [41].Doddapattar P, Radović B, Patankar JV, Obrowsky S, Jandl K, Nusshold C, Kolb D, Vujić N, Doshi L, Chandak PG, Goeritzer M, Ahammer H, Hoefler G, Sattler W, Kratky D, Mol Nutr Food Res, 2013, 57, 1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Hanske L, Hussong R, Frank N, Gerhäuser C, Blaut M, Braune A, Mol Nutr Food Res, 2005, 49, 868. [DOI] [PubMed] [Google Scholar]
- [43].Rozalski M, Micota B, Sadowska B, Stochmal A, Jedrejek D, Wieckowska-Szakiel M, Rozalska B, Biomed Res Int, 2013, 2013, 101089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Stevens JF, Maier CS, Phytochem Rev, 2016, 15, 425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Gerhäuser C, Mol Nutr Food Res, 2005, 49, 827. [DOI] [PubMed] [Google Scholar]
- [46].Stompor M, Żarowska B, Molecules, 2016, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Cermak P, Olsovska J, Mikyska A, Dusek M, Kadleckova Z, Vanicek J, NYC O, Sigler K, Bostikova V, Bostik P, APMIS, 2017, 125, 1033. [DOI] [PubMed] [Google Scholar]
- [48].Nair AB, Jacob S, J Basic Clin Physiol Pharmacol, 2016, 7, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Kuribayashi H, Miyata M, Yamakawa H, Yoshinari K, Yamazoe Y, Eur J Pharmacol, 2012, 697, 132. [DOI] [PubMed] [Google Scholar]
- [50].Czyzewski BK, Wang D-N, Nature. 2012, 483, 494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Gerritsen J, Hornung B, Renckens B, van Hijum SAFT, Martins Dos Santos VAP, Rijkers GT, Schaap PJ, de Vos WM, Smidt H, PeerJ, 2017, 5, e3698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Ridlon JM, Kang D-J, Hylemon PB, J Lipid Res, 2006, 47, 241. [DOI] [PubMed] [Google Scholar]
- [53].Vlahcevic ZR, Pandak WM, Stravitz RT, Gastroenterol Clin North Am, 1999, 28: 1. [DOI] [PubMed] [Google Scholar]
- [54].Li T, Chiang JYL, Curr Opin Gastroenterol, 2015, 31, 159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Makki K, Froguel P, Wolowczuk I, ISRN Inflamm, 2013, 2013, 139239. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.