

Abstract
颅锁骨发育不全是一种罕见的常染色体显性遗传疾病,以骨骼及牙齿发育异常为特征。本文对1例颅锁骨发育不全病例进行报道,并经基因检测,证实了一个新的移码突变。
Keywords: 颅锁骨发育不全, 常染色体显性遗传, 基因突变
Abstract
Cleidocranial dysplasia is a rare autosomal dominant hereditary disease characterized by abnormal skeletal and dental development. In this work, a case of cleidocranial dysplasia is reported, and a new frameshift mutation is confirmed by gene detection.
Keywords: cleidocranial dysplasia, autosomal dominant inheritance, gene mutation
颅锁骨发育不全(cleidocranial dysplasia,CCD)也称Scheuthauer-Marie-Sainton综合征或颅锁发育不良,1871年由Scheuthauer首先准确地描述该综合征,其特征为发育异常的骨骼及牙齿,是一种常染色体显性遗传疾病。CCD的发生率约为1︰1 000 000,无种族及性别差异[1]。国内关于CCD的报道大多是关于临床表现和影像学诊断,有关基因突变检测的研究很少报道。本文描述1例CCD患者,经基因检测证实了一个新的移码突变,现报道如下。
1. 病例报告
患者,男,12岁,2018年2月以换牙迟缓5年为主诉于南昌大学附属口腔医院口腔正畸科就诊。经正畸科医生检查,要求拔除埋伏多生牙及滞留乳牙以促进恒牙萌出,转口腔颌面外科就诊。
患者身高142 cm,体重51.3 kg,智力正常。囟门迟闭(7~8岁闭合),前额突出,眼距宽,前倾鼻,鼻上翘,鼻梁塌陷,肩部窄小,胸部呈圆锥形,锁骨短小,仅在近胸骨处可扪及,双肩下垂并可在胸前并拢。口腔检查:双侧颜面部对称,直面型,腭穹窿高,混合牙列,恒牙仅16、26、36、46、31、41萌出,61脱落,51-55、62-65、72-75、82-85滞留,82松动Ⅱ度,前牙对刃关系,双侧后牙反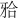 (图1)。
(图1)。
图 1. 口内照.

Fig 1 Pictures inside the mouth
A:正面观;B:右侧观;C:左侧观;D:上颌观;E:下颌观。
全景片示:11–15、21–25、32–35、42–45牙埋伏阻生,17、27、37、47牙未萌,4个象限可见多颗埋伏多生牙,滞留乳牙牙根未见明显吸收,下颌升支缩窄,前后缘几乎平行,异常细而尖(图2)。
图 2. 全景片.

Fig 2 Panoramic radiograph
头颅正侧位片:头颅增大增宽,颅缝增宽,面中部发育不足,直面型,测得SNA角69.7°,SNB角78.6°,ANB角−8.9°(图3)。胸片示:双侧胸廓对称,胸廓呈锥形,锁骨中远段缺如(图4)。
图 3. 头颅正位片(左)及侧位片(右).

Fig 3 Posteroanterior cephalogram (left) and lateral cephalogram (right)
图 4. 胸片.

Fig 4 Chest radiograph
高通量测序检测显示:Chr6(hg19)45459710位置Runx2基因c.722_725delTGTT;p.L241Sfs*8杂合变异,为新发突变(图5)。用先证者查证法对该CCD患者家系各成员进行全身健康及口腔专科检查,家系各成员未见明显异常。
图 5. 患者(上)与健康人(下)Runx2序列电泳图谱.

Fig 5 Electropherogram of the Runx2 sequence of the CCD patient (up) and healthy person (down)
口腔颌面外科医生和正畸科医生共同讨论手术方案:术前拍摄锥形束CT(图6),精确定位多生牙的数量及位置,确定4个象限多生牙一共16个,且多数位于恒牙胚的舌/腭侧,由于多生牙数量较多,且与恒牙胚联系紧密,为尽可能避免损伤恒牙胚,建议患者分次拔除埋伏多生牙及滞留乳牙,术后观察恒牙萌出情况,如仍无法萌出,考虑正畸牵引。手术过程:首先沿36—46牙舌侧龈缘切开翻瓣,充分暴露手术区,去骨,暴露埋伏多生牙,分牙,分块取出,依次拔除下颌9个埋伏多生牙,同时拔除松动的82牙;然后沿16—26牙腭侧牙龈缘切开翻瓣,去骨,暴露埋伏多生牙,共拔除5个埋伏多生牙及2个松动乳牙51、52(图7)。另外2个埋伏多生牙因与恒牙胚联系紧密,为避免损伤恒牙胚,考虑择期拔除。行牙龈成形术,严密缝合伤口,下颌舌侧切口处留置负压引流管,术后第3天拔除负压引流管,术后1周拆线,伤口愈合良好。术后半年随访,12、42牙已自行萌出。
图 6. 锥形束CT.

Fig 6 Cone-beam computed tomography
A:正面观;B:右侧观;C:左侧观。
图 7. 术中拔除的多生牙(左)及滞留乳牙(右).

Fig 7 Extracted supernumerary teeth (left) and retained deciduous teeth (right)
2. 讨论
CCD是一种遗传性的骨骼发育不良性疾病,可影响骨膜及软骨成骨。临床表现为身材矮小,囟门闭合延迟,前额突出,牙齿发育异常,双肩缩窄下垂,肩胛骨过度活动,肩膀能不同程度的靠近胸部。临床表现即使是在同一个家族中表型也各异,最轻的仅仅表现为牙齿的异常,严重的表现为骨骼明显异常。
CCD影像学表现为:锁骨缩短或缺失,颅骨以及盆骨延迟骨化[2]。锁骨完全缺失或发育不全,表现为一侧或双侧。10%的病例表现为锁骨完全缺失。CCD牙齿的影像学表现为乳牙滞留以及多个阻生的恒牙及多生牙。阻生牙可偶发含牙囊肿。尽管乳牙的发育很少会受影响,但乳牙的牙根吸收及脱落容易延迟。
CCD是一种发生于骨骼的常染色体显性遗传疾病,具有高外显率和不同的表型,20%~40%的病例为偶发[2]。位于6号染色体6p21.1位点的成骨细胞特异性转录因子Runx2基因突变是CCD的致病原因[3]。Runx2基因是一个骨骼主转录因子,对于成骨细胞的分化和骨骼发育具有关键作用[4]。研究[5]还表明,Runx2通过参与成牙本质细胞的分化、成釉器的形成及牙板的增殖,在牙齿发育中扮演着重要的角色。这些功能的破坏从而导致牙本质异常。研究[6]表明,Runx2基因突变形式超过90种,包括缺失、插入、易位、错义、移码和剪接突变。本研究中先证者为722位点出现移码突变,722cDNA位点T的缺失导致241密码子亮氨酸残基顺延移码,从而导致249密码子异常地终止,而非正常的密码子。这是在中国人CCD患者中检测到的新发突变。本研究家系中只有先证者本人受累,家系其他成员表型正常,考虑基因突变为偶发性,偶发后呈常染色体显性遗传。
对于CCD患者的治疗需要长期、详细的治疗计划及多学科团队协作。对于青少年CCD患者,应该拔除乳牙及多生牙,对阻生恒牙进行外科开窗,正畸导萌是首选治疗。覆盖在恒牙表面的骨质也要去除,因为以往的研究[1],[7]表明CCD患者牙槽骨有异常致密的骨小梁。牙根发育完成2/3对于促进牙齿的萌出较为合适。CCD患者的牙齿发育年龄与患者实足年龄间有近3年的差异。外科手术前拍摄CBCT有利于精确定位阻生的多生牙。正畸治疗使用微型种植螺钉牵引阻生牙能够减少CCD患者的治疗时间[8]。对于颌骨已经完全发育的成年人,种植牙及固定义齿是成年CCD患者在拔除多个牙齿之后的首选治疗方法。面中部的缺陷通过正颌外科能够得到很好的纠正[9]。
尽管CCD伴有各种骨骼的发育异常,然而CCD患者常常因牙齿问题首先在口腔科就诊。因此,口腔科医生必须能够及时诊断CCD,给出一个多学科综合治疗的计划并加以实施,从而提高CCD患者的生活质量。
Footnotes
作者声明本文无利益冲突。
References
- 1.Becker A, Shteyer A, Bimstein E, et al. Cleidocranial dysplasia: part 2—treatment protocol for the orthodontic and surgical modality[J] Am J Orthod Dentofac Orthop. 1997;111(2):173–183. doi: 10.1016/s0889-5406(97)70213-0. [DOI] [PubMed] [Google Scholar]
- 2.McNamara CM, O'Riordan BC, Blake M, et al. Cleidocranial dysplasia: radiological appearances on dental panoramic radiography[J] Dentomaxillofac Radiol. 1999;28(2):89–97. doi: 10.1038/sj/dmfr/4600417. [DOI] [PubMed] [Google Scholar]
- 3.Lee B, Thirunavukkarasu K, Zhou L, et al. Missense mutations abolishing DNA binding of the osteoblast-specific transcription factor OSF2/CBFA1 in cleidocranial dysplasia[J] Nat Genet. 1997;16(3):307–310. doi: 10.1038/ng0797-307. [DOI] [PubMed] [Google Scholar]
- 4.Jaruga A, Hordyjewska E, Kandzierski G, et al. Cleidocranial dysplasia and Runx2-clinical phenotype-genotype correlation[J] Clin Genet. 2016;90(5):393–402. doi: 10.1111/cge.12812. [DOI] [PubMed] [Google Scholar]
- 5.Camilleri S, McDonald F. Runx2 and dental development[J] Eur J Oral Sci. 2006;114(5):361–373. doi: 10.1111/j.1600-0722.2006.00399.x. [DOI] [PubMed] [Google Scholar]
- 6.Shen Z, Zou CC, Yang RW, et al. Cleidocranial dysplasia: report of 3 cases and literature review[J] Clin Pediatr (Phila) 2009;48(2):194–198. doi: 10.1177/0009922808323107. [DOI] [PubMed] [Google Scholar]
- 7.Becker A, Lustmann J, Shteyer A. Cleidocranial dysplasia: part 1—general principles of the orthodontic and surgical treatment modality[J] Am J Orthod Dentofacial Orthop. 1997;111(1):28–33. doi: 10.1016/s0889-5406(97)70298-1. [DOI] [PubMed] [Google Scholar]
- 8.Kuroda S, Yanagita T, Kyung HM, et al. Titanium screw anchorage for traction of many impacted teeth in a patient with cleidocranial dysplasia[J] Am J Orthod Dentofac Orthop. 2007;131(5):666–669. doi: 10.1016/j.ajodo.2005.06.026. [DOI] [PubMed] [Google Scholar]
- 9.Hwang SM, Park B, Hwang MK, et al. Aesthetic facial correction of cleidocranial dysplasia[J] Arch Craniofac Surg. 2016 doi: 10.7181/acfs.2016.17.2.82. [DOI] [PMC free article] [PubMed] [Google Scholar]