

Abstract
Perhaps the most intriguing scientific question about mammalian prions is how these proteinaceous entities encode and propagate infectivity. Over the past decade, our laboratory has taken a reductionist biochemical approach to study this challenging question. Our studies have resulted in the identification of endogenous phospholipid and polyanionic cofactor molecules that facilitate prion formation in vitro. Using these cofactor molecules, we have been able to produce purified, chemically defined prions with high levels of specific infectivity for wild type animal hosts. Our most recent studies suggest that cofactor molecules may also play crucial roles in maintaining the infectious conformation and strain properties of mammalian prions. The ability to produce high titer prions in vitro using cofactors provides an unprecedented opportunity to study the structural mechanism of infectious prion formation directly.
Keywords: prion, cofactors, phosphatidylethanolamine, phospholipids, RNA
Introduction
Mammalian prions are unusual infectious agents known to lack a replicating nucleic acid genome. The unorthodox mechanism by which prions are able to reproduce exponentially in infected hosts has therefore attracted much scientific interest.
The conformational change of the normal, host-encoded, cellular, prion protein, PrPC, into a misfolded conformer, PrPSc, is the key in prion formation pathway, and PrPSc appears to be a critical component of infectious prions.1 Therefore, the mechanism responsible for PrPC→PrPSc conversion and the molecular composition and structure of infectious prions are important topics of investigation. This mini-review describes work relevant to these topics performed in our lab over the past decade, some of which was presented at the Prion 2013 conference in Alberta.
For many years, prion conversion could only be studied in animal and cell culture models, and early attempts to replicate mammalian prions in vitro using a variety of approaches were reportedly unsuccessful.2 In pioneering work, Caughey and colleagues developed a cell-free assay in which PrPC could be converted into PrPSc in vitro.3-5 In this system, purified, radiolabeled PrPC could be converted in PrPSc in a highly specific manner by seeding with a stoichiometric excess of unlabeled PrPSc. Later, Soto and colleagues made a critical advance with their innovative development of the protein misfolding cyclic amplification (PMCA) method, in which PrPSc and prion infectivity could be substantially amplified in vitro by mixing crude normal brain homogenates containing PrPC with sub-stoichiometric amounts of PrPSc seed, and subjecting the mixture to cycles of intermittent sonication.6,7
Struck by the vast difference in conversion efficiency between PMCA of crude brain homogenate substrates and the cell-free conversion of purified PrPC, we considered the possibility that unidentified components within the crude brain homogenate other than PrPC were required for the formation of PrPSc and prion infectivity in vitro. However, an alternative explanation for the high efficiency observed in PMCA reactions is sonication alone is responsible for promoting prion replication in vitro. To confirm that brain cofactors play a role in the efficiency of PMCA, we tested whether PrPSc could be amplified in mixtures of crude and prion-infected brain homogenates without sonication. Our results indicated that, although sonication accelerates PrPSc formation, it is not required for PrPSc amplification in mixtures of crude brain homogenates, and therefore such homogenates must contain cofactors other than PrPC, which are necessary for efficient PrPSc formation.8
Identification of RNA as an Endogenous Cofactor for Hamster PrPSc Propagation
It had been previously suggested on the basis of indirect genetic data and theoretical molecular models that prion propagation might employ a hypothetical “Protein X” catalyst.9-12 However, no protein with prion catalyzing activity has been isolated biochemically, and indeed the results of biochemical studies argue against the existence of a protein catalyst.13,14 We therefore adopted an unbiased, reductionist approach to identify the in vitro prion propagation cofactors present in brain homogenate.
To provide a source of cofactors that could to facilitate the conversion of purified PrPC, we used Prnp0/0 (PrP gene knockout) brain homogenate to reconstitute PrPSc amplification reactions. Early studies employing this reconstitution system immediately confirmed that in vitro PrPSc amplification requires the addition of factors other than PrPC, since purified native PrPC was unable to amplify PrPSc in seeded reactions, but a mixture of PrPC and Prnp0/0 brain homogenate successfully amplified PrPSc >10-fold in parallel reactions.8
To identify the cofactor molecules in Prnp0/0 brain homogenate, we subjected crude homogenate substrate to a variety of chemical and enzymatic treatments known to inactivate specific classes of biological molecules, and characterized the effects of these treatments on PrPSc amplification. To our surprise, these experiments revealed that treating brain homogenate substrate with enzymes able to degrade single-stranded RNA could abolish amplification of hamster PrPSc molecules, and addition of RNA could reconstitute PrPSc amplification in pretreated homogenates.15 These unexpected results led us to test a wide variety of polyanions for their ability to stimulate hamster PrPSc amplification in vitro. We discovered that single-stranded nucleic acids at least 40 bases in length were more potent and effective stimulators than other classes of polyanions, such as glycosaminoglycans.16,17 Additional tests using RNA preparations isolated from various animal species as well as a series of homopolymeric nucleic acids indicated that no specific nucleotide sequence is required for efficient stimulation, although some sequences (such as poly-purines) are unable to support PrPSc amplification.16
Formation of Infectious Prions from Minimal Components
Because it is known that PrPC is an essential substrate for PrPSc formation, we began our work by developing methods to solubilize and separate PrPC from the other components of the brain homogenate. An important consideration was to keep PrPC, which is inherently unstable, in its native conformation throughout the isolation process. This required identifying specific detergents and chromatographic techniques that could maintain the ability of purified PrPC to convert into PrPSc.15,16,18 The use of purified, native PrPC rather than bacterially expressed recombinant (rec)PrP as the substrate for our initial studies was advantageous because PrPC is a more robust substrate than refolded recPrP, especially in the setting of reconstitution with crude homogenate.19 The use of PrPC substrate also helped us avoid unexpected potential pitfalls associated with using a substrate lacking post-translational modifications, such as the relatively poor ability of unglycosylated hamster PrP to convert into PrPSc 19.
Our purification scheme produced hamster PrPC that was devoid of other proteins and nucleic acids, as determined by mass spectroscopy and radioactive end labeling techniques, respectively. However, unidentified species of lipid molecules containing 20-carbon fatty acids were detected by mass spectroscopy at levels stoichiometric with PrPC in the immunopurified preparation.15
In serial (s)PMCA reactions seeded in the initial round with infectious prions, purified PrPC alone could not convert into propagate the PrPSc conformation.15 However, addition of synthetic homopolymeric poly(A) RNA to the substrate cocktail enabled efficient (conversion efficiency ~75–100%) and indefinite serial PrPSc propagation, confirming that RNA is a critical cofactor for hamster prion formation in vitro. Importantly, end-point titration (bioassay of serial dilutions of inoculum) data indicated that the PrPSc molecules formed after multiple rounds of sPMCA in the minimal substrate cocktail (PrPC, synthetic poly(A) RNA, and co-purified lipid) was infectious to wild type hosts, with a specific infectivity of ~2 × 105 LD50/µg PrP.15 This result represented the first time that the composition of bona fide infectious prions could be chemically defined (and be specifically shown to lack informational nucleic acids), and therefore provided the first chemical proof against a viral etiology of prion disease.
De novo Formation of Infectious Prions In Vitro
Most cases of human prion diseases arise sporadically in the absence of germ line mutations in the PrP sequence or exposure to infectious prions. There are many potential explanations for how sporadic disease might arise, including somatic mutation or exposure to an environmental factor that is able to trigger PrP conformational change in neurons. As part of our study showing that infectious hamster prions could be formed from minimal components, we examined whether PrPSc molecules and prion infectivity could be formed spontaneously (without initial seeding) in a rigorously prion-free environment. We observed that PrPSc was generated stochastically when PrPC was mixed with poly(A) RNA (Fig. 1), and bioassays demonstrated for the first time that prions possessing significant levels of specific infectivity for wild type hosts could be produced de novo.15 Similar results were subsequently obtained with crude brain homogenate20 and recPrP/cofactor substrates.21 Collectively, these results suggest that the pathophysiology of sporadic prion diseases such as Creutzfeldt-Jakob disease (CJD) may involve the stochastic conversion of PrPC into PrPSc, possibly facilitated by interaction with endogenous cofactor molecules.

Figure 1. Formation of PrPSc molecules de novo during serial PMCA propagation of unseeded purified substrates.15 western blot showing unseeded samples containing PrPC plus poly(A) RNA substrate subjected to 16 rounds of sPMCA, serial dilution, and propagation in a rigorously prion-free environment. A sample not subjected to proteinase K digestion is shown in the first lane as a reference for comparison of electrophoretic mobility (-PK). All other samples were subjected to limited proteolysis with proteinase K. Reproduced with permission from reference 15.
Identification of Phosphatidylethanolamine as a Promiscuous Prion Propagation Cofactor
PrPSc molecules derived from many different animal species and infectious prions strains can be amplified in vitro.22 When we compared the requirements for efficient PrPSc amplification in different animal species, we were surprised to find that RNA did not appear to be necessary for the amplification of either mouse or prairie vole PrPSc molecules.23 Instead, reconstitution experiments nuclease-treated Prnp0/0 brain homogenates revealed the existence of a novel membrane-bound prion propagation cofactor for these animal species. We were able to exploit the differential detergent solubility, thermostability, nuclease-resistance, and protease-resistance to partially purify this unknown cofactor.24 Conveniently, this partially purified cofactor preparation was also able to convert mouse recPrP substrate, which can be purified more easily than native PrPC, into recrPrPSc, presumably because, unlike hamster prions, mouse prions preferentially utilize unglycosylated substrate during PrPSc formation. Because the physical and chemical characteristics of the partially purified cofactor preparation resembled those of phospholipids, we tested whether the novel cofactor activity might be attributable to an endogenous phospholipid molecule. Both normal phase chromatography and phospholipase C digestion experiments confirmed that the active component of the purified cofactor preparation must be a phospholipid.24
To identify the unknown phospholipid, we used 31P nuclear magnetic resonance (NMR) and mass spectrometry to analyze the composition of the partially purified cofactor preparation. These analyses revealed that the preparation only contained 5 phospholipids: phosphatidylethanolamine (PE), phosphatidylcholine (PC), phosphatidylinositol, lyso PE, and lyso PC. Having a relatively short list of candidate molecules allowed us to test HPLC-purified preparations of each phospholipid individually for cofactor activity. These tests revealed that PE was the only phospholipid able to facilitate PrPSc propagation in reconstituted sPMCA assays (Fig. 2). Moreover, chemically synthesized PE served as an efficient solitary cofactor for PrPSc propagation.
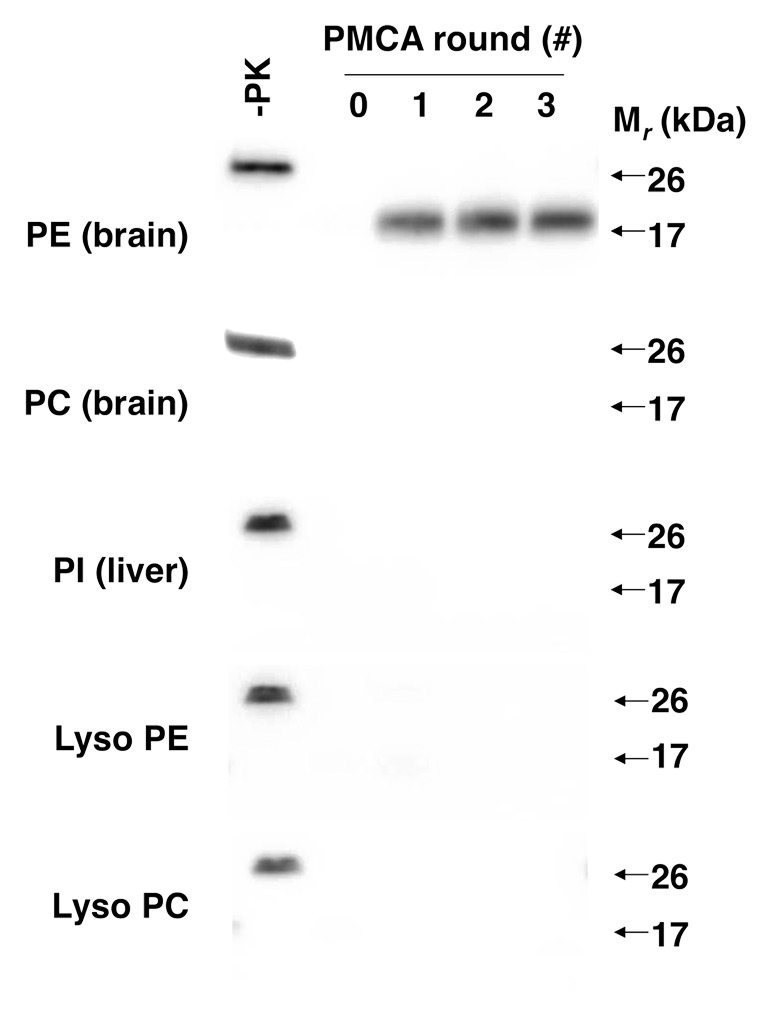
Figure 2. Effect of various phospholipids on prion formation.24 Western blots showing 3-round sPMCA reactions using recPrP substrate and seeded with recPrPSc template, supplemented with various commercial preparations of purified and synthetic phospholipids at 2.5 mM final concentration, as indicated.
Having identified PE as an endogenous cofactor for mouse PrPSc formation in vitro, we next tested whether it could facilitate PrPSc formation in other animal species. The results of these studies demonstrated that, unlike RNA, PE is a relatively promiscuous cofactor that was able to facilitate the propagation of prions from every animal species tested, including hamster.24 It is worth noting that higher concentrations of PE were required to facilitate hamster PrPSc propagation (compared with the concentration needed to facilitate mouse PrPSc propagation); therefore, there appear to be species-dependent preferences in cofactor potency.
Role of Cofactor Molecules in Forming and Maintaining Prion Infectivity
We were curious whether cofactor molecules are required for maintaining the infectious PrPSc conformation, i.e., is it possible to withdraw cofactor during serial propagation and continue to propagate PrPSc molecules and prion infectivity with only recPrP substrate? Our original expectation was that PrPSc propagation would simply cease, and this was observed in approximately half of the individual experiments. Surprisingly, there were also experiments in which PrPSc appeared to change conformation upon cofactor withdrawal (as evidenced by a ~2kDa shift in the molecular weight of the protease-resistant core), but nonetheless continued to propagate indefinitely as an autocatalytic, protease-resistant “protein-only” conformation25 (Fig. 3). Once formed, this “protein-only” PrPSc conformation did not revert to the cofactor-dependent PrPSc conformation by adding back PE in subsequent propagation cycles, suggesting that the cofactor is needed to maintain a specific meta-stable PrPSc structure.
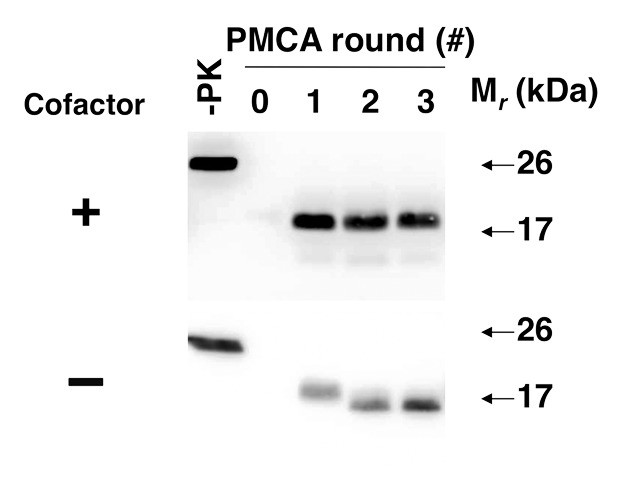
Figure 3. Adaptation of autocatalytic PrPSc molecules upon cofactor withdrawal.25 western blots of reconstituted sPMCA reactions. All reactions were initially seeded with cofactor PrPSc molecules, and subsequently propagated in substrate cocktails containing recPrP with or without cofactor, as indicated.
Our ability to generate and indefinitely propagate two alternative autocatalytic PrPSc conformers that derived from the same original template and which differ only in terms of their composition (i.e., “protein-only” or protein plus PE) enabled us to test the role played by the cofactor component in prion infectivity. We compared the specific infectivity of the two preparations by performing end-point titration bioassays of quantified amounts of protein in wild type mice. The results were striking (Table 1), and showed that recPrPSc molecules formed with PE have a specific infectivity of ~2 × 106 LD50/µg PrP, whereas even the most concentrated sample of the “protein-only” PrPSc inocula failed to cause scrapie (and therefore must have a level of specific infectivity at least 105-fold lower than cofactor-dependent PrPSc).25 We conclude from these findings that cofactor molecules likely play a crucial (and possibly structural) role in maintaining the infectious conformation of PrPSc.
Table 1. Bioassay of in vitro-generated recombinant PrPSc molecules in normal C57BL mice25.
| Inoculum | Dilution | n/n0 | IP (days)* |
|---|---|---|---|
| Cofactor PrPSc | 10−1 | 7/7 | 356 ± 12 |
| 10−2 | 3/3 | 451 ± 16 | |
| 10−3 | 4/4 | 481 ± 42 | |
| 10−4 | 4/4 | 501 ± 28 | |
| 10−5 | 1/3 | 539 | |
| 10−6 | 0/4 | >570 | |
| Protein Only PrPSc Sample A | 10−1 | 0/4 | >570 |
| 10−2 | 0/4 | >570 | |
| 10−3 | 0/4 | >570 | |
| 10−4 | 0/4 | >570 | |
| Protein Only PrPSc Sample B | 10−1 | 0/4 | >570 |
| 10−2 | 0/4 | >570 | |
| 10−3 | 0/4 | >570 | |
| 10−4 | 0/4 | >570 |
Mean incubation period (IP) of scrapie sick animals ± standard error.
Role of Cofactor Molecules in Maintaining Prion Strain Properties
Self-propagating mammalian prion strains are defined by their distinguishable clinical, neuropathological, and incubation time profiles in a single animal species host. In many cases, different strains are also associated with different conformations of PrPSc molecules.26,27 However, the underlying molecular mechanism responsible for these observations remains unknown.
The ability of PE to serve as a solitary cofactor for PrPSc propagation seeded by a variety of different prion strains provided us with a unique ability to test the hypothesis that cofactor molecules might be responsible for generating and maintaining prion strain diversity. We used three easily distinguishable mouse prion strains to seed sPMCA reactions containing only recPrP and PE substrate, and used standard strain-typing assays (scrapie incubation time, neuropathology, urea denaturation) to compare the strain properties of the input vs. output PrPSc molecules. Remarkably, the results showed that propagation in the presence of only one potential cofactor, PE, resulted in transformation of all three input strains into a single, novel strain.25 The output samples all exhibited a relatively long incubation time despite their high titer and relatively low PrPSc conformational stability. All three sets of output inocula also produced a uniform neuropathological profile that was markedly different from any of the three input strains (Fig. 4). Because it had previously been established that sPMCA reactions with crude and reconstituted brain homogenate substrates preserves the strain properties of mouse prions,22,28 the observed convergence of strain properties could be attributed to restricted cofactor availability, i.e., PE was the only cofactor available. It is worth noting that one of the input strains used (OSU) was a recombinant (i.e., recPrPSc) prion produced by sPMCA propagation with RNA and phosphatidylglycerol (PG).29 In this case, we can conclude that the chemically defined change from RNA/PG to PE alone produced an output recombinant prion with drastically different strain properties.

Figure 4. Regional neuropathology of infected mice.25 Profiles of vacuolation scores of animals inoculated with either input prions or PE PrPSc prions (produced by sPMCA propagation with recPrP and PE substrate). Prion strains: OSU, red squares; ME7, blue circles; 301C, green triangles. Brain regions: I-II, cerebral cortical layers 1 and 2; III-IV, cortical layers 3 and 4; V-VI, cortical layers 5 and 6; CC, cerebral cortex (all layers); H, hippocampus; T, thalamus; HT, hypothalamus; Mid, midbrain; BS, brain stem; Cb, cerebellum. The mean values are shown ± SEM.
Based on these experiments, we have proposed a “cofactor selection” hypothesis as a potential molecular mechanism for prion strain diversity and selective neurotropism.25 According to this hypothesis, each prion strain is associated with a unique PrPSc conformation that requires a specific set of cofactors to propagate efficiently. The ability of various types of cofactor molecules to propagate a given PrPSc conformation would presumably be based on how each cofactor specifically interacts with influences the folding pathway of PrP. Variation in the expression levels of such cofactors in different cell types and parts of the brain could explain the specific patterns of neurotropism exhibited by prion strains. In this scenario, each strain would naturally replicate most efficiently in cells that expressed high levels of the particular cofactor molecules preferred by that strain. This explanation is also consistent with experiments in which RNA depletion alters prion strain properties following brain homogenate PMCA30,31
Potential Relevance to Other Neurodegenerative Diseases
It has been suggested that prion-like mechanisms may be involved in the local spread of misfolded proteins in more commonly occurring neurodegenerative diseases, such as Alzheimer and Parkinson disease.32 However, it is unlikely that such diseases are medically infectious in the same manner as prion diseases because (1) intracerebral inoculation experiments in primates did not transmit any neurodegenerative disease other than prion disease,33 (2) epidemics of iatrogenic prion disease have not been accompanied by increases in the incidence of other neurodegenerative diseases (despite the higher overall prevalence of these diseases).34 It is an intriguing possibility that the unique infectivity of mammalian prions may result from their interactions with specific cofactor molecules.
Future Directions
Our in vitro prion propagation studies suggest that a wide variety of endogenous cofactor molecules (other than RNA and PE) are needed to encode the structural diversity of prion strains. It would be helpful to identify additional endogenous prion cofactors; especially those that are able facilitate the formation of medically relevant strains such as variant CJD.
The discovery that PE can be used to form recombinant prions in vitro with high specific infectivity will facilitate efforts to determine the structure of infectious PrPSc molecules using techniques such as solid-state nuclear magnetic resonance spectroscopy. Furthermore, it should also be possible to study the conversion pathway using a combination of biophysical techniques.
In summary, we already have learned much about the mechanism of infectious prion formation by applying a reductionist approach in vitro, including the essential role of cofactor molecules in maintaining prion infectivity and strain properties. Future studies should also provide interesting new details about the nature of prion cofactor molecules and the structural basis of prion formation.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgements
This work was funded by a research grants from the National Institutes of Health (2R01 NS046478).
References
- 1.Prusiner SB. . Prions. Proc Natl Acad Sci U S A 1998; 95:13363 - 83; http://dx.doi.org/ 10.1073/pnas.95.23.13363; PMID: 9811807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Raeber AJ, Borchelt DR, Scott M, Prusiner SB. . Attempts to convert the cellular prion protein into the scrapie isoform in cell-free systems. J Virol 1992; 66:6155 - 63; PMID: 1356161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bessen RA, Kocisko DA, Raymond GJ, Nandan S, Lansbury PT, Caughey B. . Non-genetic propagation of strain-specific properties of scrapie prion protein. Nature 1995; 375:698 - 700; http://dx.doi.org/ 10.1038/375698a0; PMID: 7791905 [DOI] [PubMed] [Google Scholar]
- 4.Kocisko DA, Come JH, Priola SA, Chesebro B, Raymond GJ, Lansbury PT, Caughey B. . Cell-free formation of protease-resistant prion protein. Nature 1994; 370:471 - 4; http://dx.doi.org/ 10.1038/370471a0; PMID: 7913989 [DOI] [PubMed] [Google Scholar]
- 5.Kocisko DA, Priola SA, Raymond GJ, Chesebro B, Lansbury PT Jr., Caughey B. . Species specificity in the cell-free conversion of prion protein to protease-resistant forms: a model for the scrapie species barrier. Proc Natl Acad Sci U S A 1995; 92:3923 - 7; http://dx.doi.org/ 10.1073/pnas.92.9.3923; PMID: 7732006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Saborio GP, Permanne B, Soto C. . Sensitive detection of pathological prion protein by cyclic amplification of protein misfolding. Nature 2001; 411:810 - 3; http://dx.doi.org/ 10.1038/35081095; PMID: 11459061 [DOI] [PubMed] [Google Scholar]
- 7.Castilla J, Saá P, Hetz C, Soto C. . In vitro generation of infectious scrapie prions. Cell 2005; 121:195 - 206; http://dx.doi.org/ 10.1016/j.cell.2005.02.011; PMID: 15851027 [DOI] [PubMed] [Google Scholar]
- 8.Lucassen R, Nishina K, Supattapone S. . In vitro amplification of protease-resistant prion protein requires free sulfhydryl groups. Biochemistry 2003; 42:4127 - 35; http://dx.doi.org/ 10.1021/bi027218d; PMID: 12680767 [DOI] [PubMed] [Google Scholar]
- 9.Telling GC, Scott M, Mastrianni J, Gabizon R, Torchia M, Cohen FE, DeArmond SJ, Prusiner SB. . Prion propagation in mice expressing human and chimeric PrP transgenes implicates the interaction of cellular PrP with another protein. Cell 1995; 83:79 - 90; http://dx.doi.org/ 10.1016/0092-8674(95)90236-8; PMID: 7553876 [DOI] [PubMed] [Google Scholar]
- 10.Kaneko K, Zulianello L, Scott M, Cooper CM, Wallace AC, James TL, Cohen FE, Prusiner SB. . Evidence for protein X binding to a discontinuous epitope on the cellular prion protein during scrapie prion propagation. Proc Natl Acad Sci U S A 1997; 94:10069 - 74; http://dx.doi.org/ 10.1073/pnas.94.19.10069; PMID: 9294164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zulianello L, Kaneko K, Scott M, Erpel S, Han D, Cohen FE, Prusiner SB. . Dominant-negative inhibition of prion formation diminished by deletion mutagenesis of the prion protein. J Virol 2000; 74:4351 - 60; http://dx.doi.org/ 10.1128/JVI.74.9.4351-4360.2000; PMID: 10756050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Perrier V, Kaneko K, Safar J, Vergara J, Tremblay P, DeArmond SJ, Cohen FE, Prusiner SB, Wallace AC. . Dominant-negative inhibition of prion replication in transgenic mice. Proc Natl Acad Sci U S A 2002; 99:13079 - 84; http://dx.doi.org/ 10.1073/pnas.182425299; PMID: 12271119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee CI, Yang Q, Perrier V, Baskakov IV. . The dominant-negative effect of the Q218K variant of the prion protein does not require protein X. Protein Sci 2007; 16:2166 - 73; http://dx.doi.org/ 10.1110/ps.072954607; PMID: 17766375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Geoghegan JC, Miller MB, Kwak AH, Harris BT, Supattapone S. . Trans-dominant inhibition of prion propagation in vitro is not mediated by an accessory cofactor. PLoS Pathog 2009; 5:e1000535; http://dx.doi.org/ 10.1371/journal.ppat.1000535; PMID: 19649330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Deleault NR, Harris BT, Rees JR, Supattapone S. . Formation of native prions from minimal components in vitro. Proc Natl Acad Sci U S A 2007; 104:9741 - 6; http://dx.doi.org/ 10.1073/pnas.0702662104; PMID: 17535913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Deleault NR, Geoghegan JC, Nishina K, Kascsak R, Williamson RA, Supattapone S. . Protease-resistant prion protein amplification reconstituted with partially purified substrates and synthetic polyanions. J Biol Chem 2005; 280:26873 - 9; http://dx.doi.org/ 10.1074/jbc.M503973200; PMID: 15917229 [DOI] [PubMed] [Google Scholar]
- 17.Geoghegan JC, Valdes PA, Orem NR, Deleault NR, Williamson RA, Harris BT, Supattapone S. . Selective incorporation of polyanionic molecules into hamster prions. J Biol Chem 2007; 282:36341 - 53; http://dx.doi.org/ 10.1074/jbc.M704447200; PMID: 17940287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nishina K, Deleault NR, Lucassen RW, Supattapone S. . In vitro prion protein conversion in detergent-solubilized membranes. Biochemistry 2004; 43:2613 - 21; http://dx.doi.org/ 10.1021/bi035889l; PMID: 14992599 [DOI] [PubMed] [Google Scholar]
- 19.Nishina KA, Deleault NR, Mahal SP, Baskakov I, Luhrs T, Riek R, Supattapone S. . The stoichiometry of host PrPC glycoforms modulates the efficiency of PrPSc formation in vitro. Biochemistry 2006; 45:14129 - 39; http://dx.doi.org/ 10.1021/bi061526k; PMID: 17115708 [DOI] [PubMed] [Google Scholar]
- 20.Barria MA, Mukherjee A, Gonzalez-Romero D, Morales R, Soto C. . De novo generation of infectious prions in vitro produces a new disease phenotype. PLoS Pathog 2009; 5:e1000421; http://dx.doi.org/ 10.1371/journal.ppat.1000421; PMID: 19436715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang F, Wang X, Ma J. . Conversion of bacterially expressed recombinant prion protein. Methods 2011; 53:208 - 13; http://dx.doi.org/ 10.1016/j.ymeth.2010.12.013; PMID: 21176786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Castilla J, Morales R, Saá P, Barria M, Gambetti P, Soto C. . Cell-free propagation of prion strains. EMBO J 2008; 27:2557 - 66; http://dx.doi.org/ 10.1038/emboj.2008.181; PMID: 18800058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Deleault NR, Kascsak R, Geoghegan JC, Supattapone S. . Species-dependent differences in cofactor utilization for formation of the protease-resistant prion protein in vitro. Biochemistry 2010; 49:3928 - 34; http://dx.doi.org/ 10.1021/bi100370b; PMID: 20377181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Deleault NR, Piro JR, Walsh DJ, Wang F, Ma J, Geoghegan JC, Supattapone S. . Isolation of phosphatidylethanolamine as a solitary cofactor for prion formation in the absence of nucleic acids. Proc Natl Acad Sci U S A 2012; 109:8546 - 51; http://dx.doi.org/ 10.1073/pnas.1204498109; PMID: 22586108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Deleault NR, Walsh DJ, Piro JR, Wang F, Wang X, Ma J, Rees JR, Supattapone S. . Cofactor molecules maintain infectious conformation and restrict strain properties in purified prions. Proc Natl Acad Sci U S A 2012; 109:E1938 - 46; http://dx.doi.org/ 10.1073/pnas.1206999109; PMID: 22711839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bruce ME. . Scrapie strain variation and mutation. Br Med Bull 1993; 49:822 - 38; PMID: 8137131 [DOI] [PubMed] [Google Scholar]
- 27.Carlson GA. . Prion strains. Curr Top Microbiol Immunol 1996; 207:35 - 47; http://dx.doi.org/ 10.1007/978-3-642-60983-1_4; PMID: 8575205 [DOI] [PubMed] [Google Scholar]
- 28.Piro JR, Harris BT, Nishina K, Soto C, Morales R, Rees JR, Supattapone S. . Prion protein glycosylation is not required for strain-specific neurotropism. J Virol 2009; 83:5321 - 8; http://dx.doi.org/ 10.1128/JVI.02502-08; PMID: 19297485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang F, Wang X, Yuan CG, Ma J. . Generating a prion with bacterially expressed recombinant prion protein. Science 2010; 327:1132 - 5; http://dx.doi.org/ 10.1126/science.1183748; PMID: 20110469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Saá P, Sferrazza GF, Ottenberg G, Oelschlegel AM, Dorsey K, Lasmézas CI. . Strain-specific role of RNAs in prion replication. J Virol 2012; 86:10494 - 504; http://dx.doi.org/ 10.1128/JVI.01286-12; PMID: 22811520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gonzalez-Montalban N, Lee YJ, Makarava N, Savtchenko R, Baskakov IV. . Changes in prion replication environment cause prion strain mutation. FASEB J 2013; 27:3702 - 10; http://dx.doi.org/ 10.1096/fj.13-230466; PMID: 23729586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fernández-Borges N, Eraña H, Elezgarai SR, Harrathi C, Gayosso M, Castilla J. . Infectivity versus Seeding in Neurodegenerative Diseases Sharing a Prion-Like Mechanism. Int J Cell Biol 2013; 2013:583498; http://dx.doi.org/ 10.1155/2013/583498; PMID: 24187553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brown P, Gibbs CJ Jr., Rodgers-Johnson P, Asher DM, Sulima MP, Bacote A, Goldfarb LG, Gajdusek DC. . Human spongiform encephalopathy: the National Institutes of Health series of 300 cases of experimentally transmitted disease. Ann Neurol 1994; 35:513 - 29; http://dx.doi.org/ 10.1002/ana.410350504; PMID: 8179297 [DOI] [PubMed] [Google Scholar]
- 34.Brown P, Preece M, Brandel JP, Sato T, McShane L, Zerr I, Fletcher A, Will RG, Pocchiari M, Cashman NR, et al. . Iatrogenic Creutzfeldt-Jakob disease at the millennium. Neurology 2000; 55:1075 - 81; http://dx.doi.org/ 10.1212/WNL.55.8.1075; PMID: 11071481 [DOI] [PubMed] [Google Scholar]