

Abstract
Prion diseases are consistently associated with prion protein (PrPC) misfolding rendering a cascade of auto-catalytic self-perpetuation of misfolded PrP in an afflicted individual. The molecular process is intriguingly similar to all known amyloid diseases both local and systemic. The prion disease is also infectious by the transfer of misfolded PrP from one individual to the next. Transmissibility is surprisingly efficient in prion diseases and given the rapid disease progression following initial symptoms the prionoses stand out from other amyloidoses, which all may be transmissible under certain circumstances. The nature of the infectious prion as well as the genotype of the host is important for transmissibility. For hitherto unexplained reasons the majority of Europeans carry a missense mutation on one or both alleles of the PrP gene (PRNP), and hence express a variant of PrP with a substitution for valine (V) instead of methionine (M) in position 129. In fact the 129M/V variant is very common in all populations except for the Japanese. Sporadic Creutzfeldt-Jakob disease is a disease rarely striking people below the age of 60, where homozygosity especially 129MM is a very strong risk factor. Paradoxically, the 129M/V polymorphism suggestive of heterozygote advantage is one of the most clear cut disease associated traits of the human population, yet prion disease is extraordinarily rare. The genetic basis for how this trait spread with such prevalence within human populations is still target to investigations and deserves attention. This short essay represents a somewhat provocative hypothetical notion of a possible ancient significance of this polymorphism.
Keywords: prion, polymorphism, 129, hyperdisease, panzootic, extinction, Pleistocene, megafauna
The prion protein (PrP) is most abundant in mammalian neurons but is ubiquitously expressed throughout various cells and tissues. The functional role of native PrP is not fully understood. PrP is associated with a number of different prionoses; sporadic, inherited, and acquired, all of which are invariably fatal. Prion diseases show a rapid progression following presentation of symptoms. Nevertheless, prion diseases can remain dormant for decades prior to any outbreak of disease. This is one of many confounding aspects of these neurodegenerative diseases.
The common molecular pathognomonic marker for prionoses is the presence of severe vacuolation within the CNS rendering a sponge like brain tissue. Concomitant with the presence of spongiosis, a conformational isoform of PrP appears. The largely helical globular protein PrPC has converted into an aggregation prone β-sheet conformation, PrPSc, that often assemble into protein deposits with obvious similarities to amyloid.1
Several inherited prion disease-causing point mutations are found in the human PrP gene (PRNP).2,3 There are in addition two nonpathogenic polymorphisms, methionine, or valine in position 129 (M129V) and glutamic acid or lysine at codon 219 (E219K).4,5 These polymorphisms are not directly pathogenic but rather on the contrary hetrozygosity at either of these positions is associated with resistance to sporadic Creutzfeldt-Jakob disease (sCJD).6,7 The polymorphism in position 129 is also associated with sCJD disease phenotype and influences the molecular type of prion strains.8 Individuals homozygous at position 129 are more susceptible to infectious prion disease such as Kuru, variant CJD (vCJD) from Bovine spongiform encephalopathy (BSE), and iatrogenic CJD (iCJD).9-14 A recent large scale assessment of the silent carrier frequency of vCJD in Great Britain from histological analysis of appendices suggests that potentially 30 000 people are infected with BSE.15 Notably a higher number of 129V homozygous cases were observed than would be expected from its distribution in the whole population.
Taken together there is abundant evidence that position 129 in PrP is a key site for prion disease susceptibility and hence by virtue with PrP misfolding.
Recently we investigated the molecular basis for the dependency for misfolding of PrP as a consequence of variations in position 129.16 The basis for selection of this site for mutagenesis was obvious from its strong genetic link to prion disease as discussed above. Our data was based on the in vitro behavior of PrP in solution and showed that position 129 was an important site for recruitment of PrP into the amyloid state. Charge mutations (M129E and M129K) showed decreased spontaneous conversion propensity (nucleation) compared with the wild type. We also observed that M129C was totally resistant toward spontaneous engulfment of PrP into the amyloid fold, most likely due to the formation of an intermolecular disulfide bridge forming in the lag phase, rendering a non-convertible disulfide linked dimer effectuated by the 129CC′ interaction.
In contrast to the rather large differences in fibrillation propensity found for extreme amino acid substitutions such as tryptophan, proline, cysteine (see above), and charge (see above), quite minor changes were observed for conservative mutations M129L and M129A. M129V was also similar to 129M albeit presenting slower fibril growth rate (Fig. 1). Cross-seeding with either pure 129M or M129V also recapitulated the notion that valine in position 129 less efficiently incorporates into the amyloid fibril fold and is a less efficient seed for 129M (Fig. 1). Our observations hence underlines that position 129 has profound influence on the molecular misfolding mechanism of PrP.
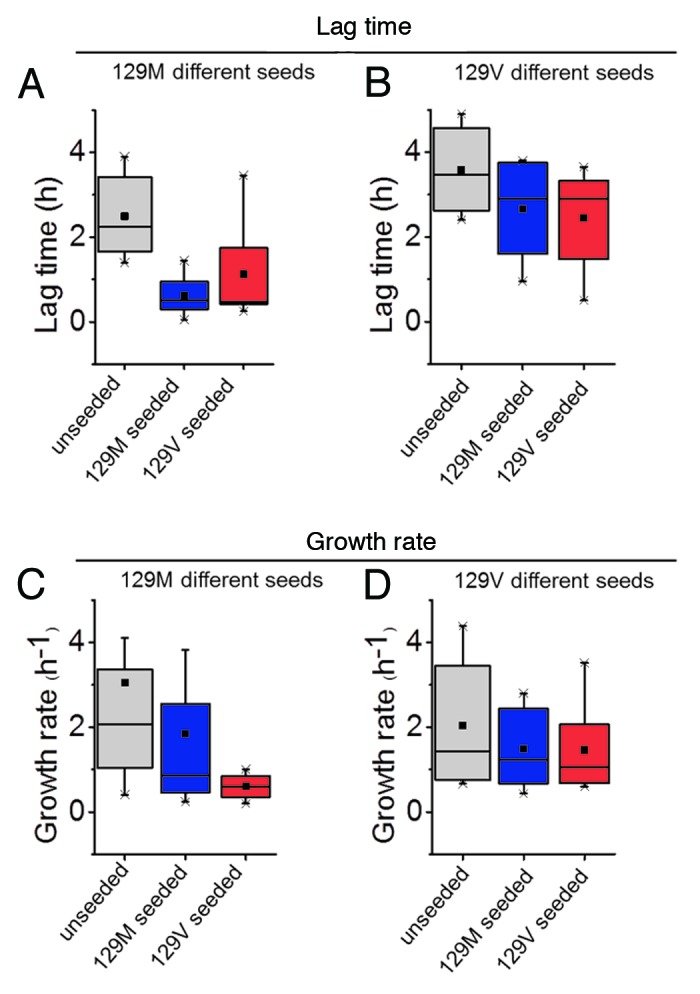
Figure 1. Modulation of fibril formation of rHuPrP90–231 by the M129V mutant. (A) Lag phase and (C) growth rate of 129M spontaneous (gray), seeded with 129M (blue) and seeded with 129V (red). (B) Lag phase and (D) Growth rate of 129V spontaneous (gray), seeded with 129V (blue) and seeded with 129M (red). All data represent ThT fibril formation kinetics in vitro (n = 6), in the native condition conversion assay described in Almstedt et al., 2009.60
Heterozygote Advantage in Infectious Disease
Heterozygote advantage has been advocated in the proliferation of diseased genes as resistance traits against infectious human diseases. One well known example within the field of protein aggregation is the Sickle cell Malaria hypothesis (reviewed in17). Carriers of the HbS trait (β globin E6V mutant which polymerizes from the native structure in low oxygen conditions) in Western Africa appear to be resistant against Malaria. In this case the selective pressure has been favoring against the invading parasite Plasmodium Falciparum (PF). In short the selective pressure has been fortunate for heterozygote carriers of HbS that carry one copy of the hemoglobin HbβE6V gene which minimizes the chance for the PF parasite to feed and proliferate on hemoglobin molecules in the erythrocytes, whereas the heterozygote carrier has near normal oxygen uptake due to the non-polymerizable wild type β globin. The Sickle Cell Malaria hypothesis is very old but has gained momentum18 and has recently gained strong support in the literature.19
The HbS case is highly informative on how genetic proliferation of a deleterious trait came about. What is most remarkable is that the HbS gene has so many carriers despite the dramatic harmful effects presented for a homozygote, which results in Sickle cell disease which is a very serious disease debilitating and shortening the life of some 300 000 people in Africa.19 Conclusively this example demonstrates that evolutionary adaptation even allows for such a deleterious trait to proliferate in order to avoid infectious disease, especially in the young.18
Heterozygote Advantage in Amyloid Disease
Regardless of the epidemiologic and genetic knowledge that can be gained from this classic example, the HbS protein does not cause a misfolding disease. Hence the molecular backdrop is somewhat different from that of a protein which changes conformation in order to be pathogenic. Are there any examples of heterozygote advantage in amyloid disease?
Familial amyloidotic polyneuropathy (FAP) is an autosomal dominant systemic amyloid disease where point mutations in gene coding for the homotetrameric protein transthyretin (TTR) afford a progressive polyneuropathy with presentations of symptoms between the ages of 20–70 depending on the point mutation. It has been shown that TTR amyloidosis is correlated with kinetic and thermodynamic conformational stability of the native protein,20 as well as with efficiency of the cellular endoplasmic reticulum folding and degradation control machinery.21 For TTR, tetramer dissociation into monomers is a prerequisite for misfolding and amyloid formation. Serendipitously the most common FAP mutation is a valine to methionine mutation in position 30, V30M. Portugal is an endemic site for FAP. Usually Portuguese patients carrying the V30M mutation present disease in their mid-30s, with an 80–90% penetrance.22 However, some particular V30M carriers did not present disease, and it was revealed that these carriers were compound heterozygote for another mutation, T119M on the second allele.23 This finding triggered molecular studies of this apparent heterozygote advantage. Because transthyretin is a homotetramer, mixed tetramers between mutants and wild type in normal heterozygote carriers as well as between V30M and T119M in compound heterozygotes form in vivo. Importantly mixed tetramers of V30M and T119M, as opposed to V30M and wild type, were not prone to misfolding and amyloidogenesis, rather the inclusion of merely one subunit of the T119M was enough to halt this process.24,25 Mechanistically it was shown that this feature was due to kinetic partitioning of T119M containing tetramers toward the native folded state which presented elevated dissociation barriers for the rate determining step for amyloidogenesis.25 TTR amyloidosis from wt protein is also a prevalent feature of cardiac amyloid in men above 60 years of age.26 A recent Danish epidemiological study further support heterozygote advantage in T119M carriers with wt TTR on the second allele, herein these carriers were at lower risk for ischemic disease.27 Hence the TTR T119M mutation represents the first molecular evidence for heterozygote advantage for two amyloid diseases where the mechanism entails shifting the kinetics toward native folded tetrameric protein.
Heterozygote Advantage in Prion Disease
The Collinge group has thoroughly shown that there has been a strong selective pressure on the 129 M/V PrP polymorph.14 The basis for negative selection away from 129MM homozygotes was suggested to originate from Kuru-like prehistoric epidemics. The rationale for this was beautifully demonstrated from the modern time experience from Kuru in Papua New Guinea where such selective pressure promoting 129MV heterozygosity was observed.14 The origin for the M129V variant in the human population was likely very early and was approximated to appear in early humans 500 000 years BP.14 What is remarkable is its current wide spread.
Position 129 in PrP is located in the center of the initial β–strand 1 in the globular domain of PrP (Fig. 2A). Our data on the respective mutations in HuPrP showed that conserved mutations (M129L, M129V) did not influence thermodynamic stability of the protein whereas introducing the β-sheet breaker proline in M129P was severely destabilizing implicating importance of β-strand 1 for conformational stability of PrP, and hence for retaining function. PrP is evolutionary well conserved but there are variable regions in the protein.28 The PrP sequence from many organisms retains a methionine in the position corresponding to 129 in the human sequence, i.e., this particular region appears to be extremely highly conserved in mammals (Table 1) and only deviates when moving toward reptilians and avians (Table 2 and Fig. 2B), while the overall fold is remarkably similar. Hence, in contrast to what is observed for humans, selective pressure in the wild appears non-prevalent on this site. Hence if this is an example of heterozygote advantage for prion disease resistance it would implicate that it is a trait of pure human breeding.
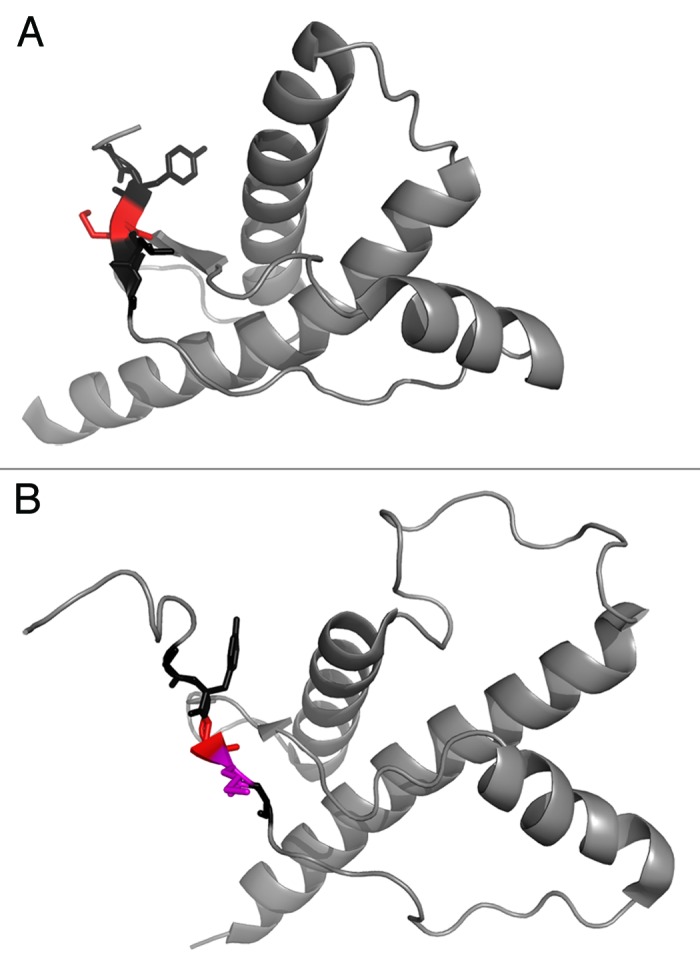
Figure 2. (A) Human PrP 90–230 structure from Zahn et al., 2000.61 PDB entry 1QM1. The sequence 127–131 is highlighted where residue 129 is colored red. (B) Chicken PrP 119–230 structure from Calzolai et al., 2005.62 PDB entry 1U3M. The sequence 127–131 is highlighted where residue 129 is colored red and residue 130 is colored in magenta. Sequence numbering according to human sequence.
Table 1. Mammals.
| Species | PrP sequence (127–131)* |
|---|---|
| Human (Homo Sapiens) | GYMLG |
| Cattle, bovine (Bos Taurus) | GYMLG |
| Black Rhinoceros (Diceros bicornis) | GYMLG |
| Elk (Alces alces) | GYMLG |
| Indian Elephant (Elephas maximus) | GYMLG |
| Mule deer (Odocoileus hemionus) | GYMLG |
| White tailed Deer (Odocoileus virginianus) | GYMLG |
Numbering according to human sequence.
Table 2. Avians.
| Species | PrP sequence (127–131)* |
|---|---|
| Chicken (Gallus gallus) | GYAMG |
| Common turkey (Meleagris gallopavo) | GYAMG |
| Green peafowl (Pavo muticus) | GYALG |
| Ostrich (Struthio camelus) | GYVMG |
Numbering according to human sequence
The molecular basis for heterozygote advantage in 129M/V is still unaccounted for.29 Nevertheless it is reasonable to assume that recruitment of dissimilar local sequences at residue of 129 or exposure of cryptic epitopes elsewhere in the PrP molecule modulated by residue 129 are operational during oligomerization of PrP in the conversion cascade. Our in vitro data implicate that M129V is less efficient as a seed and less susceptible toward seeded growth (Fig. 1) which suggest that prion replication is hampered in M129V carriers.16
This impaired molecular conversion is reminiscent of the high kinetic barriers dissociation of the native tetramer shown for TTR T119M suppressor mutant carriers resistant for FAP. Our data hence suggest that elevated kinetic barriers for PrP conversion in M129V is a plausible molecular mechanism for heterozygote advantage.
The Superprion
Is the heterozygote advantage of 129M/V the first evidence of protein-only epigenetic population based selectivity among humans? This is obviously a quite notable possibility. What was the cause for selectivity? Let us speculate that it was an early onset disease. For selective pressure to commence it cannot be on the basis for resistance against sCJD, because it is a disease of the aged. It would need to be an infectious disease striking the young, cf. Sickle Cell Malaria Hypothesis. Highly adapted prion strains within susceptible hosts can be very efficient killers, both on a dose basis and terms of kinetics.30 The only known importance of the 129M/V mutation is enhanced resistance toward prion disease, hence the wide spread prevalence of the 129M/V polymorphism as a resistance trait can hypothetically have be caused by an efficient prion strain, a superprion.
In modern times the endocannibalism in Kuru and neocannibalistic practice from iatrogenic transfer of prions in cadaveric pituitary derived growth hormone treatment or dura mater transplants, leading to iCJD, are tragic reminders of the lethal efficiency of prions. These instances show dramatic selectivity in human subjects with preference for high hit rates in 129 homozygotes, especially in 129MM individuals. An ancient superprion with short incubation times which efficiently affected 129MM individuals could have dramatically selected for heterozygote advantage if there was a vector for transmission. Hence the dependency on the host (129MM carriers) and prion strain could afford such a scenario. When in history did the selectivity for the heterozygote advantage occur? The M129V mutant is less prevalent in Asian populations and especially in Japanese14 compared with European, South American and African ethnic groups. Human migration from Africa toward Asia occurred approximately 60 kyrs BP.31 The time frame for this event is heavily debated, but Asia was likely colonized before modern humans moved on to Europe which, in turn, was at the earliest 45 kyrs BP.32 Hereafter selective pressure has been rather prominent in Europe. The Collinge group suggested that the most likely reason for M129V to proliferate in ancient Europe was due to endocannibalism,14 for which there is ample evidence in modern studies of Kuru.
A Panzootic Superprion as a Hyperdisease Agent?
To our knowledge there has been no alternative hypothesis put forward to argue against the Mead and Collinge proposal of endocannibalism to account for the selectivity of the 129M/V polymorphism as a heterozygote advantage against Kuru-like epidemics.14 Our aim with this paper is not to argue against this proposal but rather to append an alternative possibility on the actual origin of the hypothetical prion disease, which we refer to as the superprion, that drove the selective pressure. We propose that the superprion did not originate in humans but rather in another mammal(s).
During human migration and colonization throughout Europe in the late Pleisoscene (21 kyr BP) until 6 kyrs BP, large mammals sometimes referred to as the megafauna became severely deprived in numbers to the extent of extinction.33 The reason for the elimination of the megafauna is not known but climate change, human hunting, or disease have been discussed. The co-occurance of humans as hunters and climate change as the culprit is a common suggestion.33 Hypothetically one or several of these mammalian species could have been struck by a prion disease similar to chronic wasting disease (CWD) or scrapie. Both these diseases occur sporadically and can spread horizontally. The prehistoric existence of some kind of panzootic, devastating disease as a qualifier for mass extinction is referred to as the hyperdisease theory.34 The hyperdisease theory of the late Pleisocene extinction in North America is a debated topic. One of several arguments against the hyperdisease theory is that a hyperdisease agent has not yet been accounted for. A superprion could fulfill the MacPhee and Marx criteria postulated for the hypothetical hyperdisease agent.35 Let us scrutinize the hyperdisease criteria in terms of prions:
A reservoir species in which a stable carrier state for the pathogen occurs
Prions can incubate for extended periods in clinically silent mammalian carriers prior to disease outbreak. All mammals possess the ability to sustain high titers of replicating prions. Perplexing as it may seem this in particular confines to herbivores, e.g., sheep, goat, deer, through horizontal transmission.
A high potential for causing infections in susceptible species, affecting all age groups
Most mammals are susceptible species.36,37 Table 3 summarizes over 50 known mammalian species susceptible to prion disease.36,38-40 There appears to be less susceptible species such as canines compared with felines. Nonetheless few invariably fatal diseases strike as many different species as prions. In particular a zoonotic prion stain which would be transmissible both horizontally and through consumption would have the potential to affect all age groups.
Table 3. Prion infected mammals.

A capacity for hyperlethality, defined as mortality rates, in the range of 50–75%
All known prion diseases are invariably lethal. If subclinical cases are considered, strain adaptation will provide a spectrum of potential hyperlethality in apparent resistant species
Honestly there are weaknesses for prions as hyperdisease agent within all these criteria, but notwithstanding the fact remains that extinctions have occurred and no-one have so far been able to identify a plausible agent for the megafaunal extinctions. As exceptional as this hypothesis may appear at first glance, the superprion does not have to be assessed as an isolated event. One possibility for the extreme consequences from such an episode of hyperdisease which have been discussed is the co-occurrence of hyperdisease with environmental disaster/climate change, triggering the ecological disaster. Prion disease-struck animals would have harbored a number of parasitic, bacterial, and viral infections further fostering the spread of disease. Hence, multiple vectors for comorbidity could have bolstered a cascade of disease also in birds and other animals. It is particularly interesting from the lack of current evidence that Paleolithic remains of ancient material is currently being investigated in this respect. Tuberculosis and West Nile virus have been brought forth as potential candidates for zoonoses with ability to act as an omnipotent hyperdisease agents.41,42 MacPhee and Greenwood35 are arguing for investigating potential hyperdisease agents by searching paleolothic frozen specimens using modern DNA extraction methods. While DNA or RNA from putative viruses and bacteria likely have been degraded over the millennia, it would be prudent in the light of our argument herein to propose to also search for prions. Prions are known to be highly resistant to degradation43,44 and could potentially still be intact.
Natural Resistance—More Clues in Evolutionary Records
The spread of natural prion strains (scrapie and CWD) can severely affect a population and with a surprising efficiency as demonstrated by CWD in North America. Such a disease would have rendered these animals easy prey for a growing human population. Much of the ancient world was covered by megafauna up until 21 kyrs BP,33 but are extinct today. Herein these large mammals coincided with humans for millennia. Did the megafaunal depriviation culminating with extinction coincide with the heterozygote advantage evolution of the human M129V polymorphism? Given the extensive incubation time of prion diseases especially in zoonotic prion diseases, (i.e., BSE transfer causing vCJD) a sickened animal should not have been considered bad food due to prion disease. An ancient superprion strain propagating in mammals with effective transmission to humans should have been very efficient in relation to triggering the human hetrozygote advantage. If endocannibalism followed the triggering index cases the spread of the superprion would have perpetuated the heterozygote advantage. Interestingly Mead and coworkers recently reported a novel polymorphism G127V appearing in the Fore people of Papua New Guinea as a resistance trait against Kuru.45 The localization of this mutation is of course very suggestive for the importance of the region of PrP for conversion but since it is 2 residues downstream from 129 it could hypothetically argue for a different prion strain rather than Kuru to account for the ancient selection for 129V in Europeans and in fact globally, although the incidence rates are lower in most parts of Asia.
One thing which appears to argue against the zoonotic transfer hypothesis is that we should find predatory mammals with the corresponding 129M/V polymorphism. To our knowledge this has not been rigorously tested, and given the dominance of humans as the main predator and the long lifetime of humans in comparison to other mammalian predators in the wild, it is possible that it cannot be found even though it would be worth looking for.
For scrapie there are evidently protective genetics at play. Artificial selection (directed evolution) toward scrapie resistant sheep by enforced breeding programs have been rather successful in this respect. Herein selectivity has mainly been based on positions 136, 154, 171 in ovine PrP, corresponding to positions 133, 151, and 168 in the human PrP sequence. Nonetheless, the PRNP genetics within sheep and susceptibility toward scrapie is particularly complex.46 The bank vole is a species with high susceptibility to various prion strains.47-49 The bank vole exhibits a Met/Ile polymophism in position 109, corresponding to position 108 in the human PrP sequence. Heterozygosity in this position reduces susceptibility to various scrapie strains.50 The examples described above are clearly supporting the heterozygote advantage in as resistance mechanisms in prion disease.
In addition to heterozygote advantage there are several positions in the PrP sequence reported for modulation of disease susceptibility in a number of mammals. Comparison of PrP sequences between Chinese hamster and Syrian hamster delineates, among others, a Met-Val deviation at position 112 in the PrP sequence. Early work from Prusiner and coworkers demonstrate that this difference is correlated with disease susceptibility in favor of the Val carrying Chinese hamster. The Chinese hamster is less susceptible to prion infection regardless of donor.51
Are there any species with mutations in corresponding to position 129 in the human PrP sequence? Interestingly, there is one species, actually emanating from the megafauna, within the mammalian class where heterozygosity have occurred at the site corresponding to position 129 in the human PrP. Position 132 is polymorphic for Leucine in Rocky Mountain elk (Wapiti)52 and also appear to be a protective trait against chronic wasting disease (CWD),52 reflected by prolonged incubation time. In our in vitro fibrillation studies of HuPrP, the M129L and M129V mutants were indistinguishable,16 supporting the notion of also M129L being protective against PrP misfolding. If this is a case of convergent evolution driven by the same superprion strain would be interesting to delineate. A range of other polymorphic sites in cervids with different aspects of disease modification are reviewed in.53
Superprion Strain(s)
What would be the characteristics of the ancient superprion? It would need to infect a highly diverse population of animals and to be peripherally transmissible. It would be required to be neuroinvasive, stable in its reservoir (e.g., host mammal, parasitic vector, water, soil, or plant). The incubation period would optimally be long enough to allow silent carriers to be preyed or efficiently participate in horizontal transmission but short enough to intervene in the reproductive cycle. This calls for a subacute disease offered by the prionoses which would make large mammals with long lifetimes and few offspring especially vulnerable. Furthermore, the long lifetime of large mammals would allow for prion strain adaptation for facilitated cross species infection. It has been shown that prion infections undergo temporally distinct transitions comprising both replication and neurotoxic phases.54 Beringue et al. have summarized different pathways which may facilitate cross-species transmission.55 A naïve host can react to a prion strain in a number of different ways. (1) The naïve host may be totally resistant and non-transmitting hence acting as an end point for the prion. (2) The naïve host may become a silent carrier without clinical symptoms but with the ability to transfer the infection to a third species. (3) The naïve host will come down with clinical disease after a long incubation time in the primary subject and then display a shortened lag time as a consequence of strain adaptation. (4) The naïve host is a “perfect match” for the donor strain resulting in efficient transmission at the primary infection. (5) The naïve host converts the strain to a new strain with efficient transmission to a third species.
The molecular characteristics of the candidate superprion strain promoting human heterozygote advantage would likely be the ability of efficient replication to allow advantage for M129V carriers. Recent studies show that fibrillar prion aggregates are less neuroinvasive than non-fibrillar aggregates following peripheral infection.53 In addition, strains with high resistance against detergent and chaotrope denaturation result in shorter incubation times than less resistant strains.56,57
The most versatile, most tested, and likely most efficient superprion candidate found in modern time is BSE. BSE has been shown to be experimentally and naturally transmissible to a huge variety of mammalian hosts (Table 3). BSE shows impressive robustness and remains highly infectious even after 5 years buried underground.44 Obviously, BSE is particularly interesting in the view of the human PrP 129 genotype.9-14 The origin of BSE is unknown, but is most likely an adapted form of Bovine amyloidotic spongiform encephalopathy (BASE)58 and was transmitted through a rather unnatural process. In the cases of BASE and BSE these strains have been shown to undergo strain adaptation during serial passage in different species.55,58 Two prion strains which are naturally occurring and horizontally transmitted in the wild are CWD and scrapie. It is possible that both CWD and scrapie are adapted forms of the ancient superprion which have been confined by the selective processes discussed above to a few species. It is possible that there are other unknown silent hosts carrying adapted forms of the ancient superprion with the potential for human infection. It is conceivable that humans are such silent carriers of prion infection. The overrepresentation of iatrogenic cases of CJD caused by dura mater grafts in Japanese recipients being mainly 129MM homozygotes can support this notion.59
Summary
Heterozygote advantage in humans has been shown to be a population genetic modifier in response to infectious diseases (Sickle cell anemia). Heterozygote advantage is operative in a rare but clinically important amyloid disease (familial amyloidotic polyneuropathy). The PrP 129M/V polymorphism is one of the most prevalent genetic disease modifiers known, but is working in a population where disease modifiers are lacking as selective pressure. Surely sCJD being a disease of the elderly cannot account for this putative hererozygote advantage. The current wide spread of this polymorphism is hence most likely the outcome of a severe bottle neck selection for heterozygote carriers. In contrast to Sickle cell anemia (HbβE6V) there is no negative trait associated with the PrP129V allele which enables sustained prevalence of the gene despite lack of selective pressure in modern times. In several populations the mutation M129V is dominating over the wild type trait. This apparent paradox rendered us to propose a hypothesis on the convergence of the prevalence of the human M129V mutation and the extinction of the Pleisocene mammalian megafauna by an ancient hyperdisease agent. We suggest that the present composition of the European population could be the outcome of a paleolithic panzootic superprion pandemic. If this notion is correct this prehistoric outbreak of a zoonotic prion disease is one reason for the small number of afflicted individuals in the BSE outbreak in Europe eons later. Occasionally, albeit rarely, when history repeats itself it is for the better.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Peter Nilsson for comments on the manuscript and we thank the Swedish Research Council for financial support (Grant #2011-5804).
References
- 1.Cobb NJ, Surewicz WK. . Prion diseases and their biochemical mechanisms. Biochemistry 2009; 48:2574 - 85; http://dx.doi.org/ 10.1021/bi900108v; PMID: 19239250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Collinge J. . Prion diseases of humans and animals: their causes and molecular basis. Annu Rev Neurosci 2001; 24:519 - 50; http://dx.doi.org/ 10.1146/annurev.neuro.24.1.519; PMID: 11283320 [DOI] [PubMed] [Google Scholar]
- 3.Mead S. . Prion disease genetics. Eur J Hum Genet 2006; 14:273 - 81; http://dx.doi.org/ 10.1038/sj.ejhg.5201544; PMID: 16391566 [DOI] [PubMed] [Google Scholar]
- 4.Owen F, Poulter M, Collinge J, Crow TJ. . A codon 129 polymorphism in the PRIP gene. Nucleic Acids Res 1990; 18:3103; http://dx.doi.org/ 10.1093/nar/18.10.3103; PMID: 1971924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Petraroli R, Pocchiari M. . Codon 219 polymorphism of PRNP in healthy Caucasians and Creutzfeldt-Jakob disease patients. Am J Hum Genet 1996; 58:888 - 9; PMID: 8644754 [PMC free article] [PubMed] [Google Scholar]
- 6.Palmer MS, Dryden AJ, Hughes JT, Collinge J. . Homozygous prion protein genotype predisposes to sporadic Creutzfeldt-Jakob disease. Nature 1991; 352:340 - 2; http://dx.doi.org/ 10.1038/352340a0; PMID: 1677164 [DOI] [PubMed] [Google Scholar]
- 7.Shibuya S, Higuchi J, Shin RW, Tateishi J, Kitamoto T. . Protective prion protein polymorphisms against sporadic Creutzfeldt-Jakob disease. Lancet 1998; 351:419; http://dx.doi.org/ 10.1016/S0140-6736(05)78358-6; PMID: 9482303 [DOI] [PubMed] [Google Scholar]
- 8.Polymenidou M, Stoeck K, Glatzel M, Vey M, Bellon A, Aguzzi A. . Coexistence of multiple PrPSc types in individuals with Creutzfeldt-Jakob disease. Lancet Neurol 2005; 4:805 - 14; http://dx.doi.org/ 10.1016/S1474-4422(05)70225-8; PMID: 16297838 [DOI] [PubMed] [Google Scholar]
- 9.Collinge J, Palmer MS, Dryden AJ. . Genetic predisposition to iatrogenic Creutzfeldt-Jakob disease. Lancet 1991; 337:1441 - 2; http://dx.doi.org/ 10.1016/0140-6736(91)93128-V; PMID: 1675319 [DOI] [PubMed] [Google Scholar]
- 10.Collinge J, Beck J, Campbell T, Estibeiro K, Will RG. . Prion protein gene analysis in new variant cases of Creutzfeldt-Jakob disease. Lancet 1996; 348:56; http://dx.doi.org/ 10.1016/S0140-6736(05)64378-4; PMID: 8691941 [DOI] [PubMed] [Google Scholar]
- 11.Zeidler M, Stewart G, Cousens SN, Estibeiro K, Will RG. . Codon 129 genotype and new variant CJD. Lancet 1997; 350:668; http://dx.doi.org/ 10.1016/S0140-6736(05)63366-1; PMID: 9288076 [DOI] [PubMed] [Google Scholar]
- 12.Hill AF, Butterworth RJ, Joiner S, Jackson G, Rossor MN, Thomas DJ, Frosh A, Tolley N, Bell JE, Spencer M, et al. . Investigation of variant Creutzfeldt-Jakob disease and other human prion diseases with tonsil biopsy samples. Lancet 1999; 353:183 - 9; http://dx.doi.org/ 10.1016/S0140-6736(98)12075-5; PMID: 9923873 [DOI] [PubMed] [Google Scholar]
- 13.Lee HS, Brown P, Cervenáková L, Garruto RM, Alpers MP, Gajdusek DC, Goldfarb LG. . Increased susceptibility to Kuru of carriers of the PRNP 129 methionine/methionine genotype. J Infect Dis 2001; 183:192 - 6; http://dx.doi.org/ 10.1086/317935; PMID: 11120925 [DOI] [PubMed] [Google Scholar]
- 14.Mead S, Stumpf MP, Whitfield J, Beck JA, Poulter M, Campbell T, Uphill JB, Goldstein D, Alpers M, Fisher EM, et al. . Balancing selection at the prion protein gene consistent with prehistoric kurulike epidemics. Science 2003; 300:640 - 3; http://dx.doi.org/ 10.1126/science.1083320; PMID: 12690204 [DOI] [PubMed] [Google Scholar]
- 15.Gill ON, Spencer Y, Richard-Loendt A, Kelly C, Dabaghian R, Boyes L, Linehan J, Simmons M, Webb P, Bellerby P, et al. Prevalent abnormal prion protein in human appendixes after bovine spongiform encephalopathy epizootic: large scale survey. BMJ (Clinical research ed 2013; 347:f5675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nyström S, Mishra R, Hornemann S, Aguzzi A, Nilsson KP, Hammarström P. . Multiple substitutions of methionine 129 in human prion protein reveal its importance in the amyloid fibrillation pathway. J Biol Chem 2012; 287:25975 - 84; http://dx.doi.org/ 10.1074/jbc.M112.372136; PMID: 22669942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Williams TN, Obaro SK. . Sickle cell disease and malaria morbidity: a tale with two tails. Trends Parasitol 2011; 27:315 - 20; http://dx.doi.org/ 10.1016/j.pt.2011.02.004; PMID: 21429801 [DOI] [PubMed] [Google Scholar]
- 18.Aidoo M, Terlouw DJ, Kolczak MS, McElroy PD, ter Kuile FO, Kariuki S, Nahlen BL, Lal AA, Udhayakumar V. . Protective effects of the sickle cell gene against malaria morbidity and mortality. Lancet 2002; 359:1311 - 2; http://dx.doi.org/ 10.1016/S0140-6736(02)08273-9; PMID: 11965279 [DOI] [PubMed] [Google Scholar]
- 19.Piel FB, Patil AP, Howes RE, Nyangiri OA, Gething PW, Williams TN, Weatherall DJ, Hay SI. . Global distribution of the sickle cell gene and geographical confirmation of the malaria hypothesis. Nat Commun 2010; 1:104; http://dx.doi.org/ 10.1038/ncomms1104; PMID: 21045822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hammarström P, Jiang X, Hurshman AR, Powers ET, Kelly JW. . Sequence-dependent denaturation energetics: A major determinant in amyloid disease diversity. Proc Natl Acad Sci U S A 2002; 99:Suppl 4 16427 - 32; http://dx.doi.org/ 10.1073/pnas.202495199; PMID: 12351683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sekijima Y, Wiseman RL, Matteson J, Hammarström P, Miller SR, Sawkar AR, Balch WE, Kelly JW. . The biological and chemical basis for tissue-selective amyloid disease. Cell 2005; 121:73 - 85; http://dx.doi.org/ 10.1016/j.cell.2005.01.018; PMID: 15820680 [DOI] [PubMed] [Google Scholar]
- 22.Sousa A, Coelho T, Barros J, Sequeiros J. . Genetic epidemiology of familial amyloidotic polyneuropathy (FAP)-type I in Póvoa do Varzim and Vila do Conde (north of Portugal). Am J Med Genet 1995; 60:512 - 21; http://dx.doi.org/ 10.1002/ajmg.1320600606; PMID: 8825887 [DOI] [PubMed] [Google Scholar]
- 23.Coelho TCR, Sousa A, Alves IL, Torres MF, Saraiva MJM. . Compound heterozygotes of transthyretin Met 30 and transthyretin Met 119 are protected from the devastating effects of familial amyloid polyneuropathy. Neuromuscul Disord 1996; 6:Suppl S20; http://dx.doi.org/ 10.1016/0960-8966(96)88826-2 [DOI] [Google Scholar]
- 24.Hammarström P, Schneider F, Kelly JW. . Trans-suppression of misfolding in an amyloid disease. Science 2001; 293:2459 - 62; http://dx.doi.org/ 10.1126/science.1062245; PMID: 11577236 [DOI] [PubMed] [Google Scholar]
- 25.Hammarström P, Wiseman RL, Powers ET, Kelly JW. . Prevention of transthyretin amyloid disease by changing protein misfolding energetics. Science 2003; 299:713 - 6; http://dx.doi.org/ 10.1126/science.1079589; PMID: 12560553 [DOI] [PubMed] [Google Scholar]
- 26.Westermark P, Sletten K, Johansson B, Cornwell GG 3rd. . Fibril in senile systemic amyloidosis is derived from normal transthyretin. Proc Natl Acad Sci U S A 1990; 87:2843 - 5; http://dx.doi.org/ 10.1073/pnas.87.7.2843; PMID: 2320592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hornstrup LS, Frikke-Schmidt R, Nordestgaard BG, Tybjærg-Hansen A. . Genetic stabilization of transthyretin, cerebrovascular disease, and life expectancy. Arterioscler Thromb Vasc Biol 2013; 33:1441 - 7; http://dx.doi.org/ 10.1161/ATVBAHA.113.301273; PMID: 23580146 [DOI] [PubMed] [Google Scholar]
- 28.Wopfner F, Weidenhöfer G, Schneider R, von Brunn A, Gilch S, Schwarz TF, Werner T, Schätzl HM. . Analysis of 27 mammalian and 9 avian PrPs reveals high conservation of flexible regions of the prion protein. J Mol Biol 1999; 289:1163 - 78; http://dx.doi.org/ 10.1006/jmbi.1999.2831; PMID: 10373359 [DOI] [PubMed] [Google Scholar]
- 29.Hedrick PW. A heterozygote advantage. Science (New York, NY 2003; 302:57. [DOI] [PubMed] [Google Scholar]
- 30.Büeler H, Raeber A, Sailer A, Fischer M, Aguzzi A, Weissmann C. . High prion and PrPSc levels but delayed onset of disease in scrapie-inoculated mice heterozygous for a disrupted PrP gene. Mol Med 1994; 1:19 - 30; PMID: 8790598 [PMC free article] [PubMed] [Google Scholar]
- 31.Appenzeller T. . Human migrations: Eastern odyssey. Nature 2012; 485:24 - 6; http://dx.doi.org/ 10.1038/485024a; PMID: 22552074 [DOI] [PubMed] [Google Scholar]
- 32.Callaway E. . Archaeology: Date with history. Nature 2012; 485:27 - 9; http://dx.doi.org/ 10.1038/485027a; PMID: 22552075 [DOI] [PubMed] [Google Scholar]
- 33.Lorenzen ED, Nogués-Bravo D, Orlando L, Weinstock J, Binladen J, Marske KA, Ugan A, Borregaard MK, Gilbert MT, Nielsen R, et al. . Species-specific responses of Late Quaternary megafauna to climate and humans. Nature 2011; 479:359 - 64; http://dx.doi.org/ 10.1038/nature10574; PMID: 22048313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.MacPhee RD, Marx PA. Humans, hyperdisease and first-contact extinctions. In: Goodman S, Patterson, B.D., ed. Natural Change and Human Impact in Madagascar. Washington D.C.: Smithsonian Press, 1997:169–217. [Google Scholar]
- 35.Macphee RD, Greenwood AD. . Infectious disease, endangerment, and extinction. Int J Evol Biol 2013; 2013:571939; http://dx.doi.org/ 10.1155/2013/571939; PMID: 23401844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sigurdson CJ, Miller MW. . Other animal prion diseases. Br Med Bull 2003; 66:199 - 212; http://dx.doi.org/ 10.1093/bmb/66.1.199; PMID: 14522860 [DOI] [PubMed] [Google Scholar]
- 37.Imran M, Mahmood S. . An overview of animal prion diseases. Virol J 2011; 8:493; http://dx.doi.org/ 10.1186/1743-422X-8-493; PMID: 22044871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gajdusek DC, ed. Nobel Lecture: Unconventional Viruses and the Origin and Disappearance of Kuru. Nobel Media AB: World Scientific Publishing Co, 1976. [Google Scholar]
- 39.Williams ES, Kirkwood JK, Miller MW. Transmissible Spongiform Encephalopathies. In: Elizabeth S. Williams, Barker IK, eds. Infectious Diseases of Wild Mammals: Iowa State University Press, Ames, 2001:292-301. [Google Scholar]
- 40.Marsh RF, Bessen RA, Lehmann S, Hartsough GR. . Epidemiological and experimental studies on a new incident of transmissible mink encephalopathy. J Gen Virol 1991; 72:589 - 94; http://dx.doi.org/ 10.1099/0022-1317-72-3-589; PMID: 1826023 [DOI] [PubMed] [Google Scholar]
- 41.Rothschild BM, Laub R. . Hyperdisease in the late Pleistocene: validation of an early 20th century hypothesis. Naturwissenschaften 2006; 93:557 - 64; http://dx.doi.org/ 10.1007/s00114-006-0144-8; PMID: 16953418 [DOI] [PubMed] [Google Scholar]
- 42.Lyons SK, Smith FA, Wagner PJ, White EP, Brown JH. . Was a 'hyperdisease' responsible for the late Pleistocene megafaunal extinction?. Ecol Lett 2004; 7:859 - 68; http://dx.doi.org/ 10.1111/j.1461-0248.2004.00643.x [DOI] [Google Scholar]
- 43.Brown P, Gajdusek DC. . Survival of scrapie virus after 3 years’ interment. Lancet 1991; 337:269 - 70; http://dx.doi.org/ 10.1016/0140-6736(91)90873-N; PMID: 1671114 [DOI] [PubMed] [Google Scholar]
- 44.Smith A, Somerville R, Fernie K. . TSE infectivity survives burial for five years with little reduction in titer. Prion 2012; 6:138 [Google Scholar]
- 45.Mead S, Whitfield J, Poulter M, Shah P, Uphill J, Campbell T, Al-Dujaily H, Hummerich H, Beck J, Mein CA, et al. . A novel protective prion protein variant that colocalizes with kuru exposure. N Engl J Med 2009; 361:2056 - 65; http://dx.doi.org/ 10.1056/NEJMoa0809716; PMID: 19923577 [DOI] [PubMed] [Google Scholar]
- 46.Baylis M, Goldmann W. . The genetics of scrapie in sheep and goats. Curr Mol Med 2004; 4:385 - 96; http://dx.doi.org/ 10.2174/1566524043360672; PMID: 15354869 [DOI] [PubMed] [Google Scholar]
- 47.Nonno R, Di Bari MA, Cardone F, Vaccari G, Fazzi P, Dell’Omo G, Cartoni C, Ingrosso L, Boyle A, Galeno R, et al. . Efficient transmission and characterization of Creutzfeldt-Jakob disease strains in bank voles. PLoS Pathog 2006; 2:e12; http://dx.doi.org/ 10.1371/journal.ppat.0020012; PMID: 16518470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Agrimi U, Nonno R, Dell’Omo G, Di Bari MA, Conte M, Chiappini B, Esposito E, Di Guardo G, Windl O, Vaccari G, et al. . Prion protein amino acid determinants of differential susceptibility and molecular feature of prion strains in mice and voles. PLoS Pathog 2008; 4:e1000113; http://dx.doi.org/ 10.1371/journal.ppat.1000113; PMID: 18654630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Heisey DM, Mickelsen NA, Schneider JR, Johnson CJ, Johnson CJ, Langenberg JA, Bochsler PN, Keane DP, Barr DJ. . Chronic wasting disease (CWD) susceptibility of several North American rodents that are sympatric with cervid CWD epidemics. J Virol 2010; 84:210 - 5; http://dx.doi.org/ 10.1128/JVI.00560-09; PMID: 19828611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cartoni C, Schininà ME, Maras B, Nonno R, Vaccari G, Di Baria MA, Conte M, Liu QG, Lu M, Cardone F, et al. . Identification of the pathological prion protein allotypes in scrapie-infected heterozygous bank voles (Clethrionomys glareolus) by high-performance liquid chromatography-mass spectrometry. J Chromatogr A 2005; 1081:122 - 6; http://dx.doi.org/ 10.1016/j.chroma.2005.04.035; PMID: 16013608 [DOI] [PubMed] [Google Scholar]
- 51.Lowenstein DH, Butler DA, Westaway D, McKinley MP, DeArmond SJ, Prusiner SB. . Three hamster species with different scrapie incubation times and neuropathological features encode distinct prion proteins. Mol Cell Biol 1990; 10:1153 - 63; PMID: 2406562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Perucchini M, Griffin K, Miller MW, Goldmann W. . PrP genotypes of free-ranging wapiti (Cervus elaphus nelsoni) with chronic wasting disease. J Gen Virol 2008; 89:1324 - 8; http://dx.doi.org/ 10.1099/vir.0.83424-0; PMID: 18420812 [DOI] [PubMed] [Google Scholar]
- 53.Robinson SJ, Samuel MD, O’Rourke KI, Johnson CJ. . The role of genetics in chronic wasting disease of North American cervids. Prion 2012; 6:153 - 62; http://dx.doi.org/ 10.4161/pri.19640; PMID: 22460693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sandberg MK, Al-Doujaily H, Sharps B, Clarke AR, Collinge J. . Prion propagation and toxicity in vivo occur in two distinct mechanistic phases. Nature 2011; 470:540 - 2; http://dx.doi.org/ 10.1038/nature09768; PMID: 21350487 [DOI] [PubMed] [Google Scholar]
- 55.Béringue V, Vilotte JL, Laude H. . Prion agent diversity and species barrier. Vet Res 2008; 39:47; http://dx.doi.org/ 10.1051/vetres:2008024; PMID: 18519020 [DOI] [PubMed] [Google Scholar]
- 56.Bett C, Kurt TD, Lucero M, Trejo M, Rozemuller AJ, Kong Q, Nilsson KP, Masliah E, Oldstone MB, Sigurdson CJ. . Defining the conformational features of anchorless, poorly neuroinvasive prions. PLoS Pathog 2013; 9:e1003280; http://dx.doi.org/ 10.1371/journal.ppat.1003280; PMID: 23637596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ayers JI, Schutt CR, Shikiya RA, Aguzzi A, Kincaid AE, Bartz JC. . The strain-encoded relationship between PrP replication, stability and processing in neurons is predictive of the incubation period of disease. PLoS Pathog 2011; 7:e1001317; http://dx.doi.org/ 10.1371/journal.ppat.1001317; PMID: 21437239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Capobianco R, Casalone C, Suardi S, Mangieri M, Miccolo C, Limido L, Catania M, Rossi G, Di Fede G, Giaccone G, et al. . Conversion of the BASE prion strain into the BSE strain: the origin of BSE?. PLoS Pathog 2007; 3:e31; http://dx.doi.org/ 10.1371/journal.ppat.0030031; PMID: 17352534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Brown P, Brandel JP, Sato T, Nakamura Y, MacKenzie J, Will RG, Ladogana A, Pocchiari M, Leschek EW, Schonberger LB. . Iatrogenic Creutzfeldt-Jakob disease, final assessment. Emerg Infect Dis 2012; 18:901 - 7; http://dx.doi.org/ 10.3201/eid1806.120116; PMID: 22607808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Almstedt K, Nyström S, Nilsson KP, Hammarström P. . Amyloid fibrils of human prion protein are spun and woven from morphologically disordered aggregates. Prion 2009; 3:224 - 35; http://dx.doi.org/ 10.4161/pri.3.4.10112; PMID: 19923901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zahn R, Liu A, Lührs T, Riek R, von Schroetter C, López García F, Billeter M, Calzolai L, Wider G, Wüthrich K. . NMR solution structure of the human prion protein. Proc Natl Acad Sci U S A 2000; 97:145 - 50; http://dx.doi.org/ 10.1073/pnas.97.1.145; PMID: 10618385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Calzolai L, Lysek DA, Pérez DR, Güntert P, Wüthrich K. . Prion protein NMR structures of chickens, turtles, and frogs. Proc Natl Acad Sci U S A 2005; 102:651 - 5; http://dx.doi.org/ 10.1073/pnas.0408939102; PMID: 15647366 [DOI] [PMC free article] [PubMed] [Google Scholar]