

Molecular simulations reveal that multiple lipid interactions are needed for high-affinity binding of protein domains to membranes.
Abstract
Association of peripheral proteins with lipid bilayers regulates membrane signaling and dynamics. Pleckstrin homology (PH) domains bind to phosphatidylinositol phosphate (PIP) molecules in membranes. The effects of local PIP enrichment on the interaction of PH domains with membranes is unclear. Molecular dynamics simulations allow estimation of the binding energy of GRP1 PH domain to PIP3-containing membranes. The free energy of interaction of the PH domain with more than two PIP3 molecules is comparable to experimental values, suggesting that PH domain binding involves local clustering of PIP molecules within membranes. We describe a mechanism of PH binding proceeding via an encounter state to two bound states which differ in the orientation of the protein relative to the membrane, these orientations depending on the local PIP concentration. These results suggest that nanoscale clustering of PIP molecules can control the strength and orientation of PH domain interaction in a concentration-dependent manner.
INTRODUCTION
Cell signaling and trafficking are regulated by peripheral membrane proteins that associate with cell membrane surfaces in a lipid-dependent fashion (1, 2). Recognition of cell membranes is brought about by lipid recognition domains (3). The pleckstrin homology (PH) domains form a large and well-characterized family present in many membrane recognition proteins including, e.g., AKT and Btk (4, 5). PH domains can bind to phosphatidylinositol phosphates (PIPs), which confer a molecular identity to the different membranes with a eukaryotic cell (1). Lipid cooperativity (i.e., interaction with more than one lipid molecule and/or species) may also play a key role in the recruitment of PH domains to cell membranes (6, 7). Furthermore, it is thought that nanoscale lipid clustering may play a key role in the interactions of membrane proteins with lipids (8, 9), in turn influencing the avidity of recognition proteins for membranes (10–13).
The structure of a PH domain consists of ~120 amino acid residues folded into an antiparallel β sheet, followed by one or two amphipathic helices. Many structures of PH domains are known, a number of which include a PIP headgroup (i.e., an inositol phosphate) bound at a canonical binding site (CA) formed by positively charged residues of the β1/β2 and β3/β4 loops (see Fig. 1A) (14, 15). A KXn(K/R)XR sequence motif in the β1/β2 loop determines contacts of the PH domain with different classes of PIP molecules. Certain PH domains, e.g., those of β-spectrin and ArhGAP9 (15, 16), lack this motif and instead have an alternative, noncanonical binding site (NCA) on the opposite face of the β1/β2 loop in between the β1/β2 and β5/β6 loops. The crystal structure of the ASAP1 PH domain reveals two bound PIP headgroups, one at the CA and one at the NCA (6). This suggests that recruitment of PH domains to cellular membranes may involve binding to multiple PIP molecules by one domain.
Fig. 1. MD simulations for calculating PMFs of PH/PIP3 interactions.
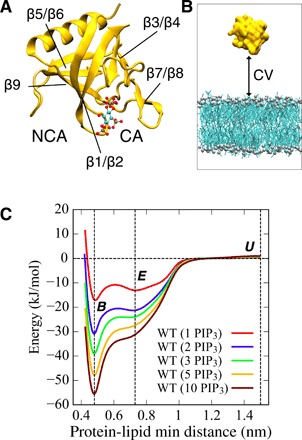
(A) Structure of the GRP1 PH domain (Protein Data Bank ID: 1FGY; yellow cartoon format) with an Ins(1,3,4,5)P4 molecule (ball and stick format) bound at the canonical site (CA). The approximate location of the noncanonical site (NCA) is indicated. (B) Collective variable (CV) based on the minimum distance between the protein and lipid as used in the REUS-MD simulations. (C) Potentials of mean force (PMFs) for the GRP1 PH domain interacting with lipid bilayers containing from 1 to 10 PIP3 molecules, showing the free energy of interaction as a function of protein-lipid minimum distance. The three vertical dashed lines correspond to the protein-lipid minimum distances of 0.48, 0.73, and 1.5 nm (see text for details).
Biophysical and computational studies have explored, in some detail, the mechanism of membrane binding by PH domains (17–20). A number of key aspects remain unresolved, in particular, the impact of PIP clustering on PH domain interactions with membranes and the influence on the mechanism and strength of binding of the presence of both canonical and alternative binding sites on the same PH domain. PIP concentrations in cell membranes are relatively low: less than 5% in the plasma membranes and about 10% in Golgi membranes (21, 22). However, a number of studies have indicated that PIP molecules can cluster in cell membranes to form nanoscale domains, which, in turn, enhance interactions with proteins (8, 9). In vitro studies have explored the effects of other phospholipids (11) and of Ca2+ ions (10) on PIP clustering and conformation. Both experimental and computational investigations have suggested clustering of PIP molecules around PH domains (23–26). The association of PH domains with cell membranes is influenced by sites distinct from the canonical PIP binding pocket (27), and a number of PH domains [e.g., that from ASAP1 (6)] have revealed multiple PIP-binding sites in crystal structures. Some PH domains have been demonstrated to bind cooperatively to PIPs [as indicated by, e.g., sigmoidal PIP dependence of binding to vesicles (6)]. It has also been shown that other anionic lipid species [e.g., phosphatidylserine (PS)] may contribute to the binding of PH domains to liposomes in a microarray-based assay (7) and that the presence of PS leads to a ~10-fold increase in the affinity of GRP1 PH for PIP3 in a bilayer (17). Analysis of 33 different yeast PH domains revealed that only 1 bound phosphoinositides with high affinity, while 6 other PH domains bound with moderate affinity and low specificity (28). Together, these data indicate that while many PH domains may have a relatively low canonical binding site affinity for PIPs, interaction of multiple PIP molecules and/or other anionic lipid species may enable overall tight binding of a PH domain to a membrane. Thus, PH domains may act via coincidence sensing, i.e., detection of (local) clusters of PIP molecules and/or other anionic lipids (2, 7). Furthermore, binding of PH domains and other lipid-binding modules to membranes may, in turn, mediate PIP clustering by modifying the local lipid environment (29). PIP clustering affects the diffusivity of PH domains on the membrane surfaces, which is likely to play a role in regulating the function of membrane-bound proteins (30).
Molecular dynamics (MD) simulations enable investigation of both protein-lipid interactions (31) and the larger-scale organization of complex cell membranes (32). Simulations of PH domains have been used to explore the structure (18, 33), dynamic mechanisms (20, 24, 26, 30), and energetics (34, 35) of PH domain/membrane interactions. Here, we exploit recent advances in replica-exchange umbrella sampling (REUS) of protein-lipid interactions (36) to explore how the binding free energy of the GRP1 PH domain changes with respect to the number of PIP3 molecules with which it interacts within a membrane. Comparison of our results with experimental estimates of the dissociation constant of PH domain from a PIP3-containing bilayer suggests that at least three PIP3 molecules interact with the PH domain. Our simulations also suggest a three-step mechanism for tight association of the PH domain with the membrane. These results have implications more generally for coincidence-sensing mechanisms of recognition of complex cell membranes by proteins containing PH domains.
RESULTS
Potentials of mean force for GRP1 PH/PIP3 interactions
To estimate potentials of mean force (PMFs) for the interaction of the GRP1 PH domain with PIP-containing lipid bilayers, we performed REUS-MD simulations (37) using a Martini coarse-grained (CG) model (38). REUS enables faster convergence relative to standard umbrella sampling (US) and also allowed us to avoid the need for a constraint on the PIP3 lipid head group as used in our previous PMF calculations (34). We used a collective variable (CV) based on minimum distance between the protein and lipid (see Methods for further details). We were able to extensively explore the free energy landscape (see below) of PH/membrane interactions, as the REUS approach allowed us to sample multiple binding and dissociation events, thereby revealing a potential binding pathway for the protein.
For the GRP1 PH domain, simulation systems with different concentrations of PIP3 in lipid bilayers were performed, i.e., with from 1 to 10 PIP3 molecules in each leaflet of the bilayer, corresponding to concentrations from 0.8 to 8%. As an initial configuration of the system, the PH domain was displaced away from the membrane surface. Simulations were performed for 15 μs for each replica, yielding a total REUS-MD simulation time of 240 μs and thus a total MD simulation time of over 1 ms for all of the systems explored.
PMFs as a function of the GRP1-PIP3 PH protein-lipid minimum distance are shown in Fig. 1C for different numbers of PIP3 molecules within the membrane. For a single PIP3 molecule in the protein-exposed leaflet, a major and a minor minimum are seen, with an overall minimum interaction energy of −17 kJ/mol relative to the unbound (U) state, in agreement with previous estimates (34). The two minima at minimum distances of 0.73 and 0.48 nm thus correspond to a loosely interacting state (subsequently to as the encounter state E; see below) and a more tightly bound state (B) of the PH domain on the membrane surface. As the PIP3 concentration is increased, the bound state B is increasingly stabilized relative to the encounter state E such that the free energy difference between these two states is 4 kJ/mol for the 1 PIP3 system, increasing to 20 kJ/mol for the 5 PIP3 system.
Free energy landscapes for interaction
To investigate the binding mechanism in more detail, exploring the orientation of the PH domain relative to the membrane surface, two-dimensional free energy surfaces were calculated for the system with three PIP3 molecules in each leaflet of the membrane. Thus, free energy surfaces (Fig. 2) were calculated as a function of (i) the protein-membrane center of mass (COM) distance versus cosθ (where θ is a tilt angle of the PH domain α helix relative to the membrane), (ii) the protein-lipid minimum distance versus cosθ, and (iii) protein-membrane COM distance versus the protein-lipid minimum distance.
Fig. 2. Free energy surfaces of the GRP1 PH domain interacting with a lipid bilayer including 3 PIP3 molecules in each leaflet.
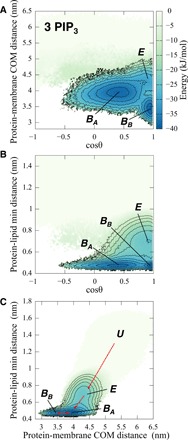
E, BA, and BB refer to the Encounter, BoundA and BoundB states of the PH domain when interacting with the membrane (see main text and Fig. 5B for further details). Three different projections of the free energy landscape are shown: (A) as a function of cosθ (where θ is the angle between a vector corresponding to the PH domain α helix and the z axis perpendicular to the membrane) and the protein-membrane COM distance, (B) as a function of cosθ and the protein-membrane minimum distance, and (C) as a function of the protein-membrane COM distance and the corresponding minimum distance.
From these free energy surfaces, it is evident that there are actually three states of the PH domain interacting with the membrane. The Encounter state (E) is characterized by a minimum distance of 0.73 nm and a COM distance of 4.3 nm. The bound state, characterized by a minimum distance of 0.48 nm, can be seen to be split into two orientational states BoundA (BA) and BoundB (BB), which differ in their COM distance such that BB is closer to the center of the bilayer. From the projection of the free energy landscape in Fig. 2C, we would suggest that the mechanism of binding is
where the BA and BB states are of comparable stability.
Examination of these maps reveals how the free energy landscape changes depending on the concentration of PIP3 within the membrane (see Fig. 3). For the membrane containing one PIP3 molecule in each leaflet, states E and BB are of similar stability. For the 2 PIP3 system, the BB state becomes more stable, whereas as seen above for the 3 PIP3 membrane, the free energy of states BA and BB are about the same, and finally for 5 PIP3 only the BA state is seen. Thus, the concentration of PIP3 in the bilayer can alter not only the overall strength of interaction of the PH domain with the membrane but also the orientation of the bound protein relative to the bilayer.
Fig. 3. Free energy surfaces of the GRP1 PH domain interacting with a lipid bilayer including 1 to 10 PIP3 molecules in each leaflet.
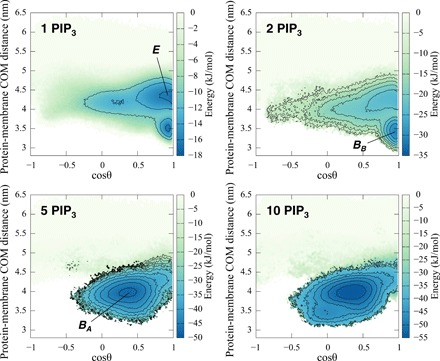
The free energy landscapes are shown as a function of cosθ and the protein-membrane COM distance.
PH bound to multiple PIP3 molecules
From the free energy surfaces, it is evident that there are two bound states of the PH domain, both with a protein-lipid minimum distance of 0.48 nm. Although the free energies of states BA and BB for the 3 PIP3 system are almost the same, they differ in their orientations relative to the bilayer and their interactions with PIP3 molecules in the membrane. In both states, the β1/β2 loop interacts with PIP3 headgroups at the membrane surface. In state BA, the α helix is away from the membrane surface, whereas in state BB, the β1/β2 loop interacts more closely with the membrane, and β3/β4, β5/β6, β9, and the α helix also approach the membrane more closely. From the probability densities of PIP3 molecules in contact with the PH domain, it can be seen that this shifts from two to three molecules bound in the 3 PIP3 system to four to five molecules bound in the 5 PIP3 system (Fig. 4). Examining the probability densities of PIP3 headgroups in the membrane plane in the vicinity of the bound PH domain, it can be seen that for, e.g., the 3 PIP3 simulation, in state BA, there are three regions of high PIP3 density, corresponding to the CA (Fig. 4), the NCA, and a third region adjacent to CA. In state BB, which penetrates more deeply into the bilayer, PIP3 molecules are largely restricted to the CA and NCA sites, with a higher density in the NCA region than for BA. In the presence of five PIP3 molecules, as noted above, state BA is preferred and PIP3 molecules are present at the CA, NCA, and third sites, and also more diffusely around the whole footprint of the bound PH domain. Thus, the PH domain can alternate between two orientations with different lipid footprints and the relative contribution of these two patterns of interaction dependent on the concentration of PIP3 molecules present in the membrane.
Fig. 4. Clustering of lipids in the bilayer (xy) plane underneath a bound GRP1 PH domain.
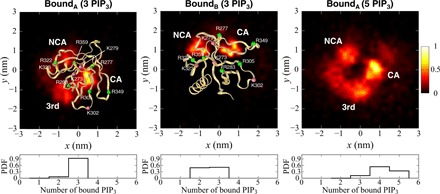
The density (unbiased density normalized by the maximum density) of phosphate headgroups of PIP3 molecules in the bilayer plane corresponding to each bound state is shown on a heat map scale from dark red to yellow. Peaks in the density corresponding to interactions with the canonical binding site (CA), the noncanonical binding site (NCA), and a third site (3rd; see main text for details) are shown. Density maps are shown for the PH domain in the BoundB and BoundA configurations interacting with a lipid bilayer including 3 PIP3 molecules in each leaflet and in the BoundA configuration interacting with a lipid bilayer including 5 PIP3 molecules in each leaflet (in this latter case, the protein is not shown in the interests of clarity). Bottom: Probability density functions (PDFs) for different numbers of PIP3 molecules bound to the PH domain in each of the states.
Binding mechanism
Experimental estimates of the dissociation constants for PIP3 from GRP1 PH range from 5 nM to 1 μM (14, 17, 20, 39–45), which depend on the experimental conditions, e.g., pH condition, temperatures, and lipid bilayer composition. This corresponds to a free energy range of −48 to −34 kJ/mol. From the PMFs in Fig. 1, we can calculate dissociation constants (see Methods for details) and hence free energies of binding. For the 1, 2, 3, 5, and 10 PIP3 systems, this yields values for Kd (dissociation constant) = 2.1 × 10−4, 3 × 10−6, 1.6 × 10−7, 6.3 × 10−9, and 3.9 × 10−10 M, respectively, corresponding to free energies ΔG of −23, −34, −42, −51, and − 58 kJ/mol, respectively. Plotting the free energy minimum in PMFs (Fig. 5A for the REUS simulations and also fig. S4 for corresponding US simulations) suggests that there is no substantive increase in well depth beyond five PIP3 molecules present in the bilayer proximal leaflet to which the PH domain is bound. Examination of the 10 PIP3 systems (both REUS and US) suggested that, on average, ~4 PIP3 molecules were bound to the PH domain. Comparing the free energies of binding as a function of the number of PIP3 molecules present with the range of experimental estimates suggests that the most likely state of the PH domain is bound to between three and five PIP3 molecules.
Fig. 5. A mechanism of PH binding proceeding via an encounter state to two bound states.
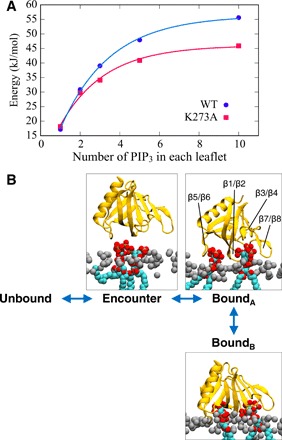
(A) Depth of minimum in the PMF (see also Fig. 1C) of the GRP1 PH domain as a function of the number of PIP3 molecules in each leaflet of the bilayer. Data points for the WT PH domain are shown for simulations using REUS and 1 to 10 PIP3 molecules in each leaflet of the bilayer (blue). The red points correspond to REUS simulations of the K273A mutant. (B) Schematic of a three-step mechanism for binding of the GRP1 PH domain to a bilayer containing multiple PIP3 molecules (see main text for details).
A single mutation K273A within the canonical PIP binding site results in loss of experimentally detectable PIP3 binding to the GRP1 PH domain (41). Our simulations suggest that the protein-lipid interactions for the mutant are reduced by up to 10 kJ/mol, depending on the local concentration of PIP3. This difference between calculations and experiment may reflect the limitations of the current CG model (46) in describing the K273A mutant and/or may reflect the sensitivity of binding experiments to the conditions used. For example, while our simulations measured the interactions of the PH domain with a PIP3-containing lipid bilayer, the experiments on the K273A mutant (41) measured binding of the PH domain to either Ins(1,3,4,5)P4 (i.e., the water-soluble head group of PIP3) or to the corresponding di-C8-phosphoinositide in aqueous solution. The second bound state, BB, is not heavily populated for the K273A mutant in the presence of three PIP3 molecules (see fig. S3) in contrast to the wild-type (WT) PH domain under similar conditions (see above). This suggests the mutation perturbs both the strength and mode of interaction of the domain with a PIP3-containing membrane.
DISCUSSION
Our simulations and PMF calculations indicate that a PH domain can bind simultaneously to multiple PIP molecules in a bilayer, via both the canonical and noncanonical sites alongside a third site and further less well spatially defined interactions. Interactions at these three binding sites increase the overall avidity of the PH domain for a PIP-containing membrane. This is seen in the dependence of the binding free energy (above), which indicates that assuming the GRP1 PH domain binds to at least three molecules of PIP3 gives good agreement with the range of measured dissociation constants for this interaction.
Inspection of the free energy landscape for this interaction suggests a three-stage mechanism for the interaction of the GRP1 PH domain with a PIP-containing membrane, proceeding via an initial Encounter state to two distinct bound states BA and BB that differ in the orientation of the PH domain and the depth of penetration of the bilayer (Fig. 5B). This may be compared with an earlier model of Lai et al. (20) who proposed that a transient membrane association state leads to the PH domain bound to PIP3, thereby enabling a two-dimensional search of the membrane surface for the target lipid. Our data suggest that this mechanism corresponds to a complex free energy landscape with two bound states, the relative occupancies of which is influenced by the (local) concentration of PIP3. Thus, the nature (strength and orientation) of the interaction of the PH domain with the bilayer surface will be influenced by nanoscale clustering of PIP3 and possibly of other molecules within the membrane. This correlates with previous studies of clustering of PIP molecules around bound PH domains (30). Our simulations should enable the design of further experiments to probe the relationship between nanoscale clustering of PIPs and local (i.e., single molecule) binding affinities of PH and related membrane recognition domains.
In addition to the influence of nanoscale clustering of PIP molecules, as noted above, the presence of anionic lipid species other than PIPs (e.g., PS) in a bilayer can influence the affinity of GRP1 PH for PIP3 (17). One can envisage competing effects of background anionic lipids on PH domain interactions with (multiple) PIP molecules in a membrane, namely, (i) a nonspecific electrostatic effect whereby an anionic membrane surface potential favors the formation of an initial encounter complex [cf. (47)] and (ii) a specific effect whereby binding of one or more anionic lipid headgroups to sites on the PH domain competes directly with PIP3 molecules for those sites. This latter effect might be expected to weaken the interaction of a PH domain with a PIP-containing membrane, relative to the interaction of the PH domain with multiple PIP molecules. Preliminary simulations suggest a complex interplay between the electrostatic environment presented by the membrane surface and the number of PIP3 molecules bound to the PH domain. A more systematic analysis of the effects of background anionic lipids on the free energy landscapes of the interaction of PH domains with a PIP-containing bilayer would be an appropriate subject for a future study. In the experimental literature, no significant GRP1 PH domain binding is seen for bilayers containing ~20% PS in the absence of PIP3 (17, 44). In simulations, a PMF calculated for interaction of GRP1 PH with a bound PS molecule (unpublished data) did not reveal any greater interaction than that for the same domain interacting with a pure phosphatidylcholine (PC) bilayer. Thus, we may conclude that the binding of PIP3 to GRP1 PH cannot be substituted for by a high concentration of a simple anionic lipid such as PS.
Overall, our studies suggest that recognition of specific cell membranes by PH domain may be achieved by coincidence detection, either of locally clustered PIP molecules or of PIP molecules alongside other anionic lipids (2), the latter as suggested by data on the effects of other anionic lipids [e.g., (7, 17)]. Thus, high-avidity interaction of a PH domain with a membrane would require a local nanoscale cluster of PIP molecules in an anionic lipid-enriched background. A quantitative mechanistic understanding of the nature of these interactions, for PH and for other membrane recognition domains, will be essential if we wish to intervene therapeutically in a rational fashion when correct membrane recognition is impaired by mutation or other disease processes.
METHODS
Simulations
To investigate of interactions of a membrane-bound protein on a membrane surface, we performed CG-MD simulations of the GRP1 PH domain interacting with a PIP3-containing lipid bilayer. For the structure of GRP1 PH domain, we used the crystal structure of the GRP1 PH domain bound to an Ins(1,3,4,5)P4 molecule (Protein Data Bank ID: 1FGY). A single mutation on the GRP1 PH domain (K273A) was modeled using MODELLER (48). The bilayer used in the simulations consisted of symmetric 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC)/PIP3 bilayers (234/2, 232/4, 230/6, 226/10, or 216/20 molecules). The systems were solvated with 6000 CG water molecules, and NaCl ions at a concentration of 150 mM were added to neutralize the system. The Martini 2.1 force field (49) was used for the CG model of the protein (residues 264 to 380, with a total charge of +3 for the resultant protein model), and the phosphates of PIP3 were assumed to be fully ionized, yielding a total change of −7 for the lipid headgroup). An elastic network model was applied to all backbone particles within a cutoff distance of 0.7 nm to model secondary and tertiary structure (50). The bond lengths were constrained to equilibrium lengths using the LINCS algorithm (51). Lennard-Jones and Coulombic interactions are cut off at 1.1 nm, with the potentials shifted to zero at the cutoff (52).
In the initial configuration of each simulation, the PH domain was displaced away from the lipid bilayer surface. All systems were subjected to steepest-descent energy minimized to remove the initial close contacts and equilibrated for 1 ns with the protein backbone particles restrained in NPT constant CG-MD simulations. A time step of 30 fs was used. The neighbor list was updated every 20 steps using the Verlet neighbor search algorithm. The systems were subject to pressure scaling to 1 bar using Parrinello-Rahman barostat (53), with temperature scaling to 323 K using velocity-rescaling method (54) with coupling times of 1.0 and 12.0 ps.
Estimation of PMFs
The last window frame of the pre-equilibrated simulation was used for initial configuration for the unbound states. For the production runs of the REUS-MD simulation, the PLUMED2 package (version 2.3.3) (55) was used to patch GROMACS 5.1.4 (56), define the CVs, and perform the biasing. REUS-MD simulations were produced using a similar protocol used in a previous study for lipid interaction with transmembrane protein within lipid membranes (36). Replica exchanges were evaluated using the Boltzmann criterion. A CV was defined by a minimum distance between protein amino acids and phosphate group of lipids. US windows were set up with 16 windows, with the CV linearly spaced distances from 0.4 to 1.5 nm, with a force constant of 1000 kJ mol−1 nm−2. Exchanges of replicas were attempted every 1000 steps. The simulations were performed for 15 μs for each replica, yielding a total REUS-MD simulation time of 240 μs. For the analysis of each simulation, data for 0 to 2 μs were discarded before collecting data from 2 to 15 μs, which yield good convergence of the PMFs (see fig. S2). Only for the system of WT (10 PIP3), 20-μs simulation was performed for each replica. Multiple binding and unbinding transitions were observed in the continuous trajectories obtained by following the replicas. The unbiased PMFs after subtracting the effect of the umbrella potentials were calculated by the weighted histogram analysis method (57, 58). The two-dimensional free energy surface for other variables, ξ2 and ξ3, was estimated (using locally written code) by reweighting the trajectories obtained by the REUS-MD simulation biasing along a single CV, ξ1 (59). Note that this assumes that the REUS-MD simulations for ξ1 provided sufficient sampling for the other variables, ξ2 and ξ3. US simulations were performed as described previously (34, 35). Molecular graphics images were generated using VMD (60).
Calculation of the density of lipid around the protein
The density of phosphate headgroups of PIP3 corresponding to each bound state was calculated with the unbiased distribution obtained from the REUS simulation. Each bound state was distinguished from the free energy surfaces. The bound state BA was defined as protein-lipid minimum distance [0.46, 0.52], protein-membrane COM distance [3.91, 3.97], and cosθ [0.36, 0.46]. The bound state BB was defined as protein-lipid minimum distance [0.45, 0.51], protein-membrane COM distance [3.45, 3.51], and cosθ [0.89, 0.99]. A cutoff distance of 0.7 nm was used for the protein-lipid contact, corresponding to a generally used definition for protein-lipid interactions for the MARTINI CG model.
Calculation of the dissociation constant
For binding of a protein to a membrane in a periodic box with the membrane perpendicular to the z axis
where [M] is the molar ratio of the protein, y is the fraction bound, A is the x-y area of the membrane, Lz is the box length in the z direction, NA is Avogadro’s number, and F(r) is the PMF for association on the membrane. F(r) should be the constant zero above the bound distance b, and then we get
Taking the limit as Lz → ∞, we get
Here, we used b = 1 nm.
Supplementary Material
Acknowledgments
Funding: E.Y. was supported by MEXT (Ministry of Education, Culture, Sports, Science and Technology) Grant-in-Aid for the “Building of Consortia for the Development of Human Resources in Science and Technology.” This work was supported by JSPS KAKENHI grant number JP18H04678. J.D. was supported by a Wellcome and NIH Four-year PhD Studentship program (grant number WT100946AIA). R.B.B. and J.D. were supported by the Intramural Research Program of the National Institute of Diabetes and Digestive and Kidney Diseases of the NIH. This work used the computational resources of the NIH HPC Biowulf cluster (http://hpc.nih.gov). Simulations were carried out, in part, on the ARCHER UK National Supercomputing Service (www.archer.ac.uk), provided by HECBioSim (www.hecbiosim.ac.uk), which is supported by the EPSRC (EP/L000253/1). Research in M.S.P.S.’s group is supported by Wellcome (208361/Z/17/Z), BBSRC (BB/R00126X/1, BB/S003339/1, and BB/N000145/1), and EPSRC (EP/R004722/1). Research in P.J.S.’s group is supported by MRC, Wellcome (208361/Z/17/Z), and BBSRC (BB/P01948X/1, BB/R002517/1, and BB/S003339/1). F.B.N. was supported by Oxford University Clarendon Fund and by Lincoln College. Author contributions: E.Y., J.D., and F.B.N. performed simulations and analysis. The research reported emerged from discussions between all authors, and all authors contributed to the writing of the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/8/eaay5736/DC1
Fig. S1. PMFs for the GRP1 PH domain with a single mutation (K273A) interacting with lipid bilayers containing 1 to 10 PIP3 molecules.
Fig. S2. Convergence of PMF calculations.
Fig. S3. Free energy maps for the GRP1 PH domain with a single mutation (K273A) interacting with a lipid bilayer including 1 to 10 PIP3 molecules in each leaflet.
Fig. S4. PMFs from US for the GRP1 PH domain interacting with lipid bilayers containing 1, 2, or 10 PIP3 molecules.
REFERENCES AND NOTES
- 1.Lemmon M. A., Membrane recognition by phospholipid-binding domains. Nat. Rev. Mol. Cell Biol. 9, 99–111 (2008). [DOI] [PubMed] [Google Scholar]
- 2.Stahelin R. V., Scott J. L., Frick C. T., Cellular and molecular interactions of phosphoinositides and peripheral proteins. Chem. Phys. Lipids 182, 3–18 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kutateladze T. G., Translation of the phosphoinositide code by PI effectors. Nat. Chem. Biol. 6, 507–513 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carpten J. D., Faber A. L., Horn C., Donoho G. P., Briggs S. L., Robbins C. M., Hostetter G., Boguslawski S., Moses T. Y., Savage S., Uhlik M., Lin A., du J., Qian Y. W., Zeckner D. J., Tucker-Kellogg G., Touchman J., Patel K., Mousses S., Bittner M., Schevitz R., Lai M. H. T., Blanchard K. L., Thomas J. E., A transforming mutation in the pleckstrin homology domain of AKT1 in cancer. Nature 448, 439–444 (2007). [DOI] [PubMed] [Google Scholar]
- 5.Baraldi E., Carugo K. D., Hyvönen M., Surdo P. L., Riley A. M., Potter B. V. L., O’Brien R., Ladbury J. E., Saraste M., Structure of the PH domain from Bruton’s tyrosine kinase in complex with inositol 1,3,4,5-tetrakisphosphate. Structure 7, 449–460 (1999). [DOI] [PubMed] [Google Scholar]
- 6.Jian X., Tang W. K., Zhai P., Roy N. S., Luo R., Gruschus J. M., Yohe M. E., Chen P. W., Li Y., Byrd R. A., Xia D., Randazzo P. A., Molecular basis for cooperative binding of anionic phospholipids to the PH domain of the Arf GAP ASAP1. Structure 23, 1977–1988 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vonkova I., Saliba A. E., Deghou S., Anand K., Ceschia S., Doerks T., Galih A., Kugler K. G., Maeda K., Rybin V., van Noort V., Ellenberg J., Bork P., Gavin A. C., Lipid cooperativity as a general membrane-recruitment principle for PH domains. Cell Rep. 12, 1519–1530 (2015). [DOI] [PubMed] [Google Scholar]
- 8.van den Bogaart G., Meyenberg K., Risselada H. J., Amin H., Willig K. I., Hubrich B. E., Dier M., Hell S. W., Grubmüller H., Diederichsen U., Jahn R., Membrane protein sequestering by ionic protein-lipid interactions. Nature 479, 552–555 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tsuji T., Takatori S., Fujimoto T., Definition of phosphoinositide distribution in the nanoscale. Curr. Opin. Cell Biol. 57, 33–39 (2019). [DOI] [PubMed] [Google Scholar]
- 10.Bilkova E., Pleskot R., Rissanen S., Sun S., Czogalla A., Cwiklik L., Róg T., Vattulainen I., Cremer P. S., Jungwirth P., Coskun Ü., Calcium directly regulates phosphatidylinositol 4,5-bisphosphate headgroup conformation and recognition. J. Am. Chem. Soc. 139, 4019–4024 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Graber Z. T., Thomas J., Johnson E., Gericke A., Kooijman E. E., Effect of H-bond donor lipids on phosphatidylinositol-3,4,5-trisphosphate ionization and clustering. Biophys. J. 114, 126–136 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Meca J., Massoni-Laporte A., Martinez D., Sartorel E., Loquet A., Habenstein B., McCusker D., Avidity-driven polarity establishment via multivalent lipid-GTPase module interactions. EMBO J. 38, e99652 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ni T., Kalli A. C., Naughton F. B., Yates L. A., Naneh O., Kozorog M., Anderluh G., Sansom M. S. P., Gilbert R. J. C., Structure and lipid-binding properties of the kindlin-3 pleckstrin homology domain. Biochem. J. 474, 539–556 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ferguson K. M., Kavran J. M., Sankaran V. G., Fournier E., Isakoff S. J., Skolnik E. Y., Lemmon M. A., Structural basis for discrimination of 3-phosphoinositides by pleckstrin homology domains. Mol. Cell 6, 373–384 (2000). [DOI] [PubMed] [Google Scholar]
- 15.Moravcevic K., Oxley C. L., Lemmon M. A., Conditional peripheral membrane proteins: Facing up to limited specificity. Structure 20, 15–27 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ceccarelli D. F. J., Blasutig I. M., Goudreault M., Li Z., Ruston J., Pawson T., Sicheri F., Non-canonical interaction of phosphoinositides with Pleckstrin Homology domains of Tiam1 and ArhGAP9. J. Biol. Chem. 282, 13864–13874 (2007). [DOI] [PubMed] [Google Scholar]
- 17.Corbin J. A., Dirkx R. A., Falke J. J., GRP1 pleckstrin homology domain: Activation parameters and novel search mechanism for rare target lipid. Biochem. 43, 16161–16173 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lumb C. N., He J., Xue Y., Stansfeld P. J., Stahelin R. V., Kutateladze T. G., Sansom M. S. P., Biophysical and computational studies of membrane penetration by the GRP1 pleckstrin homology domain. Structure 19, 1338–1346 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen H.-C., Ziemba B. P., Landgraf K. E., Corbin J. A., Falke J. J., Membrane docking geometry of GRP1 PH domain bound to a target lipid bilayer: An EPR site-directed spin-labeling and relaxation study. PLOS ONE 7, e33640 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lai C.-L., Srivastava A., Pilling C., Chase A. R., Falke J. J., Voth G. A., Molecular mechanism of membrane binding of the GRP1 PH domain. J. Mol. Biol. 425, 3073–3090 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.van Meer G., Voelker D. R., Feigenson G. W., Membrane lipids: Where they are and how they behave. Nat. Rev. Mol. Cell Biol. 9, 112–124 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Balla T., Phosphoinositides: Tiny lipids with giant impact on cell regulation. Physiol. Rev. 93, 1019–1137 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Picas L., Viaud J., Schauer K., Vanni S., Hnia K., Fraisier V., Roux A., Bassereau P., Gaits-Iacovoni F., Payrastre B., Laporte J., Manneville J. B., Goud B., BIN1/M-Amphiphysin2 induces clustering of phosphoinositides to recruit its downstream partner dynamin. Nat. Commun. 5, 5647 (2014). [DOI] [PubMed] [Google Scholar]
- 24.Yamamoto E., Kalli A. C., Akimoto T., Yasuoka K., Sansom M. S. P., Anomalous dynamics of a lipid recognition protein on a lipid membrane surface. Sci. Rep. 5, 18245 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Karandur D., Nawrotek A., Kuriyan J., Cherfils J., Multiple interactions between an Arf/GEF complex and charged lipids determine activation kinetics on the membrane. Proc. Natl. Acad. Sci. U.S.A. 114, 11416–11421 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang Q., Pechersky Y., Sagawa S., Pan A. C., Shaw D. E., Structural mechanism for Bruton’s tyrosine kinase activation at the cell membrane. Proc. Natl. Acad. Sci. U.S.A. 116, 9390–9399 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Park W. S., Do Heo W., Whalen J. H., O’Rourke N. A., Bryan H. M., Meyer T., Teruel M. N., Comprehensive identification of PIP3-regulated PH domains from C. elegans to H. sapiens by model prediction and live imaging. Mol. Cell 30, 381–392 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yu J. W., Mendrola J. M., Audhya A., Singh S., Keleti D., DeWald D. B., Murray D., Emr S. D., Lemmon M. A., Genome-wide analysis of membrane targeting by S. cerevisiae pleckstrin homology domains. Mol. Cell 13, 677–688 (2004). [DOI] [PubMed] [Google Scholar]
- 29.McLaughlin S., Murray D., Plasma membrane phosphoinositide organization by protein electrostatics. Nature 438, 605–611 (2005). [DOI] [PubMed] [Google Scholar]
- 30.Yamamoto E., Akimoto T., Kalli A. C., Yasuoka K., Sansom M. S. P., Dynamic interactions between a membrane binding protein and lipids induce fluctuating diffusivity. Sci. Adv. 3, e1601871 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Corradi V., Mendez-Villuendas E., Ingólfsson H. I., Gu R. X., Siuda I., Melo M. N., Moussatova A., DeGagné L. J., Sejdiu B. I., Singh G., Wassenaar T. A., Delgado Magnero K., Marrink S. J., Tieleman D. P., Lipid–protein interactions are unique fingerprints for membrane proteins. ACS Central Sci. 4, 709–717 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marrink S. J., Corradi V., Souza P. C. T., Ingólfsson H. I., Tieleman D. P., Sansom M. S. P., Computational modeling of realistic cell membranes. Chem. Rev. 119, 6184–6226 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yamamoto E., Kalli A. C., Yasuoka K., Sansom M. S. P., Interactions of pleckstrin homology domains with membranes: Adding back the bilayer via high-throughput molecular dynamics. Structure 24, 1421–1431 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Naughton F. B., Kalli A. C., Sansom M. S. P., Association of peripheral membrane proteins with membranes: Free energy of binding of GRP1 PH domain with phosphatidylinositol phosphate-containing model bilayers. J. Phys. Chem. Lett. 7, 1219–1224 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Naughton F., Kalli A. C., Sansom M. S. P., Modes of interaction of pleckstrin homology domains with membranes: Toward a computational biochemistry of membrane recognition. J. Mol. Biol. 430, 372–388 (2018). [DOI] [PubMed] [Google Scholar]
- 36.Domański J., Hedger G., Best R., Stansfeld P. J., Sansom M. S. P., Convergence and sampling in determining free energy landscapes for membrane protein association. J. Phys. Chem. B 121, 3364–3375 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sugita Y., Kitao A., Okamoto Y., Multidimensional replica-exchange method for free-energy calculations. J. Chem. Phys. 113, 6042–6051 (2000). [Google Scholar]
- 38.Marrink S. J., Tieleman D. P., Perspective on the Martini model. Chem. Soc. Rev. 42, 6801–6822 (2013). [DOI] [PubMed] [Google Scholar]
- 39.Kavran J. M., Klein D. E., Lee A., Falasca M., Isakoff S. J., Skolnik E. Y., Lemmon M. A., Specificity and promiscuity in phosphoinositide binding by pleckstrin homology domains. J. Biol. Chem. 273, 30497–30508 (1998). [DOI] [PubMed] [Google Scholar]
- 40.Klarlund J. K., Tsiaras W., Holik J. J., Chawla A., Czech M. P., Distinct polyphosphoinositide binding selectivities for pleckstrin homology domains of GRP1-like proteins based on diglycine versus triglycine motifs. J. Biol. Chem. 275, 32816–32821 (2000). [DOI] [PubMed] [Google Scholar]
- 41.Cronin T., DiNitto J. P., Czech M. P., Lambright D. G., Structural determinants of phosphoinositide selectivity in splice variants of Grp1 family PH domains. EMBO J. 23, 3711–3720 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Manna D., Albanese A., Park W. S., Cho W., Mechanistic basis of differential cellular responses of phosphatidylinositol 3,4-bisphosphate- and phosphatidylinositol 3,4,5-trisphosphate-binding pleckstrin homology domains. J. Biol. Chem. 282, 32093–32105 (2007). [DOI] [PubMed] [Google Scholar]
- 43.He J., Haney R. M., Vora M., Verkhusha V. V., Stahelin R. V., Kutateladze T. G., Molecular mechanism of membrane targeting by the Grp1 PH domain. J. Lipid Res. 49, 1807–1815 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Knight J. D., Falke J. J., Single-molecule fluorescence studies of a PH domain: New insights into the membrane docking reaction. Biophys. J. 96, 566–582 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pilling C., Landgraf K. E., Falke J. J., The GRP1 PH domain, like the AKT1 PH domain, possesses a sentry glutamate residue essential for specific targeting to plasma membrane PI(3,4,5)P3. Biochemistry 50, 9845–9856 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Alessandri R., Souza P. C. T., Thallmair S., Melo M. N., de Vries A. H., Marrink S. J., Pitfalls of the Martini model. J. Chem. Theor. Comput. 15, 5448–5460 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lumb C. N., Sansom M. S. P., Finding a needle in a haystack: The role of electrostatics in target lipid recognition by PH domains. PLOS Comp. Biol. 8, e1002617 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fiser A., Šali A., Modeller: Generation and refinement of homology-based protein structure models. Methods Enzymol. 374, 461–491 (2003). [DOI] [PubMed] [Google Scholar]
- 49.Monticelli L., Kandasamy S. K., Periole X., Larson R. G., Tieleman D. P., Marrink S. J., The MARTINI coarse grained force field: Extension to proteins. J. Chem. Theory Comput. 4, 819–834 (2008). [DOI] [PubMed] [Google Scholar]
- 50.Atilgan A. R., Durell S. R., Jernigan R. L., Demirel M. C., Keskin O., Bahar I., Anisotropy of fluctuation dynamics of proteins with an elastic network model. Biophys. J. 80, 505–515 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hess B., Bekker H., Berendsen H. J. C., Fraaije J. G. E. M., LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 18, 1463–1472 (1997). [Google Scholar]
- 52.de Jong D. H., Baoukina S., Ingolfsson H. I., Marrink S. J., Martini straight: Boosting performance using a shorter cutoff and GPUs. Comput. Phys. Commun. 199, 1–7 (2016). [Google Scholar]
- 53.Parrinello M., Rahman A., Polymorphic transitions in single-crystals: A new molecular-dynamics method. J. Appl. Phys. 52, 7182–7190 (1981). [Google Scholar]
- 54.Bussi G., Donadio D., Parrinello M., Canonical sampling through velocity rescaling. J. Chem. Phys. 126, 014101 (2007). [DOI] [PubMed] [Google Scholar]
- 55.Tribello G. A., Bonomi M., Branduardi D., Camilloni C., Bussi G., PLUMED 2: New feathers for an old bird. Comput. Phys. Commun. 185, 604–613 (2014). [Google Scholar]
- 56.Abraham M. J., Murtola T., Schulz R., Páll S., Smith J. C., Hess B., Lindah E., GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1–2, 19–25 (2015). [Google Scholar]
- 57.Souaille M., Roux B., Extension to the weighted histogram analysis method: Combining umbrella sampling with free energy calculations. Comput. Phys. Commun. 135, 40–57 (2001). [Google Scholar]
- 58.Hub J. S., de Groot B. L., van der Spoel D., g_wham—A free weighted histogram analysis implementation Including robust error and autocorrelation estimates. J. Chem. Theory Comput. 6, 3713–3720 (2010). [Google Scholar]
- 59.Banavali N. K., Roux B., Free energy landscape of A-DNA to B-DNA conversion in aqueous solution. J. Am. Chem. Soc. 127, 6866–6876 (2005). [DOI] [PubMed] [Google Scholar]
- 60.Humphrey W., Dalke A., Schulten K., VMD: Visual molecular dynamics. J. Mol. Graph. 14, 33–38 (1996). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/8/eaay5736/DC1
Fig. S1. PMFs for the GRP1 PH domain with a single mutation (K273A) interacting with lipid bilayers containing 1 to 10 PIP3 molecules.
Fig. S2. Convergence of PMF calculations.
Fig. S3. Free energy maps for the GRP1 PH domain with a single mutation (K273A) interacting with a lipid bilayer including 1 to 10 PIP3 molecules in each leaflet.
Fig. S4. PMFs from US for the GRP1 PH domain interacting with lipid bilayers containing 1, 2, or 10 PIP3 molecules.