

The IRE1α kinase pathway is a potential drug development target for diseases caused by misfolded CFTR and pendrin.
Abstract
The most prevalent pathogenic mutations in the CFTR (ΔF508) and SLC26A4/pendrin (p.H723R), which cause cystic fibrosis and congenital hearing loss, respectively, evoke protein misfolding and subsequent defects in their cell surface trafficking. Here, we report that activation of the IRE1α kinase pathway can rescue the cell surface expression of ΔF508-CFTR and p.H723R-pendrin through a Golgi-independent unconventional protein secretion (UPS) route. In mammalian cells, inhibition of IRE1α kinase, but not inhibition of IRE1α endonuclease and the downstream effector XBP1, inhibited CFTR UPS. Treatment with the IRE1α kinase activator, (E)-2-(2-chlorostyryl)-3,5,6-trimethyl-pyrazine (CSTMP), rescued cell surface expression and functional activity of ΔF508-CFTR and p.H723R-pendrin. Treatment with a nontoxic dose of CSTMP to ΔF508-CFTR mice restored CFTR surface expression and CFTR-mediated anion transport in the mouse colon. These findings suggest that UPS activation via IRE1α kinase is a strategy to treat diseases caused by defective cell surface trafficking of membrane proteins, including ΔF508-CFTR and p.H723R-pendrin.
INTRODUCTION
Protein folding and trafficking deficiency is a common pathogenic mechanism that disrupts the homeostasis of living systems and therefore contributes to numerous human diseases (1–3). In eukaryotic systems, secretory proteins, including transmembrane proteins, are synthesized on the ribosomes and cotranslationally translocated into the endoplasmic reticulum (ER). The ER maintains various chaperones and folding catalysts that enable correctly folded proteins to be distinguished from misfolded ones (4). Secretory proteins that satisfy the “ER quality control” mechanism are exported from the ER, transported to the Golgi, and lastly sent to the plasma membrane or extracellular medium. In contrast, incompletely folded proteins are retained in the ER and are selectively targeted to the ER-associated degradation (ERAD) process to prevent risks that result from malfunctioning proteins when they escape the ER (5). Many of the disease-causing mutations of cystic fibrosis (CF) transmembrane conductance regulator (CFTR, ABCC7) and pendrin (SLC26A4) result in defective protein folding. Consequently, these misfolded proteins fail to enter the conventional ER-to-Golgi protein secretion pathway, which ultimately cause CF and congenital hearing loss, respectively.
CFTR is a cyclic adenosine 5′-monophosphate (cAMP)–regulated transporter with anion channel activity that conducts Cl− and HCO3− at the apical surface of epithelial cells in secretory organs, including the airways, pancreas, intestines, sweat glands, and exocrine glands (6). CFTR has two N-linked glycosylation sites that are initially core-glycosylated in the ER (band B) and mediate interactions with the ER lectin chaperones as part of the ER quality control system (7). After translocation into the Golgi, these sites undergo complex glycosylation (band C), and the complex-glycosylated CFTR travels to the apical surface of epithelial tissues (8). Among nearly 2000 identified mutations, the deletion of the phenylalanine residue at the position 508 (ΔF508) is the most common disease-causing CFTR mutation, which leads to protein misfolding, ER retention, and degradation by ERAD (9). Consequently, ΔF508-CFTR proteins remain in the core-glycosylated form within the ER, and negligible quantities are expressed at the plasma membrane surface (1).
Pendrin is a transmembrane protein that transports anions, such as Cl−, I−, and HCO3− in the inner ear and thyroid follicles (10). Loss of pendrin function, resulting from gene mutations, underlies nonsyndromic deafness with an enlarged vestibular aqueduct (DFNB4) and Pendred syndrome, which is the second most common cause of congenital hearing loss worldwide and the most common cause in East Asia. The most frequent disease-causing mutation of pendrin, p.H723R (His723Arg), results in protein misfolding, ER retention, and degradation by ERAD, which is similar to the fate of ΔF508-CFTR proteins (3).
Most secretory proteins use the conventional Golgi-mediated secretion pathway to travel from the ER to the plasma membrane. In addition to this classical secretory pathway, an emerging mechanism termed “unconventional protein secretion (UPS)” mediates the cell surface transport of diverse cytosolic and transmembrane proteins (11). Prior studies have demonstrated that immature core-glycosylated CFTR and pendrin can reach the plasma membrane via a Golgi-independent UPS route under blocked ER-to-Golgi transport or ER stress conditions (12, 13). Once rescued on the cell surface, the core-glycosylated CFTR and pendrin retain their anion transporting activity, although at an attenuated level (12, 13). Therefore, selective activation of UPS without initiating substantial cellular stress is a potential therapeutic approach to treat diseases caused by defects in the folding and cell surface trafficking of membrane proteins. However, the previously reported methods to activate the UPS of CFTR and pendrin (i.e., overexpression of GRASP55 for ΔF508-CFTR and overexpression of DNAJC14 for p.H723R-pendrin) (12, 13) are not clinically relevant for the treatment of human patients with CF, DFNB4, or Pendred syndrome.
Although recent studies have identified several key molecules associated with the UPS of membrane proteins (i.e., Sec16A in ER exit and early autophagy/multivesicular body components in the vesicular pathway for UPS of CFTR) (14, 15), the entire complex of UPS regulatory mechanisms remains unknown. In general, most UPS is not constitutive but instead is stress induced (16). For example, ER-to-Golgi transport blockades induce stress-associated signals that are involved in the UPS of membrane proteins. The blockade of conventional protein secretion from the ER to the Golgi causes unfolded protein to accumulate in the ER lumen, which then induces ER stress and an adaptive cellular response called unfolded protein response (UPR) (17). In eukaryotes, there are three major branches of UPR sensors, including inositol-requiring enzyme 1α (IRE1α) and IRE1β, protein kinase RNA-like ER kinase (PERK), and activating transcription factor 6α (ATF6α) and ATF6β, which localize at the ER membrane and transduce ER stress signaling. IRE1α, the most evolutionarily conserved form of UPR signaling, appears to play an important role in the ER stress–induced UPS of membrane proteins. Depletion of IRE1α abolished the ER-to-Golgi blockade–induced UPS of CFTR and pendrin (13). Nevertheless, the specific mechanism of UPS regulation by IRE1α remains elusive.
IRE1 is a type I ER transmembrane protein that consists of an ER-luminal domain, which acts as an unfolded protein sensor during ER stress, and cytosolic domains that include the endoribonuclease domain and the Ser/Thr protein kinase domain. Activated mammalian IRE1α protein can transmit ER stress signals through endonucleolytic cleavage of the mRNAs that encode for transcription factor X-box–binding protein 1 (XBP1), resulting in active spliced XBP1 (XBP1s) that up-regulates the UPR-related genes involved in protein folding, protein quality control, and ERAD (18). Using this mechanism, the unfolded protein burden in the ER is reduced, and ER homeostasis is regained. The IRE1-dependent decay (RIDD) process is also recognized as a complementary mechanism that attenuates the ER load by degrading mRNAs of various proteins (19). In addition, the activation of IRE1 protein kinase can trigger “alarm stress pathways” by recruiting adaptor proteins, such as tumor necrosis factor receptor–associated factor 2 (TRAF2), which leads to the activation of apoptosis signal–regulating kinase 1 (ASK1) and its downstream effector, c-Jun N-terminal kinase (JNK) (20).
In the present study, we investigated the specific role of IRE1α in UPS by analyzing each signaling arm. Our data revealed that UPS of CFTR and pendrin is activated in vitro and in vivo through the IRE1α kinase–mediated signaling cascade but not XBP1- and RIDD-dependent pathways that require IRE1α ribonuclease (RNase) activity. The IRE1α kinase pathway may provide a potential drug development target for diseases caused by protein folding and trafficking deficiencies.
RESULTS
Endoribonuclease activity of IRE1α is redundant for UPS of CFTR
Mammalian IRE1α is maintained as an inactive monomer by associating with the ER chaperone protein BiP (21). Oligomerization of IRE1 monomers is the earliest step of IRE1 activation in response to the accumulation of unfolded proteins in the ER (21). Inhibition of ER-to-Golgi transport by the dominant-inhibitory form of the Arf1 guanosine triphosphatase, Arf1-Q71L, causes ER stress due to the accumulation of secretory proteins in the ER lumen (12). ER stress can be also induced by treatment with the Ca2+–adenosine triphosphatase (ATPase) inhibitor thapsigargin, which depletes calcium stores in ER lumen (22). Consistent with these mechanisms of inducing ER stress, IRE1α clustered into oligomers after Arf1-Q71L expression (48 hours) or thapsigargin treatment (12 hours) in a cross-linking assay performed in human embryonic kidney (HEK) 293 cells (fig. S1A).
Oligomerization of IRE1α opens the kinase domain to initiate its activity and activates the RNase domain for mRNA substrate processing (21). Both Arf1-Q71L and thapsigargin triggered IRE1α kinase activity in HEK293 cells and consequently induced IRE1α autophosphorylation and phosphorylation of the downstream effector ASK1 (Fig. 1, A and B). Treatment with thapsigargin (12 hours), which induced ER stress by depleting ER Ca2+, evoked XBP1 splicing, while the prolonged induction of ER stress with Arf1-Q71L overexpression (48 hours) did not (Fig. 1, A and B, and fig. S1B). Because both Arf1-Q71L and thapsigargin induced UPS of membrane proteins (12, 13), it implies that IRE1α kinase–mediated signaling, but not the IRE1α RNase substrate XBP1, is predominantly involved in ER stress–associated UPS. Surface biotinylation experiments demonstrated that the depletion of IRE1α, but not XBP1, abolished the UPS of ΔF508-CFTR, further supporting the hypothesis that ER stress–associated UPS is independent of XBP1 (Fig. 1, C and D). Immunoblot analysis and mRNA quantification for IRE1α and of XBP1 (Fig. 1C and fig. S1, B and C) confirmed the knockdown of each gene in the present study.
Fig. 1. XBP1 splicing and IRE1α endonuclease activity are not involved in UPS of ΔF508-CFTR.
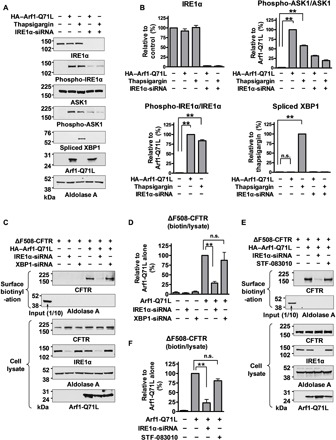
(A and B) Arf1-Q71L–induced ER-to-Golgi blockade activates ASK1 phosphorylation but not XBP1 splicing. HEK293 cells were transfected with plasmids for Arf1-Q71L (48 hours) or treated with thapsigargin (5 μM, 12 hours) to induce ER stress. Representative immunoblots are shown in (A), and the quantification results of multiple experiments are summarized in (B) (n = 3). The phosphorylation levels of IRE1α and ASK1 were calculated as a ratio of the phospho/total proteins. (C and D) IRE1α, but not XBP1, is required for the Arf1-Q71L–induced UPS of ΔF508-CFTR. Surface biotinylation assays were performed in HEK293 cells transfected with the indicated plasmids and/or small interfering RNAs (siRNAs). Representative immunoblots are shown in (C), and the results of multiple experiments are summarized in (D) (n = 4). Cell surface–specific labeling of proteins was confirmed by the absence of the cytosolic protein aldolase A in the biotinylated fraction. (E and F) Inhibition of IRE1α endonuclease activity by STF-083010 did not block UPS of ΔF508-CFTR. Surface biotinylation assays were performed with STF-083010 treatment (60 μM, 12 hours). Representative results are shown in (E), and the results of multiple experiments are summarized in (F) (n = 4). Bar graph data are shown as the means ± SEM. **P < 0.01; n.s., not significant. Data were analyzed by one-way analysis of variance followed by Tukey’s multiple comparison test.
Because of the unexpected finding that UPS of ΔF508-CFTR is independent of XBP1 splicing, we next investigated whether IRE1α could mediate UPS in the absence of its RNase activity. STF-083010 is a compound that targets the catalytic core of the IRE1α RNase domain and inhibits IRE1α endonuclease activity without affecting its kinase activity or overall oligomerization state (23). Treatment with STF-083010 (60 μM, 12 hours) abolished thapsigargin-induced XBP1s production (fig. S1D). However, STF-083010 did not affect the Arf1-Q71L–induced UPS of ΔF508-CFTR (Fig. 1, E and F). These results collectively indicate that the RNase activity of IRE1α is redundant for UPS of ΔF508-CFTR.
IRE1α kinase–ASK1 pathway is required for UPS of ΔF508-CFTR
We next focused on the role of IRE1α kinase–mediated signaling in UPS of membrane proteins. The type I kinase inhibitor APY29 inhibits IRE1α trans-autophosphorylation by competitively occupying the adenosine 5′-triphosphate (ATP)–binding pocket of IRE1α (24). In HEK293 cells, the median inhibitory concentration value of APY29 against IRE1α phosphorylation was estimated to be 9.6 μM, and treatment with 100 μM APY29 for 12 hours almost completely inhibited the Arf1-Q71L–induced phosphorylation of IRE1α and ASK1 (fig. S2, A and B). APY29 (100 μM) abolished the surface targeting of ΔF508-CFTR induced by Arf1-Q71L ectopic expression (Fig. 2, A and B). If APY29 inhibits CFTR protein synthesis, it may also cause a rapid reduction in cell surface ΔF508-CFTR because its stability is known to be lower than that of wild type (WT)–CFTR (25). Treatment with the protein synthesis inhibitor cycloheximide (0.1 mg/ml) induced an accelerated decrease in cell surface ΔF508-CFTR levels (fig. S2, C to F). However, APY29 reduced only the cell surface ΔF508-CFTR levels without affecting the total protein levels, while cycloheximide caused a rapid decay of both the cell surface and total cellular ΔF508-CFTR levels (Fig. 2, A and B). These data indicate that APY29 selectively inhibited the cell surface rescue of ΔF508-CFTR without interfering with protein synthesis or degradation.
Fig. 2. IRE1α kinase–ASK1 pathway is required for UPS of ΔF508-CFTR.
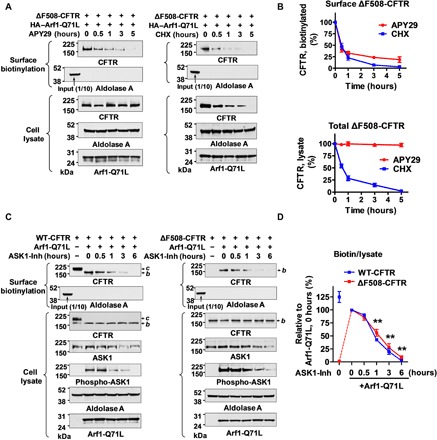
(A and B) Effects of IRE1α kinase inhibitor APY29 on the UPS of ΔF508-CFTR. Surface biotinylation assays were performed in HEK293 cells after the induction of ΔF508-CFTR UPS with Arf1-Q71L, followed by the addition of IRE1α kinase inhibitor APY29 (100 μM) or the protein synthesis inhibitor cycloheximide (0.1 mg/ml) for the indicated time periods. Representative immunoblots are shown in (A), and results of multiple experiments are summarized in (B) (n = 5). APY29 reduced surface ΔF508-CFTR in a time-dependent manner without affecting the total protein levels. (C and D) The ASK1 inhibitor MSC2032964A (ASK1-Inh) inhibits Arf1-Q71L–induced UPS of CFTR. Surface biotinylation assays were performed in HEK293 cells transfected with plasmids encoding for WT-CFTR, ΔF508-CFTR, and/or Arf1-Q71L. Some cells were treated with MSC2032964A (10 μM) for the indicated time periods. Representative surface biotinylation results of WT-CFTR and ΔF508-CFTR are presented in (C). The line graph in (D) summarizes the results of multiple experiments (n = 6). The inhibitory effect of MSC2032964A on ASK1 activity was confirmed by decreased ASK1 phosphorylation. b, core-glycosylated CFTR; c, complex-glycosylated CFTR. **P < 0.01, compared to Arf1-Q71L, 0 hours (for both WT-CFTR and ΔF508-CFTRs).
Given that IRE1α activates ASK1 through an IRE1-TRAF2-ASK1 complex formation on the ER outer membrane, and thus couples the ER stress signal to the ASK1-JNK signaling pathway (20, 26), we further investigated the role of ASK1 in unconventional CFTR secretion. MSC2032964A is a potent and selective ASK1 inhibitor that blocks lipopolysaccharide–induced ASK1 phosphorylation (27). The results in Fig. 2 (C and D) show that inhibition of ASK1 phosphorylation with MSC2032964A (ASK1-Inh, 10 μM) treatment reduced Arf1-Q71L–induced UPS of both WT- and ΔF508-CFTRs in a time-dependent manner. In addition, depletion of ASK1 by treatment with specific small interfering RNAs (siRNAs) abolished the Arf1-Q71L–induced UPS of WT- and ΔF508-CFTRs (fig. S3). Together, these results indicate that the kinase activity of IRE1α and ASK1 plays a critical role in the UPS of core-glycosylated CFTRs.
Up-regulation of IRE1α kinase induces UPS of ΔF508-CFTR
Next, we examined the effects of IRE1α kinase up-regulation in the context of rescuing trafficking defects caused by the ΔF508-CFTR mutation. As shown in Fig. 3 (A and B), overexpression of IRE1α alone resulted in surface expression of ΔF508-CFTR. Notably, overexpression of IRE1α kinase–dead mutant (K599A) (28) did not induce the cell surface expression of ΔF508-CFTR, while the IRE1α endonuclease–dead mutant (N906A) (28) did (Fig. 3, A and B), highly supporting the hypothesis that IRE1α kinase up-regulation can rescue defective ΔF508-CFTR trafficking.
Fig. 3. IRE1α kinase activation by CSTMP induces UPS of ΔF508-CFTR.
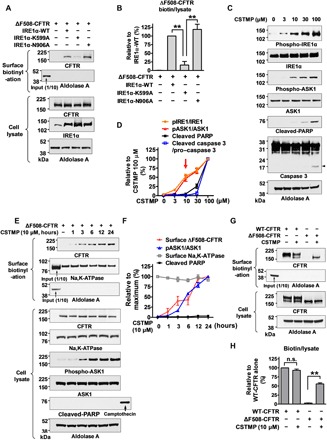
(A and B) Overexpression of IRE1α kinase activates UPS of ΔF508-CFTR. HEK293 cells expressing ΔF508-CFTR were cotransfected with plasmids encoding WT or mutant IRE1α (1 μg/ml, 48 hours). Overexpression of WT and RNase-dead (N906A) IRE1α, but not kinase-dead (K599A) IRE1α, activates UPS of ΔF508-CFTR. Representative surface biotinylation assays are presented in (A), and the results of multiple experiments (n = 3) are summarized in (B). (C and D) Effects of the IRE1α kinase activator CSTMP on ASK1 phosphorylation and cell death signals. HEK293 cells were treated with 3 to 100 μM CSTMP for 48 hours. Activation of cell death signals were analyzed by cleavage of PARP and caspase 3 (arrowhead). Representative immunoblots are shown in (C), and the results of multiple experiments are summarized in (D) (n = 5). CSTMP at a concentration of 10 μM activated ASK1 but not cell death signals (red arrow). (E and F) CSTMP induces UPS of ΔF508-CFTR. Surface biotinylation assays were performed in HEK293 cells expressing ΔF508-CFTR. Cells were treated with CSTMP (10 μM) for the indicated time periods. Representative immunoblots are presented in (E), and a summary of multiple experiments is depicted in (F) (n = 3). Cell surface–specific labeling of proteins was confirmed by the presence of the plasma membrane protein Na- and K-dependent ATPase (Na,K-ATPase) and the absence of the cytosolic protein aldolase A in the biotinylated fraction. (G and H) Comparison of surface expression levels of WT-CFTR and the CSTMP-rescued ΔF508-CFTR. HEK293 cells transfected with WT- and ΔF508-CFTR were incubated with or without CSTMP (10 μM) for 24 hours. Representative surface biotinylation results are shown in (G), and the quantification results of multiple experiments (n = 3) are summarized in (H). The cell surface levels of CFTR (biotin) were normalized to its total protein (lysate) levels as detailed in Materials and Methods. Bar and line graphs data are shown as the means ± SEM. **P < 0.01.
Small-molecule therapeutics are more desirable than gene overexpression approaches to treat human patients with diseases caused by defects in protein folding and trafficking. A tetramethylpyrazine derivative, (E)-2-(2-chlorostyryl)-3,5,6-trimethyl-pyrazine (CSTMP), has been shown to activate the IRE1α-TRAF2-ASK1 complex (29). However, CSTMP can also cause cellular toxicity at high concentrations (>50 μM), as it was reported to induce apoptosis of human non–small cell lung cancer A549 cells through JNK activation and mitochondrial dysfunction (29). Treatment of HEK293 cells with various concentrations of CSTMP for 48 hours revealed that 10 μM CSTMP sufficiently activated ASK1 without triggering apoptotic signals, while a higher dose of CSTMP (100 μM) led to cleavage of the apoptosis marker proteins poly(adenosine 5′-diphosphate–ribose) polymerase (PARP) and caspase 3 (Fig. 3, C and D).
Therefore, we treated the cells with 10 μM CSTMP and investigated whether it could rescue the trafficking defects of ΔF508-CFTR. CSTMP alone was sufficient to induce the cell surface expression of ΔF508-CFTR. Cells expressing ΔF508-CFTR began to show cell surface CFTR expression after 3-hour treatment with CSTMP (10 μM), which plateaued after 12 hours (Fig. 3, E and F). Analyses of the surface/cytosol ratio of CFTR proteins revealed that CSTMP increased the surface/cytosol ratio of ΔF508-CFTR to 59% of the level measured in WT-CFTR (Fig. 3, G and H). Knockdown of IRE1α inhibited the CSTMP-induced cell surface rescue of ΔF508-CFTR (fig. S4, A and B). Furthermore, overexpression of IRE1α kinase–dead mutant (K599A), but not its endonuclease-dead mutant (N906A), abolished the CSTMP effects (fig. S4, C and D). In addition, treatment with CSTMP evoked the cell surface expression of ΔF508-CFTR band B protein, which was inhibited by the IRE1α kinase inhibitor APY29 (fig. S4, E and F). In control experiments, the CFTR corrector VX-809 (25) induced the cell surface expression of ΔF508-CFTR band C protein, which was not affected by APY29 (fig. S4, E and F). All these results indicate that CSTMP induces the cell surface expression of ΔF508-CFTR via IRE1α kinase activation.
We next characterized the CSTMP-induced, cell surface trafficking route of ΔF508-CFTR. First, the N-glycosylation status of CFTR was analyzed by digestion with Endoglycosidase H (Endo H), which removes the ER-mediated high mannose and some hybrid types of N-linked carbohydrates but not the Golgi-mediated complex oligosaccharides (11). The results in fig. S5 (A and B) show that CSTMP-induced, cell surface–expressed band B forms of WT- and ΔF508-CFTRs were Endo H sensitive, while the band C of WT-CFTR under control condition was Endo H resistant, suggesting that CSTMP-rescued CFTRs travel through a Golgi-bypassing route to reach the cell surface. Second, we examined the Sar1 and Syntaxin 5 dependency of the CSTMP-rescued ΔF508-CFTR. Dominant-inhibitory forms of Sar1 (Sar1-T39N and Sar1-H79G) inhibit conventional COPII-mediated ER-to-Golgi trafficking (12). In addition, overexpression of Syntaxin 5, a target soluble N-ethylmaleimide–sensitive factor attachment protein receptor (SNARE) of the acceptor Golgi membrane, inhibits conventional ER-to-Golgi transport by deteriorating the stoichiometric balance between vesicular and target membrane SNAREs (12). As shown in fig. S5 (C to F), the cell surface rescued ΔF508-CFTR by CSTMP was Sar1 and Syntaxin 5 independent, while the cell surface trafficking of complex-glycosylated WT-CFTR (band C) was blocked by the Sar1 dominant-inhibitory mutants and Syntaxin 5 overexpression. These results indicate that CSTMP-rescued ΔF508-CFTR does not travel through the conventional ER-to-Golgi route on its way to the cell surface.
Next, we compared the cellular toxicity of a 10 μM CSTMP treatment with that of Arf1-Q71L overexpression. As shown in fig. S6 (A and B), transfection with Arf1-Q71L–expressing plasmids induced the cell surface rescue of ΔF508-CFTR and peaked at 48 hours. Of note, transfection with Arf1-Q71L–expressing plasmids for 72 hours, which caused a prolonged ER-to-Golgi blockade and ER stress, evoked PARP cleavage. In contrast, treatment with CSTMP (10 μM) up to 72 hours led to a sustained surface rescue of ΔF508-CFTR without triggering an apoptotic signal (fig. S6, C and D).
Morphological and functional rescue of ΔF508-CFTR by CSTMP
The effects of CSTMP on cell surface expression of ΔF508-CFTR were also examined using immunofluorescence analyses in nonpermeabilized (surface CFTR) and permeabilized (total CFTR) cells. HeLa cells instead of HEK293 cells were used for the morphological assay because they attach more firmly to coverslips and are less vulnerable to cell loss and disruption during the immunostaining procedures (15, 30). In control cells, the ΔF508-CFTR proteins were retained only within the ER (Fig. 4A, top). Treatment with CSTMP (10 μM) for 12 hours significantly induced cell surface expression of CFTR proteins in cells expressing ΔF508-CFTR [Fig. 4, A (bottom) and B]. Furthermore, treatment with CSTMP (10 μM) up to 72 hours did not induce expression of the apoptosis marker protein annexin V, while prolonged ectopic Arf1-Q71L expression for 72 hours did evoke annexin V expression (fig. S7), indicating minimal or no cellular toxicity of the CSTMP treatment.
Fig. 4. Cell surface expression and functional rescue of ΔF508-CFTR by CSTMP.
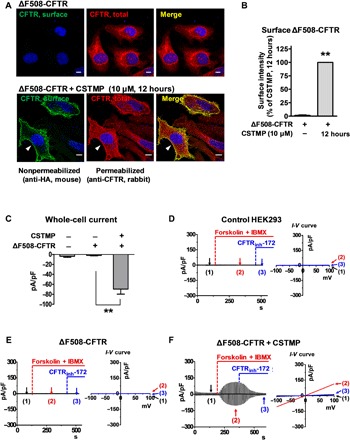
(A and B) Immunofluorescence images of ΔF508-CFTR. HeLa cells were transfected with plasmids encoding for extracellular hemagglutinin (HA)–tagged ΔF508-CFTR. Some cells were incubated with CSTMP (10 μM) for 12 hours before fixation. CFTR at the cell surface was immunostained with anti-HA antibodies before membrane permeabilization (green), and the total CFTR was stained with anti-R4 CFTR antibodies after permeabilization (red). Arrowheads indicate surface CFTR. Morphometric quantification of surface CFTR intensity is shown in (B) (n = 5, each from analyses of 20 to 30 cells). Scale bars, 10 μm. **P < 0.01, compared to no treatment (first lane). (C to F) CSTMP induced discernible CFTR currents in cells expressing ΔF508-CFTR. Whole-cell currents were recorded from HEK293 cells transfected with mock or ΔF508-CFTR plasmids for 48 hours. Currents were evoked by applying a ramp pulse from −100 to +100 mV (0.8 mV/ms; holding potential, 0 mV) at 10-s intervals. CFTR Cl− currents were activated by cAMP (5 μM forskolin and 100 μM IBMX) and inhibited by CFTRinh-172 (5 μM). A summary of current densities measured at −80 mV is shown in (C) (n = 8 to 11). In (F), cells were treated with CSTMP (10 μM) for 24 hours. Bar graph data are shown as the means ± SEM; **P < 0.01.
To examine whether the cell surface ΔF508-CFTR induced by CSTMP retains functional activity, HEK293 cells expressing ΔF508-CFTR were treated with CSTMP (10 μM) for 48 hours, and CFTR-mediated Cl− currents were measured (Fig. 4, C to F). Incubation with forskolin and 3-isobutyl-1-methylxanthine (IBMX), which elevates intracellular cAMP, did not evoke any Cl− currents in control HEK293 cells that were treated with transfection reagents alone (Fig. 4D). In addition, cells expressing ΔF508-CFTR did not show any discernible cAMP-activated Cl− currents under normal conditions (Fig. 4E). Notably, treatment with CSTMP (10 μM) for 48 hours induced a cAMP-activated Cl− current in cells expressing ΔF508-CFTR (Fig. 4F). The evoked currents exhibited typical characteristics of CFTR currents that were (i) activated by cAMP treatment (forskolin and IBMX), (ii) inhibited by the CFTR inhibitor CFTRinh-172, and (iii) had a linear current-voltage (I-V) relationship (Fig. 4F). The current amplitude induced by CSTMP (10 μM) in the ΔF508-CFTR cells was 32% of that measured in WT-CFTR cells, which is comparable to that induced by VX-809 (10 μM) (fig. S8). In control experiments, treatment with CSTMP (10 μM) for 48 hours did not significantly alter the amplitude or characteristics of WT-CFTR currents (fig. S8, A, B, and E).
Rescue of p.H723R-pendrin by CSTMP
Because IRE1α is involved in the ER stress–induced UPS of pendrin (13), we next explored the effects of the IRE1α kinase activator CSTMP on the rescue of cell surface trafficking defects caused by the p.H723R-pendrin mutant protein. PANC-1 cells derived from pancreatic ducts were used to examine the cell surface expression of pendrin (13). We first examined the surface expression of p.H723R-pendrin using surface biotinylation assays. As shown in Fig. 5 (A and B), p.H723R-pendrin is barely detectable at the plasma membrane in control cells. In comparison, treatment with CSTMP (30 μM) induced robust cell surface expression of p.H723R-pendrin. Then, we again examined the UPS nature of the CSTMP-induced, cell surface trafficking route of H723R-pendrin. As shown in fig. S9 (A and B), cell surface–rescued p.H723R-pendrin by CSTMP was Endo H sensitive, while WT-pendrin was Endo H resistant. The results in fig. S9 (C to F) demonstrate that Sar1 mutants did not block the cell surface expression of p.H723R-pendrin (band B) by CSTMP, while the same treatment inhibited the cell surface expression of WT-pendrin (band C). These results indicated that CSTMP-rescued H723R-pendrin reached the cell surface via a Golgi-bypassing route.
Fig. 5. CSTMP rescues the cell surface expression of misfolded pendrin.
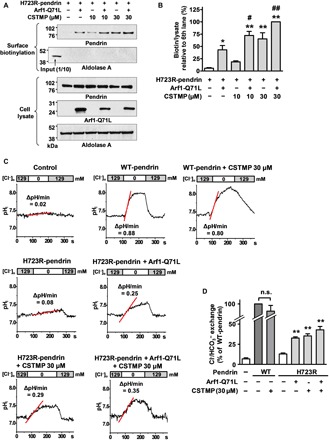
(A and B) Effects of CSTMP on the surface expression of p.H723R-pendrin. PANC-1 cells stably expressing p.H723R-pendrin were transfected with or without plasmids encoding for Arf1-Q71L and incubated with CSTMP (10 or 30 μM) for 48 hours. Representative surface biotinylation assays are shown in (A), and the results of multiple experiments are summarized in (B) (n = 5). *P < 0.05 and **P < 0.01, compared to no treatment (first lane). #P < 0.05 and ##P < 0.01, compared to Arf1-Q71L alone (second lane). (C and D) Functional rescue of p.H723R-pendrin by CSTMP. The Cl−o/HCO3−i exchange activity was measured by recording intracellular pH (pHi) as detailed in Materials and Methods. Representative anion exchange measurements are shown in (C), and the quantitation of multiple experiments is depicted in (D) (n = 7 to 8). CSTMP (30 μM) did not affect the activity of WT-pendrin but significantly increased Cl−/HCO3− exchange activity in cells expressing p.H723R-pendrin. Bar graph data are shown as the means ± SEM. **P < 0.01, compared to p.H723R-pendrin alone.
Notably, CSTMP evoked pendrin-mediated Cl−/HCO3− exchange activity in p.H723R-pendrin–expressing cells, which was similar to the effects of Arf1-Q71L overexpression (Fig. 5, C and D). These results imply that IRE1α kinase activation has the therapeutic potential to treat not only diseases caused by trafficking-defective mutants of CFTR but also diseases caused by other mutant membrane proteins, including p.H723R-pendrin.
In vivo treatment with CSTMP restores CFTR cell surface expression and CFTR-mediated anion transport in the CftrF508del mouse colon
Last, we evaluated whether in vivo administration of CSTMP could restore the epithelial transport defects caused by the ΔF508-CFTR mutation in mice. The loss-of-function ΔF508-CFTR mutation causes defects that predominantly affect intestinal ion and fluid transport and consequently evokes intestinal obstruction in mice (31). To analyze the in vivo effects of CSTMP, we first evaluated toxicity by estimating the median lethal dose (LD50) of CSTMP in WT (CftrWT) mice. Mice were orally treated for 5 days with three different doses of CSTMP that were equivalent to 10 to 1000 μM (assuming that CSTMP is evenly distributed throughout the whole body). The LD50 value in mice was calculated as 25.9 mg/kg per day (equivalent to 100 μM) (fig. S10). Then, 6-week-old mice that were homozygous for WT-CFTR (CftrWT) or ΔF508-CFTR (CftrF508del) were treated with 1/10 of the CSTMP LD50 dose (2.59 mg/kg per day, equivalent to 10 μM, once daily) for 5 days. At this dosage, no mice died during the LD50 evaluation experiments (fig. S10).
Notably, surface biotinylation experiments using samples harvested from the mouse colonic mucosa revealed that CSTMP rescued the cell surface expression of core-glycosylated ΔF508-CFTR (Fig. 6, A and B). Immunohistochemical analysis recapitulated the rescue effects of CSTMP in CftrF508del mice (Fig. 6C). In CftrWT mice, CFTR is expressed at the apical membrane of colonic crypt cells, and the CSTMP treatment did not affect the distribution of WT-CFTR. Consistent with prior results (12, 32), the apical expression of ΔF508-CFTR was absent in the colonic crypts of CftrF508del mice. Notably, the oral administration of CSTMP rescued ΔF508-CFTR expression at the apical membrane of colonic crypt cells in CftrF508del mice (Fig. 6C).
Fig. 6. In vivo CSTMP treatment rescues ΔF508-CFTR in the CftrF508del mouse colon.
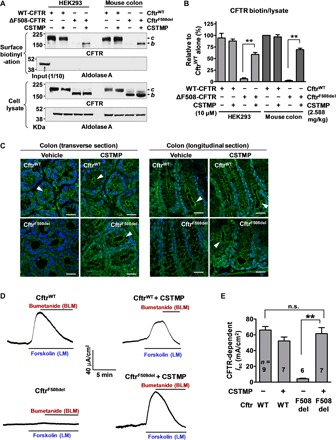
(A and B) Surface biotinylation assays were performed using epithelial cells harvested from the colonic mucosa. Protein samples from HEK293 cells were used as controls. WT-CFTR (CftrWT) or ΔF508-CFTR (CftrF508del) mice that were 6 weeks old received vehicle or CSTMP (2.59 mg/kg, per os, once daily) for 5 days. Representative surface biotinylation assay results are shown in (A), and the results of multiple experiments are summarized in (B) (n = 4). (C) Immunohistochemistry of CFTR. Transverse and longitudinal cross sections of mouse colonic crypts were immunostained with anti-CFTR R4 rabbit polyclonal antibodies. Arrowheads indicate CFTR expression at the apical membrane of colonic crypts. The CSTMP treatment induced the cell surface expression of CFTR in the CftrF508del mouse colon. Six independent experiments showed similar results. Scale bars, 10 μm. (D and E) Short-circuit current (Isc) measurements in the mouse colon. The apical side of the mouse colon was treated with amiloride (100 μM) to block epithelial Na+ channels. Application of the adenylyl cyclase activator forskolin (10 μM) to the apical side evoked a lumen-negative Isc, which was fully inhibited by the Na+-K+-2Cl− cotransporter inhibitor bumetanide (100 μM) delivered to the basolateral side. The CSTMP treatment induced the CFTR-dependent Isc in the CftrF508del mouse colon. Representative traces of Isc measurements are presented in (D), and the results of multiple experiments are summarized in (E) (n = 6 to 9). b, core-glycosylated CFTR; c, complex-glycosylated CFTR. Bar graph data are shown as the means ± SEM. BLM, basolateral membrane; LM, luminal membrane; **P < 0.01.
Furthermore, the CSTMP treatment resulted in the functional recovery of CFTR-mediated anion transport in the CftrF508del mouse colon. Increased intracellular cAMP levels induced by forskolin treatment evoked a typical CFTR-dependent short-circuit current (Isc) in the CftrWT mouse colon that was (i) lumen negative, (ii) inhibited by the blockade of basolateral Cl− uptake using the Na+/K+/2Cl− cotransporter inhibitor bumetanide, and (iii) absent in the CftrF508del mouse colon (Fig. 6, D and E). Notably, the CSTMP treatment induced a forskolin-activated, lumen-negative, and bumetanide-inhibitable Isc, with a current amplitude that reached 89% of the level measured in CftrWT mice (Fig. 6, D and E). Together, these results provide in vivo evidence supporting the therapeutic potential of CSTMP to treat diseases caused by the trafficking-defective CFTR mutation.
DISCUSSION
During the past two decades, researchers have incrementally established the concept and molecular mechanisms of the noncanonical, Golgi-independent UPS of transmembrane proteins (33, 34). Mutations that cause defective folding of membrane proteins often result in loss-of-function diseases by disrupting the normal balance between protein folding, trafficking, and degradation. Proteins with folding defects, such as ΔF508-CFTR and p.H723R-pendrin, can be alternatively transported to the plasma membrane via UPS under certain conditions (12, 13). However, substantial efforts are needed to develop UPS activating therapeutics that are clinically applicable to human patients with these diseases. For example, prolonged ER-to-Golgi blockade by Arf1-Q71L is cytotoxic as shown in the figures of this study (figs. S6 and S7). In addition, there are technical difficulties that are associated with inducing controlled gene overexpression in human participants. Here, we reported the first efficient and pharmacologically acceptable approach to activate UPS of folding-defective membrane proteins without severe toxicity. In the present study, we demonstrated that the kinase activity of the ER stress sensor IRE1α plays a pivotal role in UPS of folding-defective CFTR. Furthermore, we found that the small-molecule CSTMP can activate IRE1α kinase and its downstream effectors to enhance UPS and rescue defects caused by ΔF508-CFTR and p.H723R-pendrin mutations both in vitro and in vivo.
Under physiological conditions, to compensate for ER stress induced by excessive amounts of unfolded protein, UPR is activated. UPR reduces the overloaded protein burden in the ER by reducing transcription and translation of secretory proteins and by directing misfolded proteins into ERAD (5). In addition to these classical mechanisms, cells trigger UPS to diminish the protein burden when unfolded proteins are accumulated in the ER (12) and cytosol (35). The three essential transducers of UPR—IRE1, PERK, and ATF6—are all ER transmembrane proteins and share a common process of transducing the ER stress signal by activating transcription factors, such as XBP1s, ATF4, and cleaved ATF6, respectively (17). It has been demonstrated that IRE1α is an upstream signal that regulates the UPS of CFTR and pendrin in response to ER stress (12, 13). However, the mechanism of IRE1α-mediated UPS is not well understood.
In the present study, both ectopic expression of Arf1-Q71L and thapsigargin treatment evoked IRE1α oligomerization (fig. S1A) and kinase activation (Fig. 1). The most well-known downstream effect of IRE1α signaling is the excision of a 26-nucleotide intron of XBP1 mRNA into its active form, XBP1s, in the cytosol (18, 36). The active XBP1s functions as a transcription factor and controls the expression of a series of genes, which ultimately leads to a protective remodeling of ER homeostasis. Unexpectedly, ectopic expression with Arf1-Q71L, which is one of the most effective activators of CFTR UPS, did not induce XBP1 splicing, while thapsigargin did (Fig. 1, A and B). The RNA interference–based depletion of XBP1 also verified that XBP1 is dispensable for UPS of ΔF508-CFTR (Fig. 1, C and D). It is unclear at present why Arf1-Q71L cannot induce XBP1 splicing. Notably, it has been shown that clustering of IRE1α (formation of larger dynamic oligomers) is dispensable for and does not predict its RNase activity; in contrast, IRE1α clustering highly correlates with its kinase activity (37). Furthermore, mild forms of ER stress do not efficiently evoke XBP1 splicing but can still induce IRE1α clustering (37). An ER-to-Golgi blockade by Arf1-Q71L is anticipated to slowly induce ER stress. Therefore, it appears that slow and mild ER stress evoked by Arf1-Q71L cannot activate IRE1α endonuclease, but it can still induce IRE1α oligomerization and activate its kinase activity. The regulated RIDD process, which is an XBP1-independent UPR output of IRE endonuclease activity by reducing the ER load of proteins through rapid degradation of specific mRNAs (19), could be an alternative candidate signaling pathway for UPS induction. However, experiments using the IRE1α endonuclease inhibitor STF-083010 and endonuclease-dead mutant (IRE1α-N906A) indicated that signaling pathways that originated from endonuclease activity, including RIDD, are not involved in UPS of CFTR (Figs. 1 and 3 and fig. S4).
In addition to indirect evidence that an IRE1α component other than its endonuclease activity mediates UPS, the following three pieces of direct evidence support the notion that the IRE1α kinase–ASK1 pathway is critically involved in UPS of membrane proteins: (i) blockade of IRE1α kinase function using APY29 (Fig. 2) or IRE1α kinase–dead mutant (IRE1α-K599A; Fig. 3 and fig. S4) abolished the Arf1-Q71L–or CSTMP-induced UPS of CFTR, (ii) chemical inhibition or knockdown of ASK1 reduced UPS of CFTR (Fig. 2, C and D, and fig. S3), and (iii) stimulation with IRE1α kinase by CSTMP evoked UPS of ΔF508-CFTR and p.H723R-pendrin (Figs. 3 to 5). Although the complete mechanism remains to be established, recent studies have provided a framework to understand how IREα-mediated signaling may contribute to the UPS of membrane proteins. The IRE1α kinase–ASK1 cascade was suggested to play a role in the initiation of autophagosome formation by modulating Beclin-1 (38). Components of the early autophagosome formation, such as autophagy related 5 (ATG5) and autophagy related 7 (ATG7), participate in the vesicular structures involved in ER stress–associated UPS of CFTR (14). In addition, IRE1α is an upstream signal that regulates the generation and anchoring of Sec16A at the ER exit sites, which increases the ER export of core-glycosylated CFTRs during UPS in response to ER stress (15). It has been reported that knockdown of IRE1α/ERN1 induced a small increase (10 to 20%) in cell surface trafficking of the ΔF508-CFTR band C protein in an RNA interference screening of kinases, suggesting that IRE1α inhibits the Golgi maturation of ΔF508-CFTR (39). Although the mechanisms of this finding are unknown, it is partially explained by the present results that IRE1α directs ΔF508-CFTR into a Golgi-bypass route.
IRE1α is a platform for activating the proapoptotic JNK pathway under unmitigated stress conditions (20, 26), which implies that severe IRE1α activation could switch the life-to-death decisions of IRE1α kinase–ASK1 and evoke cellular toxicity. Excessive IRE1α kinase activation by >100 μM CSTMP (Fig. 3) and prolonged ER-to-Golgi blockade by Arf1-Q71L over 72 hours (fig. S6) both induced the apoptotic signals of PARP and caspase 3 cleavages in HEK293 cells. However, a nontoxic level of IRE1α kinase activation by 10 μM CSTMP selectively activated ASK1 without activating those apoptotic signals (Fig. 3). Furthermore, oral administration of CSTMP (2.59 mg/kg per day) for 5 days in mice, which rescued 89% of the cAMP-stimulated transepithelial Cl− secretion (Isc) in the CftrF508del colon, caused no identifiable toxicity (Fig. 6 and fig. S10). Therefore, CSTMP appears to have curative effects on CF by promoting the cell surface expression of folding-defective CFTR in mice, without severe toxicity. In the present pilot study, we empirically selected 1/10 of the LD50 of CSTMP as a dose and showed >50% of efficacy in restoring CFTR-mediated Isc, implying that therapeutic index (LD50/ED50 (half-maximal effective dose)] of CSTMP in mice was at least >10. Although this value minimally satisfies the safety of a drug, development of a safer and more potent drug using a large-scale screening of small molecules that can selectively activate IRE1α kinase, but not IRE1α endonuclease and apoptotic signals, would increase the clinical feasibility of an IRE1α kinase activator. Epithelial cells in patients with CF are exposed to various stressful conditions. These cellular stresses may activate IRE1α kinase and partially induce the UPS of ΔF508-CFTR, which could affect disease severity. Therefore, individual variations in the inherent capacity of the UPR and subsequent UPS responses may also contribute to some of wide variation in the apparent severity of CF with ΔF508 mutation.
In whole-cell current measurements, the combination of CSTMP and VX-809 did not show additive effects (fig. S8E). The current amplitude induced by high concentrations of VX-809 (10 μM) and CSTMP (10 μM) in ΔF508-CFTR cells was ~30% of the value measured in WT-CFTR cells, and this would be the maximum level that can be achieved in ΔF508-CFTR cells considering their accelerated degradation and intrinsically low ion channel activity. More sophisticated in vitro and in vivo evaluations are required to fully analyze the additive or potentiation effects between the IRE1α kinase activator and VX-809. The levels of CSTMP-induced rescue of ΔF508-CFTR differed in each experiment. For example, in HEK293 cells, CSTMP increased the surface/cytosol ratio of ΔF508-CFTR to 59% of the level measured in WT-CFTR in the surface biotinylation experiments (Fig. 3, G and H), while it induced an average of 32% recovery in Cl− channel activity in the whole-cell current measurements (fig. S8). However, CSTMP rescued 89% of cAMP-stimulated transepithelial Cl− secretion in the CftrF508del mouse colon (Fig. 6). Because basolateral anion (Cl− and HCO3−) uptake is the rate-limiting step in most secretory epithelia (6, 40), a ~30% recovery of the CFTR-mediated anion efflux at the apical membrane appears to be sufficient for restoring overall transepithelial ion transport in the colonic epithelia.
The present results indicate the possibility of applying IREα kinase activators to in vivo human trials. Regarding the in vivo effective dose, pharmacokinetic profiles of CSTMP including concentrations in the target tissues should be analyzed in future studies. Because we administered CSTMP per os, the local concentration of CSTMP in the target tissue, the mouse intestinal epithelium, might be higher than that of deep visceral tissues, which would result in strong therapeutic effects without systemic toxicity. A local aerosol-based inhalation therapy would be an alternative approach for human patients with CF who show predominantly respiratory phenotypes, in case CSTMP concentrations in the target human airway tissues do not reach effective levels through systemic CSTMP administration. In conclusion, we present here a UPS activation via the IRE1α kinase–ASK1 pathway as a promising therapeutic strategy for diseases caused by folding- and trafficking-defective transmembrane proteins, including CF and Pendred syndrome.
MATERIALS AND METHODS
Cell culture, plasmids, siRNA, and mice
HEK293 and HeLa cells were maintained in Dulbecco’s modified Eagle’s medium–high glucose (Gibco no. 11995-065, Carlsbad, CA) supplemented with 10% fetal bovine serum and 1% 100× antibiotic-antimycotic (100 units/ml penicillin, 100 units/ml streptomycin, and 250 ng/ml amphotericin B) (Gibco no. 15240062). Cells were grown at 37°C in a 5% CO2 incubator. The mammalian expression plasmid encoding human IRE1α-pcDNA3.EGFP was purchased from Addgene (gene ID: 2081). The plasmids encoding human pCMV-ΔF508-CFTR, pCMV-WT-CFTR (pCMVNot6.2), pCMV-GRASP55-Myc, and extracellular-tagged hemagglutinin (HA)–ΔF508-CFTR, pClneo-HA–Syntaxin 5, pClneo-Myc-Sar1-T39N, pClneo-Myc-Sar1-H79G, and pcDNA3-HA–Arf1-Q71L were described previously (12, 30). ON-TARGETplus Human ERN1 siRNA (IRE1a, gene ID: 2081) and human XBP1 siRNA (gene ID: 7494) were purchased commercially (SMARTpool; Dharmacon, Lafayette, CO, USA). Transfection with both plasmid DNA and siRNA into HEK293 or HeLa cells was performed using the TransIT-X2 Dynamic Delivery System (Mirus Bio LLC, Madison, WI, USA) according to the manufacturer’s protocol. CftrF508del mice were obtained from K. R. Thomas (University of Utah, Salt Lake City, UT, USA) (31) as previously reported (12). The mice were bred and maintained according to the Yonsei Medical Center Animal Research Requirements. All animal experiments were approved by the Institutional Laboratory Animal Resources and the Committee of Animal Research at Yonsei Biomedical Research Center (approval no. 2018-0139).
Chemicals and antibodies
Thapsigargin (Sigma-Aldrich, T9033), STF-083010 (TOCRIS, 4509), APY29 (TOCRIS, 4865), MSC 2032964A (TOCRIS, 5641), VX-809 (Selleckchem, Chemical Abstracts Service registry no. 936727-05-8), VX-770 (Selleckchem, CAS registry no. 873054-44-5), and cycloheximide (Sigma-Aldrich, C4859) were purchased commercially. CSTMP was custom-synthesized by Cayman Chemical (Ann Arbor, MI, USA) (CAS registry no. 1000672-89-8).
The following antibodies were acquired commercially: anti-CFTR M3A7 (Millipore, Billerica, MA, USA), anti-IRE1α (Cell Signaling Technology, 3294), anti–phospho-S724 IRE1α (Abcam, ab48187), anti-XBP1 (Abcam, ab198999), anti-ASK1 (Cell Signaling Technology, 3762), anti–phospho-Thr845 ASK1 (Cell Signaling Technology, 3765), anti-BiP (Cell Signaling Technology, 3177), anti-CHOP (Cell Signaling Technology, 2895), anti-pro/p17–caspase 3, anti–cleaved PARP1 (Abcam, ab136812), anti–Na- and K-dependent ATPase (Na,K-ATPase) α1 (Cell Signaling Technology, 23565), anti-HA (Cell Signaling Technology, 2367), anti-Myc (Cell Signaling Technology, 2276), anti–aldolase A (Abcam, ab78339) anti–β-actin (Santa Cruz Biotechnology, sc47778), and anti-pendrin (Santa Cruz Biotechnology, sc23779). The anti-R4 polyclonal antibody was raised against peptides corresponding to amino acids 1458 to 1471 of human CFTR, as described previously (12).
Quantitative polymerase chain reaction analysis
Total RNA was extracted from HEK293 cells using the Tri-RNA reagent (Favorgen Biotech Corp, Taiwan) according to the manufacturer’s protocol. To reverse-transcribe the RNA into complementary DNA (cDNA), purified RNA samples were combined with the RNA to cDNA EcoDry Premix (Takara Bio Inc., Shiga, Japan), and the mixtures were incubated at 42°C for 1 hour, followed by 70°C for 10 min.
Quantitative polymerase chain reaction (qPCR) was performed using the Applied Biosystem StepOne system (Applied Biosystems, Foster City, CA, USA). The real-time PCR reaction was measured by detecting the binding of fluorescent SYBR Green dye to double-stranded DNA. For PCR amplification, the total reaction volume was adjusted to 20 μl with RNase-free water after mixing with 100 ng of cDNA, 2 μl of primer sets, 10 μl of 2× SYBR Premix Ex Taq, and 0.4 μl of 50× ROX reference dye (Takara, RR420L). Amplification was performed with the following cycling conditions: 95°C for 15 min, followed by 40 cycles of 95°C for 15 s and 60°C for 40 s. Analyses were performed in triplicate for each cDNA. The relative mRNA expression levels were calculated using the comparative threshold cycle (Ct) method by normalizing the gene expression with the housekeeping gene GAPDH, and the ΔCt value was calculated as follows: ΔCt = Ct(GAPDH) − Ct(target gene). The average ΔCt was then subtracted from each experimental condition described to yield the ΔΔCt value. The fold change in gene expression was normalized to GAPDH, and relative to control samples was calculated as 2−ΔΔCt. The primer sequences used for the qPCR analysis were as follows: hIRE1α, forward primer 5′-CGG GAG AAC ATC ACT GTC CC-3′ and reverse primer 5′-CCC GGT AGT GGT GCT TCT TA-3′; hXBP1 (total), forward primer 5′-TTG TCA CCC CTC CAG AAC ATC-3′ and reverse primer 5′-TCC AGA ATG CCC AAC AGG AT-3′; hXBP1s (spliced), forward primer 5′-TGC TGA GTC CGC AGC AGG TG-3′ and reverse primer 5′-GCT GGC AGG CTC TGG GGA AG-3′; GAPDH (glyceraldehyde phosphate dehydrogenase), forward primer 5′-AAT CCC ATC ACC ATC TTC CA-3′ and reverse primer 5′-TGG ACT CCA CGA CGT ACT CA-3′.
Surface biotinylation, glycosidase digestion, cross-linking assay, and immunoblot analysis
For biotinylation, HEK293 cells grown in six-well plates (1 × 106) were incubated for 5 min at 4°C and washed three times with chilled phosphate-buffered saline (PBS). For biotinylation in mouse colonic mucosa, colon tissues were opened longitudinally and stripped of the connective tissues and muscles. The transmembrane proteins in the plasma membrane of cultured cells or colonic mucosa were then biotinylated by gentle shaking incubations with 1 ml of biotin solution [0.3 mg/ml Sulfo-NHS-SS-Biotin in chilled PBS (Thermo Pierce, 21331)] for 30 min at 4°C in the dark. The cells or colon tissues were then incubated with a quenching buffer containing 0.5% bovine serum albumin (BSA) in PBS for 10 min at 4°C to remove excess biotin and then washed three times with PBS. Next, the surface-biotinylated cells were harvested in lysis buffer containing 25 mM tris (pH 7.4), 1% (v/v) NP-40, 150 mM NaCl, 10% glycerol, and 1 mM EDTA-Na2 supplemented with protease inhibitor cocktail (Roche, Germany). The cell lysates were homogenized with sonication for 20 s (1-s pulse) followed by centrifugation at 16,000g for 20 min at 4°C. The resulting supernatants, containing 400 μg of total protein, were incubated with 200 μl of 10% streptavidin agarose (Thermo Pierce, 20347). The streptavidin-bound, biotinylated proteins were centrifuged and washed five times with lysis buffer. Biotinylated proteins were eluted in 2× SDS sample buffer supplemented with dithiothreitol (DTT) (0.02 g/ml) and separated by SDS–polyacrylamide gel electrophoresis (PAGE). The separated proteins were electrotransferred to a nitrocellulose membrane and blotted with the appropriate primary and secondary antibodies (in 5% skim milk). Protein bands were detected by enhanced chemiluminescence, and the densities of each protein band were quantified using imaging software (Multi Gauge version 3.0; Fujifilm, Tokyo, Japan). For quantification of cell surface CFTR and pendrin, we simultaneously performed surface biotinylation and immunoblot experiments using samples from all designed experimental conditions. The amount of cell surface protein (biotin) was normalized to each total cellular protein (lysate) by analyzing band intensities in the same surface biotinylation and lysate blots.
Digestion of N-glycosylated proteins by peptide N-glycosidase F (PNGase F) (New England Biolabs, no. P0704L) and Endo H (New England Biolabs, no. P0702L) was performed with slight modification based on the manufacturer’s manual (12). Briefly, 50-μg protein samples were first denatured by adding 0.5% SDS and 40 mM DTT and incubated at 37°C for 10 min. The denatured protein samples were then treated with PNGase F (500 U per reaction, with 1% NP-40) in a sodium phosphate–containing solution (50 mM, pH 7.5) or treated with Endo H (1000 U per reaction) in a sodium acetate–containing solution (50 mM, pH 6.0) at 37°C for 2 hours.
The cross-linking assay was performed as described previously (30). HEK293 cells were washed three times with PBS and incubated with 500 μM ethylene glycol succinimidyl succinate (EGS; Thermo Scientific, 21565) in PBS (pH 8.2) for 30 min at 37°C. After cross-linking, the cells were subsequently incubated with 20 mM tris (pH 7.4) for 10 min to stop the reaction by quenching the EGS. Cells were lysed, homogenized, sampled with 2× SDS sample buffer, separated by SDS-PAGE, electrotransferred, and immunoblotted with an anti-IRE1α antibody.
Immunocytochemistry, immunohistochemistry, and morphometric analysis
Immunofluorescence staining was performed by slightly modifying previously reported protocols (12, 15). For immunohistochemistry in the colonic mucosa, tissues from mice were embedded in optimal cutting temperature compound (Miles, Elkhart, IN, USA), frozen in liquid nitrogen, and cryosectioned into 4-μm sections. Then, the mouse colon cryosections were fixed and permeabilized by incubating with cold methanol for 5 min at −20°C. For immunocytochemistry, HeLa cells were cultured on 18-mm round coverslips and fixed with 3.7% formaldehyde for 6 min at room temperature. Cells were then permeabilized with 0.1% Triton X-100 in PBS at room temperature for 5 min. Tissue sections on glass slides or cells grown on coverslips were washed three times with PBS and then incubated with PBS containing 1% BSA and 5% serum of the appropriate species (horse/donkey/goat serum) for 1 hour at room temperature to block nonspecific binding sites. After blocking, the tissue sections or cells were stained by incubation with the appropriated primary antibodies, followed by staining with secondary fluorophore-conjugated antibodies. To achieve surface-specific labeling of CFTR, cells without permeabilization after fixation were incubated with blocking solution and stained with anti-HA antibody to detect the extracellular HA epitope of CFTR. Cells on the coverslips were mounted to the slide glasses with fluorescence mounting medium (S3025; Dako, USA). Fluorescence images were captured using a laser scanning confocal microscope (LSM 780; Carl Zeiss, Berlin, Germany) with a 63× 1.4 numerical aperture oil objective lens.
Morphometric analysis of the captured confocal images was performed using the MetaMorph microscopy analysis software (version 7.1; Molecular Devices, Sunnyvale, CA, USA) as described previously (15). Briefly, for image quantification under each condition, 24-bit confocal images including red, green, and blue components were converted into three 8-bit monochannel images. For the quantification of surface CFTR intensity, pixels above a threshold level of 60 were defined as CFTR positive. The intensity profile of each single cell was presented as the SD around the mean of the average intensity value for the entire region.
Measurements of Cl− channel activity and short-circuit current (Isc)
Whole-cell recordings were performed on CFTR-transfected HEK293 cells as described previously (12). Cells were transferred into a bath mounted on the stage of an inverted microscope (Ti2, Nikon), and the whole-cell patch was achieved by rupturing the membrane after gigohm sealing. The bath solution was perfused at 5 ml/min. The voltage and current recordings were performed at room temperature (22° to 25°C). Patch pipettes with a resistance of 2 to 4 megohm were connected to the head stage of a patch-clamp amplifier (Axopatch-200B; Molecular Devices, Sunnyvale, CA, USA). The bath solution contained 140 mM N-methyl-d-glucamine chloride (NMDG-Cl), 1 mM CaCl2, 1 mM MgCl2, 10 mM glucose, and 10 mM Hepes (pH 7.4). The pipette solution contained 140 mM N-methyl-d-glucamine chloride, 5 mM EGTA, 1 mM MgCl2, 3 mM MgATP, and 10 mM Hepes (pH 7.2). To determine the I-V relationship, the voltage clamp mode and I-V curve were obtained by applying ramp pulses from −100 to 100 mV (0.8 mV/ms; holding potential, 0 mV). CFTR currents were activated by cAMP (5 μM forskolin and 100 μM IBMX). The current generated by the CFTRs was confirmed by applying the CFTR inhibitor CFTRinh-172 (10 μM). To acquire data and apply command pulses, pClamp 10.2 and Digidata 1550B (Molecular Devices) were used. Voltage and current traces were stored and analyzed using pClamp 10.2 and Origin 8.0 (OriginLab Corp., Northampton, MA, USA). The currents were filtered at 5 kHz and sampled at 1 kHz. All data were normalized to the whole-cell capacitance (pF).
The mouse colon Isc was measured using slight modifications to previously reported protocols (12). Mice were anesthetized with CO2 gas and euthanized by cervical dislocation. Colon specimens from cecum to rectum were retrieved and opened along the mesenteric border. The luminal contents were washed away with a HCO3−-buffered solution, and the specimens were mounted on the Ussing chamber (World Precision Instruments, Stevenage, UK) with an exposed surface area of 12.6 mm2. The tissues were bathed on each side with 10 ml of HCO3−-buffered solution containing 120 mM NaCl, 5 mM KCl, 1 mM MgCl2, 1 mM CaCl2, 10 mM d-glucose, 5 mM Hepes, and 25 mM NaHCO3 (pH 7.4) at 37°C and 95% O2–5% CO2. Tissues were voltage-clamped at 0 mV using an EVC-4000 voltage clamp (World Precision Instruments), and Isc was continuously recorded using the PowerLab data acquisition system (AD Instruments, Castle Hill, Australia).
Measurement of Cl−/HCO3− exchange activity
Measurements of intracellular pH (pHi) in PANC-1 cells were performed with the pH-sensitive fluorescent probe 2′,7′-bis-(2-carboxyethyl)-5-(and-6)-carboxyfluorescein (BCECF) according to previously reported protocols (13). Briefly, cells were incubated with 2 μM BCECF acetoxy-methylester for 5 min and then perfused with a HCO3−-buffered solution [containing 120 mM NaCl, 5 mM KCl, 1 mM MgCl2, 1 mM CaCl2, 10 mM d-glucose, 5 mM Hepes, and 25 mM NaHCO3 (pH 7.4)]. BCECF fluorescence was recorded at excitation wavelengths of 490 and 440 nm at a resolution of 2/s on a recording setup (Delta Ram; PTI Inc., Edison, NJ, USA). The Cl−/HCO3− exchange activities were estimated from the initial rate of pHi increase as a result of Cl− removal from the HCO3−-containing buffer (25 mM HCO3− with 5% CO2). The pHi calibration was performed with standard pH solutions containing 150 mM KCl and 5 μM nigericin. The intrinsic buffer capacity (βi) was calculated by measuring ΔpHi in response to 5 to 40 mM NH4Cl pulses in Na+-free solutions. Because the βi values were not substantially affected by transfection with the plasmids encoding for WT-pendrin or H723R-pendrin, the Clˉ/HCO3ˉ exchange activities were expressed as ΔpH unit/min without compensating for the buffer capacity.
Statistics
The results of multiple experiments were presented as the means ± SEM. Statistical analysis was performed using two-sided Student’s t tests or with one-way analysis of variance followed by Tukey’s multiple comparison test, as appropriate, using GraphPad Prism 5 (GraphPad Software Inc., La Jolla, CA, USA). P < 0.05 was considered statistically significant.
Supplementary Material
Acknowledgments
We thank the Yonsei-Carl Zeiss Advanced Imaging Center for technical assistance. Funding: This work was supported by grants 2013R1A3A2042197 from the National Research Foundation, the Ministry of Science and ICT, Republic of Korea. Author contributions: H.P. designed and carried out the biochemistry and immunofluorescence experiments of CFTR. H.P. also wrote the manuscript. D.H.S. performed patch-clamp and Isc measurements. J.-R.S. performed surface biotinylation and functional pendrin assays. S.A. contributed to animal data acquisition. M.G.L. designed and conceived all experiments and wrote and edited the manuscript. Competing interests: H.P. and M.G.L. are inventors on a pending patent related to this work filed by Yonsei University Office of Research Affairs/University Industry Foundation (no. 10-2019-0062327, filed 28 May 2019). The authors declare no other competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/8/eaax9914/DC1
Fig. S1. XBP1 splicing and IRE1α endonuclease activity are not involved in UPS of ΔF508-CFTR (control experiments of Fig. 1).
Fig. S2. IRE1α kinase–ASK1 pathway is required for UPS of ΔF508-CFTR (control experiments of Fig. 2).
Fig. S3. Depletion of ASK1 inhibits Arf1-Q71L–induced UPS of CFTR.
Fig. S4. CSTMP stimulates the cell surface expression of ΔF508-CFTR by activating IRE1α kinase.
Fig. S5. CSTMP induces the cell surface expression of ΔF508-CFTR via UPS.
Fig. S6. CSTMP (10 μM) does not evoke apoptotic signal.
Fig. S7. CSTMP induces the cell surface expression of ΔF508-CFTR without evoking cellular apoptosis.
Fig. S8. Measurements of CFTR Cl− channel activity (control experiments of Fig. 4, C to F).
Fig. S9. CSTMP induces the cell surface expression of p.H723R-pendrin via UPS.
Fig. S10. The LD50 value of CSTMP in mice (per os) is 25.9 mg/kg per day.
REFERENCES AND NOTES
- 1.Cheng S. H., Gregory R. J., Marshall J., Paul S., Souza D. W., White G. A., O’Riordan C. R., Smith A. E., Defective intracellular transport and processing of CFTR is the molecular basis of most cystic fibrosis. Cell 63, 827–834 (1990). [DOI] [PubMed] [Google Scholar]
- 2.Thomas P. J., Qu B. H., Pedersen P. L., Defective protein folding as a basis of human disease. Trends Biochem. Sci. 20, 456–459 (1995). [DOI] [PubMed] [Google Scholar]
- 3.Yoon J. S., Park H.-J., Yoo S.-Y., Namkung W., Jo M. J., Koo S. K., Park H.-Y., Lee W.-S., Kim K. H., Lee M. G., Heterogeneity in the processing defect of SLC26A4 mutants. J. Med. Genet. 45, 411–419 (2008). [DOI] [PubMed] [Google Scholar]
- 4.Hammond C., Helenius A., Quality control in the secretory pathway. Curr. Opin. Cell Biol. 7, 523–529 (1995). [DOI] [PubMed] [Google Scholar]
- 5.Meusser B., Hirsch C., Jarosch E., Sommer T., ERAD: The long road to destruction. Nat. Cell Biol. 7, 766–772 (2005). [DOI] [PubMed] [Google Scholar]
- 6.Lee M. G., Ohana E., Park H. W., Yang D., Muallem S., Molecular mechanism of pancreatic and salivary gland fluid and HCO3- secretion. Physiol. Rev. 92, 39–74 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chang X.-b., Mengos A., Hou Y.-x., Cui L., Jensen T. J., Aleksandrov A., Riordan J. R., Gentzsch M., Role of N-linked oligosaccharides in the biosynthetic processing of the cystic fibrosis membrane conductance regulator. J. Cell Sci. 121, 2814–2823 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Amaral M. D., CFTR and chaperones: Processing and degradation. J. Mol. Neurosci. 23, 41–48 (2004). [DOI] [PubMed] [Google Scholar]
- 9.Ward C. L., Omura S., Kopito R. R., Degradation of CFTR by the ubiquitin-proteasome pathway. Cell 83, 121–127 (1995). [DOI] [PubMed] [Google Scholar]
- 10.Alper S. L., Sharma A. K., The SLC26 gene family of anion transporters and channels. Mol. Aspects Med. 34, 494–515 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gee H. Y., Kim J., Lee M. G., Unconventional secretion of transmembrane proteins. Semin. Cell Dev. Biol. 83, 59–66 (2018). [DOI] [PubMed] [Google Scholar]
- 12.Gee H. Y., Noh S. H., Tang B. L., Kim K. H., Lee M. G., Rescue of ∆F508-CFTR trafficking via a GRASP-dependent unconventional secretion pathway. Cell 146, 746–760 (2011). [DOI] [PubMed] [Google Scholar]
- 13.Jung J., Kim J., Roh S. H., Jun I., Sampson R. D., Gee H. Y., Choi J. Y., Lee M. G., The HSP70 co-chaperone DNAJC14 targets misfolded pendrin for unconventional protein secretion. Nat. Commun. 7, 11386 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Noh S. H., Gee H. Y., Kim Y., Piao H., Kim J., Kang C. M., Lee G., Mook-Jung I., Lee Y., Cho J. W., Lee M. G., Specific autophagy and ESCRT components participate in the unconventional secretion of CFTR. Autophagy 14, 1761–1778 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Piao H., Kim J., Noh S.-H., Kweon H.-S., Kim J. Y., Lee M. G., Sec16A is critical for both conventional and unconventional secretion of CFTR. Sci. Rep. 7, 39887 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Giuliani F., Grieve A., Rabouille C., Unconventional secretion: A stress on GRASP. Curr. Opin. Cell Biol. 23, 498–504 (2011). [DOI] [PubMed] [Google Scholar]
- 17.Hetz C., Papa F. R., The unfolded protein response and cell fate control. Mol. Cell 69, 169–181 (2018). [DOI] [PubMed] [Google Scholar]
- 18.Calfon M., Zeng H., Urano F., Till J. H., Hubbard S. R., Harding H. P., Clark S. G., Ron D., IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 415, 92–96 (2002). [DOI] [PubMed] [Google Scholar]
- 19.Hollien J., Weissman J. S., Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science 313, 104–107 (2006). [DOI] [PubMed] [Google Scholar]
- 20.Nishitoh H., Matsuzawa A., Tobiume K., Saegusa K., Takeda K., Inoue K., Hori S., Kakizuka A., Ichijo H., ASK1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats. Genes Dev. 16, 1345–1355 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Korennykh A. V., Egea P. F., Korostelev A. A., Finer-Moore J., Zhang C., Shokat K. M., Stroud R. M., Walter P., The unfolded protein response signals through high-order assembly of Ire1. Nature 457, 687–693 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pahl H. L., Signal transduction from the endoplasmic reticulum to the cell nucleus. Physiol. Rev. 79, 683–701 (1999). [DOI] [PubMed] [Google Scholar]
- 23.Hetz C., Chevet E., Harding H. P., Targeting the unfolded protein response in disease. Nat. Rev. Drug Discov. 12, 703–719 (2013). [DOI] [PubMed] [Google Scholar]
- 24.Wang L., Perera B. G. K., Hari S. B., Bhhatarai B., Backes B. J., Seeliger M. A., Schürer S. C., Oakes S. A., Papa F. R., Maly D. J., Divergent allosteric control of the IRE1α endoribonuclease using kinase inhibitors. Nat. Chem. Biol. 8, 982–989 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Meng X., Clews J., Kargas V., Wang X., Ford R. C., The cystic fibrosis transmembrane conductance regulator (CFTR) and its stability. Cell. Mol. Life Sci. 74, 23–38 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Urano F., Wang X., Bertolotti A., Zhang Y., Chung P., Harding H. P., Ron D., Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 287, 664–666 (2000). [DOI] [PubMed] [Google Scholar]
- 27.Guo X., Harada C., Namekata K., Matsuzawa A., Camps M., Ji H., Swinnen D., Jorand-Lebrun C., Muzerelle M., Vitte P. A., Ruckle T., Kimura A., Kohyama K., Matsumoto Y., Ichijo H., Harada T., Regulation of the severity of neuroinflammation and demyelination by TLR-ASK1-p38 pathway. EMBO Mol. Med. 2, 504–515 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Han D., Lerner A. G., Vande Walle L., Upton J. P., Xu W., Hagen A., Backes B. J., Oakes S. A., Papa F. R., IRE1α kinase activation modes control alternate endoribonuclease outputs to determine divergent cell fates. Cell 138, 562–575 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang J., Liang Y., Lin Y., Liu Y., YouYou, Yin W., IRE1α-TRAF2-ASK1 pathway is involved in CSTMP-induced apoptosis and ER stress in human non-small cell lung cancer A549 cells. Biomed. Pharmacother. 82, 281–289 (2016). [DOI] [PubMed] [Google Scholar]
- 30.Kim J., Noh S. H., Piao H., Kim D. H., Kim K., Cha J. S., Chung W. Y., Cho H.-S., Kim J. Y., Lee M. G., Monomerization and ER relocalization of GRASP is a requisite for unconventional secretion of CFTR. Traffic 17, 733–753 (2016). [DOI] [PubMed] [Google Scholar]
- 31.Zeiher B. G., Eichwald E., Zabner J., Smith J. J., Puga A. P., McCray P. B. Jr., Capecchi M. R., Welsh M. J., Thomas K. R., A mouse model for the delta F508 allele of cystic fibrosis. J. Clin. Invest. 96, 2051–2064 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Namkung W., Kim K. H., Lee M. G., Base treatment corrects defects due to misfolding of mutant cystic fibrosis transmembrane conductance regulator. Gastroenterology 129, 1979–1990 (2005). [DOI] [PubMed] [Google Scholar]
- 33.Rabouille C., Pathways of Unconventional Protein Secretion. Trends Cell Biol. 27, 230–240 (2017). [DOI] [PubMed] [Google Scholar]
- 34.Kim J., Gee H. Y., Lee M. G., Unconventional protein secretion—New insights into the pathogenesis and therapeutic targets of human diseases. J. Cell Sci. 131, jcs213686 (2018). [DOI] [PubMed] [Google Scholar]
- 35.Xu Y., Cui L., Dibello A., Wang L., Lee J., Saidi L., Lee J.-G., Ye Y., DNAJC5 facilitates USP19-dependent unconventional secretion of misfolded cytosolic proteins. Cell Discov. 4, 11 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hetz C., Glimcher L. H., Fine-tuning of the unfolded protein response: Assembling the IRE1α interactome. Mol. Cell 35, 551–561 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ricci D., Marrocco I., Blumenthal D., Dibos M., Eletto D., Vargas J., Boyle S., Iwamoto Y., Chomistek S., Paton J. C., Paton A. W., Argon Y., Clustering of IRE1α depends on sensing ER stress but not on its RNase activity. FASEB J. 33, 9811–9827 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pattingre S., Bauvy C., Carpentier S., Levade T., Levine B., Codogno P., Role of JNK1-dependent Bcl-2 phosphorylation in ceramide-induced macroautophagy. J. Biol. Chem. 284, 2719–2728 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Trzcinska-Daneluti A. M., Chen A., Nguyen L., Murchie R., Jiang C., Moffat J., Pelletier L., Rotin D., RNA interference screen to identify kinases that suppress rescue of ∆F508-CFTR. Mol. Cell. Proteomics 14, 1569–1583 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Barrett K. E., Keely S. J., Chloride secretion by the intestinal epithelium: Molecular basis and regulatory aspects. Annu. Rev. Physiol. 62, 535–572 (2000). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/8/eaax9914/DC1
Fig. S1. XBP1 splicing and IRE1α endonuclease activity are not involved in UPS of ΔF508-CFTR (control experiments of Fig. 1).
Fig. S2. IRE1α kinase–ASK1 pathway is required for UPS of ΔF508-CFTR (control experiments of Fig. 2).
Fig. S3. Depletion of ASK1 inhibits Arf1-Q71L–induced UPS of CFTR.
Fig. S4. CSTMP stimulates the cell surface expression of ΔF508-CFTR by activating IRE1α kinase.
Fig. S5. CSTMP induces the cell surface expression of ΔF508-CFTR via UPS.
Fig. S6. CSTMP (10 μM) does not evoke apoptotic signal.
Fig. S7. CSTMP induces the cell surface expression of ΔF508-CFTR without evoking cellular apoptosis.
Fig. S8. Measurements of CFTR Cl− channel activity (control experiments of Fig. 4, C to F).
Fig. S9. CSTMP induces the cell surface expression of p.H723R-pendrin via UPS.
Fig. S10. The LD50 value of CSTMP in mice (per os) is 25.9 mg/kg per day.