

Large and labile surface reservoirs control air mixing ratios of many indoor species.
Abstract
Human health is affected by indoor air quality. One distinctive aspect of the indoor environment is its very large surface area that acts as a poorly characterized sink and source of gas-phase chemicals. In this work, air-surface interactions of 19 common indoor air contaminants with diverse properties and sources were monitored in a house using fast-response, on-line mass spectrometric and spectroscopic methods. Enhanced-ventilation experiments demonstrate that most of the contaminants reside in the surface reservoirs and not, as expected, in the gas phase. They participate in rapid air-surface partitioning that is much faster than air exchange. Phase distribution calculations are consistent with the observations when assuming simultaneous equilibria between air and large weakly polar and polar absorptive surface reservoirs, with acid-base dissociation in the polar reservoir. Chemical exposure assessments must account for the finding that contaminants that are fully volatile under outdoor air conditions instead behave as semivolatile compounds indoors.
INTRODUCTION
For the outdoor atmosphere, the volatility of individual chemical compounds is framed relative to the mass loading of airborne particles present, where a species vapor pressure describes its propensity to be in the gaseous state. Shifts in the airborne particle loading alter the tendency of compounds to partition between the gas phase and particles, according to the physicochemical properties of compounds and the chemical composition of the particles (1). Indoors, the volatility of compounds is more pertinently related to the sorption capacity of static indoor surfaces. Although indoor particle levels may be similar to those outdoors, the much larger abundances of fixed sorptive surfaces in the indoor environment favor partitioning to these surfaces (2). Past studies exploring this partitioning uptake have primarily been conducted in chambers, often configured with genuine indoor wall, furnishing, or window components (3–6). A notable early study by Singer et al. (7) demonstrated the dynamic, time-resolved nature of the sorption of select organic compounds in genuine indoor spaces; a limitation of that study was the use of relatively slow time-response, off-line chemical analysis. A more recent study of the dynamic response of semivolatile organic compounds (SVOCs) reported measurements every hour (8). In one ventilation experiment, the total SVOC signal declined during an enhanced-ventilation (EV) period in a home, with the signal reverting back to steady state with a response time scale of a few hours after the end of the EV period.
There are many condensed-phase materials and types of chemical interactions that occur between semivolatile species and indoor surface reservoirs. Here, we define a surface reservoir as a condensed-phase material containing chemical constituents that undergo exchange with the gas phase. In particular, molecules can strongly adsorb to the chemical interfaces of clean glass (9) and other building materials, dissolve into thin surface organic films formed from uptake of gaseous species or particle deposition (10), or undergo slower diffusion into the underlying building materials and furnishings, such as wallboard and paint (11). These substrates are collectively referred to as surface reservoirs. We note that condensed-phase water may modulate these interactions by competing for adsorption sites or by changing species diffusivity in organic matrices (12). In addition, there is the potential for sorbed molecules to partition to a water-rich phase present on surfaces, to water sorbed in different building materials (e.g., hydrated gypsum in wallboard), or even to bulk water present, e.g., in kitchens and bathrooms (13).
The partitioning of organic compounds in indoor environments has been conceptually modeled by Weschler and Nazaroff (2), where it was predicted that SVOCs that partition strongly to the organic films on indoor surfaces will more likely reside in a condensed phase than in air. Using octanol as a thermodynamic surrogate for organic films, and depending on the thickness of the film present, they predict that species with log Koa values (where Koa is the octanol-air partition coefficient) larger than about 5 or 6 will reside mostly on surfaces. This feature of the indoor environment affects the fate, transport, and human exposure pathways for SVOCs. For example, species such as flame retardants, pesticides, and plastic contaminants readily partition to indoor surfaces (10, 14). In addition, it has been shown that a wide range of building materials sorbs volatile organics, often taking many hours to attain equilibrium partitioning (15, 16).
In a recent study, dedicated ventilation-manipulation experiments were conducted in a residence, which demonstrated that an inorganic species, nitrous acid (HONO), exhibits highly dynamic partitioning behavior between indoor surfaces and air (17). The pronounced effects of dynamic partitioning to indoor surfaces occur, despite HONO being commonly viewed as too volatile to exhibit this behavior. In particular, indoor HONO mixing ratios decreased during EV through opening of doors and windows in the home, and rapidly returned to the high steady-state mixing ratios commonly encountered in indoor environments upon closing the home. It was hypothesized that partitioning with nitrite in surface reservoirs sustains gas-phase HONO mixing ratios prevalent indoors, providing a buffer to counteract rapid changes in ventilation rates.
Here, we report the first study of the dynamics of gas-surface partitioning across a broad suite of species using fast time-response, on-line instrumentation in a well-characterized indoor environment. In particular, during controlled EV experiments, conducted within the House Observations of Microbial and Environmental Chemistry (HOMEChem) field campaign, we made gas-phase measurements of a diverse range of organic and inorganic compounds, the majority of which have log Koa values below 5. Prompted, in part, by the HONO partitioning study, a motivation of this work is to study compounds that are usually viewed as volatile by the outdoor atmospheric chemistry community (in the absence of clouds) (18). We examine whether these species act in a semivolatile manner indoors.
We report results for isocyanic acid (HNCO), a toxic compound with combustion sources (19); HONO, an indoor source of hydroxyl radicals (20); formic acid (HCOOH), an abundant indoor acid with both primary and secondary sources (21); and other small monocarboxylic acids (C2 to C9), which arise from cooking, off-gassing from building materials, and oxidation reactions (13, 21). These acids are abundant in indoor environments (13, 21), and elevated exposures may pose health risks to occupants (19, 22). The study also included nonacidic molecules: furfural, a species that can originate from decomposition of cellulosic materials (23); isoprene and terpenes that are commonly observed indoors with many sources (24, 25); D5-siloxane, present in personal care products (26); oxygenated VOCs such as ethanol and phenol, originating from cooking, cleaning, and human breath (25); and ammonia (NH3), emitted from occupants and their activities (27).
To explore the dynamic response of these species to changing conditions, we conducted two types of perturbation experiments: (i) EV and (ii) surface cleaning with acidic and basic cleaners. During the EV experiments, the house was repeatedly flushed at intervals with outside air, and then the indoor air composition was allowed to rebound under normal ventilation (house closed) conditions. The EV experiments were described in the HOMEChem overview paper (28) as “sequential ventilation” experiments. In this work, we use “enhanced-ventilation” to describe that the house ventilation rate was enhanced during each of these sequential experiments. The application of vinegar and ammonia-based cleaning agents, which are popular low-cost, household products, changed the pH of indoor surfaces. We were interested to determine whether changes in the surface pH would affect the extent of gas-surface partitioning of acidic or basic molecules via either enhanced protonation or deprotonation. These effects have previously been demonstrated in dedicated chamber experiments involving carbon dioxide and ammonia (29).
Overall, both the EV and surface cleaning experiments yield evidence that indoor surfaces act as labile reservoirs of all the gas-phase species measured. Comprehensive high time-resolution measurements provide the first characterization of the kinetics of gas-surface dynamic partitioning in a real indoor environment.
RESULTS
Sequential EV experiments
Indoor air mixing ratios
Time-of-flight (TOF) chemical ionization mass spectrometers (CIMSs) measured the mixing ratios of gas-phase acids during the HOMEChem campaign at the UTest house in Austin, Texas. A proton transfer reaction–TOF–MS (PTR-TOF-MS) measured nonacidic organic molecules; ammonia mixing ratios were determined by cavity ring-down spectroscopy (see Materials and Methods). The signals of each species decreased immediately upon enhancing the rate of ventilation by opening windows and doors and rapidly returned to the vicinity of their starting values upon shutting the windows and doors. Figure 1 illustrates this behavior for several acidic gases; other carboxylic acids, nonacidic species, and ammonia exhibit similar responses (fig. S1). Outdoor mixing ratios of all the measured species (the orange dots in Fig. 1) were lower than indoor levels, consistent with the observed decrease of indoor levels during EV. This evidence confirms that these chemicals are all emitted or formed indoors.
Fig. 1. Indoor mixing ratios of several acids during EV experiments.
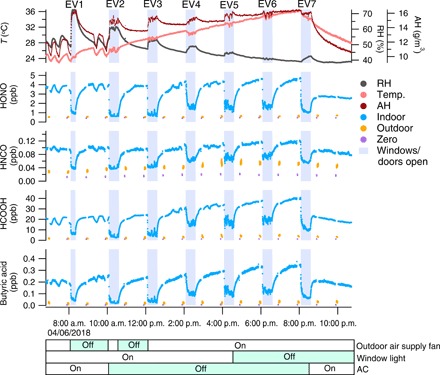
The top panel shows the measured house temperature (T; left axis), relative humidity (RH; right axis), and absolute humidity (AH; second right axis). Temperature and RH were measured in the kitchen within 1 m of the CIMS inlet. The shaded areas indicate when doors and windows were open to increase the ventilation rate of the house. The hourly 2-min background measurement (measuring zero air) is shown with purple dots, followed by a 5-min outdoor measurement (orange dots). The average air exchange rate (AER) when the house is closed following each EV experiment is summarized in table S2. The color bars at the bottom of the plot show the state of outdoor air supply fan (on/off), window light (with/without), and air conditioning (AC) (on/off) during the experiment, with the green shaded periods showing when the fan, window light, and AC were off.
A full list of compounds and their physicochemical properties is provided in table S1. Table S2 summarizes the average mixing ratios of individual species in indoor air during the EV and closed-house periods and in outdoor air. For example, the indoor mixing ratio of HONO at steady state after the first six EV periods (EV1 to EV6) was between 3.7 and 4.3 parts per billion (ppb), while the average outdoor mixing ratio was 0.57 ± 0.07 ppb. These abundances are similar to those previously reported (17). The mixing ratios of HONO during the EV period reflect the influence of outdoor air dilution on indoor HONO levels. In particular, indoor HONO decreased to ~1 ppb during the first EV period (EV1) and was slightly higher during subsequent experiments, especially in experiments EV4 to EV6. This observed increase in levels during EV is likely due to less indoor-outdoor air exchange during the last three experiments, arising from slower local wind speeds (table S2). Similarly, mixing ratios of other compounds during the EV periods in EV4 to EV6 were higher than those in EV1 to EV3. The steady-state indoor mixing ratio of HNCO varied between 0.09 and 0.11 ppb, larger than the average outdoor mixing ratio of 0.04 ± 0.01 ppb. These HNCO measurement results are similar to a median mixing ratio of 0.15 ppb reported in a Toronto residence (30). There is a relatively high indoor mixing ratio of formic acid (21 to 38 ppb) compared to a lower outdoor level (1.7 to 4.3 ppb).
The steady-state mixing ratios for all species during the recovery period after each EV episode are similar across the course of the day, with small upward trends associated with and possibly attributable to increased house temperature with the air conditioning (AC) off. The increasing steady-state signal with increasing temperature, as reported previously for phthalates in the same test house (14), is likely due to a shift in partitioning between air and condensed-phase surface reservoirs. The nature of the surface reservoirs is discussed below. Note that the decrease of the steady-state concentrations for the small acids in Fig. 1 and fig. S1A after experiment EV7 is attributed to the operation of AC, i.e., through removal of water-soluble organic gases (WSOGs) by dissolution into condensing water during dehumidification. A similar decrease was observed for NH3 and some neutral oxygenated species with relatively large water solubility, including phenol, ethanol, and furfural, and the effects were less pronounced for sparingly water-soluble species such as isoprene and monoterpenes (fig. S1B). These observations support the inference that AC can act as a sink for indoor WSOGs under humid conditions, as was demonstrated in another recent study (13).
The operation of the outdoor supply air fan was intended to change the air exchange rate (AER) by either mechanical ventilation (fan on) or infiltration (fan off) (see Materials and Methods). With the outdoor supply air fan on for EV3 to EV6, there is only a slightly higher AER (on average 0.53 ± 0.06 hour−1 through mechanical ventilation) compared to EV1 and EV2 when the fan was off (on average 0.45 ± 0.02 hour−1, due to infiltration) (table S2). The steady-state mixing ratio of measured species is not sensitive to the small difference in AER when outdoor supply air fan was on or off. Note also that the presence or absence of light in the house had no clear effect on the steady-state mixing ratios (compare EV1 to EV4 versus EV5 to EV7).
Time constants for return to steady state
Until recently, indoor VOC and SVOC concentrations were largely made via off-line methods with limited time resolution. A key advantage of high time-resolution data from CIMS, PTR-TOF-MS, and on-line spectroscopic methods is the ability to quantify dynamic response times needed to reestablish steady-state conditions after ventilation perturbations. We have assessed response times by fitting exponential growth curves to mixing ratio data when the house was closed after periods of EV. Because of the temperature dependence of the observed indoor mixing ratios as the house warmed during the day, the original data were detrended to minimize the influence of temperature (see details in text S2 and fig. S2).
The response time constants for all small acids and for phenol (Fig. 2) were similar, on average, between 700 and 1000 s. Several other measured species exhibit a longer response time: NH3, sesquiterpenes, monoterpenes, isoprene, D5 siloxane, ethanol, and furfural have response times from 1600 to 2700 s. While the AC operation had some effects on trace gas mixing ratios as described above, the AC (on or off), including its effects on temperature and relative humidity, showed little influence on the response times of any of the gases after periods of EV (EV1 versus EV2 to EV6). For all species, there was no pronounced dependence of the response times on light levels (dark versus illuminated indoors) or the outdoor supply air fan operation (on or off).
Fig. 2. Box and whisker plot of response time constants.
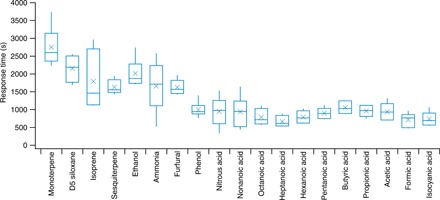
Only the first six EV experiments (EV1 to EV6) are shown. Response times for EV7 are not shown due to the significant change of house conditions, i.e., dehumidification associated with air conditioner operation. The horizontal line and the cross symbol inside each box denote the median and the mean, respectively. The top and bottom boundaries of each box show the third and first quartiles. The top and bottom of the vertical line (i.e., the whisker) show the maximum and minimum data points, respectively. The acids were measured with CIMS (acetic acid measured with iodide CIMS and the others measured with acetate CIMS), and the nonacidic compounds were measured with the PTR-TOF-MS, except for NH3, which was measured by cavity ring-down spectroscopy. Data for D5 siloxane in EV1 and EV2 and for acetic acid in EV5 were excluded due to interferences.
When ventilation is the controlling removal process for indoor air constituents emitted from a constant source, the indoor species should return to a steady-state mixing ratio after an EV period with a characteristic response time equal to the reciprocal of the AER.However, in this study, the establishment of steady-state indoor concentrations after EV occurs much more rapidly than 1/AER independent of the outdoor supply air fan operation status. This observation combines with the consistency of the response signals to demonstrate that there must be one or more large and responsive replenishment reservoirs in the studied indoor environment. Given the rapid response times, these reservoirs are almost certainly associated with either surface films or the outer layers of building materials and furnishings that are able to participate in rapid gas exchange. The repetitive behavior for each EV experiment indicates that the size of the reservoir is substantial, as it is not depleted after the house has been repetitively flushed.
Direct evidence from analysis of surface nitrite on glass plates deployed during HOMEChem substantiates the hypothesis that indoor surfaces act as a reservoir for HONO (text S1), in agreement with similar measurements in a previous study (17). Other studies have also indirectly provided evidence for indoor surfaces being likely sources or reservoirs of carboxylic acids, e.g., elevated indoor carboxylic acid mixing ratios were observed in a university classroom when ventilation was off, likely due to off-gassing from indoor surfaces (21).
It is well recognized that organic molecules can partition to indoor surfaces (2), and past work using off-line measurements has illustrated a rebound effect for a variety of hydrocarbons and oxygenated VOCs after a period of EV in simulated and real residential environments (3, 7). In the present study, volatile small acids (HONO, HNCO, and C1 to C9 monocarboxylic acids) and many small nonacidic species (ammonia, monoterpenes, isoprene, sesquiterpenes, D5 siloxane, furfural, phenol, and ethanol) all display rapid and repeatable rebounding, evidence of substantial dynamic partitioning of a diverse suite of species between indoor air and surfaces.
The fast response times for the small acids and phenol suggest that their airborne abundances are governed by rapid dynamic partitioning with a surface reservoir that does not have a significant mass transfer constraint. Given the similarity of the response times across a wide range of molecular weights, mass transfer through the air-side boundary layer is likely the rate-determining step in the replenishment process. Other species with considerably longer response times (e.g., ammonia, terpenes, isoprene, furfural, and D5 siloxane) may be more strongly sorbed to a surface or dissolved in a reservoir through which mass transfer is slower, e.g., the organic binder of a paint layer. It is plausible that the matrix may be sufficiently viscous and/or thick to impede the mass transfer rate to a value lower than the air-side mass transfer would yield. In addition, we cannot entirely rule out that relatively inaccessible air volumes slowly supply molecules, leading to their slower response times. However, this explanation for the slower response times seems unlikely, given the open design of the house and the very high internal mixing rates (see Materials and Methods). A response time of several hours was observed for SVOCs in a residence in California (8). The longer response time for the SVOCs may suggest that, for these SVOCs, gas-phase mass transfer is not rate limiting for air-surface exchange. Instead, diffusion in the indoor surface reservoirs may be the rate-limiting step, different from the behavior of many of the VOCs observed in the present study.
We can assess the expected response time for molecules that desorb rapidly from a surface reservoir. In particular, when gas-phase mass transfer is the rate-limiting process, the characteristic time (τ) for a gas to reach the steady-state concentration depends on AER (hour−1), the deposition velocity (vd, m hour−1) to indoor surfaces, and the surface area to volume ratio (S/V) (2)
| (1) |
The average closed-house AER (for EV1 to EV6) was measured to be 0.5 hour−1, and vd is estimated to be 1 m hour−1 for a typical SVOC (2, 31). Using a typical value of 3 m−1 for S/V (2, 32), the expected value of τ, in this case, is estimated to be approximately 1000 s. Given the simplicity of the method and uncertainties in S/V and vd, this response time estimate is consistent with the values measured for small acids. For the conditions in this study, the τ values are predominantly determined by the product vd × S/V, with only minor dependence on AER. We note that there are considerable uncertainties in these assumed values, including the dependence of S/V on the degree of room furnishing and on room size. Uncertainty is also present in how vd varies with the physicochemical characteristics of both the gas and the surface to which uptake/desorption is occurring. During the experiments, the study site was more sparsely furnished than might be common; in addition, there was some instrumentation installed indoors that influenced surface abundance.
Vinegar and ammonia cleaning experiments
On another day during the HOMEChem campaign (26 June 2018), the house floor was mopped with vinegar solutions four times (see Materials and Methods). This activity presumably alters indoor surface reservoir pH through the partitioning of enhanced levels of gas-phase acetic acid. The gas-phase mixing ratios of HONO, HNCO, and HCOOH increased during and following vinegar mopping; there was either only a small increase or no obvious change for weaker acids and for nonacidic species (Fig. 3 and fig. S3). The ratios of the concentrations during mopping to the average steady-state concentrations during 15 min before mopping are shown in fig. S4. A ratio above 1.0 indicates an increase attributable to the mopping procedure. The increase of acid signals occurred during all four experiments when different amounts of vinegar were used (table S3).
Fig. 3. Indoor air acid mixing ratios during vinegar and ammonia cleaning.
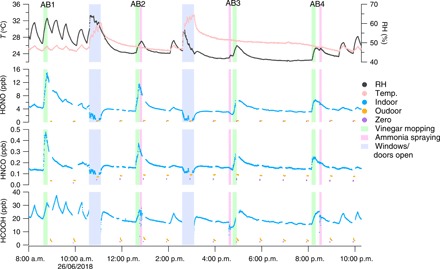
AB1 to AB4 represent four acid-base experiments. The shaded areas show when doors and windows were open to enhance house ventilation (blue). Times of mopping the floor with vinegar solution (green) and spraying ammonia on indoor surfaces (pink) are also indicated. Mixing ratios measured in outdoor air and for zero air are denoted by orange and purple dots. The top panel shows the RH and temperature measured in the kitchen. The AC system was turned off during the two ventilation periods.
The increase of gas-phase acids during vinegar mopping suggests that indoor surface reservoirs, specifically those with a substantial condensed water component, act as a transient source of these small acids. In particular, the data are consistent with an inference that acetic acid (pKa = 4.75) from the vinegar affects the pH of indoor surfaces by the uptake of gas-phase acetic acid (see fig. S5), resulting in acidified polar reservoirs. There may also be comparable effects arising directly from the application of acetic acid to the surface being mopped. The elevated HONO, HNCO, and HCOOH signals during vinegar mopping are consistent with their low pKa values (pKa < 4) as compared with the other acids (pKa > 4.8) (table S1). These stronger sorbed acids (HONO, HNCO, and HCOOH) are likely in the dissociated form before vinegar mopping. Decreasing surface pH leads to their protonation, shifting the air-surface partitioning toward the gas phase.
After the first mopping experiment, HONO and HNCO increased substantially from 4.5 and 0.13 ppb (average for 15 min before mopping starts) to maxima of 15 and 0.48 ppb, respectively. The degree of increase became progressively smaller from experiment AB1 to AB4 for HONO and HNCO (fig. S4), although an increasing amount of vinegar was used and acetic acid mixing ratios in the air did not decrease (table S3 and fig. S5). It is possible that the surface pH and composition changed after mopping repetitively. Formic acid did not exhibit the same trend, possibly because it has a larger Henry’s law constant and is less volatile than HONO and HNCO (table S1). In addition, formic acid was influenced considerably by the AC system, for example, during the first mopping experiment, making it difficult to accurately quantify the influence of vinegar mopping separate from the influence of condensed water on the AC cooling coils.
Increasing surface pH by ammonia spraying, attributable to uptake of elevated gas-phase NH3 (fig. S5), had an opposite yet less pronounced effect on the air-surface partitioning, which may be due to the fact that many of the acids are already dissociated on the surface. In experiments AB2 and AB4, the ammonia cleaner was used immediately after vinegar mopping, and the effects of ammonia spraying were attenuated. In experiment AB3, the mixing ratios were influenced when the ammonia cleaner was applied before vinegar mopping as compared to the vinegar mopping case alone. We note that the observed rebound dynamics after EV periods are different on the cleaning day than on the EV day, potentially affected by the different nature of the surfaces, the role of AC, or the residual presence of cleaning agents.
Distribution of indoor volatile species between air and surface reservoirs
There are many surface reservoirs into which gas-phase compounds might partition. For example, organic films on indoor surfaces have been identified as temporary storage reservoirs for SVOCs (2, 10). For more water-soluble species, such as the small acids included in this work, a water-rich surface film would similarly enhance the uptake of water-soluble gases from indoor air. Situated below organic and water-rich surface films exist indoor finishing materials, such as paint, wallboard, flooring, and furnishings, which provide additional potential reservoirs. It is possible that bulk water, commonly present in the kitchen, bathroom, or AC system, could also participate as a sorptive substrate.
To display the distribution of chemicals in indoor environments and to assist in interpreting experimental observations, we apply two-dimensional chemical partitioning space plots, previously used to assess the phase distribution of organic compounds in the atmosphere (see text S3 for details) (18, 33). A few key assumptions are applied in the analysis. (i) We assume that polar and weakly polar partitioning media of variable volumes are present indoors; their geometry is irrelevant for the present purposes. We use liquid water and octanol as surrogates for these media, recognizing that this description simplifies a much more chemically complex system. Octanol is a surrogate for a weakly polar sorbing material, such as paint. Water is a surrogate for a more polar medium, such as hydrated gypsum in wallboard. (ii) Partitioning among indoor air, the polar reservoir and the weakly polar reservoir are assumed to be the only processes affecting gas-phase concentrations. We allow acid-base dissociation to occur in polar phase as in a liquid-water phase. (iii) The analysis assumes equilibrium partitioning between indoor air and surface reservoirs. The partitioning space plot is a useful graphical tool to display where a chemical resides under thermodynamic equilibrium conditions.
In Fig. 4, all chemical species are placed in the partitioning space plot based on their respective values of log Kwa (water-air partitioning coefficient, i.e., Henry’s law constant) and log Koa (table S1). The dashed lines in each plot indicate boundaries for different fractions of compounds, with their location dependent on the relative volume of weakly polar, polar, and gas phases. Chemicals located on the boundary lines between two different color regions exist with equal abundance in the two phases. Chemicals located in the darkest pink, green, and blue regions of the plot have >99% abundance in one specific phase. Those areas more gently shaded pink, green, and blue have between 50 and 99% abundance in one specific phase, as indicated in the plot. The white central triangle shows the transition region where less than 50% is present in each of the three phases.
Fig. 4. Partitioning space plots.
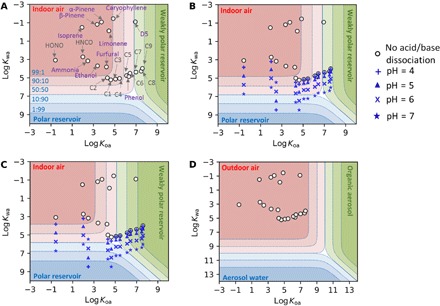
(A) An indoor environment with 1.5 × 10−7 m3 of weakly polar and polar reservoirs each per cubic meter of air, without acid and base dissociation; (B) same volume of polar and weakly polar reservoirs as in (A), but considering acid and base dissociation in the polar reservoir; (C) 7.5 × 10−6 m3 and 1.5 × 10−6 m3 of weakly polar and polar reservoir per cubic meter of air, respectively, with acid and base dissociation; and (D) a highly polluted outdoor environment with 100 mg m–3 each for liquid water and organic content in the ambient aerosol [phase separated into organic (represented by octanol) and water phase (51)], with acid and base dissociation. Names of the individual species are labeled in (A), with C1 to C9 representing monocarboxylic acids with one to nine carbons; their locations shift slightly in (B) and (C) relative to the boundaries, but the ordering does not change. The dashed lines in the space indicate boundaries for different fractions of compounds in each phase, and the blue text shows the fraction in gas versus polar phase for the dashed line below the blue label. The different symbols in (B) to (D) indicate the assumed pH of the polar phase, as indicated in the legend. Note that, in (D), the range of log Kwa and log Koa is expanded to 13, as compared to 10 in (A) to (C).
The results in Fig. 4A predict that many species would be in the gas phase indoors under the assumed conditions, whereas the sequential EV perturbation experiments indicate instead that these molecules reside primarily in surface reservoirs. Whereas in Fig. 4A all molecules were assumed to be in their undissociated state, in both Fig. 4, B and C, we allow acids to dissociate at the specified pH of the polar reservoir and for ammonia to become protonated, according to the effective Henry’s law constant [Keff,wa = Kwa(1 + 10pH-pKa) for acids, and Keff,wa = Kwa(1 + 10pKa-pH) for bases, where pKa is for the protonated form of the base, e.g., ]. Figure 4C assumes much larger reservoir volumes than in Fig. 4 (A and B). Overall, the predictions in Fig. 4C are more consistent with the experimental observations as to whether a molecule is predicted to be in the gas phase or in a surface reservoir, with notable exceptions of isoprene and the monoterpenes.
The influence of acid dissociation on phase distribution is particularly large for the more acidic compounds with smaller values of Koa, i.e., those not predominantly in the weakly polar, organic phase. The overall effect is to move the location of the chemicals much closer to the polar phase portion of the plot, depending on the assumed pH of surface water. The effect is stronger for HONO, HNCO, and HCOOH than for the C3 to C9 acids, because they are located toward the left region of the partitioning space plots (in the polar phase component of the plot) and they have lower pKa values than the C3 to C9 acids (table S1). Trends displayed in the partitioning plots are consistent with findings from the vinegar mopping experiments, during which the largest changes in gas-phase concentration with mopping were observed for these lower pKa species. By contrast, the C3 to C9 acids are more likely partitioned to the weakly polar, organic reservoirs in the house and thus experience less change upon vinegar mopping. Including the effect of protonation for NH3 shifts its distribution notably into the polar reservoir with pH between 4 and 7. Other polar phase interactions, such as hydration, hydrolysis, salt effects, and house conditions (e.g., temperature change), could also influence the air-surface partitioning (34–37).
If the polar reservoir consisted of uniformly thick liquid water films existing throughout the house (with an assumed S/V of 3 m−1), then they would be 50 nm thick in Fig. 4, A and B and 500 nm thick in Fig. 4C. The corresponding total volume is equivalent to 35 and 350 cm3 of aqueous water in the studied house (235 m3 volume), respectively. Aqueous surface films as thick as 50 and 500 nm are improbable. On the other hand, some water will certainly be sorbed to hygroscopic building materials such as gypsum wallboard, sorbed on furnishing fabrics, condensed in the AC system, and present in other forms of bulk water such as on sink surfaces and in sink drains. Regardless of its precise location, the results in Fig. 4 suggest that a large amount of condensed water (or other polar media) must exist in order for more water-soluble/polar molecules with low Koa values, such as NH3, HONO, HNCO, and HCOOH, to reside in a partitioning reservoir that exhibits rapid reversibility of the type exhibited in these experiments.
Similarly, if the weakly polar reservoir consisted entirely of a uniform organic film over all indoor surfaces, the film would be thick, specifically 2500 nm for the conditions in Fig. 4C. Current estimates for the thickness of deposited organic films on indoor surfaces are much smaller, on the order of a few tens of nanometers or less (10). Thus, under the assumptions of our model, there must be a larger organic reservoir prevalent indoors. This organic reservoir could be thicker organic films in the kitchen associated with cooking or it may be present in the building materials and surfaces that underlie condensed organic films (38, 39). A potential candidate is the painted walls and ceiling, ubiquitous in indoor environments, consisting of a viscous matrix of paint binder and pigment. Previous studies measured drywall paint thickness to be on the order of 30 to 50 μm (40), i.e., representing an organic reservoir volume much larger than assumed in modeling above. If only a fraction of that paint layer is available for rapidly reversible surface partitioning, then it could represent an important reservoir for compounds with low water solubility and large Koa values. Predictions for partitioning to a 50-μm-thick organic film demonstrate the maximum potential effect (see fig. S6), and in this case, most chemicals (except for NH3, HONO, HNCO, ethanol, β-pinene, and isoprene) would be predominantly present in the large organic, weakly polar reservoir.
An overall conclusion of this modeling exercise is that substantial effective volumes of both polar (e.g., water-rich) and weakly polar (e.g., organic-rich) surface reservoirs could explain the observed behavior during the EV experiments. At this point, the effective volumes of these reservoirs are highly uncertain. However, it is interesting that the model with large weakly polar and polar reservoirs (Fig. 4C) cannot accurately predict the phase distribution of highly volatile molecules such as isoprene and the monoterpenes, which have low polarity. Note that the chemical interactions with the surface reservoirs may not be fully captured by the conventional measures of partitioning, specifically the Koa and Kwa values. As one example, in the case of limonene, strong hydrogen bonding occurs to glass surfaces (9). These interactions are not included by the two conventional partitioning parameters used here.
DISCUSSION
Our sequential EV experiments indicate that easily accessible indoor surface matrices, potentially including both surface films and indoor building materials and furnishings, provide an important transient reservoir of many gas-phase VOCs, NH3, and HONO. The very short response times to return to steady state after EV suggest that surface-sorbed species readily volatilize to the gas phase without surface side mass transfer limitations. Additional support for the presence of large surface reservoirs for acidic, water-soluble molecules is provided by the vinegar and ammonia cleaning experiments, in which rapid change of gas-phase mixing ratios of acidic chemicals was observed upon altering surface pH. Although less volatile species, such as phthalate and flame-retardant SVOCs, are known to substantially partition to indoor surfaces (41–43), this study uses fast time-response instrumentation to illustrate that a wide array of volatile, small molecules also participate in dynamic surface-gas partitioning.
The chemical interactions of the sorbing species with the surface reservoirs are expected to be complex, given the heterogeneous character of indoor surface materials. As a step toward a more complete description, we present a new application of two-dimensional chemical partitioning space calculations for indoor gas-surface interactions in which we model both weakly polar (e.g., organic-rich) and polar (e.g., water-rich) surface reservoirs, represented by octanol and water, respectively. Despite the simplicity imposed by these assumptions, the partitioning space analysis is informative by illustrating that some species (e.g., NH3, HONO, HNCO, and HCOOH) are much more likely to be sorbed to a polar reservoir than to a weakly polar organic-rich reservoir, provided that the reservoir is not highly acidic (with the exception of ammonia). However, other species that have only somewhat lower water solubility and larger pKa values, such as the C3 to C9 acids, will likely sorb to an organic reservoir instead. The model results are also valuable for illustrating how unlikely it is for certain molecules (e.g., isoprene and monoterpenes) to largely partition to a thin-film organic reservoir. This finding implies that the time scale for the rebound effect observed for these species is controlled by a different process, such as mass transfer within indoor building materials (e.g., latex paint) or release from more strongly bound surface states.
An important conclusion is that many compounds that are fully volatile under typical outdoor air conditions exhibit semivolatile behavior indoors with a higher abundance on the surface than in the gas phase. This outcome is driven by the very high surface areas (and thus large sorptive surface reservoir volumes) prevalent in the spaces in which we live and work. The contrast to indoor behavior is demonstrated in Fig. 4D, a partitioning space plot for highly polluted conditions outdoors, in which partitioning is assumed to occur between the gas phase and either the liquid water content or the organic content (represented by octanol) of ambient aerosol. Despite the assumption in that analysis of high aerosol loading (100 μg m−3 each for liquid water and organic content), it is nevertheless predicted that all the studied molecules are predominantly in the gas phase for outdoor conditions, whereas this study illustrates that they are largely partitioned to condensed phases indoors.
The partitioning distribution between indoor air and surfaces influences indoor chemical exposure pathways. In particular, the fast response times of many studied species indicate that temporary window opening may be less effective than expected in reducing time-averaged exposures, because many species rapidly return to high mixing ratios within a brief period after ventilation. In addition, cleaning with products containing acidic and basic species may lead to temporary elevated exposures of other acidic and basic compounds, respectively. Future studies should strive to measure the concentrations of sorbed molecules within the different surface reservoirs. It is important to know whether the compounds are sorbing to thin surface films, in which case there will likely not be mass transfer limitations in their response to ventilation perturbations, or whether mass transfer will limit the response of trace gas mixing ratios because they reside in a more strongly bound condensed phase or a deeper, viscous reservoir.
A better understanding of how chemicals respond indoors to a change of state (e.g., EV and change of surface pH) and the importance of surface storage reservoirs is important for accurately interpreting experimental data measured in a dynamic environment, for mathematical modeling, and, ultimately, for mitigating exposure to pollutants. The surface-gas interactions of indoor pollutants revealed in this study are likely to be common in many indoor environments, given the universally high indoor surface areas. More complex modeling studies that take account of human behavior and the diversity of indoor environments are needed to better understand the impact of occupant behavior on chemical concentrations and exposure (44, 45).
MATERIALS AND METHODS
Site and experiment description
This work presents data collected during the HOMEChem field campaign that took place in June 2018 at the UTest house situated at the J. J. Pickle Research Campus of the University of Texas at Austin. An overview of HOMEChem experiments and instrumentation is described in (28). In brief, the test house is a 110-m2 (volume of 250 m3, operated at 235 m3 because the bathroom doors were closed), single-story manufactured home with three bedrooms and two bathrooms. The house is equipped with two independent heating ventilation and AC (HVAC) systems with separate ceiling and floor air distribution systems. During HOMEChem, only the ceiling HVAC system was used, and the floor HVAC was sealed to limit leakage. A refrigeration cycle AC system cooled and dehumidified indoor air based on a thermostat signal. The internal recirculation fan of this cooling system was continuously on, independent of the thermostat, providing continuous indoor air mixing throughout the house equivalent to eight house volumes per hour. There were no filters in the HVAC system during these experiments. The house was equipped with a dedicated ventilation system that was set to supply outdoor air at a supply rate of 0.5 house volumes per hour when the outdoor air supply fan is on. Because of infiltration of outdoor air through uncontrolled leaks in the building envelope, the AER was measured to be 0.45 ± 0.02 hour−1 when the outdoor air supply fan was off during the experiments.
Sequential EV experiments
Seven EV experiments (EV1 to EV7) were performed on 4 June 2018. Four windows (one each in the kitchen, living room, and two front bedrooms) and two exterior doors (front and back) were opened for either 15 min (EV1) or 30 min (all other experiments) to flush the house with outdoor air. Other windows were closed at all times. The interior doors separating the living room from the two bedrooms were kept open, and the doors to two bathrooms were kept closed. The air inside the portions of the house coupled through open doors was expected to mix rapidly (46). The house was closed for 105 min after EV1 and for 90 min after experiments EV2 to EV6. After EV7, the house was kept closed throughout the following night. The thermostat (which controls indoor temperature and humidity, on or off), light (all windows blocked with blinds or aluminum foil or unblocked to alter light intensity), and ventilation condition (outdoor air supply fan on or off, i.e., mechanical ventilation versus infiltration) were altered deliberately during different experiments. The AC system was off between EV2 and EV6. The blinds of all windows were closed for EV5 and EV6 to avoid any direct or diffuse sunlight entering the house. There were no interior lights on during any experiments. The outdoor air supply fan was turned off during the period when the house was closed after the first two EV experiments, and the fan was on during all other periods. The details of the experimental conditions are summarized in table S4.
Vinegar and ammonia cleaning experiments
Four vinegar-based cleaning and three ammonia-based cleaning experiments (AB1 to AB4) were performed on 26 June 2018. For each vinegar cleaning experiment, two volunteers entered the house to prepare the mopping solution (10-min preparation), mopped the living room and kitchen floors continuously for 10 min, and then left the house. A vinegar solution consisting of approximately 3.8 liters of water and 240 ml of a commercial vinegar cleaner (4 to 6% by weight acetic acid) was prepared in a bucket. The next mopping experiment started 4 hours after the start of a previous mopping experiment. Indoor surfaces (other than the mopped floors, such as countertops, tables, and refrigerator) in the living room and kitchen were cleaned with an ammonia cleaner (1 to 3% by weight ammonia) either before or after a vinegar mopping activity. Two hours after the start of the first two mopping experiments, the house was ventilated by opening the windows and doors for 30 min, during which the AC system was turned off. The AC was controlled by the thermostat during all other times. The outdoor air supply fan was kept on at all times, providing an AER of 0.5 hour−1. A detailed timeline of activities and the estimated mass of acetic acid and ammonia used during cleaning are provided in table S3.
Measurements
A CIMS (Aerodyne) with acetate as the reagent ion was used to detect gas-phase organic and inorganic acids (17, 47). The CIMS was located in an air-conditioned trailer beside the UTest house (direct distance: ~5 m). The sampling inlet was in the kitchen of the house, with perfluoroalkoxy (PFA) tubing used as the inlet line [outside diameter (OD), 0.635 cm; length, 10 and 8 m for indoor and outdoor air, respectively]. A detailed description of the CIMS is available elsewhere (17, 47). Briefly, nitrogen gas (20 ml/min) (Air Gas, NI UHP200LT230) passed over the headspace of acetic anhydride (≥99%, Sigma-Aldrich) contained in a stainless steel bottle (Swagelok) at room temperature. This flow was subsequently diluted by nitrogen (2.2 liters/min) and then passed through a 210Po radioactive source (NRD, model P-2031) at a flow of 2 liters/min set by a critical orifice to generate the ionization reagent ions (acetate ions). Acetate ions ionize analyte molecules through either proton transfer or clustering reactions, followed by declustering before detection by TOF-MS (47). Acetic acid was measured using another CIMS with iodide as the reagent ion (48) located in the same trailer and shared the indoor and outdoor air inlet line (7 and 5 m for indoor and outdoor air, respectively) with the acetate CIMS. Instrument backgrounds were collected for 2 min of every hour by overflowing the CIMS inlet with ultrapure zero air (Air Gas, AI UZ300). Outdoor measurements were collected for 5 min every hour after the 2-min background measurement. A three-way solenoid isolation valve (NResearch Inc.) automated the sampling air switching using a custom-built LabVIEW control interface (National Instruments).
The nonacidic species were measured with PTR-TOF-MS (PTR-TOF 8000, IONICON Analytik GmbH, Austria), which uses H3O+ as the reagent ion. The PTR-TOF-MS was situated in the same trailer as the CIMS, and its indoor inlet (PFA; OD, 0.635 cm; length, 8.4 m) was located at the same location in the kitchen of the house. Air was continuously drawn through both the indoor and outdoor sample lines at 1.5 liters/m, and an automated flow-through valve (NResearch Inc., 648T091; PTFE inner contact surfaces) was used to switch from indoor to outdoor sampling (PFA; OD, 0.635 cm; length, 8.4 m) with a 30-min cycle (25 min indoors and 5 min outdoors). Details of calibration and data analysis for CIMS and PTR-TOF-MS are described in texts S4 and S5. Indoor NH3 was measured with cavity ring-down spectroscopy (Picarro G2103 Analyzer), located in the kitchen (49).
The AER was measured by means of continuous release of a tracer gas (butane-d3, Cambridge Isotope Laboratories). A detailed description of this method can be found in the study of Liu et al. (50). Indoor air and surface temperatures at multiple locations in the house, and relative humidity in supply, return air, and kitchen area were measured during all experiments.
Three glass sheets (26 cm by 20 cm each) were mounted vertically in the kitchen. Surface samples were collected daily by wiping the glass with nylon membrane filters (47 mm; Whatman 7404-004). The filters were subsequently extracted and analyzed for surface nitrite concentration using an ultraviolet-visible (UV-vis) spectrophotometric technique (text S1) (17).
Supplementary Material
Acknowledgments
We thank the HOMEChem science team for collaboration throughout the field campaign. Special acknowledgement is given to S. Bourne for running the UTest house and providing house parameters, to L. H. Ruiz and her group members at the University of Texas at Austin for experimental support, and to C. Weschler and G. Morrison for helpful discussions. C.W. acknowledges J. Murphy and Y. Tao for the use of the Ion Chromatography (IC) for the calibration of HNCO, and R. Hems, Q. Shi, and A. Hrdina for helpful discussions on data analysis at the University of Toronto. Funding: Funding for this study was provided by the Chemistry of Indoor Environments program of the Alfred P. Sloan Foundation (grants G-2016-7049, G-2016-7050, and G-2017-9944). Author contributions: C.W., D.B.C., and J.P.D.A. planned the study. C.W. and J.P.D.A. analyzed the data and wrote the manuscript. C.W. performed acetate CIMS measurements. C.A. and A.H.G. performed PTR-TOF-MS measurements and associated data analysis. J.M.M. and D.K.F. performed I-CIMS measurements and designed sampling and calibration setup. L.A. and P.F.D. performed ammonia measurements. D.K.F. and M.E.V. led the HOMEChem campaign. All authors provided comments and edited the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/8/eaay8973/DC1
Text S1. Surface nitrite analysis during HOMEChem.
Text S2. Detrending data and response time calculation.
Text S3. Phase distribution calculation.
Text S4. CIMS calibration and data analysis.
Text S5. PTR-TOF-MS calibration and data analysis.
Fig. S1. Time-series of different species during EV experiments.
Fig. S2. Detrending data during the EV experiments.
Fig. S3. Indoor air signals during vinegar and ammonia cleaning.
Fig. S4. Comparison of mixing ratios before and during vinegar cleaning.
Fig. S5. Indoor acetic acid and ammonia mixing ratio during vinegar and ammonia cleaning.
Fig. S6. Partitioning space plot for an indoor environment.
Table S1. Physical and chemical properties of different compounds at 25°C.
Table S2. Time-averaged steady-state mixing ratios (parts per billion).
Table S3. Vinegar and ammonia cleaning experiment details.
Table S4. EV experiment details.
REFERENCES AND NOTES
- 1.Pankow J. F., An absorption-model of gas-particle partitioning of organic-compounds in the atmosphere. Atmos. Environ. 28, 185–188 (1994). [Google Scholar]
- 2.Weschler C. J., Nazaroff W. W., Semivolatile organic compounds in indoor environments. Atmos. Environ. 42, 9018–9040 (2008). [Google Scholar]
- 3.Singer B. C., Revzan K. L., Hotchi T., Hodgson A. T., Brown N. J., Sorption of organic gases in a furnished room. Atmos. Environ. 38, 2483–2494 (2004). [Google Scholar]
- 4.Tichenor B. A., Guo Z., Dunn J. E., Sparks L. E., Mason M. A., The interaction of vapour phase organic compounds with indoor sinks. Indoor Air 1, 23–35 (1991). [Google Scholar]
- 5.Won D., Corsi R. L., Rynes M., Sorptive interactions between VOCs and indoor materials. Indoor Air 11, 246–256 (2001). [DOI] [PubMed] [Google Scholar]
- 6.Jørgensen R. B., Bjørseth O., Malvik B., Chamber testing of adsorption of volatile organic compounds (VOCs) on material surfaces. Indoor Air 9, 2–9 (1999). [DOI] [PubMed] [Google Scholar]
- 7.Singer B. C., Hodgson A. T., Hotchi T., Ming K. Y., Sextro R. G., Wood E. E., Brown N. J., Sorption of organic gases in residential rooms. Atmos. Environ. 41, 3251–3265 (2007). [Google Scholar]
- 8.Kristensen K., Lunderberg D. M., Liu Y., Misztal P. K., Tian Y., Arata C., Nazaroff W. W., Goldstein A. H., Sources and dynamics of semivolatile organic compounds in a single-family residence in northern California. Indoor Air 29, 645–655 (2019). [DOI] [PubMed] [Google Scholar]
- 9.Fang Y., Lakey P. S. J., Riahi S., McDonald A. T., Shrestha M., Tobias D. J., Shiraiwa M., Grassian V. H., A molecular picture of surface interactions of organic compounds on prevalent indoor surfaces: Limonene adsorption on SiO2. Chem. Sci. 10, 2906–2914 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weschler C. J., Nazaroff W. W., Growth of organic films on indoor surfaces. Indoor Air 27, 1101–1112 (2017). [DOI] [PubMed] [Google Scholar]
- 11.Chang J. C. S., Tichenor B. A., Guo Z., Krebs K. A., Substrate effects on VOC emissions from a latex paint. Indoor Air 7, 241–247 (1997). [Google Scholar]
- 12.Shiraiwa M., Ammann M., Koop T., Pöschl U., Gas uptake and chemical aging of semisolid organic aerosol particles. Proc. Natl. Acad. Sci. U.S.A. 108, 11003–11008 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Duncan S. M., Tomaz S., Morrison G., Webb M., Atkin J., Surratt J. D., Turpin B. J., Dynamics of residential water-soluble organic gases: Insights into sources and sinks. Environ. Sci. Technol. 53, 1812–1821 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bi C., Liang Y., Xu Y., Fate and transport of phthalates in indoor environments and the influence of temperature: A case study in a test house. Environ. Sci. Technol. 49, 9674–9681 (2015). [DOI] [PubMed] [Google Scholar]
- 15.Meininghaus R., Gunnarsen L., Knudsen H. N., Diffusion and sorption of volatile organic compounds in building materials−Impact on indoor air quality. Environ. Sci. Technol. 34, 3101–3108 (2000). [Google Scholar]
- 16.Cox S. S., Zhao D., Little J. C., Measuring partition and diffusion coefficients for volatile organic compounds in vinyl flooring. Atmos. Environ. 35, 3823–3830 (2001). [Google Scholar]
- 17.Collins D. B., Hems R. F., Zhou S., Wang C., Grignon E., Alavy M., Siegel J. A., Abbatt J. P. D., Evidence for gas–surface equilibrium control of indoor nitrous acid. Environ. Sci. Technol. 52, 12419–12427 (2018). [DOI] [PubMed] [Google Scholar]
- 18.Wang C., Yuan T., Wood S. A., Goss K. U., Li J., Ying Q., Wania F., Uncertain Henry’s law constants compromise equilibrium partitioning calculations of atmospheric oxidation products. Atmos. Chem. Phys. 17, 7529–7540 (2017). [Google Scholar]
- 19.Roberts J. M., Veres P. R., Cochran A. K., Warneke C., Burling I. R., Yokelson R. J., Lerner B., Gilman J. B., Kuster W. C., Fall R., de Gouw J., Isocyanic acid in the atmosphere and its possible link to smoke-related health effects. Proc. Natl. Acad. Sci. U.S.A. 108, 8966–8971 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gómez Alvarez E., Amedro D., Afif C., Gligorovski S., Schoemaecker C., Fittschen C., Doussin J.-F., Wortham H., Unexpectedly high indoor hydroxyl radical concentrations associated with nitrous acid. Proc. Natl. Acad. Sci. U.S.A. 110, 13294–13299 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu S., Thompson S. L., Stark H., Ziemann P. J., Jimenez J. L., Gas-phase carboxylic acids in a university classroom: Abundance, variability, and sources. Environ. Sci. Technol. 51, 5454–5463 (2017). [DOI] [PubMed] [Google Scholar]
- 22.Rasmussen T. R., Brauer M., Kjaergaard S., Effects of nitrous acid exposure on human mucous membranes. Am. J. Respir. Crit. Care Med. 151, 1504–1511 (1995). [DOI] [PubMed] [Google Scholar]
- 23.Horn W., Ullrich D., Seifert B., VOC emissions from cork products for indoor use. Indoor Air 8, 39–46 (1998). [Google Scholar]
- 24.de Blas M., Navazo M., Alonso L., Durana N., Gomez M. C., Iza J., Simultaneous indoor and outdoor on-line hourly monitoring of atmospheric volatile organic compounds in an urban building. The role of inside and outside sources. Sci. Total Environ. 426, 327–335 (2012). [DOI] [PubMed] [Google Scholar]
- 25.Goodman N. B., Wheeler A. J., Paevere P. J., Selleck P. W., Cheng M., Steinemann A., Indoor volatile organic compounds at an Australian university. Build. Environ. 135, 344–351 (2018). [Google Scholar]
- 26.Tang X., Misztal P. K., Nazaroff W. W., Goldstein A. H., Siloxanes are the most abundant volatile organic compound emitted from engineering students in a classroom. Environ. Sci. Technol. Lett. 2, 303–307 (2015). [Google Scholar]
- 27.Atkins D. H. F., Lee D. S., Indoor concentrations of ammonia and the potential contribution of humans to atmospheric budgets. Atmos. Environ. Part A 27, 1–7 (1993). [Google Scholar]
- 28.Farmer D. K., Vance M. E., Abbatt J. P. D., Abeleira A., Alves M. R., Arata C., Boedicker E., Bourne S., Cardoso-Saldaña F., Corsi R., DeCarlo P. F., Goldstein A. H., Grassian V. H., Hildebrandt Ruiz L., Jimenez J. L., Kahan T. F., Katz E. F., Mattila J. M., Nazaroff W. W., Novoselac A., O'Brien R. E., Or V. W., Patel S., Sankhyan S., Stevens P. S., Tian Y., Wade M., Wang C., Zhou S., Zhou Y., Overview of HOMEChem: House observations of microbial and environmental chemistry. Environ. Sci.: Processes Impacts 21, 1280–1300 (2019). [DOI] [PubMed] [Google Scholar]
- 29.Ongwandee M., Morrison G. C., Influence of ammonia and carbon dioxide on the sorption of a basic organic pollutant to carpet and latex-painted gypsum board. Environ. Sci. Technol. 42, 5415–5420 (2008). [DOI] [PubMed] [Google Scholar]
- 30.Hems R. F., Wang C., Collins D. B., Zhou S., Borduas-Dedekind N., Siegel J. A., Abbatt J. P. D., Sources of isocyanic acid (HNCO) indoors: A focus on cigarette smoke. Environ. Sci.: Processes Impacts 21, 1334–1341 (2019). [DOI] [PubMed] [Google Scholar]
- 31.Grøntoft T., Raychaudhuri M. R., Compilation of tables of surface deposition velocities for O3, NO2 and SO2 to a range of indoor surfaces. Atmos. Environ. 38, 533–544 (2004). [Google Scholar]
- 32.Manuja A., Ritchie J., Buch K., Wu Y., Eichler C. M. A., Little J. C., Marr L. C., Total surface area in indoor environments. Environ. Sci.: Processes Impacts 21, 1384–1392 (2019). [DOI] [PubMed] [Google Scholar]
- 33.Wania F., Lei Y. D., Wang C., Abbatt J. P. D., Goss K.-U., Using the chemical equilibrium partitioning space to explore factors influencing the phase distribution of compounds involved in secondary organic aerosol formation. Atmos. Chem. Phys. 15, 3395–3412 (2015). [Google Scholar]
- 34.Borduas N., Place B., Wentworth G. R., Abbatt J. P. D., Murphy J. G., Solubility and reactivity of HNCO in water: Insights into HNCO’s fate in the atmosphere. Atmos. Chem. Phys. 16, 703–714 (2016). [Google Scholar]
- 35.DeCarlo P. F., Avery A. M., Waring M. S., Thirdhand smoke uptake to aerosol particles in the indoor environment. Sci. Adv. 4, eaap8368 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang C., Lei Y. D., Endo S., Wania F., Measuring and modeling the salting-out effect in ammonium sulfate solutions. Environ. Sci. Technol. 48, 13238–13245 (2014). [DOI] [PubMed] [Google Scholar]
- 37.Wang C., Lei Y. D., Wania F., Effect of sodium sulfate, ammonium chloride, ammonium nitrate, and salt mixtures on aqueous phase partitioning of organic compounds. Environ. Sci. Technol. 50, 12742–12749 (2016). [DOI] [PubMed] [Google Scholar]
- 38.Or V. W., Alves M. R., Wade M., Schwab S., Corsi R. L., Grassian V. H., Crystal clear? Microspectroscopic imaging and physicochemical characterization of indoor depositions on window glass. Environ. Sci. Technol. Lett. 5, 514–519 (2018). [Google Scholar]
- 39.Grant J. S., Zhu Z., Anderton C. R., Shaw S. K., Physical and chemical morphology of passively sampled environmental films. ACS Earth Space Chem. 3, 305–313 (2019). [Google Scholar]
- 40.Corsi R. L., Lin C.-C., Emissions of 2,2,4-trimethyl-1,3-pentanediol monoisobutyrate (TMPD-MIB) from latex paint: A critical review. Crit. Rev. Environ. Sci. Technol. 39, 1052–1080 (2009). [Google Scholar]
- 41.Wu Y., Eichler C. M. A., Leng W., Cox S. S., Marr L. C., Little J. C., Adsorption of phthalates on impervious indoor surfaces. Environ. Sci. Technol. 51, 2907–2913 (2017). [DOI] [PubMed] [Google Scholar]
- 42.Wensing M., Uhde E., Salthammer T., Plastics additives in the indoor environment—Flame retardants and plasticizers. Sci. Total Environ. 339, 19–40 (2005). [DOI] [PubMed] [Google Scholar]
- 43.Lunderberg D. M., Kristensen K., Liu Y., Misztal P. K., Tian Y., Arata C., Wernis R., Kreisberg N., Nazaroff W. W., Goldstein A. H., Characterizing airborne phthalate concentrations and dynamics in a normally occupied residence. Environ. Sci. Technol. 53, 7337–7346 (2019). [DOI] [PubMed] [Google Scholar]
- 44.Chenari B., Dias Carrilho J., Gameiro da Silva M., Towards sustainable, energy-efficient and healthy ventilation strategies in buildings: A review. Renew. Sustain. Energy Rev. 59, 1426–1447 (2016). [Google Scholar]
- 45.Yan D., O'Brien W., Hong T. Z., Feng X. H., Gunay H. B., Tahmasebi F., Mahdavi A., Occupant behavior modeling for building performance simulation: Current state and future challenges. Energ. Buildings 107, 264–278 (2015). [Google Scholar]
- 46.Ferro A. R., Klepeis N. E., Ott W. R., Nazaroff W. W., Hildemann L. M., Switzer P., Effect of interior door position on room-to-room differences in residential pollutant concentrations after short-term releases. Atmos. Environ. 43, 706–714 (2009). [Google Scholar]
- 47.Brophy P., Farmer D. K., Clustering, methodology, and mechanistic insights into acetate chemical ionization using high-resolution time-of-flight mass spectrometry. Atmos. Meas. Tech. 9, 3969–3986 (2016). [Google Scholar]
- 48.Brophy P., Farmer D. K., A switchable reagent ion high resolution time-of-flight chemical ionization mass spectrometer for real-time measurement of gas phase oxidized species: Characterization from the 2013 southern oxidant and aerosol study. Atmos. Meas. Tech. 8, 2945–2959 (2015). [Google Scholar]
- 49.Ampollini L., Katz E. F., Bourne S., Tian Y., Novoselac A., Goldstein A. H., Lucic G., Waring M. S., DeCarlo P., Observations and contributions of real-time indoor ammonia concentrations during HOMEChem. Environ. Sci. Technol. 53, 8591–8598 (2019). [DOI] [PubMed] [Google Scholar]
- 50.Liu Y., Misztal P. K., Xiong J., Tian Y., Arata C., Nazaroff W. W., Goldstein A. H., Detailed investigation of ventilation rates and airflow patterns in a northern California residence. Indoor Air 28, 572–584 (2018). [DOI] [PubMed] [Google Scholar]
- 51.Wang C., Goss K.-U., Lei Y. D., Abbatt J. P. D., Wania F., Calculating equilibrium phase distribution during the formation of secondary organic aerosol using COSMOtherm. Environ. Sci. Technol. 49, 8585–8594 (2015). [DOI] [PubMed] [Google Scholar]
- 52.Ridnour L. A., Sim J. E., Hayward M. A., Wink D. A., Martin S. M., Buettner G. R., Spitz D. R., A spectrophotometric method for the direct detection and quantitation of nitric oxide, nitrite, and nitrate in cell culture media. Anal. Biochem. 281, 223–229 (2000). [DOI] [PubMed] [Google Scholar]
- 53.Holzinger R., PTRwid: A new widget tool for processing PTR-TOF-MS data. Atmos. Meas. Tech. 8, 3903–3922 (2015). [Google Scholar]
- 54.Zhao J., Zhang R., Proton transfer reaction rate constants between hydronium ion (H3O+) and volatile organic compounds. Atmos. Environ. 38, 2177–2185 (2004). [Google Scholar]
- 55.Sander R., Compilation of Henry’s law constants (version 4.0) for water as solvent. Atmos. Chem. Phys. 15, 4399–4981 (2015). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/8/eaay8973/DC1
Text S1. Surface nitrite analysis during HOMEChem.
Text S2. Detrending data and response time calculation.
Text S3. Phase distribution calculation.
Text S4. CIMS calibration and data analysis.
Text S5. PTR-TOF-MS calibration and data analysis.
Fig. S1. Time-series of different species during EV experiments.
Fig. S2. Detrending data during the EV experiments.
Fig. S3. Indoor air signals during vinegar and ammonia cleaning.
Fig. S4. Comparison of mixing ratios before and during vinegar cleaning.
Fig. S5. Indoor acetic acid and ammonia mixing ratio during vinegar and ammonia cleaning.
Fig. S6. Partitioning space plot for an indoor environment.
Table S1. Physical and chemical properties of different compounds at 25°C.
Table S2. Time-averaged steady-state mixing ratios (parts per billion).
Table S3. Vinegar and ammonia cleaning experiment details.
Table S4. EV experiment details.