

Abstract
Background and Aims:
Alcohol-associated liver disease is a leading indication for liver transplantation and leading cause of mortality. Alterations of the gut microbiota contribute to pathogenesis of alcohol-associated liver disease. Patients with alcohol-associated liver disease have increased proportions of Candida spp. in the fecal mycobiome. However, little is known about the effect of intestinal Candida on alcohol-associated liver disease. Here we evaluated the contributions of Candida albicans and its exotoxin Candidalysin on alcoholic liver disease.
Methods:
C. albicans and ECE1 were analyzed in fecal samples from controls, patients with alcohol use disorder and alcoholic hepatitis. Mice colonized with different and genetically manipulated C. albicans strains were subjected to the chronic-plus-binge ethanol diet model. Primary hepatocytes were isolated and incubated with Candidalysin.
Results:
The percentages of subjects carrying ECE1 are 0%, 4.76% and 30.77% in non-alcoholic controls, alcohol use disorder patients and alcoholic hepatitis patients, respectively. Candidalysin exacerbates ethanol-induced liver disease and is associated with increased mortality in mice. Candidalysin enhances ethanol-induced liver disease independent of the β-glucan receptor CLEC7A on bone-marrow derived cells, and Candidalysin does not alter gut barrier function. Candidalysin can damage primary hepatocytes in a dose-dependent manner in vitro and is associated with liver disease severity and mortality in patients with alcoholic hepatitis.
Conclusions:
Candidalysin is associated with progression of ethanol-induced liver disease in preclinical models and worse clinical outcomes in patients with alcoholic hepatitis.
Keywords: mycobiome, microbiota, microbiome
Graphical Abstract
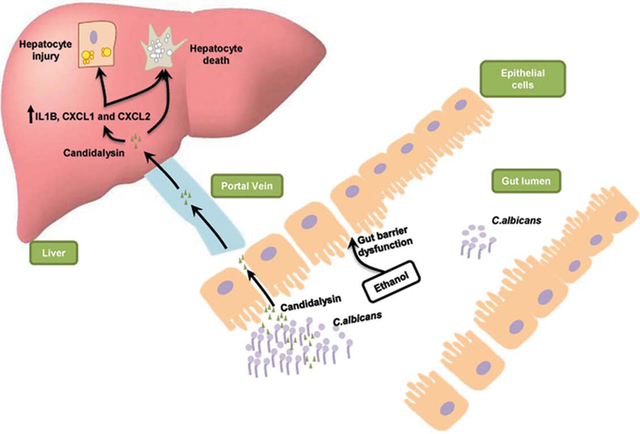
Lay summary
Candidalysin is a peptide toxin secreted by the commensal gut fungus Candida albicans. Candidalysin enhances alcohol-associated liver disease independent of the β-glucan receptor CLEC7A on bone-marrow derived cells in mice without affecting intestinal permeability. Candidalysin is cytotoxic to primary hepatocytes, indicating a direct role of Candidalysin on ethanol-induced liver disease. Candidalysin might be an effective target for therapy in patients with alcohol-associated liver disease.
Introduction
Alcohol-associated liver disease is one of the most prevalent liver diseases worldwide1, and the leading cause of liver transplantation in the U.S.2. Alcohol-related liver disease is associated with changes in the intestinal microbiota. Gut dysbiosis induces intestinal inflammation and gut barrier dysfunction, which allows viable bacteria, bacterial (such as lipopolysaccharides (LPS)) and fungal products (such as β-glucan) to translocate to the liver. Bacteria and microbial products bind to pathogen recognition receptors causing an inflammatory response of resident Kupffer cells and an infiltration of macrophages. Although many efforts were made to evaluate the role of the bacterial microbiota in alcohol-associated liver disease, the interaction between fungi and their host, and especially their contribution to alcohol-associated liver disease remains poorly understood. We have shown that β-glucan, a cell wall component of many commensal fungi, binds to C-type lectin domain family 7 member A (CLEC7A; also known as DECTIN1) on hepatic macrophages to release interleukin 1 beta (IL-1β) and increase ethanol-induced liver disease in mice3.
The yeast Candida albicans (C. albicans) normally resides as a harmless commensal in the human intestinal tract, but can translocate from the gut into the bloodstream in predisposed individuals such as immunocompromised patients4. Its virulence partly depends on the transition from yeast to hyphal growth forms5. Recent studies revealed that Ece1, the product of the gene ECE1 (extent of cell elongation 1), is critical for mucosal pathogenesis of C. albicans6. Ece1 is a polyprotein consisting of eight distinct peptides processed by the sequential activities of the serine proteases Kex2 and Kex16 (Supplementary Fig. 1). Among them, Ece1-III 62–92K, also named Candidalysin, is a secreted cytolytic peptide (exotoxin) that is important for epithelial cell damage in the host6. Candidalysin can damage epithelial mucosa directly, but also induces a danger response signaling pathway via c-Fos and mitogen-activated protein kinase (MAPK) signaling and triggers inflammatory cytokine responses (including IL-1α, IL-1β, and IL-8)6–8. We have previously demonstrated that patients with alcohol use disorder had increased fecal proportions of Candida spp, including C. albicans, compared to non-alcoholic controls. Whether intestinal overgrowth of C. albicans contributes to alcohol-associated liver disease beyond increasing systemic β-glucan level is not known. In this study, we evaluate the effect of Candidalysin on promoting alcohol-associated liver disease.
Methods
Patients.
We evaluated 11 subjects without alcohol use disorder (controls; social drinkers consuming less than 20g/day), 42 patients with alcohol use disorder, and 91 patients with alcoholic hepatitis. Patients with alcohol use disorder fulfilling the DSM IV criteria9 of alcohol dependence and with active alcohol consumption (self-reported > 60g/day) presented with various stages of liver disease (15% had advanced F3/4 fibrosis based on fibrosis-4 index (FIB-4)10; Supplementary Table 1). Patients with alcohol use disorder were recruited from an alcohol withdrawal unit where they followed a detoxification and rehabilitation program. Controls or patients with alcohol use disorder did not take antibiotics or immunosuppressive medication during the two months preceding enrollment. Other exclusion criteria were diabetes, inflammatory bowel disease, known liver disease of any other etiology, and clinically significant cardio-vascular, pulmonary or renal co-morbidities. Alcoholic hepatitis patients were enrolled from the InTeam Consortium (ClinicalTrials.gov identifier number: ) from 12 participating centers in the US, Mexico, Canada, United Kingdom, France and Spain. Inclusion criteria were: 1. Active alcohol abuse (> 50 g/day for men and > 40 g/day for women) in the last 3 months, 2. Aspartate aminotransferase (AST) > alanine aminotransferase (ALT), and total bilirubin > 3 mg/dl in the past 3 months, 3. Liver biopsy and/or clinical picture consistent with alcoholic hepatitis. Exclusion criteria were: 1. Autoimmune liver disease (ANA > 1/320), 2. Chronic viral hepatitis, 3. Hepatocellular carcinoma, 4. Complete portal vein thrombosis, 5. Extrahepatic terminal disease, 6. Pregnancy, and 7. Lack of signed informed consent11. In all patients, the clinical picture was consistent with alcoholic hepatitis and in patients who underwent liver biopsy, the histology was in line with the diagnosis of alcoholic hepatitis. Liver biopsies were only done if clinically indicated as part of routine clinical care for diagnostic purposes of alcoholic hepatitis. For 3 patients who underwent liver transplantation, the transplantation date was considered as date of death. Patients were censored at the time point they were last seen alive. Baseline characteristics are shown in Supplementary Table 1 and 2. Differences in BMI are likely explained by the presence of ascites in 68% (n=61) of alcoholic hepatitis patients. Fresh feces from healthy controls or patients were collected, immediately frozen and stored in −80°C freezer until experiments were performed. Fungal culture, quantitative PCR (qPCR) for total fungi, C. albicans and ECE1 was performed from fecal samples. The model for end-stage liver disease (MELD) score was calculated from all alcoholic hepatitis patients from whom bilirubin level, international normalized ratio (INR), and creatinine level was available. The protocol was designed according to the ethical guidelines of the 1975 Declaration of Helsinki as reflected in a priori approval by the Ethics Committee of each participating center and patients were enrolled after written informed consent was obtained from each patient.
Mice.
C57BL/6 male mice were purchased from Charles River and used in all described experiments. Clec7a−/− mice on a C57BL/6 background have been described3. Heterozygous mice were used for breeding to generate wild type and Clec7a deficient littermates.
Male mice (8–12weeks) were subjected to either chronic-plus-binge ethanol diet model (NIAAA model)12 or a chronic Lieber-DeCarli diet model for 8 weeks as previously described3. For the chronic-plus-binge ethanol diet model, mice were fed the Lieber-DeCarli diet for 15 days. The caloric intake from ethanol was 0% on days 1–5 and 36% from day 6 until the end. At day 16, mice were gavaged with one dose of ethanol (5g/kg bodyweight) in the morning and sacrificed 9 hours later. Pair-fed control mice received a diet with an isocaloric substitution of dextrose. For the chronic Lieber-DeCarli feeding model, the caloric intake from ethanol was 0 on day 1, 10% of total calories on days 2 and 3, 20% on days 4 and 5, 30% from day 6 until the end of 6 weeks, and 36% for the last 2 weeks. Control mice received an isocaloric amount of iso-maltose instead of ethanol. Antibiotics treatment was started at day 1 of ethanol feeding, and mice were gavaged daily until harvesting. The composition of antibiotics mixture has been described (Polymyxin B (150mg/kg BW) and Neomycin (200mg/kg BW))13.
To evaluate the effect of Candidalysin on liver disease, C57BL/6 male mice (Control diet: n=5–8/group; Ethanol diet: n=12–14/group) were gavaged with 108 colony forming units (CFUs) of four different C. albicans strains (wild type, M1477; ece1Δ/Δ, M2057; ece1Δ/Δ + ECE1Δ184–279, M2174; ece1Δ/Δ + ECE1, M2059)6 or phosphate-buffered saline (PBS) as vehicle control every third day starting from day 6 to day 15 of the chronic-plus-binge ethanol diet model. As described6, C.albicans was deleted for both copies of ECE1 (ece1Δ/Δ) and restored with one full-length allele (ece1Δ/Δ+ECE1) or one mutant allele lacking the Candidalysin-encoding region of ECE1 (ece1Δ/Δ+ECE1Δ184–279).
Bone marrow chimeric mice were generated as previously described3. In brief, C57BL/6 recipient male mice were given lethal doses of radiation (650 rad) twice, using a 137Cs source. Clec7a−/− male mice were used as bone marrow donors. Two weeks after bone marrow transplantation, mice were given intraperitoneal injections of 200 μl clodronate liposomes (5 mg/ml; Vrije Universiteit, Amsterdam, Netherlands) to deplete radio-resistant Kupffer cells. Chimeric mice (male, n=10–11/group) were subjected to the chronic-plus-binge ethanol diet model 4 weeks after bone marrow transplantation.
To determine the effect of Candidalysin on intestinal permeability, C57BL/6 male mice (Control diet: n=5–6/group; Ethanol diet: n=5–6/group) were gavaged with PBS as vehicle control, 108 CFUs of wild type C. albicans or ece1Δ/Δ C. albicans every third day starting from day 6 to day 15 using the chronic-plus-binge ethanol diet model. At day 16, fluorescein isothiocyanate (FITC)-dextran (4 kDa, Sigma) (200μl, 100mg/ml) was gavaged to all the mice 1 hour after binge and blood was harvested after 4 hours.
All mice were housed under specific pathogen-free (SPF) conditions in a standard environment with a 12-hour light–dark cycle at the animal facilities of University of California, San Diego under protocol S09042. Animal studies were reviewed and approved by the Institutional Animal Care and Use Committee of the University of California, San Diego.
Quantitative PCR (qPCR) and reverse transcription quantitative PCR (RT-qPCR).
For qPCR, fungal genomic DNA was extracted from human stool samples as described3. Published primer sequences for fungal 18S rRNA gene14, C. albicans internal transcribed spacer (ITS) gene15, ECE1 gene6 and SEL1 gene16 were used (Supplementary Table 3). For RT-qPCR to determine AML12 cellular and hepatic gene expression, AML12 cellular and mouse liver RNA was extracted using Trizol (Invitrogen) and cDNA was then synthesized using High Capacity cDNA Reverse Transcription kit (ABI). Primer sequences for mouse genes were originally obtained from NIH qPrimerDepot (Supplementary Table 3). qPCR and RT-qPCR were performed with iTaq Universal SYBR Green Supermix (Bio-Rad) using a StepOnePlus thermocycler (ABI) real-time PCR system. The RT-qPCR values of cellular and mouse genes were normalized to 18S. Cellular gene expression data was expressed relative to the level of blank control-treated cells. Mouse gene expression data was expressed relative to the level of control-fed PBS-treated mice.
Fecal fungi.
To determine the number of fungal CFUs in human stool samples, 50 to 300 mg of feces was resuspended into 500 μl PBS and serial dilutions were then made, followed by plating 100 μl onto a YPD agar plate with antibiotics (1% yeast extract, 2% peptone, 2% D-glucose, 1.5% agar, 100 μg/ml gentamicin and 100 μg/ml chloramphenicol). Plates were incubated at 30°C for 48 hours. Colony numbers of each sample were then counted and CFUs were normalized to weight of feces. Each colony was assessed for the presence of fungus or C. albicans by qPCR using specific primers against fungal 18S rRNA gene or C. albicans ITS gene, respectively. Each colony was also assessed by qPCR to determine ECE1 positivity using specific primers.
To determine the number of fungal CFUs in mouse feces, 10 to 30 mg of feces were resuspended into 500 μl PBS and serial dilutions were made, followed by plating 100 μl onto YPD agar plate with antibiotics. Plates were incubated at 30°C for 48 hours. Colony numbers of each sample were then counted and CFUs were normalized to weight of feces. Fungi, C. albicans,ECE1 and SEL1 presence were assessed by qPCR.
C. albicans culture.
C. albicans strains were grown in YPD broth with antibiotics (1% yeast extract, 2% peptone, 2% D-glucose, 100 μg/ml gentamicin and 100 μg/ml chloramphenicol) and incubated at 30°C with shaking at 150 rpm for 20 hours. Cell density was analyzed by measuring optical density at 600 nm in a microplate reader (Molecular Devices). Cells from YPD broth were collected by centrifugation, washed twice with PBS and adjusted to the required cell density.
Biochemical assays.
Serum levels of ALT were measured using the Infinity ALT kit (Thermo Scientific). Hepatic triglyceride levels were measured using the Triglyceride Liquid Reagents Kit (Pointe Scientific). Serum levels of ethanol were measured using the Ethanol Assay Kit (BioVision). Serum levels of intestinal fatty-acid binding protein (IFABP) were measured using the iFABP ELISA kit (MyBioSource). Serum levels of zonulin were measured using the Human Zonulin ELISA Kit (MyBioSource)17.
Staining procedures. To determine the accumulation of hepatic lipids, liver sections were embedded in OCT compound.
8 μm frozen sections were then cut and stained with Oil Red O (Sigma-Aldrich). Representative pictures from each group of mice are shown in each figure.
FITC-dextran permeability assay.
Intestinal permeability was assessed by oral gavage of FITC-dextran (4 kDa; Sigma), a non-metabolizable macromolecule that is used as a permeability probe18, 19. Serum concentration of FITC-dextran was determined using a fluorimeter with an excitation wavelength at 490 nm and an emission wavelength at 530 nm, followed by calculations using serial dilutions of FITC-dextran as standards
Mouse cell culture studies.
Primary hepatocytes were isolated from C57BL/6 male mice (n=4–7/group) as described20. Cells were seeded on plates coated with rat collagen type I and cultured for 4 hours in DMEM-F12 (Thermo Fisher Scientific) with 1% (v/v) insulin-transferrin-selenium (Thermo Fisher Scientific), 40 ng/ml dexamethasone (MP Biomedicals), 10% (v/v) fetal bovine serum (FBS; Gemini Bio-Products), and an antibiotic-antimycotic mix (CoreBio). Following an overnight starvation in medium without FBS, hepatocytes were stimulated with different concentrations (0.6 μM and 3 μM) of Candidalysin for 24 hours6. Ece1-VII was used as negative control (0.6 μM and 3 μM). Culture medium was used as blank control. AML12 (immortalized mouse hepatocyte) cells were seeded and cultured in the same medium as primary hepatocytes (as described above) 24 hours prior to transfection. AML12 cells were transfected with siRNA for mouse toll-like receptor 2 (TLR2) and TLR4 (25 nM; set of 4 siRNAs, Dharmacon) or control siRNA (siGLO Green Transfection Indicator, Dharmacon), using Lipofectamine 2000 (Invitrogen, USA) according to the manufacturer’s instructions. 48 hours after transfection, cells were serum starved for 2 hours and treated with Candidalysin (15 μM), negative control (15 μM) or blank control for 24 hours.
Hepatocyte cytotoxicity was measured using the Pierce lactate dehydrogenase (LDH) Cytotoxicity Detection Kit (Thermo Fisher Scientific). Hepatocyte viability was determined by adding 3-(4,5-dimethylthiazol-2-yl)-2,5′-diphenyltetrazolium bromide solution (MTT; Sigma-Aldrich, final concentration 0.3 mg/ml) into cell culture plates and incubated in dark for 2 hours. The medium was then removed and dimethyl sulfoxide (Sigma-Aldrich) was added to dissolve the formazan. Formazan concentration was determined by absorbance at 550 nm and the survival percentage was calculated accordingly (the mean survival rate of hepatocytes incubated with culture medium alone was set as 100%).
Statistical analysis.
All data were expressed as mean ± SE unless otherwise specified. In mouse and cell culture experiments, for comparisons of > 2 groups within one experimental setting, one-way analysis of variance (ANOVA) was used followed by the Tukey’s post-hoc test. For comparisons of >2 groups within two experimental settings, a two-way ANOVA followed by the Tukey’s post-hoc test was used. For the human cohort, comparisons of baseline characteristics between the three groups were conducted using one-Way ANOVA with the Tukey’s post-hoc test for multiple comparisons for continuous variables and Chi-squared tests for categorical variables. A Kruskal-Wallis test with the Dunn’s post-hoc test was used to compare the mean number of fungal colonies in feces among different groups. A two-proportion z-test followed by FDR procedures was used to evaluate the percentage of individuals carrying fungi or ECE1 among different groups. Multivariate Cox Proportional Hazard regression analysis was performed to detect associations of Candidalysin positivity with 90-day mortality in alcoholic hepatitis patients. Patients that were lost to follow-up were censored at the day they were last seen alive. Univariate logistic regression analysis was performed to determine associations of Candidalysin and clinical parameters.
Statistical analyses were performed using the statistical software R (V.3.5.1, 2018 the R Foundation for Statistical Computing) and GraphPad Prism (V.6.01). A P value <0.05 was considered significant.
Results
Patients with alcoholic hepatitis have increased fecal numbers of Candidalysin positive C. albicans
C. albicans is a commensal fungus of the human gut flora. Using internal transcribed spacer (ITS) sequencing we showed that the relative proportions of C. albicans increased in patients with alcohol use disorder3. To determine quantitative differences of viable fungi in the fecal mycobiome, we performed fungal cultures of stool samples from non-alcoholic controls (controls, n=11), patients with alcohol use disorder (AUD, n=42) and patients with alcoholic hepatitis (AH, n=91). Each colony was further assessed for the presence of fungi or C. albicans by quantitative PCR (qPCR) using specific primers against fungi or C. albicans. Fecal samples from patients with alcoholic hepatitis grew significantly more colonies of fungi or C. albicans as compared with alcohol use disorder patients and controls (Fig. 1A and B). Furthermore, a significantly higher proportion of alcoholic hepatitis patients was culture positive for fungi or C. albicans than patients with alcohol use disorder (Fig. 1C and D).
Fig. 1. Candidalysin is related with the development of alcoholic liver disease.

Fecal samples from controls (n=11), patients with alcohol use disorder (AUD) (n=42) and alcoholic hepatitis (n=91) were cultured on YPD agar plates with antibiotics. Each colony was then assessed by qPCR to confirm as fungus, and to determine positivity for C. albicans or ECE1. (A) Colony forming units (CFUs) of total fungi in fecal samples. (B) Colony forming units (CFUs) of C. albicans in fecal samples. (C) Percentage of subjects with fecal samples positive for fungi. (D) Percentage of subjects with fecal samples positive for C. albicans. (E) Percentage of subjects with fecal samples positive for ECE1. Results are expressed as mean ± s.e (A and B). P values were determined by Kruskal-Wallis test with Dunn’s post-hoc test (A and B), and Z test followed by false discovery rate (FDR) procedures (C - E). *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001.
We next sought to determine if ECE1, the gene encoding the C. albicans exotoxin Candidalysin, was also increased in alcoholic hepatitis patients. 30.77% of alcoholic hepatitis patients were tested positive for ECE1, while in patients with alcohol use disorder, this proportion was significantly lower (Fig. 1E). Taken together, ECE1, the gene encoding Candidalysin, was more abundant in patients with alcoholic hepatitis.
Candidalysin exacerbates ethanol-induced liver disease
We confirmed that chronic ethanol administration is associated with an increase of intestinal fungi in mice3 (Supplementary Fig. 2). However, we could not detect C. albicans or ECE1 in mice in the chronic-plus-binge ethanol diet model12 or the chronic Lieber-DeCarli model for 8 weeks3, consistent with a published report21. This indicates that C. albicans does not normally colonize the gastrointestinal mucosa of mice, or that the level of C. albicans cells or ECE1 genes was below the detection limit of fungal cultures or nucleic acid amplification.
To study the role of Candidalysin positive C. albicans for alcohol-associated liver disease, we colonized mice by repeated oral gavage of C. albicans (108 CFUs every third day) during ethanol feeding. The average amount of C. albicans in mouse feces is 1.5 × 105 CFU/g feces (24 hours following gavage) (Supplementary Fig. 3), which is similar to the C. albicans amount in feces from alcoholic hepatitis patients (4.7 × 105 CFU/g feces) (Fig. 1B). Compared to vehicle (PBS) treated mice, mice colonized with wild type C. albicans displayed more severe ethanol-associated liver injury, indicated by elevated level of serum alanine amino-transferase (ALT) (Fig. 2A) and increased hepatic steatosis (Fig. 2B and 2C). Those animals also showed more liver inflammation, with higher expression of genes Il1b, chemokine (C-X-C motif) ligand 1 (Cxcl1) and Cxcl2, which encode inflammatory cytokines and chemokines (Fig. 2D – 2F). To determine if this disease exacerbation effect depends on ECE1, we colonized mice with C. albicans strain carrying an ECE1 gene deletion (ece1Δ/Δ). These mice had less severe ethanol-associated liver injury, steatosis and inflammation (Fig. 2A – 2F). Re-introducing ECE1 back into the ece1Δ/Δ strain (ece1Δ/Δ + ECE1) and colonizing mice with this strain, resulted in comparable levels of liver injury, steatosis and inflammation compared to mice colonized with wild type C. albicans following ethanol administration (Fig. 2A – 2F). These results indicate that the gene product of ECE1 is important for C. albicans to enhance ethanol-induced liver disease.
Fig. 2. Effects of Candidalysin on ethanol-induced liver disease.

C57BL/6 mice were fed an oral isocaloric (control) diet (1–2 technical replicates) or chronic-plus-binge ethanol diet (3–4 technical replicates), and gavaged with vehicle (PBS), wild type C. albicans (Wild type), ECE1 deleted C. albicans (ece1Δ/Δ), only Ece1-III62–93 deleted C. albicans (ece1Δ/Δ + ECE1Δ184–279) or ECE1 re-integrant C. albicans (ece1Δ/Δ + ECE1) with an amount of 108 colony forming units (CFUs) every third day. (A) Serum levels of alanine aminotransferase (ALT). (B) Hepatic triglycerides levels. (C) Representative images of Oil Red O stained liver tissue. (D) Hepatic levels of Il1b mRNA. (E) Hepatic levels of Cxcl1 mRNA. (F) Hepatic levels of Cxcl2 mRNA. (G) Serum levels of ethanol in mice fed chronic-plus-binge ethanol diet. (H) Hepatic levels of Adh1 mRNA. (I) Hepatic levels of Cyp2e1 mRNA. (Control diet: PBS, n=8; Wild type, n=5; ece1Δ/Δ, n=5; ece1Δ/Δ + ECE1Δ184–279, n=5; ece1Δ/Δ + ECE1, n=5; Ethanol diet: PBS, n=14; Wild type, n=14; ece1Δ/Δ, n=14; ece1Δ/Δ + ECE1Δ184–279, n=14; ece1Δ/Δ + ECE1, n=12). Scale bars = 100 μm. Images were taken at ×100 magnification. Results are expressed as mean ± s.e (A, B, D - I). P values were determined by one-way ANOVA with Tukey’s post-hoc test (A, B, D - I). *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001.
ECE1 encodes for a polyprotein composed of eight different peptides6 (Supplementary Fig. 1). Candidalysin is among them and is a secreted cytolytic peptide exotoxin6. To test whether Candidalysin is responsible for increased ethanol-induced disease mediated by ECE1, we colonized mice with a C. albicans strain carrying a deletion of the Ece1-III62–93 Candidalysin-encoding region (ece1Δ/Δ + ECE1Δ184–279), thus not producing Candidalysin. These mice showed similar ethanol-induced liver disease as compared with mice colonized with the ece1Δ/Δ C. albicans strain (Fig. 2A – 2F), indicating that Candidalysin is necessary for exacerbation of ethanol-induced liver disease by C. albicans.
In addition, Candidalysin is associated with increased mortality. Mice colonized with Candidalysin positive strains had a significant higher mortality rate as compared with mice colonized with Candidalysin negative strains (Supplementary Figure 4). Importantly, all C. albicans strains did not affect intestinal absorption or hepatic metabolism of ethanol, as serum levels of ethanol as well as expression of hepatic alcohol dehydrogenase 1 (Adh1) and cytochrome P450 family 2 subfamily E polypeptide 1 (Cyp2e1) (two genes encoding main hepatic enzymes that metabolize ethanol and convert it to acetaldehyde) did not differ significantly between mouse groups (Fig. 2G – 2I).
Bacteria and fungi actively interact with each other in the gut22. To test whether the effects of C. albicans on liver disease are affected by its interaction with bacteria, mice were treated with antibiotics to reduce the amount of intestinal bacteria, and gavaged wild type C. albicans or vehicle (PBS). Compared with antibiotics plus vehicle treated mice, mice receiving antibiotics plus wild type C. albicans showed more severe ethanol-induced liver injury (Supplementary Fig. 5A) and steatosis (Supplementary Fig. 5B and 5C), indicating that bacteria-fungi interaction is not the major contributor to liver damage caused by C. albicans.
Taken together, these results indicate that C. albicans producing Candidalysin can enhance ethanol-associated liver disease and increase mortality in mice.
Candidalysin exacerbates ethanol-induced steatohepatitis independent of the receptor CLEC7A on bone marrow derived cells
β-glucan is the most abundant cell wall polysaccharide in most fungal species including C. albicans23. CLEC7A is a pattern recognition receptor that recognizes a variety of β-glucans and thus activates host immune response24. CLEC7A is primarily expressed on myeloid cells25, 26 and only expressed on Kupffer cells and macrophages in the liver3. We have previously shown that loss of CLEC7A from Kupffer cells decreases ethanol-induced steatohepatitis3. To investigate whether the disease promoting effect of C. albicans is mediated by CLEC7A signaling, we generated CLEC7A bone marrow chimeric mice using a combination of clodronate-mediated Kupffer cell depletion, irradiation, and bone marrow transplantation3. Wild type mice were transplanted with bone marrow cells from CLEC7A-deficient mice, which results in full reconstitution of Kupffer cells3, 27. Chimeric mice were colonized with wild type C. albicans, ece1Δ/Δ C. albicans (ece1Δ/Δ) or vehicle (PBS) and subjected to the chronic-plus-binge ethanol diet model. Chimeric mice with bone-marrow derived cells that lacked CLEC7A and were colonized with wild type C. albicans showed more severe ethanol-induced liver injury (Fig. 3A), steatosis (Fig. 3B and 3C) and inflammation (Fig. 3D – 3F) as compared with PBS treated mice and mice colonized with C. albicans strain carrying an ECE1 gene deletion. No significant difference in serum ethanol level and hepatic expression of Adh1 and Cyp2e1 was observed among all groups (Fig. 3G – 3I). These results indicate that C. albicans and Candidalysin induce liver disease independent of CLEC7A signaling on Kupffer cells and other bone-marrow derived cells following ethanol administration.
Fig. 3. Effects of Candidalysin on ethanol-induced liver disease in mice lacking CLEC7A in bone marrow derived cells.

C57BL/6 mice underwent transplantation of Clec7a−/− bone marrow (Clec7aΔBM) and were fed an oral chronic-plus-binge ethanol diet (3 technical replicates). Mice were gavaged with vehicle (PBS), wild type C. albicans (Wild type) or ECE1 deleted C. albicans (ece1Δ/Δ) with an amount of 108 CFUs every third day. (A) Serum levels of ALT. (B) Hepatic triglycerides levels. (C) Representative images of Oil Red O stained liver tissue. (D) Hepatic levels of Il1b mRNA. (E) Hepatic levels of Cxcl1 mRNA. (F) Hepatic levels of Cxcl2 mRNA. (G) Serum levels of ethanol. (H) Hepatic levels of Adh1 mRNA. (I) Hepatic levels of Cyp2e1 mRNA. (PBS, n=10; Wild type, n=11; ece1Δ/Δ, n=11). Scale bars = 100 μm. Images were taken at ×100 magnification. Results are expressed as mean ± s.e. (A, B, D - I). P values were determined by one-way ANOVA with Tukey’s post-hoc test (A, B, D - I). *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001.
Candidalysin does not affect intestinal permeability
In a cell culture system, Candidalysin causes necrosis of intestinal epithelial cells, which is a key factor in subsequent fungal translocation of C. albicans through intestinal layers28. To determine if Candidalysin induces gut barrier dysfunction in vivo, we determined intestinal permeability by measuring recovery of indigested dextran. In line with our previous report29, intestinal permeability increased in mice fed with ethanol diet (Fig. 4A). However, no significant difference was observed among mice colonized with wild type C. albicans, ece1Δ/Δ C. albicans (ece1Δ/Δ) and vehicle (PBS) following ethanol administration (Fig. 4A). Intestinal fatty-acid binding protein (IFABP) is specifically expressed in intestinal epithelial cells and released to the systemic circulation upon enterocyte damage30. Intestinal epithelial damage was not different between groups as measured by ELISA for serum IFABP (Fig. 4B). These results indicate that Candidalysin does not cause measurable intestinal barrier disruption or enterocyte death in ethanol-fed mice.
Fig. 4. Candidalysin does not affect intestinal permeability in mice fed ethanol.

C57BL/6 mice were fed an oral isocaloric (control) diet (1–2 technical replicates) or chronic-plus-binge ethanol diet (3–4 technical replicates) and gavaged with vehicle (PBS), wild type C. albicans (Wild type) or ECE1 deleted C. albicans (ece1Δ/Δ) with an amount of 108 CFUs every third day. (A) Serum levels of FD4. Mice were gavage-fed FITC labeled dextran (200μl, 100mg/ml) 1 hour after binge on the last day and then sacrificed 4 hours later, and fluorescence was measured in the serum. (Control diet: PBS, n=6; Wild type, n=6; ece1Δ/Δ, n=5; Ethanol diet: PBS, n=5; Wild type, n=6; ece1Δ/Δ, n=6). (B) Serum levels of intestinal fatty-acid binding protein (IFABP) (Control diet: PBS, n=7; Wild type, n=5; ece1Δ/Δ, n=5; Ethanol diet: PBS, n=14; Wild type, n=13; ece1Δ/Δ, n=10). Results are expressed as mean ± s.e (A and B). P values were determined by two-way ANOVA with Tukey’s post-hoc test (A and B). ****P<0.0001.
Candidalysin is cytotoxic to primary hepatocytes
To further define a mechanism by which Candidalysin enhances ethanol-induced liver disease, primary hepatocytes were isolated from mice fed an isocaloric control diet or subjected to the chronic-plus-binge ethanol feeding model. Cultured hepatocytes were incubated with pure bioactive Candidalysin. Candidalysin dose-dependently caused death of cultured hepatocytes as compared with hepatocytes incubated with control peptide (Fig. 5). Hepatocytes from ethanol-fed mice showed similar cytotoxicity as compared to hepatocytes isolated from control diet fed mice. However, incubation with ethanol increased the Candidalysin-induced cytotoxicity of hepatocytes (Figure 5A and 5B). These results indicate that Candidalysin can damage hepatocyte directly and additional ethanol stimulation in vitro can increase the cytotoxic effect.
Fig. 5. Candidalysin induces death of cultured primary hepatocytes.

Primary hepatocytes were isolated from mice fed an oral isocaloric (control) diet (A and C) or from mice subjected to the chronic-plus-binge ethanol diet model (B and D). Hepatocytes were incubated with different concentrations (0.6 μM and 3 μM) of Candidalysin or negative control peptide (Ece1-VII), as well as blank control (equal volume of culture medium), without (−) or with (+) ethanol (25 mM) for 24 hours (3 independent experiments performed in 4–7 replicates). (A and B) Cytotoxicity was determined by measuring LDH release in the supernatant. (C and D) Hepatocyte survival was measured using the MTT assay. Results are expressed as mean ± s.e. P values were determined by two-way ANOVA with Tukey’s post-hoc test. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001.
Sel1, a small secreted cysteine-rich protein of C. albicans, is critical in mediating the effects of C. albicans on innate immune cells via activation of NF kappa B and mitogen-activated protein kinases (MAPK) signaling16. Since Candidalysin can also activate MAPK/c-Fos/MAP kinase phosphatase 1 (MKP1) signaling pathway in epithelial cells8, we tested whether the effects of Candidalysin on liver injury and hepatocyte damage are modulated by Sel1. We first tested the presence of SEL1 in the four strains of C. albicans used in this study. All strains (wild type, ece1Δ/Δ, ece1Δ/Δ + ECE1Δ184–279, and ece1Δ/Δ + ECE1) carried SEL1 (Supplementary Fig. 6A). We further assessed the presence of SEL1 in mouse feces after C. albicans gavage; SEL1 is detected in both groups of mice (treated with either wild type or ece1Δ/Δ + ECE1Δ184–279 C. albicans) (Supplementary Fig. 6B). A recent study reported that SEL1 can trigger inflammatory response through TLR2/416. To determine whether Candidalysin causes hepatocyte damage via Sel1-TLR2/4 pathway, we performed TLR2/4 knockdown using siRNA in a mouse hepatocyte cell line (AML12 cells) (Supplementary Fig. 7A), and incubated these cells with pure bioactive Candidalysin. Candidalysin caused a cytotoxic effect on AML12 cells independent of TLR2/4 (Supplementary Fig. 7B and 7C). Our in vivo and in vitro studies indicate that Candidalysin exerts its effects on hepatocytes and liver independent of Sel1.
Candidalysin is related with the severity and mortality of alcoholic hepatitis
Alcohol-associated liver disease can progress to alcoholic hepatitis, a severe form of liver disease with considerable morbidity and mortality. We therefore analyzed clinical parameters and outcomes in patients with alcoholic hepatitis, who were positive for the Candidalysin coding gene ECE1 in feces. Patients with fecal samples containing genetic material for Candidalysin had higher serum levels of zonulin (a surrogate marker for gut barrier dysfunction) compared with ECE1 negative patients (Supplementary Fig. 8). Compared to ECE1 negative patients, fecal patient samples containing genetic material for Candidalysin had a higher model for end-stage liver disease (MELD) score (Fig. 6A). In the univariate logistic regression analysis, the MELD score was significantly associated with detection of ECE1 (Table 1). The detection of ECE1 was further associated with an increased 90 day mortality in alcoholic hepatitis patients with an adjusted hazard ratio of 3.01 (1.02–8.87, P=0.046) (Table 2). Importantly, 28.42% of ECE1 – positive patients with alcoholic hepatitis died within 90 days after admission compared with 11.71% of ECE1 – negative patients (Log-rank P=0.044) (Fig. 6B).
Fig. 6. Presence of Candidalysin associates with disease severity and mortality in patients with alcoholic hepatitis.

(A) MELD score for patients with alcoholic hepatitis. (Candidalysin positive, n=27; Candidalysin negative, n=62). (B) Kaplan-Meier curve of 90-day mortality for patients with alcoholic hepatitis. (Candidalysin positive, n=25; Candidalysin negative, n=61). Patients were grouped according to their ECE1 status in stool. Patients lost to follow-up were censored at the time they were last seen alive. Results are expressed as median with range. P value was determined by Mann-Whitney-Wilcoxon test (A) or Log-rank test (B).
Table 1:
Logistic regression analysis of clinical parameters associated with Candidalysin
| Candidalysin | Negative | Positive | OR, (95% CI, P value) |
|---|---|---|---|
| MELD | 24 (5.5) | 27.8 (7.7) | 1.09 (1.02–1.18, P=0.017) |
| Antibiotic use | 14 (23.0) | 10 (35.7) | 1.87 (0.69–4.96, P=0.211) |
| Steroids | 23 (37.7) | 10 (35.7) | 0.92 (0.35–2.30, P=0.857) |
Data in second and third column presented as mean (SD) or n (%). Bold font indicates significance (P value <0.05). All variables shown in Supplementary Table S1 and S2 were included in the logistic regression analysis. Shown are only the most relevant.
OR, odds ratio; CI, confidence interval; MELD, Model for End-stage Liver Disease.
Table 2:
Cox regression analysis of Candidalysin and MELD associated with mortality
| Dependent: 90 Day Mortality | HR (multivariable), (95% CI, P value*) |
|---|---|
| Candidalysin positive | 3.01 (1.02–8.87, P=0.046) |
| MELD | 1.08 (1.01–1.15, P=0.035) |
P-value adjusted for steroid and antibiotic use. Bold font indicates significance (P value <0.05).
HR, hazard ratio; CI, confidence interval; MELD, Model for End-stage Liver Disease.
Discussion
Our previous study showed that patients with alcohol use disorder have higher intestinal proportions of C. albicans. Whether this leads to more severe alcohol-associated liver disease and the precise mechanism of C. albicans promoting alcohol-associated liver disease remains poorly understood. Our current results link a specific fungal exotoxin with the progression of alcohol-associated liver disease. Candidalysin, a cytotoxic peptide secreted by C. albicans, causes direct hepatocyte damage and exacerbates ethanol-induced liver disease in mice. Fecal positivity for the Candidalysin coding gene ECE1 is associated with more severe disease and mortality in alcoholic hepatitis patients.
Different forms of cell death contribute to the pathogenesis of alcoholic liver disease31. Apoptosis of hepatocytes can be induced by organelle stress and cytokine receptor activation31, 32. It has also been shown that receptor-interacting protein 1 (RIP1), RIP3 and mixed lineage kinase domain-like protein (MLKL) trigger necroptosis of hepatocytes in alcohol-associated liver disease31, 33. Furthermore, hepatocytes undergo gasdermin-D mediated pyroptosis in alcoholic hepatitis patients34, indicating that different forms of hepatocellular death may exist/coexist based on the different microenvironment of cells. Candidalysin was found to activate MAPK/c-Fos/MAP kinase phosphatase 1 (MKP1) signaling pathway resulting in the production of pro-inflammatory cytokines such as IL-1α, IL-1β and IL-6 in epithelial cells6–8. Our study confirmed that Candidalysin increases hepatic levels of Il1b, Cxcl1 and Cxcl2 mRNAs in mice following ethanol administration. These pro-inflammatory cytokines may further recruit immune cells and hepatocytes injury. This might contribute to direct Candidalysin-induced hepatocyte death. Taken together, Candidalysin might be able to induce multiple forms of hepatocellular death through different mechanisms, which deserve further investigation in the future.
Our data indicate that Candidalysin does not increase intestinal permeability or intestinal epithelial cell damage in mice fed ethanol. C. albicans is larger in size than bacteria35 and seems to translocate predominantly via a transcellular route, and maybe to a lesser extent through the paracellular space of intestinal epithelial cells28. Chronic ethanol diet is associated with increased intestinal permeability36. Unlike what has been shown with bacteria, we have not been able to detect C. albicans in the liver of ethanol-fed mice by culture or by amplification of genetic material via qPCR. We have confirmed that Candidalysin positive C. albicans could increase liver injury, steatosis and inflammation in mice fed ethanol diet, but this injurious effect is absent in mice fed control diet. Our clinical data showed that patients with fecal samples containing genetic material for Candidalysin had increased gut permeability, higher MELD score and 90 day mortality compared with ECE1 negative patients. Thus, most likely Candidalysin produced in the intestinal lumen reaches the liver via increased intestinal permeability and exerts its effects on the liver. A direct interaction between Candidalysin and hepatocytes needs to be confirmed in future in vivo studies.
Importantly, we have discovered a link between the abundance of the Candidalysin gene ECE1 and severity of disease in alcoholic hepatitis patients. There is a positive correlation between ECE1 detection and MELD, and between ECE1 detection and patient mortality. Although an independent and larger patient cohort is required to confirm our findings, fecal ECE1 detection might be useful as non-invasive biomarker for predicting mortality in alcoholic hepatitis patients. Personalized diagnostics could be applied to identify patients colonized with Candidalysin-positive C. albicans.
In summary, our study shows that Candidalysin produced by C. albicans ethanol-induced liver disease in mice, is associated with severity and mortality in alcoholic hepatitis patients. Effective methods such as antifungal drugs targeting Candidalysin expressing C. albicans should be explored and considered as therapeutic approach for patients with alcoholic hepatitis.
Supplementary Material
Highlights.
Fecal levels of C. albicans and ECE1 are increased in patients with alcoholic hepatitis.
Candidalysin enhances ethanol-induced liver disease and is associated with higher mortality in mice.
Candidalysin damages hepatocytes in a dose–dependent manner.
Candidalysin is associated with the severity of liver disease and mortality of patients with alcoholic hepatitis.
Acknowledgements
The authors appreciate the technical support from Drs. Weizhong Sun, Tim Hendrikx, Katrin Hochrath, Jun Xu, and Lirui Wang.
Financial support: This study was supported in part by Science Research Foundation (02.03.2018-239) from Union Hospital, Tongji Medical College, Huazhong University of Science and Technology (to H.C.) and NIH grants R01 AA020703, R01 AA24726, U01 AA026939, by Award Number BX004594 from the Biomedical Laboratory Research & Development Service of the VA Office of Research and Development (to B.S.), the Deutsche Forschungsgemeinschaft (DFG) through the CRC/TR124 FungiNet project C1 and the Excellence Cluster “Balance of the Microverse” (to B.H.). The study was also supported by the NIDDK-funded San Diego Digestive Diseases Research Center (P30 DK120515) and by the NIAAA-funded Southern California Research Center for ALPD and Cirrhosis (P50AA011999).
Abbreviations:
- Adh1
alcohol dehydrogenase 1
- AH
alcoholic hepatitis
- ALT
alanine aminotransferase
- ANOVA
analysis of variance
- AST
aspartate aminotransferase
- AUD
alcohol use disorder
- C. albicans
Candida albicans
- CFUs
colony forming units
- Clec7a
C-type lectin–like receptor 7a
- Cxcl
chemokine (C-X-C motif) ligand
- Cyp2e1
cytochrome P450 family 2 subfamily E polypeptide 1
- ECE1
extent of cell elongation 1
- FBS
fetal bovine serum
- FDR
false discovery rate
- FITC
fluorescein isothiocyanate
- HR
hazard ratio
- I-FABP
intestinal fatty-acid binding protein
- IL-1β
interleukin 1 beta
- INR
international normalized ratio
- ITS
internal transcribed spacer
- LDH
lactate dehydrogenase
- MAPK
mitogen-activated protein kinase
- MELD
model for end-stage liver disease
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5′-diphenyltetrazolium bromide
- OR
odds ratio
- PBS
phosphate-buffered saline
- qPCR
quantitative PCR
- RIP1
receptor-interacting protein 1
- RT-qPCR
reverse transcription quantitative PCR
- TLR
Toll-like receptor; YPD broth, yeast extract, peptone and D-glucose broth
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest: B.S. is consulting for Ferring Research Institute.
References
- [1].WHO. Global status report on alcohol and health 2018. World Health Organization website; https://www.who.int/substance_abuse/publications/global_alcohol_report/en/. Accessed March 27, 2019. [Google Scholar]
- [2].Lee BP, Vittinghoff E, Dodge JL, Cullaro G, Terrault NA. National Trends and Long-term Outcomes of Liver Transplant for Alcohol-Associated Liver Disease in the United States. JAMA Intern Med. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Yang AM, Inamine T, Hochrath K, Chen P, Wang L, Llorente C, et al. Intestinal fungi contribute to development of alcoholic liver disease. J Clin Invest. 2017;127(7):2829–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Wang Y. Looking into Candida albicans infection, host response, and antifungal strategies. Virulence. 2015;6(4):307–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Jacobsen ID, Wilson D, Wachtler B, Brunke S, Naglik JR, Hube B. Candida albicans dimorphism as a therapeutic target. Expert review of anti-infective therapy. 2012;10(1):85–93. [DOI] [PubMed] [Google Scholar]
- [6].Moyes DL, Wilson D, Richardson JP, Mogavero S, Tang SX, Wernecke J, et al. Candidalysin is a fungal peptide toxin critical for mucosal infection. Nature. 2016;532(7597):64–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Moyes DL, Runglall M, Murciano C, Shen C, Nayar D, Thavaraj S, et al. A biphasic innate immune MAPK response discriminates between the yeast and hyphal forms of Candida albicans in epithelial cells. Cell Host Microbe. 2010;8(3):225–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Richardson JP, Willems HME, Moyes DL, Shoaie S, Barker KS, Tan SL, et al. Candidalysin Drives Epithelial Signaling, Neutrophil Recruitment, and Immunopathology at the Vaginal Mucosa. Infect Immun. 2018;86(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Beck AT, Guth D, Steer RA, Ball R. Screening for major depression disorders in medical inpatients with the Beck Depression Inventory for Primary Care. Behav Res Ther. 1997;35(8):785–91. [DOI] [PubMed] [Google Scholar]
- [10].Sterling RK, Lissen E, Clumeck N, Sola R, Correa MC, Montaner J, et al. Development of a simple noninvasive index to predict significant fibrosis in patients with HIV/HCV coinfection. Hepatology. 2006;43(6):1317–25. [DOI] [PubMed] [Google Scholar]
- [11].Brandl K, Hartmann P, Jih LJ, Pizzo DP, Argemi J, Ventura-Cots M, et al. Dysregulation of serum bile acids and FGF19 in alcoholic hepatitis. J Hepatol. 2018;69(2):396–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Bertola A, Mathews S, Ki SH, Wang H, Gao B. Mouse model of chronic and binge ethanol feeding (the NIAAA model). Nat Protoc. 2013;8(3):627–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Chen P, Starkel P, Turner JR, Ho SB, Schnabl B. Dysbiosis-induced intestinal inflammation activates tumor necrosis factor receptor I and mediates alcoholic liver disease in mice. Hepatology. 2015;61(3):883–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Liu CM, Kachur S, Dwan MG, Abraham AG, Aziz M, Hsueh PR, et al. FungiQuant: a broad-coverage fungal quantitative real-time PCR assay. BMC Microbiol. 2012;12:255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Heisel T, Podgorski H, Staley CM, Knights D, Sadowsky MJ, Gale CA. Complementary amplicon-based genomic approaches for the study of fungal communities in humans. PLoS One. 2015;10(2):e0116705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Wang W, Deng Z, Wu H, Zhao Q, Li T, Zhu W, et al. A small secreted protein triggers a TLR2/4-dependent inflammatory response during invasive Candida albicans infection. Nat Commun. 2019;10(1):1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Lang S, Duan Y, Liu J, Torralba MG, Kuelbs C, Ventura-Cots M, et al. Intestinal fungal dysbiosis and systemic immune response to fungi in patients with alcoholic hepatitis. Hepatology. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Brandl K, Rutschmann S, Li X, Du X, Xiao N, Schnabl B, et al. Enhanced sensitivity to DSS colitis caused by a hypomorphic Mbtps1 mutation disrupting the ATF6-driven unfolded protein response. Proc Natl Acad Sci U S A. 2009;106(9):3300–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Wang L, Llorente C, Hartmann P, Yang AM, Chen P, Schnabl B. Methods to determine intestinal permeability and bacterial translocation during liver disease. Journal of immunological methods. 2015;421:44–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Llorente C, Jepsen P, Inamine T, Wang L, Bluemel S, Wang HJ, et al. Gastric acid suppression promotes alcoholic liver disease by inducing overgrowth of intestinal Enterococcus. Nat Commun. 2017;8(1):837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Conti HR, Huppler AR, Whibley N, Gaffen SL. Animal models for candidiasis. Curr Protoc Immunol. 2014;105:19 6 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Lemoinne S, Kemgang A, Ben Belkacem K, Straube M, Jegou S, Corpechot C, et al. Fungi participate in the dysbiosis of gut microbiota in patients with primary sclerosing cholangitis. Gut. 2019;pii: gutjnl-2018–317791. [DOI] [PubMed] [Google Scholar]
- [23].Theel ES, Doern CD. beta-D-glucan testing is important for diagnosis of invasive fungal infections. J Clin Microbiol. 2013;51(11):3478–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Netea MG, Brown GD, Kullberg BJ, Gow NA. An integrated model of the recognition of Candida albicans by the innate immune system. Nature reviews Microbiology. 2008;6(1):67–78. [DOI] [PubMed] [Google Scholar]
- [25].Dambuza IM, Brown GD. C-type lectins in immunity: recent developments. Curr Opin Immunol. 2015;32:21–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Taylor PR, Brown GD, Reid DM, Willment JA, Martinez-Pomares L, Gordon S, et al. The beta-glucan receptor, dectin-1, is predominantly expressed on the surface of cells of the monocyte/macrophage and neutrophil lineages. J Immunol. 2002;169(7):3876–82. [DOI] [PubMed] [Google Scholar]
- [27].Hartmann P, Haimerl M, Mazagova M, Brenner DA, Schnabl B. Toll-like receptor 2-mediated intestinal injury and enteric tumor necrosis factor receptor I contribute to liver fibrosis in mice. Gastroenterology. 2012;143(5):1330–40 e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Allert S, Forster TM, Svensson CM, Richardson JP, Pawlik T, Hebecker B, et al. Candida albicans-Induced Epithelial Damage Mediates Translocation through Intestinal Barriers. MBio. 2018;9(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Hartmann P, Chen P, Wang HJ, Wang L, McCole DF, Brandl K, et al. Deficiency of intestinal mucin-2 ameliorates experimental alcoholic liver disease in mice. Hepatology. 2013;58(1):108–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Sandler NG, Koh C, Roque A, Eccleston JL, Siegel RB, Demino M, et al. Host response to translocated microbial products predicts outcomes of patients with HBV or HCV infection. Gastroenterology. 2011;141(4):1220–30, 30 e1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Wang S, Pacher P, De Lisle RC, Huang H, Ding WX. A Mechanistic Review of Cell Death in Alcohol-Induced Liver Injury. Alcohol Clin Exp Res. 2016;40(6):1215–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Kurose I, Higuchi H, Miura S, Saito H, Watanabe N, Hokari R, et al. Oxidative stress-mediated apoptosis of hepatocytes exposed to acute ethanol intoxication. Hepatology. 1997;25(2):368–78. [DOI] [PubMed] [Google Scholar]
- [33].Bailey SM, Cunningham CC. Contribution of mitochondria to oxidative stress associated with alcoholic liver disease. Free Radic Biol Med. 2002;32(1):11–6. [DOI] [PubMed] [Google Scholar]
- [34].Khanova E, Wu R, Wang W, Yan R, Chen Y, French SW, et al. Pyroptosis by caspase11/4-gasdermin-D pathway in alcoholic hepatitis in mice and patients. Hepatology. 2018;67(5):1737–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Shirtliff ME, Peters BM, Jabra-Rizk MA. Cross-kingdom interactions: Candida albicans and bacteria. FEMS Microbiol Lett. 2009;299(1):1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Bluemel S, Williams B, Knight R, Schnabl B. Precision medicine in alcoholic and nonalcoholic fatty liver disease via modulating the gut microbiota. Am J Physiol Gastrointest Liver Physiol. 2016;311(6):G1018–G36. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.