

Abstract
Understanding the mechanisms underlying the selective susceptibility to ischemia of the CA3 region is very important to explain the neuropathology of memory loss after brain ischemia. We used a rat model to study changes in gene expression of the amyloid protein precursor and its cleaving enzymes and tau protein in the hippocampal CA3 sector, after transient 10-min global brain ischemia with survival times of 2, 7, and 30 days. The expression of the α-secretase gene was below control values at all times studied. But, the expression of the β-secretase gene was below the control values at 2–7 days after ischemia and the maximal increase in its expression was observed on day 30. Expression of the presenilin 1 gene was significantly elevated above the control values at 2–7 days after ischemia and decreased below the control values at day 30. Expression of the presenilin 2 gene showed an opposite trend to the expression of presenilin 1. Expression of the amyloid protein precursor gene after ischemia was at all times above the control values with a huge significant overexpression on day 7. Additionally, the expression of the tau protein gene was below the control values 2 days after ischemia, but the significant increase in its expression was observed on days 7–30. Data show that brain ischemia activates neuronal changes and death in the CA3 region of the hippocampus in a manner dependent on amyloid and tau protein, thus determining a new and important way to regulate the survival and/or death of ischemic neurons.
Keywords: Brain ischemia, Alzheimer’s disease, Amyloid protein precursor, α-Secretase, β-Secretase, Presenilin 1 and 2, Tau protein
Introduction
The hippocampus is an important part of the brain due to its anatomical structure and physiological functions, especially in the area related to learning and memory under normal conditions and dementia in Alzheimer’s disease and brain injury after ischemia [1–7]. The hippocampus plays an important role in the consolidation of information from short-term memory to long-term memory and spatial orientation, and this important transmission is disturbed inter alia in Alzheimer’s disease [3, 4]. In addition, various areas of pyramidal neurons in the hippocampus also show a rather specialized response to pathological factors, as exemplified by similar neurodegenerative changes in Alzheimer’s disease and cerebral ischemia [5, 8–10].
Alzheimer’s disease is a complex, chronic neurodegenerative disorder that leads to the death of neurons primarily in the hippocampus, brain atrophy and dementia [5, 10]. It is evident that the first structure affected by Alzheimer’s disease is the hippocampus, especially its CA1 region, where a significant decrease in neuron density is observed compared with the less significant one in the CA3 region [10]. Transient experimental brain ischemia also causes early death of pyramidal neurons in the CA1 region, while neurons in the nearby CA3 region remain relatively spared [11–13]. In our previous studies, we found gene expression changes associated with Alzheimer’s disease in the hippocampal CA1 region, such as β-secretase, presenilin 1 and 2, amyloid protein precursor, and tau protein during 2–7 days after brain ischemia [8, 9, 14, 15]. In addition, morphological observations have shown an increased number of disrupted pyramidal neurons in the CA3 region with acute and chronic lesions [11, 12] during 2 years of survival following brain ischemia [9] as one of the characteristic features for the development of dementia in Alzheimer’s disease [10]. It seems that changes in pyramidal neurons, in particular in the hippocampus, such as the CA3 region, may play a significant role in memory deficits that herald the onset of Alzheimer’s disease [5] and the development of Alzheimer’s disease dementia after brain injury due to ischemia-reperfusion [2, 4].
The hippocampal regions, including the CA3 region, are currently the main focus of research on gene expression [14, 15] due to its involvement in learning and memory and participation in the development of similar neurodegenerative changes in dementia resulting from Alzheimer’s disease or brain ischemia [2–5, 10, 16]. Mechanisms affecting different susceptibility to ischemia of pyramidal neurons in the hippocampus have recently been extensively studied in the CA1 region, while in the CA3 area, there is no new data in this regard. To date, only one experimental article in the literature has investigated differences in gene expression between the CA1 and CA3 areas of the hippocampus after focal brain ischemia with hypoxia in mice [17]. The most surprising result of the above study is that the strong transcriptional stimulus like ischemia with hypoxia leads to a significant reduction in pre-existing cell-specific gene expression patterns instead of generating new or increasing existing differences [17]. On the other hand, examination of the brains of very old men with rare senile plaques and neurofibrillary tangles showed increased expression of the amyloid protein precursor and α-synuclein in CA3 area neurons compared with CA1 [18]. Additionally, it was noted that neurodegeneration of the hippocampal CA1 region after experimental cerebral ischemia was somewhat dependent on amyloid and tau protein [14, 15]. Thus, studies on differences in the modulation of the amyloid and tau protein gene may provide key functional elements of the molecular landscape during neurodegeneration of CA1 and CA3 in the brain after cerebral ischemia and Alzheimer’s disease.
In the last years, more and more evidence points to the role of brain ischemia in the pathogenesis of Alzheimer’s disease [1, 6, 19–23]. The above suggestion is confirmed by epidemiological observations indicating that ischemic stroke is a factor contributing to the development of Alzheimer’s disease and vice versa [20]. Interestingly, this is also confirmed by similar neuropathological features observed after experimental ischemic brain damage as well as in Alzheimer’s disease [5, 6, 9, 22, 23]. However, the mechanism by which brain ischemia can lead to the development of full-blown Alzheimer’s disease remains still unclear. Understanding the genomic and proteomic effects of ischemia on the pathogenesis of Alzheimer’s disease will be of great importance in explaining the etiology of this debilitating disorder. Perhaps the ischemic regional selectivity of neuronal death in the CA1 and CA3 areas may be an important factor in understanding the mechanisms associated with the development of Alzheimer’s disease. Understanding the mechanisms underlying the selective vulnerability to ischemia of the CA3 area is very important to explain the neuropathology of memory loss after brain ischemia with reperfusion. This report is part of a continuous series of studies that focus on quantitative analysis, using RT-PCR protocol, selected genes associated with Alzheimer’s disease in various rat brain structures, such as α-secretase, β-secretase, presenilin 1 and 2, amyloid protein precursor, and tau protein to assess the selective susceptibility to ischemia of pyramidal neurons in the CA3 region of the hippocampus during development of neurodegeneration and dementia after ischemia.
Materials and Methods
In female Wistar rats (n = 30, 2 months old, weight 160–180 g), 10-min brain ischemia was induced by cardiac arrest [24]. Briefly, rats were anesthetized with 2.0% isoflurane carried by O2. Shortly before starting the cardiac arrest procedure, anesthesia was discontinued. A special blunt L-shaped hook made of a straight stiff steel needle was inserted through the right parasternal line through the third intercostal space into the chest cavity. At this stage, the hook was positioned parallel to the right parasternal line, and the closing part of the hook was in a vertical position inside the chest. Then, the shaft of the hook was moved to a vertical position. At that time, the short end of the hook was completely inside the chest and plural cavity. Then, the hook was gently pressed down to the vertebral column until slight resistance was found. Then, the hook was tilted slightly 10–20° in the tail direction. The hook was next rotated counterclockwise approximately 135–140° under the inferior vena cava. In this position, the closing part of the hook was placed under the heart vessel bundle. In the last stage, the hook was pulled up to the sternum, which caused the heart vessel bundle to be compressed by the sternum. The end of the closing part of the hook in the above arrangement was in the left parasternal line in the second intercostal space. To prevent chest movement and ensure complete vascular occlusion, the index and middle finger pressure was applied to the sternum, resulting in complete hemostasis, followed by cardiac arrest. The trachea was not closed during the whole procedure. After 3.5 min, cardiac arrest was noted and then the hook was removed from the chest in reverse order and the rats remained in this state until resuscitation began. The resuscitation procedure consisted of external heart massage and artificial ventilation until spontaneous heart activity was restored and breathing appeared. At this time, air was pumped through a polyethylene tube inserted into the trachea, which was connected to a respirator. External heart massage was performed with the index and middle finger, rapidly hitting the chest at the level of the fourth intercostal area with a frequency of 150–240/min continuously. The ratio of strokes to the frequency of ventilation was 6:1 or 8:1. The 10 min of cerebral ischemia consisted of 3.5 min of heart vessel bundle compression by the hook and 6.5 min after removing the hook from the chest [24].
The animals were kept in pairs in cages in a room at a controlled temperature of 23 ± 1 °C, humidity 55 ± 5%, and a cycle of 12-h light/dark. They had unlimited access to commercial granular laboratory chow and tap water. The experiments were carried out in a light phase and the animals were treated according to the NIH Guide on the care and use of laboratory animals and a directive of the Council of the European Communities 142. The Local Ethical Committee approved all planned experimental procedures. After brain ischemia, the survival time of the animals was 2 (n = 14), 7 (n = 8), and 30 (n = 8) days. Sham-operated rats (n = 30) with a survival time of 2 days (n = 14), 7 days (n = 8), and 30 days (n = 8) were subjected to the same experimental procedures without global cerebral ischemia and served as appropriate control groups.
Before the hippocampal sample from the CA3 region was taken, the brains were perfused with cold 0.9% NaCl through the left ventricle to rinse the blood vessels from the blood. Then, the brains were removed from the skulls and transferred to an ice-cooled Petri dish. Samples were collected from the CA3 region of the ischemic and control hippocampus with a narrow scalpel of approximately 1 mm3 on both sides and immediately placed in the RNAlater solution (Life Technologies, USA) [14].
The isolation of total cellular RNA was made using the method developed by Chomczyński and Sacchi [25]. The NanoDrop 2000 spectrophotometer (Thermo Scientific, USA) was used to assess the quality and quantity of RNA [14, 15, 26–29]. The isolated RNA was stored in 80% ethanol at – 20 °C for further analyses [14, 15, 26–29]. In further studies, 1 μg of total RNA was reverse transcribed into cDNA using a high-capacity cDNA kit for reverse transcription according to the manufacturer’s instructions (Applied Biosystems, USA). The cDNA synthesis was performed on Veriti Dx (Applied Biosystems, USA) under the following conditions: stage I 25 °C, 10 min; stage II 37 °C, 120 min; stage III 85 °C, 5 min; stage IV 4 °C. The cDNA, obtained by this procedure, was amplified by real-time gene expression analysis (qPCR) on a 7900HT Real-Time Fast System (Applied Biosystems, USA) with the Master Mix SYBR-green PCR power mix reagent, using the manufacturer’s SDS software [14]. The amplification protocol included the following cycles: initial denaturation 95 °C, 10 min, and 40 cycles, each at two different temperatures 95 °C, 15 s, and 60 °C, 1 min. The monitoring and calculation of the number of DNA copies was carried out in the 7900HT Real-Time Fast System (Applied Biosystems, USA) in each amplification cycle. The number of cycles of PCR at which the fluorescence level exceeded the specific relative expression of the threshold cycle (CT) was applied to the research software (Applied Biosystems, USA) to calculate the number of DNA molecules present in the mixture at the beginning of the reaction. Normalization was achieved against the endogenous control gene (Rpl13a) [30], and the relative amount (RQ) of the gene expression was analyzed based on the ΔCT method and the results were calculated as RQ = 2−ΔΔCT [14, 31]. The RQ values were finally analyzed after their logarithmic conversion to the RQ logarithm (LogRQ) [14]. LogRQ = 0 indicates that gene expression in control and ischemic samples is not different. LogRQ < 0 means that we have reduced gene expression in the ischemic sample, while LogRQ > 0 indicates increased gene expression in the ischemic sample compared with the control one.
Statistical evaluation of the results was carried out using the Statistica v. 12 software with the help of non-parametric Kruskal-Wallis test with the “z” test—multiple analyses of differences between groups. Data are presented as means ± SD. Statistical significance was adopted at p ≤ 0.05.
Results
Expression of the α-Secretase Gene after Ischemia
In the CA3 region of the hippocampus, the expression of the α-secretase (ADAM10) gene after 10-min brain ischemia with a survival of 2, 7, and 30 days was below the control values. On the second day after ischemia, the minimum was − 0.524-fold change and maximum − 0.004-fold change with median − 0.139-fold change. On the seventh day after ischemia, the minimum was − 0.344-fold change and maximum − 0.044-fold change with median − 0.136-fold change. On the thirtieth day after ischemia, the minimum was − 0.250-fold change and maximum − 0.009-fold change with median − 0.197-fold change. Figure 1 illustrates changes in the mean level of expression of the α-secretase gene. The changes were not statistically significant at all the times after ischemia (Fig. 1).
Fig. 1.
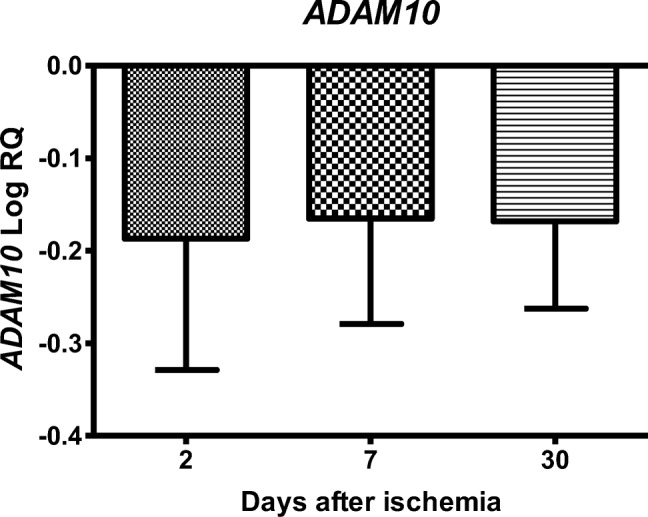
The mean gene expression levels of α-secretase (ADAM10) in the hippocampus CA3 area 2 (n = 14), 7 (n = 8), and 30 (n = 8) days after 10-min global brain ischemia. Marked SD, standard deviation. No statistical significance at all times after ischemia (Kruskal-Wallis test)
Expression of the β-Secretase Gene after Ischemia
In the CA3 region of the hippocampus, the expression of the β-secretase gene (BACE1) after ischemia with 2- and 7-day survival was lower than the control values, but on day 30, it was higher than the control values. On the second day after ischemia, the minimum was − 1.086-fold change and maximum − 0.001-fold change with median − 0.382-fold change. On the seventh day after ischemia, the minimum was − 0.457-fold change and maximum − 0.136-fold change with median − 0.334-fold change. On the thirtieth day after ischemia, the minimum was 0.043-fold change and maximum 0.336-fold change with median 0.189-fold change. Figure 2 illustrates changes in the mean level of expression of the β-secretase gene. The changes were statistically significant between 2 and 30 days after ischemia (Fig. 2).
Fig. 2.
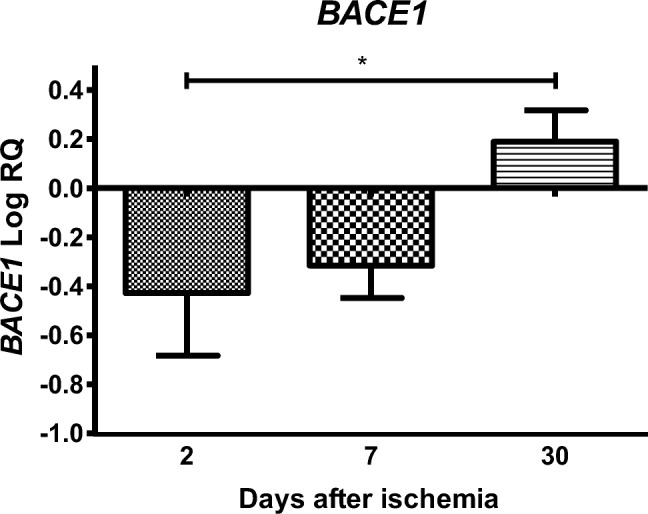
The mean gene expression levels of β-secretase (BACE1) in the hippocampus CA3 area in rats 2 (n = 14), 7 (n = 8), and 30 (n = 8) days after 10-min global brain ischemia. Marked SD, standard deviation. Indicated statistically significant differences in levels of gene expression between 2 and 30 (z = 3.368, p = 0.0022) days after ischemia (Kruskal-Wallis test). *p ≤ 0.01
Expression of the Presenilin 1 Gene after Ischemia
In the studied region, the expression of the presenilin 1 gene (PSEN1) after ischemia with a survival of 2 and 7 days was higher than the control values and lower than the control values at day 30. On the second day after ischemia, the minimum was 0.004-fold change and maximum 0.430-fold change with median 0.131-fold change. On the seventh day after ischemia, the minimum was 0.029-fold change and maximum 0.584-fold change with median 0.356-fold change. On the thirtieth day after ischemia, the minimum was − 0.105-fold change and maximum − 0.019-fold change with median − 0.062-fold change. Figure 3 illustrates changes in the mean level of expression of the presenilin 1 gene. The changes were statistically significant between 2 and 30 days and between 7 and 30 days after ischemia (Fig. 3).
Fig. 3.
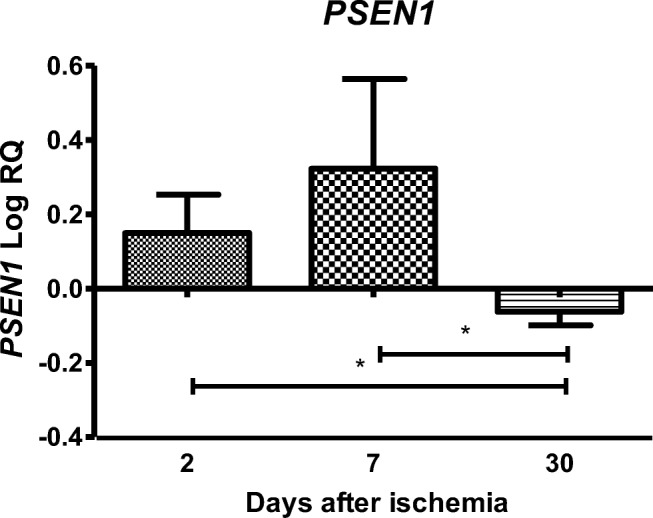
The mean gene expression levels of presenilin 1 (PSEN1) in the hippocampus CA3 area in rats 2 (n = 14), 7 (n = 8), and 30 (n = 8) days after 10-min global brain ischemia. Marked SD, standard deviation. Indicated statistically significant differences in levels of gene expression between 2 and 30 (z = 3.825, p = 0.0003) and between 7 and 30 (z = 3.920, p = 0.0002) days after ischemia (Kruskal-Wallis test). *p ≤ 0.001
Expression of the Presenilin 2 Gene after Ischemia
In the CA3 area, the expression of the presenilin 2 gene (PSEN2) after ischemia-reperfusion injury with a survival of 2 and 7 days was lower than the control values and higher than the control values at day 30. On the second day after ischemia, the minimum was − 0.498-fold change and maximum − 0.003-fold change with median − 0.177-fold change. On the seventh day after ischemia, the minimum was − 0.695-fold change and maximum − 0.034-fold change with median − 0.465-fold change. On the thirtieth day after ischemia, the minimum was 0.035-fold change and maximum 1.135-fold change with median 0.358-fold change. Figure 4 illustrates changes in the mean level of expression of the presenilin 2 gene. The changes were statistically significant between 2 and 30 days and between 7 and 30 days after ischemia (Fig. 4).
Fig. 4.
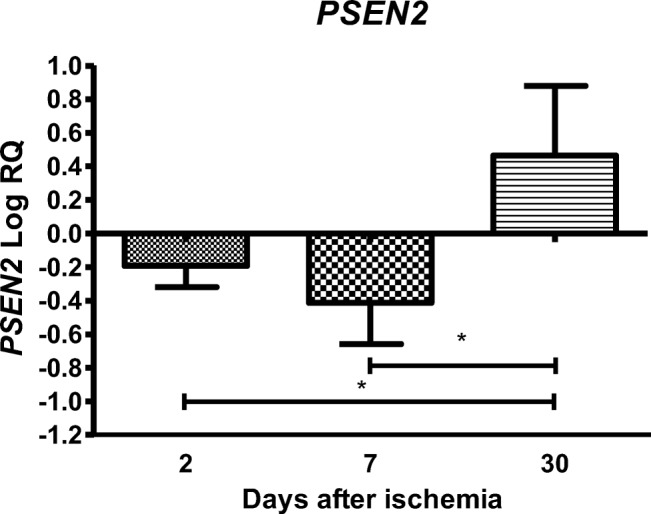
The mean gene expression levels of presenilin 2 (PSEN2) in the hippocampus CA3 area in rats 2 (n = 14), 7 (n = 8), and 30 (n = 8) days after 10-min global brain ischemia. Marked SD, standard deviation. Indicated statistically significant differences in levels of gene expression between 2 and 30 (z = 4.257, p = 0.00006) and between 7 and 30 (z = 4.550, p = 0.00001) days after ischemia (Kruskal-Wallis test). *p ≤ 0.0001
Expression of the Amyloid Protein Precursor Gene after Ischemia
After ischemia in the CA3 region of the hippocampus, the expression of the amyloid protein precursor gene during 2, 7, and 30 days of survival was above the control values. On the second day after ischemia, the minimum was 0.029-fold change and maximum 0.698-fold change with median 0.213-fold change. On the seventh day after ischemia, the minimum was 1.164-fold change and maximum 1.718-fold change with median 1.279-fold change. On the thirtieth day after ischemia, the minimum was 0.055-fold change and maximum 0.223-fold change with median 0.156-fold change. Figure 5 illustrates changes in the mean level of expression of the amyloid protein precursor gene. The changes were statistically significant between 2 and 7 days and between 7 and 30 days after ischemia (Fig. 5).
Fig. 5.
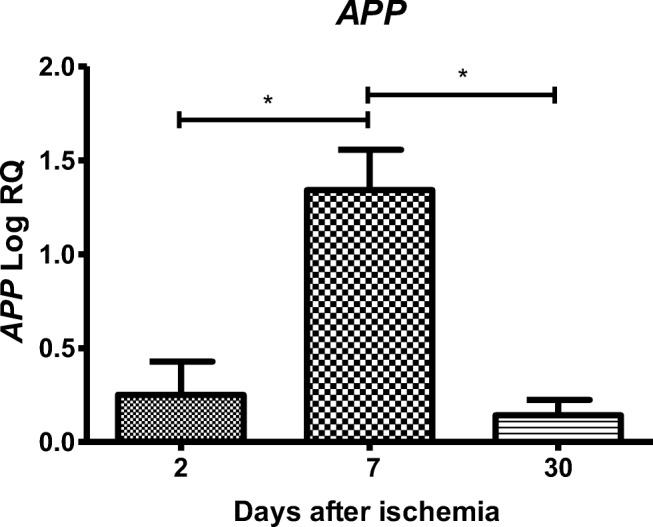
The mean gene expression levels of amyloid protein precursor (APP) in the hippocampus CA3 area in rats 2 (n = 14), 7 (n = 8), and 30 (n = 8) days after 10-min global brain ischemia. Marked SD, standard deviation. Indicated statistically significant differences in levels of gene expression between 2 and 7 (z = 3.498, p = 0.0014) and between 7 and 30 (z = 3.467, p = 0.0015) days after ischemia (Kruskal-Wallis test). *p ≤ 0.01
Expression of the Tau Protein Gene after Ischemia
In the CA3 region, the expression of the tau protein gene (MAPT) after ischemic damage with a survival of 2 days was lower than the control values and higher than the control values on days 7–30. On the second day after ischemia, the minimum was − 0.622-fold change and maximum − 0.001-fold change with median − 0.176-fold change. On the seventh day after ischemia, the minimum was 0.100-fold change and maximum 0.567-fold change with median 0.182-fold change. On the thirtieth day after ischemia, the minimum was 0.028-fold change and maximum 0.339-fold change with median 0.182-fold change. Figure 6 illustrates changes in the mean level of expression of the tau protein gene. The changes were statistically significant between 2 and 7 days and between 2 and 30 days after ischemia (Fig. 6).
Fig. 6.
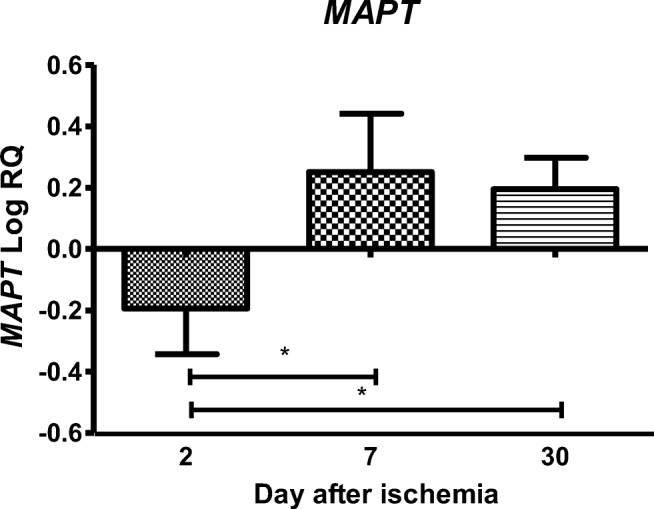
The mean gene expression levels of tau protein (MAPT) in the hippocampus CA3 area in rats 2 (n = 14), 7 (n = 8), and 30 (n = 8) days after 10-min global brain ischemia. Marked SD, standard deviation. Indicated statistically significant differences in levels of gene expression between 2 and 7 (z = 3.599, p = 0.0009) and between 2 and 30 days (z = 4.398, p = 0.00003) after ischemia (Kruskal-Wallis test). *p ≤ 0.001
Discussion
In this study, we showed for the first time the time course of changes in gene expression: α- and β-secretase, presenilin 1 and 2, amyloid protein precursor, and tau protein associated with Alzheimer’s disease in the CA3 area of the hippocampus after 10-min brain ischemia in rats with 2, 7, and 30 days of survival. Our data provide the first known role of in vivo dysregulated selected genes related with Alzheimer’s disease in neuronal changes or death in the CA3 region of the hippocampus after ischemia-reperfusion injury. In addition, our data show that brain ischemia with recirculation activates neuronal changes and death in the CA3 region of the hippocampus in a manner dependent on amyloid and tau protein, thus determining a new and important way to regulate the survival and/or death of post-ischemic neurons. These data may support the theory that amyloid generated by ischemia and changes in the tau protein may be involved in the development of neuropathology of Alzheimer’s disease associated with ischemia [14, 15, 26, 27, 32–35].
In the present study, the expression of the α-secretase gene was below control values at all times studied. On the other hand, the expression of the β-secretase gene was below the control values 2–7 days after ischemia and the maximal increase in its expression was observed on day 30. Expression of the presenilin 1 gene was significantly elevated above the control values 2–7 days after ischemia and decreased below the control values at day 30 of recirculation. Expression of the presenilin 2 gene demonstrated an opposite trend to the expression of presenilin 1. Expression of the amyloid protein precursor gene was at all times above the control values after ischemia with a huge significant overexpression on day 7. Additionally, the expression of the tau protein gene was below the control values 2 days after ischemia, but the significant increase in its expression was observed on days 7–30. This study suggests amyloidogenic processing of the amyloid protein precursor in the CA3 region of the hippocampus, which is likely to end 30 days after the ischemic event, and this observation clearly contrasts with the amyloid generation in the CA1 region [14]. In the CA1 area, amyloid production was associated with the appearance of necrotic neuronal lesions on the second day after ischemia [36–39]. Membrane integrity appears to have been maintained in most CA3 pyramidal neurons during this time [11, 12]. Our results show that there is a discrepancy in time between the amyloidogenic processing of the amyloid protein precursor in the neurons of the CA3 and CA1 regions by approximately 28–30 days [14].
The evidence available to date indicates that at least three factors are indispensable for irreversible damage to the CA3 hippocampal area after ischemia during short-term survival (2–30 days), which is independent of both ischemia and reperfusion time. These include glutamate neurotoxicity, intraneuronal calcium loading [40], and tissue acidosis [9]. Overexpression of the amyloid protein precursor and dysregulation of both β- and γ-secretases induced by ischemia provides an excess of soluble form of amyloid [8, 9, 19, 32, 33, 36, 37], which in turn can act as an additional neurotoxin that interferes with neuronal function and increases neuronal death through multiple mechanisms, including additionally impaired calcium homeostasis [40], initiation of neuroinflammatory processes [41], loss of nervous system integrity [42, 43], and by affecting the permeability of the blood-brain barrier [9, 44–47]. Some evidence indicates that amyloid reduces neuroplasticity and contributes to increased neuronal susceptibility to ischemia [41, 48]. In addition, changes in the tau protein gene expression (7–30 days post-ischemia) trigger disturbances in neuronal microtubule transport, e.g., the amyloid protein precursor, and this observation clearly contrast with these types of changes in CA1 area on day 2 [15]. On the other hand, the overexpression of genes associated with Alzheimer’s disease during recirculation may be a self-sustaining vicious cycle that leads to progressive neurodegeneration beginning in CA1 and then passing to the CA3 region of the hippocampus [10, 16] and finally spreading to other brain regions. The pathological feature of the Alzheimer’s disease is the accumulation of amyloid plaques at first in the hippocampus and spreading gradually throughout the brain [9, 10, 41]. It is likely that the accumulation is caused by both increased generation and impaired amyloid clearance and/or the transfer of amyloid from the blood to the CA3 region of the hippocampus [49–51]. These data support previous immunohistochemical observations in both animals and humans after brain ischemia suggesting a direct relationship between ischemia and increased amyloidogenic processing of the amyloid protein precursor in the hippocampus [9, 36, 52–58]. These findings are consistent with previous studies in semi-quantitative analysis of secretases in local cerebral ischemia models and additionally confirm the participation of β-secretase [59] and presenilin 1 and 2 in ischemia-induced β-amyloid peptide generation [60, 61]. In total, upregulation of genes found in our study suggests that β-secretase (after 30 days), presenilin 1 (after 2–7 days) and 2 (after 30 days) are activated at different times after ischemia in the CA3 region of the hippocampus. These data indicate that in ischemic pyramidal neurons, the amyloid protein precursor, β-secretase, and γ-secretase interact to promote an additional pathological sequence that causes neuronal death. Activation of β-secretase may then trigger further apoptotic signaling by inducing caspase activation [28, 29] and DNA fragmentation. It should be emphasized that both events are also evident in the brains of Alzheimer’s disease patients [62]. In addition, the opposite trend in the expression of presenilin 1 and 2 indicates that two presenilins can play different roles under rat brain ischemia [21, 26]. Our model of brain ischemia confirmed that presenilin 2 probably played a role in the modulation of apoptosis [21, 28, 62], because neurons in the ischemic region of the CA3 began to be under the influence of generated amyloid and dysfunctional tau protein in 7–30 days after ischemia [21, 26]. Although presenilins seem to complement each other’s functions, presenilin 1 appears to provide a fundamental constitutive function, whereas presenilin 2 appears to act as an emergency aid in ischemic conditions [14, 21].
The presented results allow to understand progressive ischemic changes in the different areas of the hippocampus, delayed amyloid accumulation, and long-term ischemic pathogenesis of Alzheimer’s disease. Current data may partly help to explain the molecular mechanism of slower occurrence of neuronal damage and death in the area of the ischemic CA3 hippocampus than in CA1 [11, 12]. The above changes are associated with the atrophy of the hippocampus [8, 9, 63] and the development of dementia of the Alzheimer’s disease type, as it was previously observed [2, 32, 33, 64–66]. These results suggest that both aberrant gene expression associated with Alzheimer’s disease and the accumulation of their products due to ischemia-reperfusion brain injury may play a key role in the neurodegeneration of CA3 pyramidal neurons [32, 33, 36, 37, 47, 54–58, 67]. It also suggests that the above processes may be directly related to the death of neurons in the ischemic CA3 area. Our study shows that gene transcription associated with Alzheimer’s disease correlates positively with the state of ischemic neurons within 7–30 days after ischemia [8, 9, 11, 12]. Expression of the studied genes in the CA3 region did not decrease in late times (30 days) after ischemia, with the exception of presenilin 1 and α-secretase. This may be a reflection of the onset of the disappearance of pyramidal neurons in the CA3 region of the hippocampus [8, 9, 11, 12]. According to our data and the results of other authors, it can be concluded that focal and/or global brain ischemia with reperfusion interferes with the metabolism of the amyloid protein precursor and tau protein at both the gene and protein levels, and leads to amyloid accumulation and tau protein dysfunction [15, 23, 34, 36, 67–75]. The conclusions that can be drawn from the CA3 area study of genes in the ischemic hippocampus indicate that many genes contributing to neuronal death and the formation of amyloid, may be important for developing therapeutic targets in the treatment of dementia in Alzheimer’s disease. Therefore, of course, further research is needed on this topic. It seems that in our rat model of ischemic brain injury in vivo, neurodegenerative symptoms are less severe in CA3 compared with the CA1 area of hippocampus during 30 days of survival. Finally, damage to the CA3 area of the hippocampus is associated with irreversible memory impairment [2, 64–66]. Because memory impairment is the earliest symptom of Alzheimer’s disease, we have found that loss of neurons in the CA3 area of the hippocampus due to ischemia-reperfusion episode is sufficient and contributes to memory impairment in a manner dependent on amyloid and tau protein at very early stages after brain ischemia and Alzheimer’s disease.
It is noteworthy that the hippocampus may also play an important role in other neurological conditions, such as epilepsy, which often accompanies Alzheimer’s disease. For example, during the induction of kindled seizures in rats, the increased convulsive response on day 25 was clearly associated with increased expression of a number of plasticity-regulating proteins [76]. It has also been documented that the kindling caused a huge increase in the number of neuronal cells possessing doublecortin (migration marker) in the dorsal hippocampus [77]. Acute seizures, 1 h after a single injection of pentylenetetrazol in rats, induced a hippocampal response reflected in a significant increase in c-fos mRNA [78]. Finally, after cardiac arrest in rats that caused cerebral ischemia, spontaneous seizure activity was observed in 68% of rats, and these animals were then susceptible to the induction of audiogenic seizures [79]. In the light of the presented data, the hippocampus probably takes part in these phenomena.
Conclusion
Our research indicates the key role of brain ischemia in the development of Alzheimer’s disease through primary neurodegeneration of various hippocampal regions, including CA3 area, and cognitive decline that progress with prolonged reperfusion times. Thus, bilateral damage to the abovementioned region results in the deterioration of short-term memory, which leads to the inability to create new memories. Changes in the CA3 region in the brain after hippocampal ischemia are particularly important in memory impairment, including Alzheimer’s disease. However, further research is needed to determine whether the damage and death of pyramidal neurons in the CA3 region are causative events or independent consequences of ischemia occurring in parallel and leading to the development of post-ischemic dementia. Apparently, prevention of brain ischemia and early treatment of ischemic stroke may have important implications for the development of Alzheimer’s disease and merit further research. Finally, the rat model used in this study seems to be a useful experimental approach to determining the role of genes associated with Alzheimer’s disease. Through in-depth research into the common genetic mechanism associated with these two neurological disorders, our findings can accelerate the current understanding of neurobiology in Alzheimer’s disease and cerebral ischemia, and lead future research into Alzheimer’s disease or cerebral ischemia in new directions.
Funding Information
The authors acknowledge the financial support from the following institutions: the Mossakowski Medical Research Centre, Polish Academy of Sciences, Warsaw, Poland (T3-RP), and the Medical University of Lublin, Lublin, Poland (DS 475/19-SJC).
Compliance with Ethical Standards
The experiments were carried out in a light phase and the animals were treated according to the NIH Guide on the care and use of laboratory animals and a directive of the Council of the European Communities 142. The Local Ethical Committee approved all planned experimental procedures.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Ryszard Pluta and Marzena Ułamek-Kozioł contributed equally to this work.
References
- 1.Pluta R. From brain ischemia-reperfusion injury to possible sporadic Alzheimer’s disease. Curr Neurovasc Res. 2004;1:441–453. doi: 10.2174/1567202043361839. [DOI] [PubMed] [Google Scholar]
- 2.Kiryk A, Pluta R, Figiel I, Mikosz M, Ułamek M, Niewiadomska G, Jabłoński M, Kaczmarek L. Transient brain ischemia due to cardiac arrest causes irreversible long-lasting cognitive injury. Behav Brain Res. 2011;219:1–7. doi: 10.1016/j.bbr.2010.12.004. [DOI] [PubMed] [Google Scholar]
- 3.Bature F, Pang D, Robinson A, Polson N, Pappas Y, Guinn B. Identifying patterns in signs and symptoms preceding the clinical diagnosis of Alzheimer’s disease: retrospective medical record review study and a nested case-control design. Curr Alzheimer Res. 2018;15:723–730. doi: 10.2174/1567205015666180404155358. [DOI] [PubMed] [Google Scholar]
- 4.Kim JH, Lee Y. Dementia and death after stroke in older adults during a 10-year follow-up: results from a competing risk model. J Nutr Health Aging. 2018;22:297–301. doi: 10.1007/s12603-017-0914-3. [DOI] [PubMed] [Google Scholar]
- 5.Kumar A, Tsao JW (2019) Alzheimer disease. StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing 2019 Jan-.2018 Dec 18.
- 6.Pluta R, Ułamek-Kozioł M, Januszewski S, Czuczwar SJ. From brain ischemia to Alzheimer-like neurodegeneration. Neuropsychiatry (London) 2018;8:1708–1714. [Google Scholar]
- 7.Knierim JJ. The hippocampus. Curr Biol. 2015;25:R1116–R1121. doi: 10.1016/j.cub.2015.10.049. [DOI] [PubMed] [Google Scholar]
- 8.Pluta R. The role of apolipoprotein E in the deposition of β-amyloid peptide during ischemia-reperfusion brain injury. A model of early Alzheimer’s disease. Ann N Y Acad Sci. 2000;903:324–334. doi: 10.1111/j.1749-6632.2000.tb06383.x. [DOI] [PubMed] [Google Scholar]
- 9.Pluta R, Ułamek M, Jabłoński M. Alzheimer’s mechanisms in ischemic brain degeneration. Anat Rec. 2009;292:1863–1881. doi: 10.1002/ar.21018. [DOI] [PubMed] [Google Scholar]
- 10.Padurariu M, Ciobica A, Mavroudis I, Fotiou D, Baloyannis S. Hippocampal neuronal loss in the CA1 and CA3 areas of Alzheimer’s disease patients. Psychiatr Danub. 2012;24:152–158. [PubMed] [Google Scholar]
- 11.Krajewski S, Mai JK, Krajewska M, Sikorska M, Mossakowski MJ, Reed JC. Upregulation of bax protein levels in neurons following cerebral ischemia. J Neurosci. 1995;15:6364–6376. doi: 10.1523/JNEUROSCI.15-10-06364.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sadowski M, Wisniewski HM, Jakubowska-Sadowska K, Tarnawski M, Lazarewicz JW, Mossakowski MJ. Pattern of neuronal loss in the rat hippocampus following experimental cardiac arrest-induced ischemia. J Neurol Sci. 1999;168:13–20. doi: 10.1016/s0022-510x(99)00159-8. [DOI] [PubMed] [Google Scholar]
- 13.Yin B, Barrionuevo G, Batinic-Haberle I, Sandberg M, Weber SG. Differences in reperfusion-induced mitochondrial oxidative stress and cell death between hippocampal CA1 and CA3 subfields are due to the mitochondrial thioredoxin system. Antioxid Redox Signal. 2017;27:534–549. doi: 10.1089/ars.2016.6706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kocki J, Ułamek-Kozioł M, Bogucka-Kocka A, Januszewski S, Jabłoński M, Gil-Kulik P, Brzozowska J, Petniak A, Furmaga-Jabłońska W, Bogucki J, Czuczwar SJ, Pluta R. Dysregulation of amyloid precursor protein, β-secretase, presenilin 1 and 2 genes in the rat selectively vulnerable CA1 subfield of hippocampus following transient global brain ischemia. J Alzheimers Dis. 2015;47:1047–1056. doi: 10.3233/JAD-150299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pluta R, Bogucka-Kocka A, Ułamek-Kozioł M, Bogucki J, Januszewski S, Kocki J, Czuczwar SJ. Ischemic tau protein gene induction as an additional key factor driving development of Alzheimer’s phenotype changes in CA1 area of hippocampus in an ischemic model of Alzheimer’s disease. Pharmacol Rep. 2018;70:881–884. doi: 10.1016/j.pharep.2018.03.004. [DOI] [PubMed] [Google Scholar]
- 16.Llorens-Martín M, Blazquez-Llorca L, Benavides-Piccione R, Rabano A, Hernandez F, Avila J, DeFelipe J (2014) Selective alterations of neurons and circuits related to early memory loss in Alzheimer’s disease. Front Neuroanat Vol 8, Article 38. [DOI] [PMC free article] [PubMed]
- 17.Newrzella D, Pahlavan PS, Kruger C, Boehm C, Sorgenfrei O, Schröck H, Eisenhardt G, Bischoff N, Vogt G, Wafzig O, Rossner M, Maurer MH, Hiemisch H, Bach A, Kuschinsky W, Schneider A. The functional genome of CA1 and CA3 neurons under native conditions and in response to ischemia. BMC Genomics. 2007;8:370. doi: 10.1186/1471-2164-8-370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ginsberg SD, Che S. Expression profile analysis within the human hippocampus: Comparison of CA1 and CA3 pyramidal neurons. J Comp Neurol. 2005;487:107–118. doi: 10.1002/cne.20535. [DOI] [PubMed] [Google Scholar]
- 19.Salminen A, Kauppinen A, Kaarniranta K. Hypoxia/ischemia activate processing of amyloid precursor protein: impact of vascular dysfunction in the pathogenesis of Alzheimer’s disease. J Neurochem. 2017;140:536–549. doi: 10.1111/jnc.13932. [DOI] [PubMed] [Google Scholar]
- 20.Cui P, Ma X, Li H, Lang W, Hao J. Shared biological pathways between Alzheimer’s disease and ischemic stroke. Front Neurosci. 2018;12:605. doi: 10.3389/fnins.2018.00605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jiang H, Jayadev S, Lardellia M, Newman MA. Review of the familial Alzheimer’s disease locus PRESENILIN 2 and its relationship to PRESENILIN 1. J Alzheimers Dis. 2018;66:1323–1339. doi: 10.3233/JAD-180656. [DOI] [PubMed] [Google Scholar]
- 22.Pluta R (2019) Brain ischemia: Alzheimer’s disease mechanisms. Nova Sciences Publishers, Ltd. New York, USA.
- 23.Pluta R, Ułamek-Kozioł M. The role of degenerative pathways in the development of irreversible consequences after brain ischemia. Neural Regen Res. 2019;14:982–983. doi: 10.4103/1673-5374.250574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pluta R, Lossinsky AS, Mossakowski MJ, Faso L, Wiśniewski HM. Reassessment of new model of complete cerebral ischemia in rats. Method of induction of clinical death, pathophysiology and cerebrovascular pathology. Acta Neuropathol. 1991;83:1–11. doi: 10.1007/BF00294424. [DOI] [PubMed] [Google Scholar]
- 25.Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 26.Pluta R, Kocki J, Ułamek-Kozioł M, Bogucka-Kocka A, Gil-Kulik P, Januszewski S, Jabłoński M, Petniak A, Brzozowska J, Bogucki J, Furmaga-JabłońskaW CSJ. Alzheimer-associated presenilin 2 gene is dysregulated in rat medial temporal lobe cortex after complete brain ischemia due to cardiac arrest. Pharmacol Rep. 2016;68:155–161. doi: 10.1016/j.pharep.2015.08.002. [DOI] [PubMed] [Google Scholar]
- 27.Pluta R, Kocki J, Ułamek-Kozioł M, Petniak A, Gil-Kulik P, Januszewski S, Bogucki J, Jabłoński M, Brzozowska J, Furmaga-Jabłońska W, Bogucka-Kocka A, Czuczwar SJ. Discrepancy in expression of β-secretase and amyloid-β protein precursor in Alzheimer-related genes in the rat medial temporal lobe cortex following transient global brain ischemia. J Alzheimers Dis. 2016;51:1023–1031. doi: 10.3233/JAD-151102. [DOI] [PubMed] [Google Scholar]
- 28.Ułamek-Kozioł M, Kocki J, Bogucka-Kocka A, Petniak A, Gil-Kulik P, Januszewski S, Bogucki J, Jabłoński M, Furmaga-Jabłońska W, Brzozowska J, Czuczwar SJ, Pluta R. Dysregulation of autophagy, mitophagy, and apoptotic genes in the medial temporal lobe cortex in an ischemic model of Alzheimer's disease. J Alzheimers Dis. 2016;54:113–121. doi: 10.3233/JAD-160387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ułamek-Kozioł M, Kocki J, Bogucka-Kocka A, Januszewski S, Bogucki J, Czuczwar SJ, Pluta R. Autophagy, mitophagy and apoptotic gene changes in the hippocampal CA1 area in a rat ischemic model of Alzheimer’s disease. Pharmacol Rep. 2017;69:1289–1294. doi: 10.1016/j.pharep.2017.07.015. [DOI] [PubMed] [Google Scholar]
- 30.Yao L, Chen X, Tian Y, Lu X, Zhang P, Shi Q, Zhang J, Liu Y. Selection of housekeeping genes for normalization of RT-PCR in hypoxic neural stem cells of rat in vitro. Mol Biol Rep. 2012;39:569–576. doi: 10.1007/s11033-011-0772-8. [DOI] [PubMed] [Google Scholar]
- 31.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real time quantitative PCR and the 2-ΔΔCT method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 32.Pluta R, Furmaga-Jabłońska W, Maciejewski R, Ułamek-Kozioł M, Jabłoński M. Brain ischemia activates β- and γ-secretase cleavage of amyloid precursor protein: Significance in sporadic Alzheimer’s disease. Mol Neurobiol. 2013;47:425–434. doi: 10.1007/s12035-012-8360-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pluta R, Jabłoński M, Ułamek-Kozioł M, Kocki J, Brzozowska J, Januszewski S, Furmaga-Jabłońska W, Bogucka-Kocka A, Maciejewski R, Czuczwar SJ. Sporadic Alzheimer’s disease begins as episodes of brain ischemia and ischemically dysregulated Alzheimer’s disease genes. Mol Neurobiol. 2013;48:500–515. doi: 10.1007/s12035-013-8439-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pluta R, Ułamek-Kozioł M, Januszewski S, Czuczwar SJ. Tau protein dysfunction after brain ischemia. J Alzheimers Dis. 2018;66:429–437. doi: 10.3233/JAD-180772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pluta R, Ułamek-Kozioł M, Januszewski S, Czuczwar SJ (2019) Amyloid pathology in the brain after ischemia. Folia Neuropathol in press [DOI] [PubMed]
- 36.Pluta R, Kida E, Lossinsky AS, Golabek AA, Mossakowski MJ, Wisniewski HM. Complete cerebral ischemia with short-term survival in rats induced by cardiac arrest. I. Extracellular accumulation of Alzheimer’s β-amyloid protein precursor in the brain. Brain Res. 1994;649:323–328. doi: 10.1016/0006-8993(94)91081-2. [DOI] [PubMed] [Google Scholar]
- 37.Lin B, Schmidt-Kastner R, Busto R, Ginsberg MD. Progressive parenchymal deposition of β-amyloid precursor protein in rat brain following global cerebral ischemia. Acta Neuropathol. 1999;97:359–368. doi: 10.1007/s004010050999. [DOI] [PubMed] [Google Scholar]
- 38.Müller GJ, Hasseldam H, Rasmussen RS, Johansen FF. Dexamethasone enhances necrosis-like neuronal death in ischemic rat hippocampus involving μ-calpain activation. Exp Neurol. 2014;261:711–719. doi: 10.1016/j.expneurol.2014.08.009. [DOI] [PubMed] [Google Scholar]
- 39.Groemer TW, Thiel CS, Holt M, Riedel D, Hua Y, Hüve J, Wilhelm BG, Klingauf J. Amyloid precursor protein is trafficked and secreted via synaptic vesicles. PLoS One. 2011;6:e18754. doi: 10.1371/journal.pone.0018754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pluta R, Salińska E, Puka M, Stafiej A, Łazarewicz JW. Early changes in extracellular amino acids and calcium concentrations in rabbit hippocampus following complete 15-min cerebral ischemia. Resuscitation. 1988;16:193–210. doi: 10.1016/0300-9572(88)90046-9. [DOI] [PubMed] [Google Scholar]
- 41.Sekeljic V, Bataveljic D, Stamenkovic S, Ułamek M, Jabłoński M, Radenovic L, Pluta R, Andjus PR. Cellular markers of neuroinflammation and neurogenesis after ischemic brain injury in the long-term survival rat model. Brain Struct Funct. 2012;217:411–420. doi: 10.1007/s00429-011-0336-7. [DOI] [PubMed] [Google Scholar]
- 42.Kamide T, Kitao Y, Takeichi T, Okada A, Mohri H, Schmidt AM, Kawano T, Munesue S, Yamamoto Y, Yamamoto H, Hamada J, Hori O. RAGE mediates vascular injury and inflammation after global cerebral ischemia. Neurochem Int. 2012;60:220–228. doi: 10.1016/j.neuint.2011.12.008. [DOI] [PubMed] [Google Scholar]
- 43.Weil ZM. Ischemia-induced hyperglycemia: consequences, neuroendocrine regulation, and a role for RAGE. Horm Behav. 2012;62:280–285. doi: 10.1016/j.yhbeh.2012.04.001. [DOI] [PubMed] [Google Scholar]
- 44.Pluta R. Blood-brain barrier dysfunction and amyloid precursor protein accumulation in microvascular compartment following ischemia-reperfusion brain injury with 1-year survival. Acta Neurochir Suppl. 2003;86:117–122. doi: 10.1007/978-3-7091-0651-8_26. [DOI] [PubMed] [Google Scholar]
- 45.Pluta R. Pathological opening of the blood-brain barrier to horseradish peroxidase and amyloid precursor protein following ischemia reperfusion brain injury. Chemotherapy. 2005;51:223–226. doi: 10.1159/000086924. [DOI] [PubMed] [Google Scholar]
- 46.Pluta R, Ułamek M, Januszewski S. Micro-blood-brain barrier openings and cytotoxic fragments of amyloid precursor protein accumulation in white matter after ischemic brain injury in long lived rats. Acta Neurochir Suppl. 2006;96:267–271. doi: 10.1007/3-211-30714-1_57. [DOI] [PubMed] [Google Scholar]
- 47.Pluta R, Januszewski S, Jabłoński M, Ułamek M. Factors in creepy delayed neuronal death in hippocampus following brain ischemia-reperfusion injury with long-term survival. Acta Neurochir Suppl. 2010;106:37–41. doi: 10.1007/978-3-211-98811-4_5. [DOI] [PubMed] [Google Scholar]
- 48.Pluta R, Jabłoński M, Czuczwar SJ. Postischemic dementia with Alzheimer phenotype: selectively vulnerable versus resistant areas of the brain and neurodegeneration versus β-amyloid peptide. Folia Neuropathol. 2012;50:101–109. [PubMed] [Google Scholar]
- 49.Pluta R, Barcikowska M, Januszewski S, Misicka A, Lipkowski AW. Evidence of blood-brain barrier permeability/leakage for circulating human Alzheimer’s β-amyloid-(1-42)-peptide. NeuroReport. 1996;7:1261–1265. doi: 10.1097/00001756-199605170-00008. [DOI] [PubMed] [Google Scholar]
- 50.Lee PH, Bang OY, Hwang EM, Lee JS, Joo US, Mook-Jung I, Huh K. Circulating beta amyloid peptide is elevated in patients with acute ischemic stroke. J Neurol Transm. 2005;112:1371–1379. doi: 10.1007/s00702-004-0274-0. [DOI] [PubMed] [Google Scholar]
- 51.Zetterberg H, Mörtberg E, Song L, Chang L, Provuncher GK, Patel PP, Ferrell E, Fournier DR, Kan CW, Campbell TG, Meyer R, Rivnak AJ, Pink BA, Minnehan KA, Piech T, Rissin DM, Duffy DC, Rubertsson S, Wilson DH, Blennow K. Hypoxia due to cardiac arrest induces a time dependent increase in serum amyloid β levels in humans. PLoS One. 2011;6:e28263. doi: 10.1371/journal.pone.0028263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Qi J, Wu H, Yang Y, Wand D, Chen Y, Gu Y, Liu T. Cerebral ischemia and Alzheimer’s disease: the expression of amyloid-β and apolipoprotein E in human hippocampus. J Alzheimers Dis. 2007;12:335–341. doi: 10.3233/jad-2007-12406. [DOI] [PubMed] [Google Scholar]
- 53.Maślińska D, Laure-Kamionowska M, Taraszewska A, Deregowski K, Maśliński S. Immunodistribution of amyloid beta protein (Aβ) and advanced glycation endproduct receptors (RAGE) in choroid plexus and ependymal of resuscitated patients. Folia Neuropathol. 2011;49:295–300. [PubMed] [Google Scholar]
- 54.Ishimaru H, Ishikawa K, Haga S, Shoji M, Ohe Y, Haga C, Sasaki A, Takashashi A, Maruyama Y. Accumulation of apolipoprotein E and β-amyloid-like protein in a trace of the hippocampal CA1 pyramidal cell layer after ischaemic delayed neuronal death. NeuroReport. 1996;17:3063–3067. doi: 10.1097/00001756-199611250-00054. [DOI] [PubMed] [Google Scholar]
- 55.Wiśniewski HM, Maślińska D. Beta-protein immunoreactivity in the human brain after cardiac arrest. Folia Neuropathol. 1996;34:65–71. [PubMed] [Google Scholar]
- 56.Jendroska K, Hoffmann OM, Patt S. Amyloid β peptide and precursor protein (APP) in mild and severe brain ischemia. Ann N Y Acad Sci. 1997;826:401–405. doi: 10.1111/j.1749-6632.1997.tb48492.x. [DOI] [PubMed] [Google Scholar]
- 57.Sinigaglia-Coimbra R, Cavalheiro EA, Coimbra CG. Postischemic hypertermia induces Alzheimer-like pathology in the rat brain. Acta Neuropathol. 2002;103:444–452. doi: 10.1007/s00401-001-0487-3. [DOI] [PubMed] [Google Scholar]
- 58.Van Groen T, Puurunen K, Maki HM, Sivenius J, Jolkkonen J. Transformation of diffuse beta-amyloid precursor protein and beta-amyloid deposits to plaques in the thalamus after transient occlusion of the middle cerebral artery in rats. Stroke. 2005;36:1551–1556. doi: 10.1161/01.STR.0000169933.88903.cf. [DOI] [PubMed] [Google Scholar]
- 59.Wen Y, Onyewuchi O, Yang S, Liu R, Simpkins JW. Increased beta-secretase activity and expression in rats following transient cerebral ischemia. Brain Res. 2004;1009:1–8. doi: 10.1016/j.brainres.2003.09.086. [DOI] [PubMed] [Google Scholar]
- 60.Tanimukai H, Imaizumi K, Kudo T, Katayama T, Tsuda M, Takagi T, Tohyama M, Takeda M. Alzheimer associated presenilin-1 gene is induced in gerbil hippocampus after transient ischemia. Mol Brain Res. 1998;54:212–218. doi: 10.1016/s0169-328x(97)00337-9. [DOI] [PubMed] [Google Scholar]
- 61.Pennypacker KR, Hernandez H, Benkovic S, Morgan DG, Willing AE, Sanberg PR. Induction of presenilins in the rat brain after middle cerebral arterial occlusion. Brain Res Bull. 1999;48:539–543. doi: 10.1016/s0361-9230(99)00031-3. [DOI] [PubMed] [Google Scholar]
- 62.Suh YH, Checler F. Amyloid precursor protein, presenilins, and alpha-synuclein: molecular pathogenesis and pharmacological applications in Alzheimer’s disease. Pharmacol Rev. 2002;54:469–525. doi: 10.1124/pr.54.3.469. [DOI] [PubMed] [Google Scholar]
- 63.Jabłoński M, Maciejewski R, Januszewski S, Ułamek M, Pluta R. One year follow up in ischemic brain injury and the role of Alzheimer factors. Physiol Res. 2011;60(Suppl 1):S113–S119. doi: 10.33549/physiolres.932186. [DOI] [PubMed] [Google Scholar]
- 64.De la Tremblaye PB, Plamondon H. Impaired conditioned emotional response and object recognition are concomitant to neuronal damage in the amygdale and perirhinal cortex in middle-aged ischemic rats. Behav Brain Res. 2011;219:227–233. doi: 10.1016/j.bbr.2011.01.009. [DOI] [PubMed] [Google Scholar]
- 65.Li J, Wang YJ, Zhang M, Fang CQ, Zhou HD. Cerebral ischemia aggravates cognitive impairment in a rat model of Alzheimer’s disease. Life Sci. 2011;89:86–92. doi: 10.1016/j.lfs.2011.04.024. [DOI] [PubMed] [Google Scholar]
- 66.Cohan CH, Neumann JT, Dave KR, Alekseyenko A, Binkert M, Stransky K, Lin HW, Barnes CA, Wright CB, Perez-Pinzon MA. Effect of cardiac arrest on cognitive impairment and hippocampal plasticity in middle-aged rats. PLoS One. 2015;10:e0124918. doi: 10.1371/journal.pone.0124918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Badan I, Dinca I, Buchhold B, Suofu Y, Walker L, Gratz M, Platt D, Kessler CH, Popa-Wagner A. Accelerated accumulation of N- and C-terminal beta APP fragments and delayed recovery of microtubule-associated protein 1B expression following stroke in aged rats. Eur J Neurosci. 2004;19:2270–2280. doi: 10.1111/j.0953-816X.2004.03323.x. [DOI] [PubMed] [Google Scholar]
- 68.Wen Y, Yang SH, Liu R, Perez EJ, Brun-Ziukemagel AM, Koulen P, Simpkins JW. Cdk5 is involved in NFT-like tauopathy induced by transient cerebral ischemia in female rats. Biochim Biophys Acta. 2007;1772:473–483. doi: 10.1016/j.bbadis.2006.10.011. [DOI] [PubMed] [Google Scholar]
- 69.Mörtberg E, Zetterberg H, Nordmark J, Blennow K, Catry C, Decraemer H, Vanmechelen E, Rubertsson S. Plasma tau protein in comatose patients after cardiac arrest treated with therapeutic hypothermia. Acta Anaesthesiol Scand. 2011;55:1132–1138. doi: 10.1111/j.1399-6576.2011.02505.x. [DOI] [PubMed] [Google Scholar]
- 70.Randall J, Mörtberg E, Provuncher GK, Fournier DR, Duffy DC, Rubertsson S, Blennow K, Zetterberg H, Wilson DH. Tau proteins in serum predict neurological outcome after hypoxic brain injury from cardiac arrest: results of a pilot study. Resuscitation. 2013;84:351–356. doi: 10.1016/j.resuscitation.2012.07.027. [DOI] [PubMed] [Google Scholar]
- 71.Majd S, Power JH, Koblar SA, Grantham HJ. Early glycogen synthase kinase-3 and protein phosphatase 2A independent tau dephosphorylation during global brain ischaemia and reperfusion following cardiac arrest and the role of the adenosine monophosphate kinase pathway. Eur J Neurosci. 2016;44:1987–1997. doi: 10.1111/ejn.13277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bi M, Gladbach A, van Eersel J, Ittner A, Przybyla M, van Hummel A, Chua SW, van der Hoven J, Lee WS, Müller J, Parmar J, Jonquieres GV, Stefen H, Guccione E, Fath T, Housley GD, Klugmann M, Ke YD, Ittner LM. Tau exacerbates excitotoxic brain damage in an animal model of stroke. Nat Commun. 2017;8:473. doi: 10.1038/s41467-017-00618-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fujii H, Takahashi T, Mukai T, Tanaka S, Hosomi N, Maruyama H, Sakai N, Matsumoto M. Modifications of tau protein after cerebral ischemia and reperfusion in rats are similar to those occurring in Alzheimer’s disease - Hyperphosphorylation and cleavage of 4- and 3-repeat tau. J Cereb Blood Flow Metab. 2017;37:2441–2457. doi: 10.1177/0271678X16668889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Basurto-Islas G, Gu JH, Tung YC, Liu F, Iqbal K. Mechanism of tau hyperphosphorylation involving lysosomal enzyme asparagine endopeptidase in a mouse model of brain ischemia. J Alzheimers Dis. 2018;63:821–833. doi: 10.3233/JAD-170715. [DOI] [PubMed] [Google Scholar]
- 75.Khan S, Yuldasheva NY, Batten TFC, Pickles AR, Kellett KAB, Saha S. Tau pathology and neurochemical changes associated with memory dysfunction in an optimized murine model of global cerebral ischaemia – a potential model for vascular dementia? Neurochem Int. 2018;118:134–144. doi: 10.1016/j.neuint.2018.04.004. [DOI] [PubMed] [Google Scholar]
- 76.Schmoll H, Badan I, Grecksch G, Walker L, Kessler C, Popa-Wagner A. Kindling status in sprague-dawley rats induced by pentylenetetrazole. Am J Pathol. 2003;162:1027–1034. doi: 10.1016/S0002-9440(10)63897-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Buga A-M, Vintilescu R, Balseanu AT, Pop OT, Streba C, Toescu E, Popa-Wagner A. Repeated PTZ treatment at 25-day intervals leads to a highly efficient accumulation of doublecortin in the dorsal hippocampus of rats. PLoS One. 2012;7:e39302. doi: 10.1371/journal.pone.0039302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Retchkiman I, Fischer B, Platt D, Popa-Wagner A. Seizure induced C-Fos mRNA in the rat brain: comparison between young and aging animals. Neurobiol Aging. 1996;17:41–44. doi: 10.1016/0197-4580(95)02022-5. [DOI] [PubMed] [Google Scholar]
- 79.Ułamek-Kozioł M, Kocki J, Bogucka-Kocka A, Januszewski S, Czuczwar SJ, Pluta R. Occurrence of spontaneous and audiogenic seizures following global brain ischaemia due to cardiac arrest. Folia Neuropathol. 2015;53:245–249. doi: 10.5114/fn.2015.54425. [DOI] [PubMed] [Google Scholar]