

Summary
Recent advancements in single-cell RNA sequencing (scRNA-seq) have facilitated the classification of thousands of cells through transcriptome profiling, wherein accurate cell type identification is critical for mechanistic studies. In most current analysis protocols, cell type-based cluster annotation is manually performed and heavily relies on prior knowledge, resulting in poor replicability of cell type annotation. This study aimed to introduce a single-cell Cluster-based Automatic Annotation Toolkit for Cellular Heterogeneity (scCATCH, https://github.com/ZJUFanLab/scCATCH). Using three benchmark datasets, the feasibility of evidence-based scoring and tissue-specific cellular annotation strategies were demonstrated by high concordance among cell types, and scCATCH outperformed Seurat, a popular method for marker genes identification, and cell-based annotation methods. Furthermore, scCATCH accurately annotated 67%–100% (average, 83%) clusters in six published scRNA-seq datasets originating from various tissues. The present results show that scCATCH accurately revealed cell identities with high reproducibility, thus potentially providing insights into mechanisms underlying disease pathogenesis and progression.
Subject Areas: Quantitative Genetics, Cell Biology, Bioinformatics, Automation in Bioinformatics
Graphical Abstract
Highlights
-
•
Construction of a comprehensive tissue-specific reference database of cell markers
-
•
Paired comparisons to identify potential marker genes for clusters to ensure accuracy
-
•
Evidence-based scoring and annotation for clustered cells from scRNA-seq data
-
•
Accurate and replicable annotation on cell types of clusters without prior knowledge
Quantitative Genetics; Cell Biology; Bioinformatics; Automation in Bioinformatics
Introduction
Recent advancements in single-cell RNA sequencing (scRNA-seq) have furthered the understanding of heterogeneous cell compositions in complex tissues through the characterization of different cell types based on gene expression levels, thus facilitating our understanding on spatiotemporal biological phenomena or disease pathogeneses, cellular lineages or differentiation trajectories, or cell-cell communication (Haque et al., 2017, Macosko et al., 2015, Potter, 2018, Regev et al., 2017). In the data processing protocols of scRNA-seq experiments, cell type identification is a vital step for subsequent analysis, and two types of strategies have been reported, e.g., cell-based and cluster-based annotation (Abdelaal et al., 2019). For cell-based strategy, the similarities between cell-based data and reference cell databases are taken to determine potential cellular identities. Several methods including SingleR (Aran et al., 2019), CellAssign (Zhang et al., 2019a), Garnett (Pliner et al., 2019), scMap (Kiselev et al., 2018), and CHETAH (de Kanter et al., 2019) belong to this category. Cluster-based strategies perform cell type identification using differentially expressed marker genes at the level of pre-computed clusters. Experimentally validated cell markers through fluorescence-activated cell sorting (FACS), in situ hybridization, and immunohistochemistry (IHC) are often used as reference.
The major challenge of cell-based strategy lies in the determination of cell types on each cluster as multiple cells with different types are present in one cluster. As shown in Figure S1, cellular composition in each cluster could vary a lot. According to cell type annotation by SingleR, cluster 3 of Chen dataset was composed of 31.6% proximal tubule cells, 36.8% intercalated cells, and 31.6% principle cells. In this case, it is rather difficult to assign an accurate cell label to this cluster. For cluster-based analysis, the selection of cluster marker genes is critical for the sensitivity and selectivity of cell type determination. In Seurat (Butler et al., 2018), a widely used data processing pipeline of scRNA-seq studies, one-against-all methods are used to derive cluster marker genes. Inevitably, in this list, a bunch of pseudo marker genes (significantly upregulated in at least two clusters rather than in one cluster) may occur, which would lead to incorrect cell type annotation. Furthermore, prior knowledge on known cell markers is needed during manual match with cluster marker genes derived in previous step. Another level of uncertainty is introduced by the fact that one cell type is commonly associated with multiple cell markers and one cell marker can be linked with multiple cell types (Zhang et al., 2019b). Replicability of this cell annotation protocol could be further reduced with increased number of clusters and multiple selections of cluster marker genes.
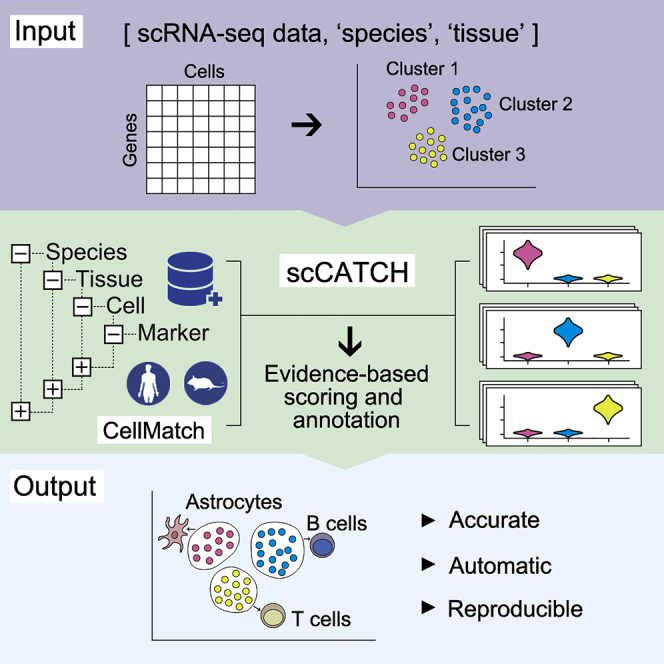
To address these issues, a single-cell Cluster-based automatic Annotation Toolkit for Cellular Heterogeneity (scCATCH) is introduced here, in which cell types are annotated through the tissue-specific cellular taxonomy reference database (CellMatch) and the evidence-based scoring (ES) protocol (workflow presented in Figure 1). The performance of scCATCH was evaluated by cell identity benchmark datasets originating from three different tissues. We further validated the accuracy of scCATCH with six independent scRNA-seq datasets. Results indicated that scCATCH facilitates analysis on scRNA-seq data and provides novel insights into the mechanisms underlying disease pathogenesis and progression.
Figure 1.

Automatic Annotation on Cell Types of Clusters from scRNA-Seq Data Using scCATCH
(A) Paired comparison of clusters to identify the potential marker genes for each cluster. Compared with every other cluster, genes significantly upregulated in only one cluster (log10 fold change ≥0.25, p < 0.05) and expressed in more than a quarter of cells (≥25%) would be considered marker genes. p values were obtained through the Wilcoxon test. ∗ indicates p < 0.05.
(B) Construction of tissue-specific cell taxonomy reference databases (CellMatch) with tissue-specific cell markers reported in the literature from humans or mice.
(C) Evidence-based score and annotation. For each cluster, cell types were scored on the basis of validated marker genes and their supporting literature, and the cell type with the highest score (top 1) was determined for the cluster.
Results
Validation of scCATCH Using the Benchmark scRNA-Seq Datasets
Knowledge in CellMatch reference database was derived from various resources, such as CellMarker (Zhang et al., 2019b), MCA (Han et al., 2018), CancerSEA (Yuan et al., 2019), and the CD Marker Handbook. In this reference database, cells were classified into three levels of subtypes in accordance with histological origin, expression of specific markers, or degrees of differentiation. Accordingly, a panel of 353 cell types and related 686 subtypes associated with 184 tissue types, 20,792 cell-specific marker genes, and 2,097 references of humans and mice were introduced into scCATCH as the reference database.
To validate the results of scCATCH, three independent scRNA-seq datasets, which were not recorded in the CellMatch database, were used, and cell types in these three datasets were identified or validated via FACS, in situ hybridization, or IHC. In particular, the Chen dataset (Chen et al., 2017) includes 203 mouse kidney cells and 3 cell types, namely intercalated cells, principal cells, and proximal tubule cells. The Xin dataset (Xin et al., 2016) includes 1,600 human pancreatic islet cells and 4 cell types, namely beta cells, alpha cells, delta cells, and pancreatic polypeptide (PP)-secreting cells. The Gierahn dataset (Gierahn et al., 2017) includes 3,694 human peripheral blood cells, namely B cells, T cells, dendritic cells (DCs), natural killer (NK) cells, and monocytes.
The cell types annotated by scCATCH were highly concordant with those verified from the literature for kidney cells, pancreatic islet cells, and peripheral blood cells (Figure 2). For the Chen dataset, scCATCH analysis identified intercalated cells and principal cells as collecting duct intercalated cells and collecting duct principal cells (Figure 2A), respectively, which is consistent with the organ origin of Chen dataset as renal collecting duct. For pancreatic islet cells in the Xin dataset, scCATCH accurately assigned cell identities for alpha cells, beta cells, delta cells, and PP cells (Figure 2B). scCATCH not only annotated the actual cell type but also identified the potential subtype of cells in each cluster, which are concordantly present among peripheral blood cells in the Gierahn dataset (Figure 2C). For example, scCATCH analysis annotated DCs as plasmacytoid DCs owing to significant upregulation of plasmacytoid DC marker genes including GPR183, SEC61B, and TBC1D4 (Villani et al., 2017) when compared with other clusters (Figure 2D). Moreover, our results marked T cells in the Gierahn dataset as regulatory T cells according to highly expressed IL7R and CD52 in this cluster (Figure 2E). These two genes were proposed as marker genes for regulatory T cells (Haase et al., 2015, Sinha et al., 2018, Wang et al., 2013). In addition, the performance of scCATCH on annotation remains stable with varied number of total cells and clusters.
Figure 2.

Validation of scCATCH
(A) Validation of scCATCH and identification of cluster marker genes upon Seurat in combination with evidence-based scoring in scCATCH (Seurat + scCATCH) for 203 mouse kidney cells from the Chen dataset.
(B) Validation of scCATCH and Seurat + scCATCH for 1,600 human pancreatic islet cells from the Xin dataset.
(C) Validation of scCATCH and Seurat + scCATCH for 3,694 human peripheral blood cells from Gierahn dataset.
(D) The violin plot of expression levels (log10) for cluster 3 marker genes GPR183, SEC61B, and TBC1D4 identified through scCATCH on Gierahn dataset.
(E) The violin plot for the expression levels (log10) of cluster 2 marker genes IL7R and CD52 identified via scCATCH on Gierahn dataset. DC, dendritic cell. NK cell, natural killer cell.
Potential marker gene selection by scCATCH is indeed interesting. Seurat, a widely used software package for scRNA-seq analysis, was applied herein to identify potential marker genes in the cluster, and the ES protocol was determined for annotation. Interestingly, cell types in the Chen and Xin datasets were still accurately labeled, whereas those in the Gierahn dataset were only partially concurrent with the results of scCATCH analysis (Figure 2A), indicating that the ES protocol is a robust identifier of cell identity.
Comparison of scCATCH with Other Methods
Cluster potential marker genes markedly contributed to the accuracy of annotation in the cluster-based method. For scCATCH analysis, we carried out paired comparisons to identify differentially expressed genes in only one specific cluster to ensure accuracy in matching the CellMatch database. On the contrary, Seurat uses a one-against-all approach, potentially generating a set of pseudo cluster potential marker genes (highly expressed in at least two clusters). Under this condition, cluster potential marker genes identified through scCATCH analysis usually were a subset of genes determined via Seurat (Figure 3A). However, an increased number of cluster potential marker genes did not benefit cell annotation. Although Seurat accurately annotated cell types common between the Chen and Xin datasets upon scCATCH analysis, Seurat accurately annotated the cell types of only two clusters (40% consistency, Figure 2C) in the Gierahn dataset, namely cluster 2 (T cells) and cluster 5 (monocytes). Apparently, the method of identifying cluster potential marker genes did not differ with a limited number of clusters. On increasing the total number of clusters, scCATCH analysis displayed better performance than Seurat in the identification of actual cluster potential marker genes present in the Gierahn dataset (Figure 2C). For example, CCL22, SWAP70, and KLRF1 were identified as cluster potential marker genes via Seurat, with a maximal fold change among the unshared marker genes between Seurat and scCATCH for clusters 1, 3, and 4, respectively. Evidently, CCL22 and SWAP70 were upregulated in multiple clusters, whereas KLRF1 was expressed in some cells in clusters 4 and 5, deterring the differentiation of actual cell types from other clusters (Figure 3B).
Figure 3.

Comparison of scCATCH with Other Methods
(A) Identification of cluster potential marker genes via Seurat (black number beside the circle) and scCATCH (red numbers beside the circle) in three validation datasets in each cluster. The black number inside the circle represents the number of overlapped genes.
(B) The violin plot for the expression levels of cluster 1 marker gene CCL22, cluster 3 marker gene SWAP70, and cluster 4 marker gene KLRF1 across 5 clusters identified in the Gierahn dataset via Seurat.
(C) Cell type annotation in the Chen dataset via scCATCH and cell-based annotation including CellAssign, SingleR, Garnett, scMap, and CHETAH.
(D) Cell type annotation in the Xin dataset via scCATCH, CellAssign, SingleR, Garnett, scMap, and CHETAH.
(E) Cell type annotation of the Gierahn dataset. The representative cell types were labeled for CellAssign and SingleR. Nodes 1, 2, 3, 4, 5, 6, 10, 69, and 70 represent intermediate cell types with a low confidence score.
Furthermore, validation datasets were used to compare scCATCH with cell-based annotation methods including CellAssign, Garnett, SingleR, scMap, and CHETAH. CellAssign, SingleR, and scMap were able to assign the accurate cell label for most cells, especially pancreatic islet cells in Xin dataset, whereas Garnett and CHETAH barely identified the actual identity of each cell (Figures 3C–3E; Table 1). The consistent rate of Garnett and CHETAH was as low as 0% on the Gierahn dataset, indicating that none of the cells were accurately identified by these two methods.
Table 1.
Comparison of scCATCH with Other Methods for Cell Type Annotation
| Dataset | Cluster | Cell Type | Cell Sum | Consistent Rate |
|||||
|---|---|---|---|---|---|---|---|---|---|
| scCATCH | CellAssign | Garnett | SingleR | scMap | CHETAH | ||||
| Chen | 1 | Intercalated cell | 110 | √ | 13% | 28% | 98% | 90% | 0% |
| 2 | Principle cell | 74 | √ | 96% | 0% | 69% | 66% | 0% | |
| 3 | Proximal tubule cell | 19 | √ | 47% | 0% | 32% | 21% | 0% | |
| All | NA | 203 | 100% | 46% | 15% | 81% | 75% | 0% | |
| Xin | 1 | Beta cell | 503 | √ | 98% | 47% | 95% | 94% | 0% |
| 2 | Alpha cell | 946 | √ | 100% | 5% | 99% | 98% | 0% | |
| 3 | Delta cell | 58 | √ | 93% | 48% | 67% | 9% | 0% | |
| 4 | PP cell | 93 | √ | 100% | 45% | 72% | 16% | 0% | |
| All | NA | 1,600 | 100% | 99% | 22% | 95% | 89% | 0% | |
| Gierahn | 1 | B cell | 376 | √ | 75% | 8% | 1% | 64% | 0% |
| 2 | T cell | 903 | √ | 70% | 0% | 9% | 45% | 0% | |
| 3 | DC | 104 | √ | 38% | 0% | 0% | 68% | 0% | |
| 4 | NK cell | 471 | √ | 10% | 0% | 55% | 37% | 0% | |
| 5 | Monocyte | 1,840 | √ | 28% | 0% | 1% | 90% | 0% | |
| All | NA | 3,694 | 100% | 41% | 0.9% | 10% | 69% | 0% | |
NA, not applicable. √ indicates scCATCH annotates the accurate cell type of the cluster.
Owing to cell heterogeneity in the clusters, cell-based strategies could assign multiple cell type labels to one cluster. Our analysis indicated that only 31.6% of proximal tubule cells in cluster 3 of Chen dataset were assigned as proximal tubule cells by SingleR, whereas 36.8% and 31.6% cells in this cluster were assigned as intercalated cells and principle cells, respectively (Figure S1). Besides, for some clusters, most cells' labels (>50%) in the cluster were not consistent with the actual cell type, which presents in all clusters of three validation datasets annotated by Garnett and CHETAH; clusters 1 and 3 of Chen dataset and clusters 3, 4, and 5 of Gierahn dataset annotated by CellAssign; cluster 3 of Chen dataset and most clusters of Gierahn dataset by SingleR; and clusters 3 and 4 of Xin dataset as well as clusters 2 and 4 of Gierahn dataset by scMap (Figures 3C–3E; Table 1). Under this condition, it is hard to assign an accurate cell label to this cluster.
Reference dataset plays a key role in cell type annotation. We next tested the effect of CellMatch on the performance of SingleR and CHETAH. For SingleR, the databases of the Immunological Genome Project (ImmGen) and the mouse RNA-seq were used as the reference list for mouse, whereas the databases HPCA as well as Encode and Blueprint Epigenomics transcriptomes were used as the reference list for human. For CHETAH, a dataset of head and neck was used as the reference. As shown in Tables S1 and S2, using CellMatch as the underlying reference, SingleR performed better on annotating the cells of three validation datasets, especially on non-blood cells, compared with using the ImmGen, mouse RNA-seq, HPCA and Encode, and Blueprint Epigenomics transcriptomes reference lists. Consequently, the consistent rate with CellMatch database by SingleR improved from 0% to 80%–90%. However, CHETAH showed no difference in the consistent rate with CellMatch.
The Performance of scCATCH during Analysis of the scRNA-Seq Dataset
scCATCH was employed to annotate six known scRNA-seq datasets to assess the performance of scCATCH with three recorded in the CellMatch database and three unrecorded. On assessing the internal datasets recorded in the CellMatch database, the Enge dataset (Enge et al., 2017) included 2,281 human pancreatic cells and 6 cell types, namely alpha cells, beta cells, delta cells, acinar cells, ductal cells, and mesenchymal cells, whereas the Wu dataset (Wu et al., 2017) included 20,679 mouse brain cells and 7 cell types including oligodendrocyte precursor cells (OPCs), astrocytes, oligodendrocytes, neurons, microglial cells, endothelial cells, and mural cells. The Lindsey dataset (Plasschaert et al., 2018) included 2,970 human lung cells and 7 cell types including basal cells, brush cells/pulmonary neuroendocrine cells (PNECs), ciliated cells, FOXN4+ cells, ionocytes, secretory cells, and SLC16A7+ cells. On assessing external datasets that were not recorded in the reference database, the Zheng dataset (Zheng et al., 2017b) included 2,638 human peripheral blood cells and 9 cell types, namely CD8+ cells, naive and memory CD4+ T cells, CD14+ and FCGR3A+ monocytes, B cells, NK cells, DCs, and platelets. Moreover, the Zeisel (Zeisel et al., 2015) and Heng (Heng et al., 2019) datasets included 2,915 (7 cell types) and 3,918 (12 cell types) mouse brain cells including neurons, oligodendrocytes, microglia, endothelial cells, mural cells, astrocytes, ependymal cells, neuronal progenitor cells, OPCs, pericytes, and fibroblasts.
In general, scCATCH detected most cell identities and accurately annotated the cluster consistent with the pre-defined cell type in the literature. Both internal datasets and external datasets displayed an average consistency rate of 83% (Table 2). For 6 cell types in the Enge dataset, scCATCH consistently identified 4 cell types, namely alpha cells, beta cells, delta cells, and acinar cells, and identified clusters 5 and 6 as epithelial cells and acinar cells or beta cells, which were ideally considered as ductal cells and mesenchymal cells (Figure 4A) on the basis of their known marker genes PROM1 and THY1, respectively. However, PROM1 encodes a pentaspan transmembrane glycoprotein that localizes to membrane protrusions and is expressed on stem cells (Oshima et al., 2007), whereas the protein encoded by THY1 is expressed in numerous cell types and widely considered as a hematopoietic stem cell marker. As marker genes EPCAM (Cheng et al., 2010, Seeberger et al., 2009), KRT19 (Seeberger et al., 2009), and CDH1 (Seeberger et al., 2009) were confirmed as marker genes of pancreatic epithelial cells, scCATCH expectedly annotated cluster 5 with epithelial cells instead of ductal cells. Moreover, cluster 6 was identified as acinar or beta cells owing to their equal ESs, probably because of the limited cell number in cluster 6, comprising only 54 among 2,811 cells. Regarding the annotation of brain cells in another internal dataset, scCATCH identified all cell types in accordance with the literature, except for mural cells, which were marked as pericytes by scCATCH (Figure 4A). Moreover, pericytes were also considered mural cells, thus indicating 100% accuracy of scCATCH in annotating cell types in the Wu dataset. Among the 7 cell types in the Lindsey dataset, scCATCH accurately identified all cell types consistent with the literature, including basal cells, brush cells/PNECs, ciliated cells, FOXN4+ cells, ionocytes, secretory cells, and SLC16A7+ cells (Figure 4A). Interestingly, cluster 2, identified as brush cells/PNECs in the literature, was concurrently annotated as brush or neuroendocrine cells owing to their similar ESs.
Table 2.
Evaluation of scCATCH with Internal and External Datasets
| Dataset | Recorded in CellMatch | Species | Tissue | Cell Sum | Cluster Sum | Consistent Rate |
|---|---|---|---|---|---|---|
| Enge | Yes | Human | Pancreas | 2,281 | 6 | 4/6 |
| Wu | Yes | Mouse | Brain | 20,679 | 7 | 6/7 |
| Lindsey | Yes | Human | Lung | 2,970 | 7 | 7/7 |
| Zheng | No | Human | PBMCs | 2,638 | 9 | 7/9 |
| Zeisel | No | Mouse | Brain | 2,915 | 10 | 9/10 |
| Heng | No | Mouse | Brain | 3,918 | 12 | 9/12 |
PBMCs, peripheral blood mononuclear cells.
Figure 4.

Evaluation of scCATCH
(A) Cell types were annotated via scCATCH in three internal and three external datasets. Cluster numbers are provided with the corresponding cells. Cell types are listed in each cluster. OPC, oligodendrocyte precursor cell; PNEC, pulmonary neuroendocrine cell; DC, dendritic cell; VSMC, vascular smooth muscle cell; NPC, neuronal progenitor cell.
(B) The violin plot for the expression levels (log10) of CD4 and IL7R across 9 cell clusters in the Zheng dataset.
For the Zheng dataset, scCATCH identified the actual cell identities of most clusters along with marker genes of the cluster, such as B cells, CD8+ T cells, CD14+ and FCGR3A+ monocytes, and DCs (Figure 4A). Cluster 1 and 2 were notably labeled as naive and regulatory T cells, which were actually naive CD4+ and memory CD4+ T cells. However, CD4 was not upregulated in either cluster 1 or 2 (Figure 4B), thus lacking evidence regarding clusters 1 and 2 to be annotated on the basis of CD4 expression. Moreover, the cluster 2 marker gene IL7R common between scCATCH and the Zheng dataset was considered a potential marker gene of regulatory T cells (Haase et al., 2015, Wang et al., 2013). Cluster 7 seemed difficult to annotate on the basis of cluster marker genes GNLY and NKG7 of NK cells and cluster marker gene CD63 of basophils in peripheral blood, whereas cluster 9, containing only 14 cells, expressed both platelet (PPBP) and naive T cell (ACTN1, ARHGAP45, LDLRAP1, and R3HDM4) marker genes (Zheng et al., 2017a).
Furthermore, two scRNA-seq datasets from mouse brain were selected for scCATCH analysis. At the level of cell type identification, scCATCH accurately annotated the primary cell types including neurons, oligodendrocytes, microglia, endothelial cells, pericytes, astrocytes, and ependymal cells (Figure 4A). Notably, cluster 1 (interneurons), cluster 2 (S1 pyramidal neurons), and cluster 3 (CA1 pyramidal neurons) in the Zeisel dataset were annotated with type IC spiral ganglionic neurons, neurons, and neurons via scCATCH. These may be due to limited number of records on markers for interneurons and pyramidal neurons. Moreover, vascular smooth muscle cells (VSMCs) were annotated with mural cells, concurrent with the fact that mural cells include VMSCs in the Zeisel dataset. For Heng dataset, 9 of 12 cell types annotated via scCATCH were consistent with the literature, whereas 3 other clusters (neural progenitor cells, VSMCs, and brain fibroblasts) were labeled as neuroblasts and type I and type II spiral ganglionic neurons via scCATCH (Figure 4A). scCATCH identified marker genes Dcx, Ccnd2, Crmp1, Dbn1, Dlx2, Pfn2, Btg1, Meis2, Stmn2, and Dlx6os1 (Luo et al., 2015, Shah et al., 2018, Zhang et al., 2009) for cluster 3, leading to a high possibility as neuroblasts. However, it is difficult to determine the cell type of clusters 5 and 11 since cluster 5 includes marker genes for type I spiral ganglionic neurons such as Cdc42ep3, Mgst3, Mob2, Nexn, Rap1a, and Tpm1 for type I spiral ganglionic neurons (Shrestha et al., 2018) and cluster 11 includes marker genes for type II spiral ganglionic neurons like Adm, Bmp7, Islr, Oat, Serpinf1, and Wls. (Shrestha et al., 2018). Regarding their subtypes, numerous marker genes of type I spiral ganglionic neurons were observed in cluster 9 when compared with the original annotation of glutamatergic neurons. The GABAergic neurons in cluster 12 were annotated with type IC spiral ganglionic neurons via scCATCH owing to limited reference data and a limited number of cells.
Discussion
In this study, we developed scCATCH, a cluster-based automatic annotation toolkit for scRNA-seq analysis, which uses a tissue-specific cell taxonomy reference database (CellMatch) and ES protocol to annotate cell types. We not only validated the extremely high feasibility of ES protocol in scCATCH but also demonstrated the superiority of scCATCH over other methods of identifying marker genes, including Seurat, the cell-based annotation method CellAssign, Garnett, SingleR, scMap, and CHETAH, through three scRNA-seq validation datasets. Moreover, scCATCH was used to assess six other known scRNA-seq datasets wherein scCATCH accurately annotated most cell types.
Thus far, common cell type annotation methods primarily include the cluster-based method by matching single or several representative cluster potential marker genes with known cell markers, which is usually carried out manually; however, such a method tends to require subjective prior knowledge among investigators, and the unstable selection of cluster potential marker genes from the pool ranging from tens to hundreds of cluster potential marker genes results in poor replicability of cell type annotation. Hence, the complete CellMatch reference database and the ES protocol were introduced into scCATCH, wherein the ES protocol was primarily based on the matched number of supporting studies and validated marker genes. The cell type with the most evidence would be selected as the ultimate annotation. Together with the frequently used Seurat and ES protocol, this study indicates the high feasibility of scCATCH for the most annotated cell types concordant with the literature. Compared with manual annotation, the present method prevents manual selection of marker genes and subjective cell type determination. Without the requirement of prior knowledge, scCATCH rapidly, accurately, and reproducibly automatically annotates cell types of clusters from scRNA-seq data.
Recently, some cell-based annotation methods have increasingly emerged to identify the cell type at the single-cell level rather than single-cluster level. CellAssign, Garnett, SingleR, scMap, and CHETAH are known methods in cell-based category that mapped the expression profile of each cell with reference profiles of known cell types. However, for biomedical research, researchers usually are more interested in cell cluster(s) that show different patterns during physiological process, disease development, or drug treatment. This could be the underlying reason for the common workflow of scRNA-seq studies to perform cluster analysis first, followed by annotating cell types using marker genes. Besides, for cell-based methods, the major challenge lies in the determination of cell types on each cluster. Uncertainty may be introduced by cell heterogeneity as shown by our analysis. In addition, for the common cluster-based strategy, an accurate and reproducible toolkit for automatically annotating cells without prior knowledge was unavailable. Hence, we developed scCATCH to help biologists to address the current challenges. Moreover, scCATCH displayed extreme superiority to the cell-based annotation methods, not only upon solid tissue cell type identification but also upon blood cell type identification.
In this study, CellMatch, a comprehensive tissue-specific cell taxonomy reference database till date, was constructed for scCATCH as the underlying data. Application of CellMatch to SingleR and CellAssign resulted in significant improvement in accuracies of cell type annotations, which suggests the great utility of CellMatch as a reference database. Furthermore, as our understanding on cell types continues to be enriched, more marker gene information would be supplemented in this reference and its performance could be further enhanced. In the workflow of scRNA-Seq analysis, clustering is of key importance to the conclusions. For cell type annotation, inadequate clustering analysis also would introduce errors into this process as too many or few cells are both problematic for labeling. It is interesting to evaluate the effects of multiple clustering algorithm on cell type annotations in the future.
In summary, this study describes the development of an automatic and efficient toolkit for the identification of cluster potential marker genes on the basis of ES protocol and annotation by constructing a comprehensive tissue-specific cell taxonomy reference database (CellMatch) as the underlying data. The feasibility and availability of scCATCH were systematically validated in different datasets. The present scCATCH analysis would potentially facilitate rapid and accurate identification of actual cell identities without prior knowledge with high replicability. The present results would greatly benefit studies on scRNA-seq data through the elucidation of the cell composition in complex tissues and provide novel insights into mechanisms underlying disease pathogenesis and progression.
Limitations of the Study
The performance of scCATCH majorly depends on the reference database. The limited reference database potentially led to incorrect annotation of cell types via scCATCH.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
This work is supported by National Natural Science Foundation of China (81774153, 81973701) and National Youth Top-notch Talent Support Program (W02070098). The authors thank Dr. Weida Tong (National Center for Toxicological Research, U.S. Food and Drug Administration) for helpful discussions in the development of this project.
Author Contributions
X.F. conceived and designed the study. X.L. and R.X. collected and analyzed the single-cell RNA-seq data. X.S., J.L., and X.F. implemented the algorithm of scCATCH. X.S., J.L., and N.A. developed the toolkit of scCATCH. All authors wrote the manuscript, read, and approved the final manuscript.
Declaration of Interests
The authors declare no conflicts of interests.
Published: March 27, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.100882.
Data and Code Availability
The source codes and results are implemented in R and are freely available (https://github.com/ZJUFanLab/scCATCH; https://github.com/ZJUFanLab/scCATCH_performance_comparison). No new data were generated for this study. All data used in this study are publicly available.
Supplemental Information
References
- Abdelaal T., Michielsen L., Cats D., Hoogduin D., Mei H., Reinders M.J.T., Mahfouz A. A comparison of automatic cell identification methods for single-cell RNA sequencing data. Genome Biol. 2019;20:194. doi: 10.1186/s13059-019-1795-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aran D., Looney A.P., Liu L., Wu E., Fong V., Hsu A., Chak S., Naikawadi R.P., Wolters P.J., Abate A.R. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat. Immunol. 2019;20:163–172. doi: 10.1038/s41590-018-0276-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler A., Hoffman P., Smibert P., Papalexi E., Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018;36:411–420. doi: 10.1038/nbt.4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L., Lee J.W., Chou C.L., Nair A.V., Battistone M.A., Paunescu T.G., Merkulova M., Breton S., Verlander J.W., Wall S.M. Transcriptomes of major renal collecting duct cell types in mouse identified by single-cell RNA-seq. Proc. Natl. Acad. Sci. U S A. 2017;114:E9989–E9998. doi: 10.1073/pnas.1710964114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng K., Follenzi A., Surana M., Fleischer N., Gupta S. Switching of mesodermal and endodermal properties in hTERT-modified and expanded fetal human pancreatic progenitor cells. Stem Cell Res. Ther. 2010;1:6. doi: 10.1186/scrt6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Kanter J.K., Lijnzaad P., Candelli T., Margaritis T., Holstege F.C.P. CHETAH: a selective, hierarchical cell type identification method for single-cell RNA sequencing. Nucleic Acids Res. 2019;47:e95. doi: 10.1093/nar/gkz543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enge M., Arda H.E., Mignardi M., Beausang J., Bottino R., Kim S.K., Quake S.R. Single-cell analysis of human pancreas reveals transcriptional signatures of aging and somatic mutation patterns. Cell. 2017;171:321–330.e14. doi: 10.1016/j.cell.2017.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gierahn T.M., Wadsworth M.H., 2nd, Hughes T.K., Bryson B.D., Butler A., Satija R., Fortune S., Love J.C., Shalek A.K. Seq-Well: portable, low-cost RNA sequencing of single cells at high throughput. Nat. Methods. 2017;14:395–398. doi: 10.1038/nmeth.4179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haase D., Puan K.J., Starke M., Lai T.S., Soh M.Y., Karunanithi I., San Luis B., Poh T.Y., Yusof N., Yeap C.H. Large-scale isolation of highly pure "untouched" regulatory T cells in a GMP environment for adoptive cell therapy. J. Immunother. 2015;38:250–258. doi: 10.1097/CJI.0000000000000083. [DOI] [PubMed] [Google Scholar]
- Han X., Wang R., Zhou Y., Fei L., Sun H., Lai S., Saadatpour A., Zhou Z., Chen H., Ye F. Mapping the mouse cell atlas by microwell-seq. Cell. 2018;173:1307. doi: 10.1016/j.cell.2018.05.012. [DOI] [PubMed] [Google Scholar]
- Haque A., Engel J., Teichmann S.A., Lonnberg T. A practical guide to single-cell RNA-sequencing for biomedical research and clinical applications. Genome Med. 2017;9:75. doi: 10.1186/s13073-017-0467-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heng J.S., Rattner A., Stein-O'Brien G.L., Winer B.L., Jones B.W., Vernon H.J., Goff L.A., Nathans J. Hypoxia tolerance in the Norrin-deficient retina and the chronically hypoxic brain studied at single-cell resolution. Proc. Natl. Acad. Sci. U S A. 2019;116:9103–9114. doi: 10.1073/pnas.1821122116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiselev V.Y., Yiu A., Hemberg M. scmap: projection of single-cell RNA-seq data across data sets. Nat. Methods. 2018;15:359–362. doi: 10.1038/nmeth.4644. [DOI] [PubMed] [Google Scholar]
- Luo Y., Coskun V., Liang A., Yu J., Cheng L., Ge W., Shi Z., Zhang K., Li C., Cui Y. Single-cell transcriptome analyses reveal signals to activate dormant neural stem cells. Cell. 2015;161:1175–1186. doi: 10.1016/j.cell.2015.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macosko E.Z., Basu A., Satija R., Nemesh J., Shekhar K., Goldman M., Tirosh I., Bialas A.R., Kamitaki N., Martersteck E.M. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell. 2015;161:1202–1214. doi: 10.1016/j.cell.2015.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshima Y., Suzuki A., Kawashimo K., Ishikawa M., Ohkohchi N., Taniguchi H. Isolation of mouse pancreatic ductal progenitor cells expressing CD133 and c-Met by flow cytometric cell sorting. Gastroenterology. 2007;132:720–732. doi: 10.1053/j.gastro.2006.11.027. [DOI] [PubMed] [Google Scholar]
- Plasschaert L.W., Zilionis R., Choo-Wing R., Savova V., Knehr J., Roma G., Klein A.M., Jaffe A.B. A single-cell atlas of the airway epithelium reveals the CFTR-rich pulmonary ionocyte. Nature. 2018;560:377–381. doi: 10.1038/s41586-018-0394-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pliner H.A., Shendure J., Trapnell C. Supervised classification enables rapid annotation of cell atlases. Nat. Methods. 2019;16:983–986. doi: 10.1038/s41592-019-0535-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potter S.S. Single-cell RNA sequencing for the study of development, physiology and disease. Nat. Rev. Nephrol. 2018;14:479–492. doi: 10.1038/s41581-018-0021-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regev A., Teichmann S.A., Lander E.S., Amit I., Benoist C., Birney E., Bodenmiller B., Campbell P., Carninci P., Clatworthy M. The human cell atlas. Elife. 2017;6:e27041. doi: 10.7554/eLife.27041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeberger K.L., Eshpeter A., Rajotte R.V., Korbutt G.S. Epithelial cells within the human pancreas do not coexpress mesenchymal antigens: epithelial-mesenchymal transition is an artifact of cell culture. Lab. Invest. 2009;89:110–121. doi: 10.1038/labinvest.2008.122. [DOI] [PubMed] [Google Scholar]
- Shah P.T., Stratton J.A., Stykel M.G., Abbasi S., Sharma S., Mayr K.A., Koblinger K., Whelan P.J., Biernaskie J. Single-cell transcriptomics and fate mapping of ependymal cells reveals an absence of neural stem cell function. Cell. 2018;173:1045–1057.e9. doi: 10.1016/j.cell.2018.03.063. [DOI] [PubMed] [Google Scholar]
- Shrestha B.R., Chia C., Wu L., Kujawa S.G., Liberman M.C., Goodrich L.V. Sensory neuron diversity in the inner ear is shaped by activity. Cell. 2018;174:1229–1246.e17. doi: 10.1016/j.cell.2018.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha D., Kumar A., Kumar H., Bandyopadhyay S., Sengupta D. dropClust: efficient clustering of ultra-large scRNA-seq data. Nucleic Acids Res. 2018;46:e36. doi: 10.1093/nar/gky007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villani A.C., Satija R., Reynolds G., Sarkizova S., Shekhar K., Fletcher J., Griesbeck M., Butler A., Zheng S., Lazo S. Single-cell RNA-seq reveals new types of human blood dendritic cells, monocytes, and progenitors. Science. 2017;356 doi: 10.1126/science.aah4573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H., Daniel V., Sadeghi M., Opelz G. Plasticity and overlap of in vitro-induced regulatory T-cell markers in healthy humans. Transpl. Proc. 2013;45:1816–1821. doi: 10.1016/j.transproceed.2012.10.060. [DOI] [PubMed] [Google Scholar]
- Wu Y.E., Pan L., Zuo Y., Li X., Hong W. Detecting activated cell populations using single-cell RNA-seq. Neuron. 2017;96:313–329.e6. doi: 10.1016/j.neuron.2017.09.026. [DOI] [PubMed] [Google Scholar]
- Xin Y., Kim J., Okamoto H., Ni M., Wei Y., Adler C., Murphy A.J., Yancopoulos G.D., Lin C., Gromada J. RNA sequencing of single human islet cells reveals type 2 diabetes genes. Cell Metab. 2016;24:608–615. doi: 10.1016/j.cmet.2016.08.018. [DOI] [PubMed] [Google Scholar]
- Yuan H., Yan M., Zhang G., Liu W., Deng C., Liao G., Xu L., Luo T., Yan H., Long Z. CancerSEA: a cancer single-cell state atlas. Nucleic Acids Res. 2019;47:D900–D908. doi: 10.1093/nar/gky939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeisel A., Munoz-Manchado A.B., Codeluppi S., Lonnerberg P., La Manno G., Jureus A., Marques S., Munguba H., He L., Betsholtz C. Brain structure. Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science. 2015;347:1138–1142. doi: 10.1126/science.aaa1934. [DOI] [PubMed] [Google Scholar]
- Zhang R.L., Chopp M., Gregg S.R., Toh Y., Roberts C., Letourneau Y., Buller B., Jia L., P Nejad Davarani S., Zhang Z.G. Patterns and dynamics of subventricular zone neuroblast migration in the ischemic striatum of the adult mouse. J. Cereb. Blood Flow Metab. 2009;29:1240–1250. doi: 10.1038/jcbfm.2009.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang A.W., O'Flanagan C., Chavez E.A., Lim J.L.P., Ceglia N., McPherson A., Wiens M., Walters P., Chan T., Hewitson B. Probabilistic cell-type assignment of single-cell RNA-seq for tumor microenvironment profiling. Nat. Methods. 2019;16:1007–1015. doi: 10.1038/s41592-019-0529-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X., Lan Y., Xu J., Quan F., Zhao E., Deng C., Luo T., Xu L., Liao G., Yan M. CellMarker: a manually curated resource of cell markers in human and mouse. Nucleic Acids Res. 2019;47:D721–D728. doi: 10.1093/nar/gky900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng C., Zheng L., Yoo J.K., Guo H., Zhang Y., Guo X., Kang B., Hu R., Huang J.Y., Zhang Q. Landscape of infiltrating T cells in liver cancer revealed by single-cell sequencing. Cell. 2017;169:1342–1356.e16. doi: 10.1016/j.cell.2017.05.035. [DOI] [PubMed] [Google Scholar]
- Zheng G.X., Terry J.M., Belgrader P., Ryvkin P., Bent Z.W., Wilson R., Ziraldo S.B., Wheeler T.D., McDermott G.P., Zhu J. Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 2017;8:14049. doi: 10.1038/ncomms14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The source codes and results are implemented in R and are freely available (https://github.com/ZJUFanLab/scCATCH; https://github.com/ZJUFanLab/scCATCH_performance_comparison). No new data were generated for this study. All data used in this study are publicly available.