

Summary
NF-κB/Rel family of transcription factors plays a central role in initiation and resolution of inflammatory responses. Here, we identified a function of the NF-κB subunit c-Rel as a transcriptional repressor of inflammatory genes. Genetic deletion of c-Rel substantially potentiates the expression of several TNF-α-induced RelA-dependent mediators of inflammation. v-Rel, the viral homologue of c-Rel, but not RelB, also possesses this repressive function. Mechanistically, we found that c-Rel selectively binds to the co-repressor HDAC1 and competitively binds to the DNA mediating HDAC1 recruitment to the promoters of inflammatory genes. A specific point mutation at tyrosine25 in c-Rel's DNA-binding domain, for which a missense single nucleotide variation (Y25H) exists in humans, completely abrogated its ability to bind DNA and repress TNF-α-induced, RelA-mediated transcription. Our findings reveal that the transactivator NF-κB subunit c-Rel also plays a role as a transcriptional repressor in the maintenance of inflammatory homeostasis.
Subject Areas: Molecular Mechanism of Gene Regulation, Molecular Interaction, Immunology
Graphical Abstract
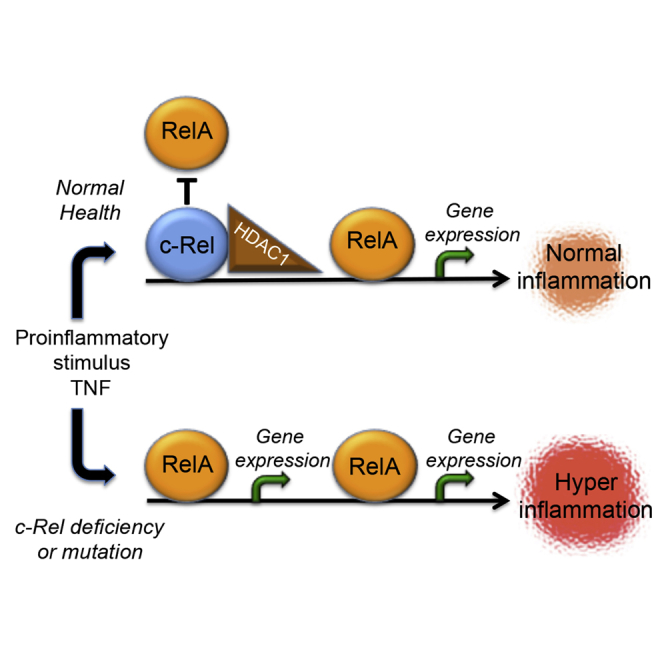
Highlights
-
•
NF-κB protein c-Rel act as a repressor of TNF-induced RelA-dependent inflammation
-
•
Mutation of Y25, an SNP site in humans, blocks repressor function of c-Rel
-
•
c-Rel recruits the co-repressor HDAC1 to the RelA-dependent promoters
-
•
Molecular mechanism of anti-inflammatory role of c-Rel
Molecular Mechanism of Gene Regulation; Molecular Interaction; Immunology
Introduction
The inflammatory response is a multi-phasic, multi-factorial cellular response to a variety of pathological stimuli; a tightly controlled inflammatory response is imperative for the maintenance of homeostasis in mammals (Matzinger, 2002, Medzhitov, 2008). Both defense against infection and cellular responses to injury involve inflammatory response in clearing the infection or to aid in the healing process (Nathan, 2002). However, if inflammation persists following recovery from damage or infection, then this host-friendly defense mechanism turns into a foe and drives a variety of chronic inflammatory and autoimmune diseases such as psoriasis, atherosclerosis, rheumatoid arthritis, and inflammatory bowel disease, as well as leads to inflammation-induced cancer (Netea et al., 2017). Thus the proper initiation, control, and resolution of inflammation is necessary for an organism's health and recovery from pathologies (Serhan et al., 2007).
One of the primary regulators of the inflammatory response is the ubiquitously expressed, inducible transcription factors of REL/NF-κB family (Ghosh et al., 1998). The NF-κB family comprises five subunits, RelA (p65), RelB, c-Rel, p105/p50, and p100/p52, that form functional homo- and heterodimers binding to conserved κB sites in the promoters of genes (Chen et al., 1998, Sen and Baltimore, 1986). In resting cells, NF-κB dimers are sequestered in the cytoplasm via their interaction with members of the Inhibitor of κB (IκB) protein family (Baeuerle and Baltimore, 1988a, Baeuerle and Baltimore, 1988b, Beg et al., 1992, Lenardo and Baltimore, 1989, Naumann et al., 1993). NF-κB activation is typically separated into two signaling pathways: the canonical pathway, which depends on the kinase IKK-β and the regulator NF-κB essential modulator (NEMO), and the alternative pathway, which depends on the kinases NIK and IKK-α (Pomerantz and Baltimore, 2002). Tumor necrosis factor alpha (TNF-α) is a proinflammatory cytokine that specifically activates the canonical NF-κB pathway (Ramakrishnan et al., 2004), leading to the expression of a number of cytokines and chemokines mediating inflammation (Sedger and McDermott, 2014). TNF-α signaling leads to IKKβ-mediated di-phosphorylation of IκB-α and its proteasomal degradation freeing the bound NF-κB (Wajant and Scheurich, 2011) (Gilmore, 1997, Gilmore, 2006). The newly freed NF-κB dimers translocate to the nucleus and bind to a loose consensus sequence (GGGRNWYYCC; R = purine, W = A or T, N = any, Y = pyrimidine) in the open chromatin, which allow binding of different dimers with varying binding strength (Chen et al., 1998).
Previous studies have separated the Rel/NF-κB family into activating subunits (RelA, RelB, and c-Rel) and repressive subunits (p50 and p52), based on functional studies and the presence or absence of the C-terminal transactivation domains (TAD) (Bours et al., 1993, Schmitz and Baeuerle, 1991). The dimers containing TADs are capable of recruiting transcriptional co-activators, which include histone acetyltransferases (HAT), such as p300/CREB-binding protein (CBP), p300/CBP associated factor (PCAF), and Tat-interacting protein 60 (Tip60), which promote gene expression (Bhatt and Ghosh, 2014, Ghizzoni et al., 2011). Homo- and heterodimers of p50 and p52 without TADs act as transcriptional repressors simply by competitively blocking or by preoccupying the κB sites of activating dimers (Schmitz and Baeuerle, 1991) or by recruiting co-repressors, such as histone deacetylases (HDACs) (Elsharkawy et al., 2010, Rocha et al., 2003).
c-Rel is one of the canonical, transactivating Rel subunits, with major roles in driving many immunological functions (Gilmore and Gerondakis, 2011). It also has a number of key functions in regards to cell cycle regulation and apoptosis across all cell types (Bash et al., 1997, Chen et al., 2003, Hsia et al., 2002, Huguet et al., 1997, Lorenz et al., 2014, Owyang et al., 2001). c-Rel has been shown to recognize a wider range of κB sites with redundancy allowed at the first and fourth positions (NGGN compared with GGGR), which increases the spectrum of target genes under its regulation (Kunsch et al., 1992). c-Rel exists as homodimer, and it also forms heterodimers with p50, p52, RelA, as well as RelB (Gilmore and Gerondakis, 2011, Marienfeld et al., 2003).
c-Rel knockout mice develop normally but show impaired response to immune challenges and infections as well as significantly reduced T regulatory cell numbers (Kontgen et al., 1995, Tumang et al., 1998, Courtine et al., 2011, Ramakrishnan et al., 2016). Observations made using c-Rel knockout mouse models suggest that c-Rel either promotes or protects from a number of inflammatory and autoimmune diseases. Deficiency of c-Rel has been shown to protect from diseases such as psoriasis (Fan et al., 2016, Fullard et al., 2013), rheumatoid arthritis (Campbell et al., 2000, Eyre et al., 2010, Fan et al., 2018), and experimental autoimmune encephalomyelitis (EAE) (Chen et al., 2011, Hilliard et al., 2002) in mouse models, likely due to the lack of c-Rel-dependent inflammatory gene expression (Gilmore and Gerondakis, 2011). In contrast, c-Rel knockout mice show accelerated pathology of diseases such as type 1 diabetes (Ramakrishnan et al., 2016) and colon inflammation (Courtine et al., 2011). However, the molecular mechanism behind accelerated inflammatory complications, in c-Rel-deficient mice, remains poorly defined.
In this study, we examined the role of c-Rel in regulating proinflammatory gene expression induced by TNF-α. Here, we show that the transcriptional activator c-Rel can act as a selective repressor of TNF-α-induced, proinflammatory gene expression. Absence of c-Rel enhances TNF-α-induced, RelA-driven transcriptional activity. We also show that c-Rel-mediated suppression is dependent on its DNA-binding ability at RelA-binding sites. Additionally, we show that c-Rel specifically binds to the co-repressor HDAC1 and mediates its recruitment to the specific promoters investigated in this study, suggesting a plausible mechanism for the increased expression of inflammatory genes in the absence of c-Rel.
Results
c-Rel Knockout Enhances TNF-α-Induced RelA-Dependent Gene Transcription
c-Rel is suggested to have both pro-and anti-inflammatory roles, and knowledge on the mechanism by which c-Rel restricts inflammation remains elusive. To have a comprehensive understanding on the requirement of c-Rel and its regulatory role in proinflammatory gene expression, we stimulated wild-type and c-Rel knockout mouse embryonic fibroblasts (MEFs) with TNF-α and then studied the expression of selected TNF-α-induced, pro-inflammatory genes. We found that several genes such as CCL2, CCL7, IP-10, CXCL1, A20, IL-6, CXCL2, CCL20, and ZFP36 showed markedly enhanced expression in the c-Rel knockout cells, while maintaining their expression kinetics (Figure 1A). This group of genes was made up largely of inflammatory cytokines and chemokines. Other genes such as ICAM1 and VCAM1 associated with cell migration proteins showed enhanced induction but displayed slower kinetics in c-Rel knockout cells (Figure 1B). The absence of c-Rel does not globally enhance or change kinetics of gene expression as we found that genes such as TNF-α, MMP10, EDN1, IFT1, IL-1β, and IκB-α showed a significantly decreased or lack of induction, indicative of dependence on c-Rel for their expression (Figure S1A). To rule out the possibility that the differences in gene expression observed were due to compensatory genetic alterations in immortalized MEFs generated from wild-type and c-Rel knockout mice, we generated c-Rel CRISPR knockout cells using our wild-type MEFs (G1 c-Rel KO) and confirmed the knockdown of c-Rel by western blotting (Figure 1C). Similar to c-Rel knockout MEFs of mouse origin, c-Rel CRISPR knockout cells also showed enhancement of selected TNF-α-induced genes (Figure 1D), demonstrating that the phenotype we observed is indeed specific to c-Rel knockout.
Figure 1.

c-Rel Knockout Enhances TNF-α-Induced RelA-Dependent Proinflammatory Gene Expression
(A) Wild-type or c-Rel knockout MEFs (3 × 105 at time of harvest) were treated in a six-well plate with 100 ng/mL TNF-α for 30 min or 3 h.
(B) Wild-type or c-Rel knockout MEFs were cultured and treated as in (A).
(C) Wild-type MEFs were transduced with lentivirus expressing three distinct CRISPR guide sequences against c-Rel, G1, G2 and G3, selected with hygromycin and examined for the knockdown of c-Rel by western blotting.
(D) Pool of four clones from c-Rel CRISPR G1 knockout MEFs was treated as above, and qPCR was performed on indicated genes.
(E) BMDM cells generated from wild-type and c-Rel knockout mice were treated as above, and qPCR were performed on indicated genes.
(F) c-Rel knockout MEFs reconstituted with human-c-Rel by lentiviral transduction (pLM), wild-type (WT), and c-Rel knockout (KO) cell lysates were analyzed by western blotting. pLCγ1 was used as a loading control for whole-cell lysates.
(G) c-Rel knockout MEFs re-expressing human c-Rel (pLM), wild-type, and c-Rel knockout MEFs (4 × 105 at time of harvest) were treated in a six-well plate with 100 ng/mL TNF-α for 30 min or 3 h. Samples were then analyzed by qPCR to determine the abundance of indicated mRNAs relative to that of ribosomal protein L32 (L32).
Data are representative of at least three independent experiments performed in triplicates, presented as mean ± standard error of mean (SEM) (n = 3). p Values were obtained by unpaired Student’s t test; ****p < 0.0001, ***p < 0.001, **p < 0.01, *p < 0.05. See also Figure S1.
To confirm that gene expression changes observed are not cell type specific or associated with transformed MEFs, we generated primary macrophages from bone marrow of wild-type and c-Rel knockout mice. We found that c-Rel knockout murine bone marrow-derived macrophages (BMDM) also showed significant enhancement in the expression of selected TNF-α-induced genes (Figure 1E). Specifically, critical inflammatory chemokines and cytokines such as CCL2, CCL7, IP-10, and CXCL1 were significantly enhanced both in TNF-α-stimulated c-Rel CRISPR knockout MEF cells and BMDM. Much like in the MEFs (Figure S1A), genes such as TNF-α and IκB-α showed markedly reduced expression in c-Rel knockout BMDM suggesting their dependence on c-Rel (Figure S1B). To further confirm that the enhanced expression of inflammatory genes in c-Rel knockout cells is not due to any secondary alteration in the cells, we re-expressed c-Rel in c-Rel knockout cells by lentiviral transduction (Figure 1F). We found that transient c-Rel expression reduced the enhanced expression of TNF-induced proinflammatory genes, CCL2, CCL7, CXCL1 and IP-10, in c-Rel knockout cells, indicating that c-Rel is directly involved in the suppression of these genes (Figure 1G). Taken together, these data show that c-Rel negatively regulates a specific subset of TNF-α-induced inflammatory genes, as well as demonstrates a physiological need for c-Rel for the expression of certain TNF-α-induced inflammatory genes.
c-Rel and Its Homologue, v-Rel, Suppresses TNF-α-Induced RelA Transcriptional Activity
Most of the proinflammatory genes we examined here were previously shown to be regulated by RelA transcriptional activity (Li et al., 2014b, Lim et al., 2007, Tourniaire et al., 2013). Consistent with this, RelA knockout decreased the expression of these inflammatory genes (Figure S2), demonstrating that c-Rel specifically targets a subset of RelA-dependent genes. To further study the molecular mechanism of c-Rel-mediated suppression of RelA activity, we utilized a reporter gene assay system using classical RelA-dependent promoters. We first examined the individual abilities of RelA and c-Rel to drive transactivation under Ig-κB, IP-10, and A20 promoters. We found that, as previously reported in several studies (Amir-Zilberstein and Dikstein, 2008, Jin et al., 2017, Kempe et al., 2005), RelA induced substantial activation of the Ig-κB, IP-10, and A20 promoters. c-Rel showed only nominal ability to activate all the three promoters tested (Figure 2A). Next, we co-transfected c-Rel with RelA and expected that the transactivating dimer complex, RelA:c-Rel, would show augmented reporter gene activity. To our surprise, we found that addition of c-Rel greatly inhibited RelA-dependent transactivation from Ig-κB, IP-10, and A20 promoters (Figure 2B). We also studied the c-Rel-mediated repression of RelA-dependent transactivation under a physiologically meaningful condition of TNF-α stimulation. We utilized TNF-α stimulation in place of RelA overexpression to activate Ig-κB and IP-10 promoters and found that c-Rel was indeed able to suppress TNF-α-induced transactivation of these promoters (Figure 2C).
Figure 2.

Both c-Rel and v-Rel Suppress TNF-α-Induced RelA-Dependent Transcriptional Activation
(A) HEK 293T cells (3 × 105) were transfected in a six-well plate with plasmids encoding FLAG-tagged RelA or c-Rel together with the indicated luciferase reporter plasmid.
(B) HEK 293T cells (3 × 105) were transfected in a six-well plate with FLAG-tagged RelA with or without FLAG tagged c-Rel plasmid and the indicated luciferase reporter plasmid.
(C) HEK 293T cells (3 × 105) were transfected in a six-well plate with the indicated luciferase reporter and c-Rel plasmids. Eighteen hours following transfection, cells were stimulated with 100 ng/mL TNF-α for 6 h.
(D) Schematic representation of c-Rel (top) and v-Rel (bottom) structures.
(E) HEK 293T cells (3 × 105) were transfected in a six-well plate with plasmids encoding FLAG-tagged RelA or v-Rel together with indicated luciferase reporter plasmid.
(F) HEK 293T cells (3 × 105) were transfected in a six-well plate with FLAG-tagged RelA and v-Rel along with luciferase reporter plasmids as indicated.
(G) HEK 293T cells (3 × 105) were transfected in a six-well plate with indicated luciferase reporter and v-Rel plasmids as indicated. Eighteen hours following transfection, cells were stimulated with 100 ng/mL TNF-α for 6 h. Luciferase activity was assessed using dual luciferase assay system. RelA alone values in the pairs Figures 2A, 2B, 2E, and 2F were from the same representative experiment for accurate comparison. Luciferase activity was assessed using dual luciferase assay system.
Data are presented as mean ± standard error of mean (SEM). p Values were obtained by unpaired Student’s t test; ***p < 0.001, **p < 0.01, *p < 0.05. (A–H) Top: Data in bar graphs are representative of three independent experiments performed in triplicates (n = 3). RelA alone values in Figures 2A and 2B were from the same representative experiment for accurate comparison. (A–H). Bottom: Western blotting analysis of total cell lysates of luciferase assay using anti-FLAG-tag antibody. See also Figure S2.
c-Rel stands for the cellular homolog of v-Rel, a protein encoded in avian reticuloendotheliosis virus strain T (Rev T) (Capobianco et al., 1990, Carrasco et al., 1996). It is thought that v-Rel was acquired by Rev T via horizontal transfer of turkey c-Rel and that subsequent mutations resulted in structural alterations in v-Rel (Figure 2D) and lent v-Rel its high oncogenic potential (Hrdlickova et al., 1994). v-Rel and c-Rel have previously been shown to possess the ability to transcriptionally repress RelA, using HIV-κB and IL-2-κB promoter sites (Ballard et al., 1990, Ballard et al., 1992). However, the suppressive abilities of v-Rel and c-Rel are not uniformly widespread across promoters as it has been shown that c-Rel activates transcription, whereas v-Rel represses transcription, under κB sites of mouse H-2Kb promoter (Inoue et al., 1991). v-Rel has been shown to act both as a suppressor and an activator of transcription in the same cell type at different differentiation stages (Walker et al., 1992). Here, we examined the ability of v-Rel to influence inflammation-associated transactivation. We overexpressed v-Rel or RelA in HEK293Ts and found that v-Rel alone had no activity at our selected promoters (Figure 2E). We then co-expressed v-Rel and RelA and observed the same trend of suppression of RelA-driven transcriptional activity as seen with c-Rel (Figure 2F). We also examined the ability of v-Rel to inhibit TNF-α-induced transactivation and found that, similar to c-Rel, v-Rel also suppressed TNF-α-induced transcriptional activity (Figure 2G). Taken together, these data show that v-Rel's specific repressive ability is likely conserved from c-Rel and the various mutations and structural changes that it acquired did not compromise its repressive function.
Suppressive Ability of c-Rel on TNF-α-Induced Signaling Is Not Conserved in RelB
Since the c-Rel and RelB subunits are structurally similar, we also tested the ability of RelB, with transactivation domain, to act as a repressor of RelA-induced transactivation. We found that RelB, like c-Rel and v-Rel, did not induce transcriptional activation under Ig-κB, IP-10, or A20 promoters (Figure 3A). Unlike c-Rel and v-Rel, RelB was unable to suppress RelA-induced transactivation of Ig-κB and A20 promoters, and it caused only a modest decrease in IP-10 promoter activation (Figure 3B). This also holds true for TNF-α-induced transcriptional activity, with RelB being unable to suppress Ig-κB or IP-10 promoters (Figure 3C).
Figure 3.

Suppressive Ability of c-Rel on TNF-α-Induced Signaling is Not Conserved in RelB
(A) HEK 293T cells (3 × 105) were transfected in a six-well plate with plasmids encoding FLAG-tagged RelA or RelB together with indicated luciferase reporter plasmid.
(B) HEK 293T cells (3 × 105) were transfected in a six-well plate with FLAG-tagged RelA with or without FLAG tagged RelB plasmid and the indicated luciferase reporter plasmid.
(C) HEK 293T cells (3 × 105) were transfected in a six-well plate with indicated luciferase reporter and RelB plasmids as indicated. Eighteen hours following transfection, cells were stimulated with 100 ng/mL TNF-α for 6 h. Luciferase activity was assessed using dual luciferase assay system. (A–C) Top: Data in bar graphs are representative of three independent experiments performed in triplicates. Bottom: Western blotting analysis of total cell lysates of luciferase assay using anti-FLAG-tag antibody. Blots are representative of three independent experiments. RelA alone values in Figures 3A and 3B were from the same representative experiment for accurate comparison.
(D) Wild-type or RelB knockout MEFs were treated with 100 ng/mL TNF-α for 30 min or 3 h. Samples were then analyzed by qPCR to determine the abundance of indicated mRNA relative to that of ribosomal protein L32 (L32). Data are presented as mean ± standard error of mean (SEM) (n = 3). p Values were obtained by unpaired Student’s t test; ***p < 0.001, **p < 0.01, *p < 0.05, ns not significant.
(E) Wild-type or RelB knockout MEFs were treated with 100 ng/mL TNF-α for 15 or 30 min. Nuclear and cytoplasmic extracts were analyzed with antibodies against the indicated proteins. hnRNPA1 was used as the loading control for nuclear fraction, and actin was used as the loading control for cytoplasmic fraction.
See also Figure S3.
We studied whether deficiency of RelB at physiological conditions affects expression of TNF-α-induced RelA-dependent genes, whose expression was increased in the absence of c-Rel (Figure 1), using wild-type and RelB KO cells. Interestingly, RelB knockout MEFs did not show enhanced TNF-α-induced gene expression, suggesting that RelB does not play an inhibitory role like c-Rel. Expressions of genes such as CCL2, CCL7, and IP-10 were found decreased in RelB knockout cells following TNF stimulation, whereas no significant effect was found on CXCL1 expression (Figure 3D). Expressions of several other inflammatory genes were compromised in RelB-deficient cells, indicating a possible role requirement of RelB in TNF-α-induced expression of these genes (Figure S3A). We found that RelB showed transient binding to Ig-κB sequences (Figure S3B top panels) and strong sustained binding to CCL2-κB site (Figure S3B middle panels) in oligonucleotide pull-down assays, which is consistent with the significant reduction of CCL2 expression in RelB knockout cells (Figure 3D). We also confirmed that the decreased gene expression in RelB knockout cells was not due to a defect in TNF-α-induced classical NF-κB activation. We found that TNF-α-induced RelA nuclear translocation and IκBα degradation occurred to a similar extent in wild-type and RelB knockout cells (Figure 3E). This shows that repression of TNF-α-induced gene expression is a unique characteristic of c-Rel, and the RelB subunit, with a conserved REL homology domain and transactivation domain, is unable to execute repressive role like c-Rel.
c-Rel Knockout Enhances TNF-α-Induced Nuclear Translocation and DNA Binding of RelA
To further dissect the molecular events that result in enhanced TNF-α-induced RelA-dependent gene expression, we examined RelA protein amounts, its nuclear translocation, and DNA binding in control and c-Rel knockout cells. First, we examined whether a deficiency of c-Rel or other NF-κB subunits affect RelA protein amounts and found comparable RelA in all the cell types tested (Figure 4A). This confirms that the observed increase in RelA-dependent gene expression in c-Rel knockout cells is not due to an increase in the total protein amounts of RelA. Next, we examined the nuclear translocation of RelA following TNF-α stimulation in control and c-Rel knockout cells and found a substantial increase in rapidly translocated RelA in the nucleus of c-Rel knockout cells (Figure 4B). To exclude the possibility that the enhanced response to TNF-α is due to a change in the surface expression of TNF receptor (TNFR), we examined the TNFR cell surface expression in control and c-Rel knockout MEFS and found comparable TNFR levels in both the cell types (Figure S4A). To confirm that the enhanced RelA activation observed in c-Rel knockout MEFs is not a cell-type-dependent phenomenon, we also studied the TNF-α-induced RelA nuclear translocation in primary BMDM. We found that, similar to MEFs, c-Rel knockout primary BMDM also showed enhanced TNF-α-induced RelA nuclear translocation (Figure 4C). However, nuclear translocation of NF-κB is insufficient to prove its transcriptional potential, which requires its binding to cognate DNA elements. To determine whether the additional RelA in the nucleus of c-Rel deficient cells was DNA binding competent, we performed oligonucleotide pull-down assays using biotinylated Ig-κB oligos. In line with the enhanced RelA-dependent gene expression observed in c-Rel-deficient cells, we found considerable increase in RelA DNA binding at the early TNF-α stimulated time point (Figure 4D). Furthermore, we also found an enhancement of p50 DNA binding along with RelA, suggesting TNF-α-induced robust activation of prototypical NF-κB dimers containing RelA and p50 subunits in c-Rel-deficient cells (Urban et al., 1991). We also examined whether the increase in RelA/p50 dimer binding to the DNA results from an increase in the preexisting RelA/p50 complex in the absence of c-Rel. We immunoprecipitated RelA and p50 from the cytoplasmic fractions of wild-type and c-Rel KO cells and found no significant changes in the proportion of RelA or p50 (Figure 4E). However, c-Rel deficiency resulted in enhanced IκBα binding of both RelA and p50 (Figure 4E). We further explored the mechanism of enhanced transactivation in c-Rel KO cells; however, we did not observe any substantial change in the binding of the co-activator p300 to the Ig-κB oligonucleotides in c-Rel knockout cells at the same time point when enhanced RelA and p50 binding was observed (Figure S4B).
Figure 4.

c-Rel Knockout Enhances TNF-α-Induced Nuclear Translocation and DNA Binding of RelA
(A) Total cell lysates of wild-type and the knockouts of RelA, RelB, c-Rel, p50, and p52 MEFs were analyzed with antibodies to each of the indicated NF-κB proteins. Actin was used as the loading control.
(B) Wild-type or c-Rel knockout MEFs were left untreated or treated with 100 ng/mL TNF-α for 15 or 30 min. Nuclear and cytoplasmic extracts were analyzed with antibodies against the indicated proteins. hnRNPA1 was used as the loading control for nuclear fraction, and actin was used as the loading control for cytoplasmic fraction.
(C). Primary bone marrow-derived macrophages generated from wild-type or c-Rel knockout mice were left untreated or stimulated with 100 ng/mL TNF-α for the indicated time points. Nuclear and cytoplasmic extracts were analyzed with antibodies against the indicated proteins.
(D) Wild-type or c-Rel knockout MEFs (3 × 106 at time of harvest) were left untreated or treated with 100 ng/mL TNF-α for 15 or 120 min. Nuclear and cytoplasmic extracts were prepared, and 100 μg of nuclear proteins per sample was utilized in an in vitro pull-down assay using biotinylated Ig-κB oligonucleotide. The precipitated proteins were separated in SDS/PAGE gel and probed for RelA, p50, and c-Rel. ns indicates non-specific band. Nuclear and cytoplasmic extracts were also probed using the indicated antibodies to examine the extent of their nuclear translocation as well as the degradation of IκB in the cytoplasm.
(E) Cytoplasmic extracts were prepared from wild-type or c-Rel knockout MEFs. Cytoplasmic lysates and immunoprecipitates of RelA and p50 antibodies were analyzed with the antibodies against indicated proteins.
Data for (A), (B), (D), and (E) are representative of four independent experiments, and C is representative of three independent experiments. See also Figure S4.
c-Rel Recruits the Co-repressor HDAC1 to the RelA-Dependent Promoters
To further validate the enhanced DNA binding of RelA in c-Rel knockout cells under physiologically relevant endogenous levels, we performed chromatin immunoprecipitation (ChIP) using anti-RelA antibody. We examined the enrichment of RelA-dependent promoters, IP-10 and CXCL1, by qPCR and found that TNF-α-induced RelA binding at both promoters was significantly enhanced in c-Rel knockout cells (Figure 5A). We also performed ChIP using anti-c-Rel antibody to confirm that c-Rel indeed binds to these RelA-dependent promoters. Our results show that c-Rel is present at the IP-10 and CXCL1 promoters at the basal state. Moreover, c-Rel occupancy at these promoters decreased, whereas RelA occupancy increased following TNF-α stimulation (Figure 5B), suggesting that c-Rel recruited to the promoters of selected RelA-dependent genes may repress transcriptional activation. Next, we studied the mechanism of c-Rel-mediated transcriptional repression by examining the binding of c-Rel to co-repressors. We found that c-Rel was bound to HDAC1 at basal state and this binding was enhanced by TNF stimulation (Figure 5C). c-Rel also showed weak binding to HDAC3, and no c-Rel binding was observed with HDAC2 and HDAC4 (Figure 5C). We performed oligonucleotide pull down with Ig-κB oligos and found that c-Rel deficiency completely blocked HDAC1 binding to the DNA (Figure 5D), suggesting that c-Rel is critical for HDAC1 recruitment to the promoter. Similar to co-immunoprecipitation experiments, oligonucleotide pull down also showed no HDAC2 and HDAC4 binding to the c-Rel containing complex. We did not observe any change in basal HDAC3 binding to the DNA in wild-type of c-Rel KO cells, and TNF stimulation decreased HDAC3 in the DNA bound complexes (Figure 5D). We also performed ChIP using anti-HDAC1 antibody to study the c-Rel-dependent HDAC1 recruitment at physiological conditions. We found that absence of c-Rel significantly decreased HDAC1 occupancy at IP-10 and CXCL1 promoters (Figure 5E), suggesting that lack of co-repressor recruitment may contribute to the enhanced transactivation of these promoters.
Figure 5.

c-Rel Recruits the Co-repressor HDAC1 to the RelA-Dependent Promoters
(A) Wild-type or c-Rel knockout MEFs (30 × 106 cells/condition at time of harvest) were left untreated or treated in 3 × 15-cm plates per condition with 100 ng/mL TNF-α for 15 min. Chromatin immunoprecipitation (ChIP) was performed using control IgG or RelA antibody.
(B) Wild-type MEFs were treated as in (A) and ChIP was performed using anti-c-Rel or anti-RelA antibodies. Enrichment of IP-10 and CXCL1 promoter regions in the ChIP samples were examined by qPCR in triplicates.
(C). Wild-type MEFs were treated with TNF-α for 15 min, and nuclear lysates were immunoprecipitated using anti-c-Rel antibody and examined for binding to the indicated HDACs.
(D). Oligonucleotide pull-down assay was performed as in Figure 4D, and the precipitated proteins were separated in SDS/PAGE gel and probed for indicated HDACs and c-Rel.
(E) Wild-type and c-Rel KO cells were treated as in (A), and ChIP was performed using anti-HDAC1 antibody.
(A)–(E) n = 4. Data are presented as mean ± standard error of mean (SEM). p Values were obtained by unpaired Student’s t test; ***p < 0.001, **p < 0.01, *p < 0.05.
c-Rel Competitively Blocks DNA Binding of RelA
All members of the NF-κB family possess the ability to bind to the DNA through their N-terminal DNA-binding domains (Ghosh and Hayden, 2008). The N-terminal half of c-Rel homodimer has been crystallized with the CD28 response element in the IL-2 promoter. It was shown that five critical, highly conserved residues in the DNA-binding domain of chicken c-Rel (arginine21, arginine23, tyrosine24, glutamic acid27, and arginine178) mediate direct base contact (Huang et al., 2001). Tyrosine24 in chicken c-Rel was also shown to mediate van der Waals interaction with the methyl groups of the DNA as well as directly interact with the DNA backbone (Huang et al., 2001). Therefore, we chose to mutate tyrosine25 in human c-Rel (the corresponding residue of tyrosine24 in chicken c-Rel), which is conserved also in v-Rel (Figure 6A), to disrupt the DNA binding of c-Rel. We introduced a conventional point mutation in the DNA-binding domain of c-Rel, tyrosine25 to phenylalanine (Y25F), and found that c-Rel Y25F failed to inhibit RelA-mediated activation of Ig-κB, A20, or IP-10 promoters (Figure 6B). We also examined the ability of c-Rel Y25F to inhibit TNF-α-induced transactivation and found that the Y25F mutation completely blocked the ability of c-Rel to suppress TNF-α-induced transcriptional activity (Figure 6C). We also found that the Y25F mutation in c-Rel completely abolishes c-Rel's DNA-binding ability (Figure 6D). To determine whether c-Rel disrupts DNA binding of RelA by competitively binding to the same κB sites, we overexpressed either wild-type or Y25F mutant c-Rel, which were expressed at similar levels, and studied RelA binding to the Ig-κB promoter. We found that overexpression of wild-type c-Rel substantially decreased DNA binding of RelA, whereas c-Rel Y25F neither bound DNA nor blocked DNA binding of RelA (Figure 6E). We confirmed that the Y25F mutation in c-Rel does not cause any gross structural defect, as it did not affect c-Rel's ability to dimerize with RelA or its ability to homodimerize with wild-type c-Rel (Figure S5A).
Figure 6.

A Point Mutation, Y25F or Y25H, Disrupting c-Rel's DNA-Binding Ability Compromises Its Repressive Function on RelA-Dependent Transactivation
(A) Amino acid sequence alignment of N-terminal DNA-binding regions of c-Rel and v-Rel, with critical residues for DNA binding highlighted.
(B) HEK 293T cells (3 × 105) were transfected in a six-well plate with plasmids encoding FLAG-tagged RelA and WT or Y25F c-Rel together with luciferase reporter plasmids as indicated in the figure.
(C) HEK 293T cells (3 × 105) were transfected in a six-well plate with FLAG-tagged Y25F c-Rel and the indicated luciferase reporter plasmid. Eighteen hours following transfection, cells were stimulated with 100 ng/mL TNF-α for 6 h. Luciferase activity was assessed using a dual luciferase assay system. Data are presented as mean ± standard error of mean (SEM). p Values were obtained by unpaired Student’s t test; ***p < 0.001, **p < 0.01, ns non significant. (B and C) Data in bar graphs are technical triplicates representative of three independent experiments (n = 3). RelA alone value in Figure 6B was from the same representative experiment in Figure 2A for accurate comparison. Bottom: Western blotting analysis of total cell lysates of luciferase assay using anti-FLAG-tag antibody. Blots are representative of three independent experiments.
(D) c-Rel knockout MEFs (3 × 106) were transfected with FLAG-tagged wild-type or Y25F c-Rel. Eighteen hours following transfection, MEFs were left untreated or treated with 100 ng/mL TNF-α for 15 min. Nuclear lysates equivalent to 200 μg of nuclear proteins per sample were utilized for in vitro pull-down assay using the Ig-κB oligonucleotide. Nuclear lysates were probed with indicated antibodies. hnRNPA1 was used as loading control.
(E) HEK 293T cells (2 × 106/10-cm plate) were transfected with FLAG-tagged wild-type or Y25F c-Rel. Cells were stimulated and analyzed as in (D).
(F) Cells were transfected with c-Rel Y25H, and experiments were performed as in (B).
(G) Cells were transfected with c-Rel Y25H, and experiments were performed as in (C).
(H) HEK 293T cells were transfected with FLAG-tagged wild-type or Y25H c-Rel as in (E). Cells were stimulated and analyzed as in (D).
Data for (D), (E), and (H) are representative of three independent experiments. See also Figure S5.
Intriguingly, a missense single nucleotide variation has been reported in human c-Rel, a change of the base T to C, that results in the codon change TAC to CAC, resulting in the amino acid substitution of tyrosine25 to histidine. This SNP was identified in the c-Rel locus in a whole-genome sequencing study of five indigenous southern African Khoisan hunter-gathers (Schuster et al., 2010); however, no clinical significance has been correlated with this SNP yet. We studied the functional effect of histidine substitution at Y25 of c-Rel by transiently expressing c-Rel Y25H in cells. We found that, similar to Y25F mutation, c-Rel Y25H mutation also compromised c-Rel's ability to suppress RelA-induced (Figure 6F) and TNF-α-induced (Figure 6G) transactivation. c-Rel Y25H neither showed the ability to bind to the DNA nor interfered with the DNA binding of RelA (Figure 6H).
We also examined the relative affinity of c-Rel to bind to the Ig-κB site in comparison with RelA. To do this, we transiently expressed FLAG-tagged wild-type c-Rel and RelA in HEK 293 cells, stimulated the cells with TNF-α to induce their nuclear translocation, and examined their affinity to bind Ig-κB promoter. We found that both c-Rel and RelA were translocated to the nucleus at comparable levels, yet c-Rel showed significantly enriched binding to the Ig-κB promoter at physiological salt concentration and its binding was retained even at high salt concentration of 250 mM, showing that c-Rel has high binding affinity as well as high avidity to bind RelA-binding sites in the DNA (Figure S5B).
c-Rel Represses TNF-α-Induced Inflammatory Gene Expression In Vivo
TNF-α injection into mice provides an experimental model to study acute inflammation in vivo (Van Bogaert et al., 2011), and it has been shown to enhance the expression of proinflammatory genes in the liver (Catrysse et al., 2016). To investigate the in vivo relevance of our findings, we injected wild-type and c-Rel knockout mice with TNF-α and studied its effect on the in vivo DNA binding of c-Rel and RelA by ChIP as well as on the expression of proinflammatory genes in the liver. We found that c-Rel indeed binds to the RelA-dependent promoters, IP-10 and CXCL1, in vivo (Figure 7A) and its binding was found increased at the time point when RelA binding was decreased at these promoters (Figure 7B). Consistent with the ex vivo cellular experiments, liver cells of c-Rel knockout mice showed enhanced and sustained RelA DNA binding at IP-10 promoter (Figure 7C left). RelA binding at CXCL1 promoter showed a difference in kinetics in c-Rel knockout liver cells, with decreased binding at early time point and substantially increased binding at later time point (Figure 7C right). We also examined the expression of IP-10 and CXCL1 in the liver cells and found that c-Rel deficiency significantly enhanced the acute in vivo expression of TNF-α-induced IP-10 (Figure 7D top). Analysis of CXCL1 expression revealed that, unlike IP-10, the early expression level of CXCL1 was not enhanced, but its expression was sustained with high levels maintained at 3 h post TNF-α injection (Figure 7D bottom).
Figure 7.

c-Rel Represses RelA DNA Binding and Transactivation In Vivo
(A–D) Wild-type or c-Rel knockout mice (n = 3) were injected with PBS or TNF-α (5 μg/mouse). Mice were euthanized at 45, 90, or 180 min after injection, and single-cell suspension of liver was processed for chromatin immunoprecipitation and qPCR. ChIP was performed using control IgG and either c-Rel (A) or RelA (B and C) antibodies. Enrichment of IP-10 and CXCL1 promoter regions in the ChIP samples were examined by qPCR in triplicate. (D) TNF-α-induced in vivo expression of IP-10 (top) and CXCL1 (bottom) relative to that of ribosomal protein L32 in the liver was examined by qPCR in triplicate. Data are presented as mean ± standard error of mean (SEM). p Values were obtained by unpaired Student’s t test; ****p < 0.0001, ***p < 0.001, **p < 0.01, ns non-significant.
(E) Hypothetical schematic model describing DNA-binding-dependent suppression of RelA-dependent transcription by c-Rel. Top. c-Rel-containing dimers with co-repressor, HDAC1, occupy certain RelA-binding sites limiting RelA-induced gene expression. Middle. Absence of c-Rel exposes sites repressed by c-Rel for RelA binding and enhanced inflammatory gene expression. Bottom. Mutation of Y25 in c-Rel blocks c-Rel's DNA binding allowing enhanced RelA binding and inflammatory gene expression.
Taken together, these results demonstrate that c-Rel is a physiologically relevant repressor of TNF-α-induced RelA-dependent transactivation and that it can recruit HDAC1 and competitively bind RelA binding regions in the DNA and thereby selectively repress proinflammatory gene expression (Schematic model, Figure 7E).
Discussion
Here, we identified a role for c-Rel as a repressor of TNF-α-induced, RelA-dependent, pro-inflammatory gene transcription. Based on the canonical function of transactivation domains, we expected that all the NF-κB subunits with transactivation potential, when overexpressed in cells, would behave promiscuously in their ability to induce transcription from prototypical NF-κB promoters. Counterintuitively, c-Rel not only showed poor transactivation potential at Ig-κB, IP-10, and A20 promoters but also significantly suppressed RelA-induced transactivation of these promoters as well as several RelA-induced proinflammatory genes. The repression by c-Rel was not a generic effect as several genes showed decreased induction in the absence of c-Rel (Figures S1A and S1B), suggesting that c-Rel has both activator and suppressor roles following TNF-α stimulation.
The suppressor role of c-Rel was found to be conserved in v-Rel as well, an ability that would be highly advantageous to the reticuloendothelial virus when trying to survive long enough to replicate in a host. Interestingly, RelB was unable to suppress RelA as well as TNF-α-induced gene expression. Both homodimer and heterodimer of RelB with p50 have been shown to bind DNA in co-crystallization studies with consensus NF-κB sequences (Huang et al., 2005, Moorthy et al., 2007). We also found RelB binding to κB sites (Figure S3B), consistent with the suggested ability of RelB containing heterodimers to possess similar DNA binding specificity like c-Rel and RelA-containing heterodimers (Siggers et al., 2011). It has also been shown that stably overexpressed RelB can inhibit RelA activity by sequestering RelA and preventing its DNA binding (Marienfeld et al., 2003). Despite its abilities to bind consensus NF-κB sequences and to inhibit RelA in an enforced expression system, why RelB does not interfere with RelA-induced transactivation under these conditions remains to be addressed. Furthermore, RelB has been shown to bind DNA poorly in vivo, most likely due to the sequestration of RelB-containing complexes by NF-κB p100 (Derudder et al., 2003) or their repression by RelA binding (Jacque et al., 2005), which may restrict its DNA binding ability. This suggests that, although c-Rel and RelB are generally classified as transactivating members of NF-κB family, there are inherent differences in the manner in which they form functional activating or repressing dimers and bind DNA, which warrants further structure-function studies.
The repressor function of c-Rel on RelA-dependent transactivation was found to be dependent on its DNA binding ability. The higher affinity of c-Rel to bind to the κB promoters (Figure S5B), along with increased RelA DNA binding in the absence of c-Rel and decreased RelA DNA binding upon c-Rel overexpression, points to a competitive blockade of RelA binding by c-Rel. However, more comprehensive studies are necessary to delineate whether the c-Rel-containing dimers preoccupy the RelA site preventing RelA's access or they competitively displace RelA complexes from the DNA based on c-Rel's higher affinity for the κB sites. In the transient co-expression experiments using FLAG-tagged proteins, despite the lower expression levels (Figures 2B and 6B), c-Rel was able to cause significant suppression at the studied promoters and showed higher binding strength to the Ig-κB-site. This is logically sound, especially outside of the lymphoid and myeloid compartments, where RelA expression has been shown to be much higher in most of these cell types than c-Rel (Oeckinghaus and Ghosh, 2009); an activated system still needs to suppress an over-stimulated cell's response even with a paucity of c-Rel.
Our results show that lack of c-Rel does not lead to an increase in RelA:p50 dimers; however, we found an increase in both RelA:IκB-α and p50:IκB-α complexes (Figure 4E). This indicates that c-Rel deficiency may result in an enhancement of RelA and p50 homodimers or other heterodimers containing RelA and p50 that may account for enhanced RelA nuclear translocation observed in c-Rel knockout cells. Our results also show that the enhanced RelA-dependent transactivation in c-Rel knockout cells is not due to increased p300 binding in the transcription complex at the minimal promoter sequence (Figure S4B). However, it is possible that p300 or other co-activators may show enhanced binding at a distal locus in the promoter region and enhance the gene expression. Whether the repressor dimer containing c-Rel is a homodimer or a heterodimer with p50 or other NF-κB subunit is an interesting question to address. Our Ig-κB oligonucleotide pull-down data show the presence of c-Rel and p50 in the wild-type cells at 120 min of TNF-α stimulation (Figure 4D). However, DNA-binding p50 is absent at the 120-min time point in c-Rel knockout cells, implying a need for c-Rel/p50 heterodimers to bind DNA at this later time point. This may very well be the dimer that represses RelA transcriptional activity.
In addition to its role in DNA binding, c-Rel also plays a role in the recruitment of co-repressor HDAC1 to an open promoter site, with no substantial effect on HDAC3 binding (Figures 5D and 5E). The increased TNF-induced NF-κB-dependent HDAC1 recruitment is consistent with previous studies, which show the enhancement of both HATs and HDACs at active transcription sites (Li et al., 2014a, Peserico and Simone, 2011, Wang et al., 2009). c-Rel contains two transactivating domains and does in fact drive transcriptional activation at several κB sites (Rao et al., 2003, Sanjabi et al., 2000, Siebenlist et al., 1994). In spite of the presence of two TADs, why c-Rel acts as an inhibitor at selected sites and whether this is a sequence-specific phenomenon remain to be determined. It appears that c-Rel is imperative for proper immune function as well as to fine-tune the quantitative output of RelA-induced transactivation by setting a threshold limiting the immune response and keeping smaller insults from generating disproportionately large inflammatory responses.
c-Rel-deficient mice have normal immunological development but poor immunological function (Gilmore and Gerondakis, 2011). c-Rel deficiency has been shown to result in an increased systemic inflammatory response as a result of polymicrobial sepsis in a cecal ligation and puncture mouse model (Courtine et al., 2011). The increased mortality in this model could stem from an increased proinflammatory cytokine storm or decreased expression of c-Rel-dependent genes regulating survival or compromised adaptive immune response. We also found an enhanced inflammatory response in a c-Rel deficient non-obese diabetic (NOD) mouse model that showed greatly accelerated diabetogenesis, a tell-tale consequence of enhanced pancreatic inflammation (Ramakrishnan et al., 2016). In contrast to these anti-inflammatory roles of c-Rel, several studies have also reported a proinflammatory role of c-Rel in systemic inflammation in collagen-induced arthritis (Campbell et al., 2000) and ovalbumin-induced pulmonary inflammation (Donovan et al., 1999). How is it possible that c-Rel displays both pro- and anti-inflammatory roles, in a cell type- or disease-dependent manner? We show here that c-Rel acts as a repressor of only a specific subset of TNF-α-induced RelA-driven proinflammatory genes (Figure 1). We and others have shown that c-Rel also positively regulates the expression of several proinflammatory genes such as IL-2, GM-CSF, IFNG, IP10, ICAM1, and TNF (Carmody et al., 2007, Chen et al., 2010, Gilmore and Gerondakis, 2011, Ramakrishnan et al., 2013); immunosuppressive FOXP3 in T regulatory cells (Isomura et al., 2009); and feedback regulators of NF-κB activation such as IκB-α (Figure S1). We show here that c-Rel is not a global suppressor of TNF-α-induced gene expression, and in fact several inflammatory genes are indeed dependent on c-Rel, including TNF-α itself (Figure S1), and several others show altered expression kinetics in the absence of c-Rel (Figure 1B). Specifically, c-Rel was found required for auto-induction of TNF-α following TNF-α stimulation (Figure S1), and this dampening of TNF-α expression may limit self-sustaining chronic inflammatory cascade. This decrease in TNF-α expression in c-Rel knockout cells is in line with the protective effect seen in c-Rel knockout mice for TNF-α-dependent inflammatory diseases such as rheumatoid arthritis (Campbell et al., 2000).
Providing a direct correlation of our findings with human disease relevance, a recent study reported a patient with a homozygous mutation causing functional knockout of c-Rel protein expression and combined immunodeficiency (Beaussant-Cohen et al., 2019). B cell responses to CD40L, IL-2 production by T cells, and myeloid-derived production of IL-12 family members IL-12, IL-21, and IL-23 were all impaired in this patient, enhancing susceptibility to opportunistic infections. These defects are consistent with those reported in c-Rel knockout mice (Gilmore and Gerondakis, 2011), and it will be interesting to study whether altered inflammatory gene expression observed in c-Rel knockout mouse recapitulates in c-Rel-deficient humans. Further studies are also required to study the striking role of c-Rel Y25H SNP in human population (Schuster et al., 2010), which compromises suppressive effect of c-Rel on RelA-dependent inflammatory gene expression (Figure 6). It also remains unknown whether Y25 is a phosphorylation site that may influence c-Rel function. Thus, our findings showing the role of c-Rel in the regulation of inflammation as a suppressor, an inducer, and a modulator of kinetics of gene expression provide a conceivable mechanism for the paradoxical observations of c-Rel-driven resistance and susceptibility to various inflammatory and autoimmune pathologies. These finding also pose c-Rel as a potential target to develop therapeutics to control the deleterious effects of uncontrolled inflammation.
Limitations of the Study
We showed a possible mechanism for the anti-inflammatory role of c-Rel in which it represses TNF-induced RelA transcriptional activity by competitive κB-site binding and HDAC1 recruitment. However, the exact c-Rel containing canonical dimer(s) (c-Rel:c-Rel homodimer or heterodimer of c-Rel with RelA, RelB, p50 or p52) mediating the repressive function is currently unknown. Additionally, we do not know whether posttranslational modification of c-Rel is involved in this process. Although we showed the repressor role of c-Rel in four cell types, i.e., mouse fibroblasts, human embryonic kidney cells, mouse primary macrophages, as well as in vivo in mouse liver cells using proinflammatory cytokine TNF, whether this happens in other cell types and in response to other inflammatory stimuli remains to be investigated.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We thank Dr. Thomas Gilmore for the gift of the v-Rel plasmid. We thank Dr. Denis Guttridge, at The Ohio State University College of Medicine, for the gift of the RelA and RelB MEFs, and Dr. George Dubyak for the J558 cells. We thank Dr. David Baltimore, California Institute of Technology for Ig-κB, IP-10, and A20 reporter gene plasmids. We also thank Drs. Shrikanth Basavarajappa, Jeffery Tomalka, Sudhanshu Shukla, and Joseph Rathkey for insightful discussions and Keman Zhang for the pLM lentiviral vector. CRISPR guides were designed using CRISPR.mit.edu. We thank Ms. Erin O'Kelly for proofreading the article. This work was supported by the National Institutes of Health, NIH/NIAID grants R01AI116730 and R21AI144264 to P.R. T.J.D was supported by Dermatology T32 pre-doctoral fellowship, National Institutes of Health, NIH/NIAMS 5T32AR007569.
Author Contributions
Conceptualization, P.R.; Methodology, T.J.D. and P.R., Investigation, T.J.D. and P.R., Validation, T.J.D., Writing – Original Draft, T.J.D., Writing – Review and Editing, P.R., Supervision, P.R., Project Administration, P.R., Funding Acquisition, P.R.
Declaration of Interests
Authors declare no competing financial interests.
Published: March 27, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.100876.
Data and Code Availability
All the data and methods necessary to reproduce this study are included in the manuscript and Supplemental Information. Reagent request will be readily fulfilled following the materials transfer policies of Case Western Reserve University.
Supplemental Information
References
- Amir-Zilberstein L., Dikstein R. Interplay between E-box and NF-kappaB in regulation of A20 gene by DRB sensitivity-inducing factor (DSIF) J. Biol. Chem. 2008;283:1317–1323. doi: 10.1074/jbc.M706767200. [DOI] [PubMed] [Google Scholar]
- Baeuerle P.A., Baltimore D. Activation of DNA-binding activity in an apparently cytoplasmic precursor of the NF-kappa B transcription factor. Cell. 1988;53:211–217. doi: 10.1016/0092-8674(88)90382-0. [DOI] [PubMed] [Google Scholar]
- Baeuerle P.A., Baltimore D. I kappa B: a specific inhibitor of the NF-kappa B transcription factor. Science. 1988;242:540–546. doi: 10.1126/science.3140380. [DOI] [PubMed] [Google Scholar]
- Ballard D.W., Walker W.H., Doerre S., Sista P., Molitor J.A., Dixon E.P., Peffer N.J., Hannink M., Greene W.C. The v-rel oncogene encodes a kappa B enhancer binding protein that inhibits NF-kappa B function. Cell. 1990;63:803–814. doi: 10.1016/0092-8674(90)90146-6. [DOI] [PubMed] [Google Scholar]
- Ballard D.W., Dixon E.P., Peffer N.J., Bogerd H., Doerre S., Stein B., Greene W.C. The 65-kDa subunit of human NF-kappa B functions as a potent transcriptional activator and a target for v-Rel-mediated repression. Proc. Natl. Acad. Sci. U S A. 1992;89:1875–1879. doi: 10.1073/pnas.89.5.1875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bash J., Zong W.X., Gelinas C. c-Rel arrests the proliferation of HeLa cells and affects critical regulators of the G1/S-phase transition. Mol. Cell. Biol. 1997;17:6526–6536. doi: 10.1128/mcb.17.11.6526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaussant-Cohen S., Jaber F., Massaad M.J., Weeks S., Jones J., Alosaimi M.F., Wallace J., Al-Herz W., Geha R.S., Chou J. Combined immunodeficiency in a patient with c-Rel deficiency. J. Allergy Clin. Immunol. 2019;144:606–608.e4. doi: 10.1016/j.jaci.2019.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beg A.A., Ruben S.M., Scheinman R.I., Haskill S., Rosen C.A., Baldwin A.S., Jr. I kappa B interacts with the nuclear localization sequences of the subunits of NF-kappa B: a mechanism for cytoplasmic retention. Genes Dev. 1992;6:1899–1913. doi: 10.1101/gad.6.10.1899. [DOI] [PubMed] [Google Scholar]
- Bhatt D., Ghosh S. Regulation of the NF-kappaB-Mediated transcription of inflammatory genes. Front. Immunol. 2014;5:71. doi: 10.3389/fimmu.2014.00071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bours V., Franzoso G., Azarenko V., Park S., Kanno T., Brown K., Siebenlist U. The oncoprotein Bcl-3 directly transactivates through kappa B motifs via association with DNA-binding p50B homodimers. Cell. 1993;72:729–739. doi: 10.1016/0092-8674(93)90401-b. [DOI] [PubMed] [Google Scholar]
- Campbell I.K., Gerondakis S., O'Donnell K., Wicks I.P. Distinct roles for the NF-kappaB1 (p50) and c-Rel transcription factors in inflammatory arthritis. J. Clin. Invest. 2000;105:1799–1806. doi: 10.1172/JCI8298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capobianco A.J., Simmons D.L., Gilmore T.D. Cloning and expression of a chicken c-rel cDNA: unlike p59v-rel, p68c-rel is a cytoplasmic protein in chicken embryo fibroblasts. Oncogene. 1990;5:257–265. [PubMed] [Google Scholar]
- Carmody R.J., Ruan Q., Liou H.C., Chen Y.H. Essential roles of c-Rel in TLR-induced IL-23 p19 gene expression in dendritic cells. J. Immunol. 2007;178:186–191. doi: 10.4049/jimmunol.178.1.186. [DOI] [PubMed] [Google Scholar]
- Carrasco D., Rizzo C.A., Dorfman K., Bravo R. The v-rel oncogene promotes malignant T-cell leukemia/lymphoma in transgenic mice. EMBO J. 1996;15:3640–3650. [PMC free article] [PubMed] [Google Scholar]
- Catrysse L., Farhang Ghahremani M., Vereecke L., Youssef S.A., Mc Guire C., Sze M., Weber A., Heikenwalder M., de Bruin A., Beyaert R. A20 prevents chronic liver inflammation and cancer by protecting hepatocytes from death. Cell Death Dis. 2016;7:e2250. doi: 10.1038/cddis.2016.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen F.E., Huang D.B., Chen Y.Q., Ghosh G. Crystal structure of p50/p65 heterodimer of transcription factor NF-kappaB bound to DNA. Nature. 1998;391:410–413. doi: 10.1038/34956. [DOI] [PubMed] [Google Scholar]
- Chen X., Kandasamy K., Srivastava R.K. Differential roles of RelA (p65) and c-Rel subunits of nuclear factor kappa B in tumor necrosis factor-related apoptosis-inducing ligand signaling. Cancer Res. 2003;63:1059–1066. [PubMed] [Google Scholar]
- Chen G., Hardy K., Bunting K., Daley S., Ma L., Shannon M.F. Regulation of the IL-21 gene by the NF-kappaB transcription factor c-Rel. J. Immunol. 2010;185:2350–2359. doi: 10.4049/jimmunol.1000317. [DOI] [PubMed] [Google Scholar]
- Chen G., Hardy K., Pagler E., Ma L., Lee S., Gerondakis S., Daley S., Shannon M.F. The NF-kappaB transcription factor c-Rel is required for Th17 effector cell development in experimental autoimmune encephalomyelitis. J. Immunol. 2011;187:4483–4491. doi: 10.4049/jimmunol.1101757. [DOI] [PubMed] [Google Scholar]
- Courtine E., Pene F., Cagnard N., Toubiana J., Fitting C., Brocheton J., Rousseau C., Gerondakis S., Chiche J.D., Ouaaz F. Critical role of cRel subunit of NF-kappaB in sepsis survival. Infect. Immun. 2011;79:1848–1854. doi: 10.1128/IAI.00021-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derudder E., Dejardin E., Pritchard L.L., Green D.R., Korner M., Baud V. RelB/p50 dimers are differentially regulated by tumor necrosis factor-alpha and lymphotoxin-beta receptor activation: critical roles for p100. J. Biol. Chem. 2003;278:23278–23284. doi: 10.1074/jbc.M300106200. [DOI] [PubMed] [Google Scholar]
- Donovan C.E., Mark D.A., He H.Z., Liou H.C., Kobzik L., Wang Y., De Sanctis G.T., Perkins D.L., Finn P.W. NF-kappa B/Rel transcription factors: c-Rel promotes airway hyperresponsiveness and allergic pulmonary inflammation. J. Immunol. 1999;163:6827–6833. [PubMed] [Google Scholar]
- Elsharkawy A.M., Oakley F., Lin F., Packham G., Mann D.A., Mann J. The NF-kappaB p50:p50:HDAC-1 repressor complex orchestrates transcriptional inhibition of multiple pro-inflammatory genes. J. Hepatol. 2010;53:519–527. doi: 10.1016/j.jhep.2010.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyre S., Hinks A., Flynn E., Martin P., Wilson A.G., Maxwell J.R., Morgan A.W., Emery P., Steer S., Hocking L.J. Confirmation of association of the REL locus with rheumatoid arthritis susceptibility in the UK population. Ann. Rheum. Dis. 2010;69:1572–1573. doi: 10.1136/ard.2009.122887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan T., Wang S., Yu L., Yi H., Liu R., Geng W., Wan X., Ma Y., Cai L., Chen Y.H. Treating psoriasis by targeting its susceptibility gene Rel. Clin. Immunol. 2016;165:47–54. doi: 10.1016/j.clim.2016.03.009. [DOI] [PubMed] [Google Scholar]
- Fan T., Zhong F., Liu R., Chen Y.H., Wang T., Ruan Q. siRNA-mediated c-Rel knockdown ameliorates collagen-induced arthritis in mice. Int. Immunopharmacol. 2018;56:9–17. doi: 10.1016/j.intimp.2018.01.010. [DOI] [PubMed] [Google Scholar]
- Fullard N., Moles A., O'Reilly S., van Laar J.M., Faini D., Diboll J., Reynolds N.J., Mann D.A., Reichelt J., Oakley F. The c-Rel subunit of NF-kappaB regulates epidermal homeostasis and promotes skin fibrosis in mice. Am. J. Pathol. 2013;182:2109–2120. doi: 10.1016/j.ajpath.2013.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghizzoni M., Haisma H.J., Maarsingh H., Dekker F.J. Histone acetyltransferases are crucial regulators in NF-kappaB mediated inflammation. Drug Discov. Today. 2011;16:504–511. doi: 10.1016/j.drudis.2011.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh S., Hayden M.S. New regulators of NF-kappaB in inflammation. Nat. Rev. Immunol. 2008;8:837–848. doi: 10.1038/nri2423. [DOI] [PubMed] [Google Scholar]
- Ghosh S., May M.J., Kopp E.B. NF-kappa B and Rel proteins: evolutionarily conserved mediators of immune responses. Annu. Rev. Immunol. 1998;16:225–260. doi: 10.1146/annurev.immunol.16.1.225. [DOI] [PubMed] [Google Scholar]
- Gilmore T.D. Introduction: the Rel/NF-kappaB signal transduction pathway. Semin. Cancer Biol. 1997;8:61–62. doi: 10.1006/scbi.1997.0056. [DOI] [PubMed] [Google Scholar]
- Gilmore T.D. Introduction to NF-kappaB: players, pathways, perspectives. Oncogene. 2006;25:6680–6684. doi: 10.1038/sj.onc.1209954. [DOI] [PubMed] [Google Scholar]
- Gilmore T.D., Gerondakis S. The c-Rel transcription factor in development and disease. Genes Cancer. 2011;2:695–711. doi: 10.1177/1947601911421925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilliard B.A., Mason N., Xu L., Sun J., Lamhamedi-Cherradi S.E., Liou H.C., Hunter C., Chen Y.H. Critical roles of c-Rel in autoimmune inflammation and helper T cell differentiation. J. Clin. Invest. 2002;110:843–850. doi: 10.1172/JCI15254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hrdlickova R., Nehyba J., Humphries E.H. v-rel induces expression of three avian immunoregulatory surface receptors more efficiently than c-rel. J. Virol. 1994;68:308–319. doi: 10.1128/jvi.68.1.308-319.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsia C.Y., Cheng S., Owyang A.M., Dowdy S.F., Liou H.C. c-Rel regulation of the cell cycle in primary mouse B lymphocytes. Int. Immunol. 2002;14:905–916. doi: 10.1093/intimm/dxf055. [DOI] [PubMed] [Google Scholar]
- Huang D.B., Chen Y.Q., Ruetsche M., Phelps C.B., Ghosh G. X-ray crystal structure of proto-oncogene product c-Rel bound to the CD28 response element of IL-2. Structure. 2001;9:669–678. doi: 10.1016/s0969-2126(01)00635-9. [DOI] [PubMed] [Google Scholar]
- Huang D.B., Vu D., Ghosh G. NF-kappaB RelB forms an intertwined homodimer. Structure. 2005;13:1365–1373. doi: 10.1016/j.str.2005.06.018. [DOI] [PubMed] [Google Scholar]
- Huguet C., Mattot V., Bouali F., Stehelin D., Vandenbunder B., Abbadie C. The avian transcription factor c-Rel is induced and translocates into the nucleus of thymocytes undergoing apoptosis. Cell Death Differ. 1997;4:413–422. doi: 10.1038/sj.cdd.4400259. [DOI] [PubMed] [Google Scholar]
- Inoue J., Kerr L.D., Ransone L.J., Bengal E., Hunter T., Verma I.M. c-rel activates but v-rel suppresses transcription from kappa B sites. Proc. Natl. Acad. Sci. U S A. 1991;88:3715–3719. doi: 10.1073/pnas.88.9.3715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isomura I., Palmer S., Grumont R.J., Bunting K., Hoyne G., Wilkinson N., Banerjee A., Proietto A., Gugasyan R., Wu L. c-Rel is required for the development of thymic Foxp3+ CD4 regulatory T cells. J. Exp. Med. 2009;206:3001–3014. doi: 10.1084/jem.20091411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacque E., Tchenio T., Piton G., Romeo P.H., Baud V. RelA repression of RelB activity induces selective gene activation downstream of TNF receptors. Proc. Natl. Acad. Sci. U S A. 2005;102:14635–14640. doi: 10.1073/pnas.0507342102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin W.J., Kim B., Kim D., Park Choo H.Y., Kim H.H., Ha H., Lee Z.H. NF-kappaB signaling regulates cell-autonomous regulation of CXCL10 in breast cancer 4T1 cells. Exp. Mol. Med. 2017;49:e295. doi: 10.1038/emm.2016.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kempe S., Kestler H., Lasar A., Wirth T. NF-kappaB controls the global pro-inflammatory response in endothelial cells: evidence for the regulation of a pro-atherogenic program. Nucleic Acids Res. 2005;33:5308–5319. doi: 10.1093/nar/gki836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kontgen F., Grumont R.J., Strasser A., Metcalf D., Li R., Tarlinton D., Gerondakis S. Mice lacking the c-rel proto-oncogene exhibit defects in lymphocyte proliferation, humoral immunity, and interleukin-2 expression. Genes Dev. 1995;9:1965–1977. doi: 10.1101/gad.9.16.1965. [DOI] [PubMed] [Google Scholar]
- Kunsch C., Ruben S.M., Rosen C.A. Selection of optimal kappa B/Rel DNA-binding motifs: interaction of both subunits of NF-kappa B with DNA is required for transcriptional activation. Mol. Cell. Biol. 1992;12:4412–4421. doi: 10.1128/mcb.12.10.4412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenardo M.J., Baltimore D. NF-kappa B: a pleiotropic mediator of inducible and tissue-specific gene control. Cell. 1989;58:227–229. doi: 10.1016/0092-8674(89)90833-7. [DOI] [PubMed] [Google Scholar]
- Li X., Yang H., Huang S., Qiu Y. Histone deacetylase 1 and p300 can directly associate with chromatin and compete for binding in a mutually exclusive manner. PLoS One. 2014;9:e94523. doi: 10.1371/journal.pone.0094523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X., Zhao Y., Tian B., Jamaluddin M., Mitra A., Yang J., Rowicka M., Brasier A.R., Kudlicki A. Modulation of gene expression regulated by the transcription factor NF-kappaB/RelA. J. Biol. Chem. 2014;289:11927–11944. doi: 10.1074/jbc.M113.539965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim C.A., Yao F., Wong J.J., George J., Xu H., Chiu K.P., Sung W.K., Lipovich L., Vega V.B., Chen J. Genome-wide mapping of RELA(p65) binding identifies E2F1 as a transcriptional activator recruited by NF-kappaB upon TLR4 activation. Mol. Cell. 2007;27:622–635. doi: 10.1016/j.molcel.2007.06.038. [DOI] [PubMed] [Google Scholar]
- Lorenz V.N., Schon M.P., Seitz C.S. c-Rel downregulation affects cell cycle progression of human keratinocytes. J. Invest. Dermatol. 2014;134:415–422. doi: 10.1038/jid.2013.315. [DOI] [PubMed] [Google Scholar]
- Marienfeld R., May M.J., Berberich I., Serfling E., Ghosh S., Neumann M. RelB forms transcriptionally inactive complexes with RelA/p65. J. Biol. Chem. 2003;278:19852–19860. doi: 10.1074/jbc.M301945200. [DOI] [PubMed] [Google Scholar]
- Matzinger P. The danger model: a renewed sense of self. Science. 2002;296:301–305. doi: 10.1126/science.1071059. [DOI] [PubMed] [Google Scholar]
- Medzhitov R. Origin and physiological roles of inflammation. Nature. 2008;454:428–435. doi: 10.1038/nature07201. [DOI] [PubMed] [Google Scholar]
- Moorthy A.K., Huang D.B., Wang V.Y., Vu D., Ghosh G. X-ray structure of a NF-kappaB p50/RelB/DNA complex reveals assembly of multiple dimers on tandem kappaB sites. J. Mol. Biol. 2007;373:723–734. doi: 10.1016/j.jmb.2007.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathan C. Points of control in inflammation. Nature. 2002;420:846–852. doi: 10.1038/nature01320. [DOI] [PubMed] [Google Scholar]
- Naumann M., Wulczyn F.G., Scheidereit C. The NF-kappa B precursor p105 and the proto-oncogene product Bcl-3 are I kappa B molecules and control nuclear translocation of NF-kappa B. EMBO J. 1993;12:213–222. doi: 10.1002/j.1460-2075.1993.tb05647.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Netea M.G., Balkwill F., Chonchol M., Cominelli F., Donath M.Y., Giamarellos-Bourboulis E.J., Golenbock D., Gresnigt M.S., Heneka M.T., Hoffman H.M. A guiding map for inflammation. Nat. Immunol. 2017;18:826–831. doi: 10.1038/ni.3790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oeckinghaus A., Ghosh S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb. Perspect. Biol. 2009;1:a000034. doi: 10.1101/cshperspect.a000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owyang A.M., Tumang J.R., Schram B.R., Hsia C.Y., Behrens T.W., Rothstein T.L., Liou H.C. c-Rel is required for the protection of B cells from antigen receptor-mediated, but not Fas-mediated, apoptosis. J. Immunol. 2001;167:4948–4956. doi: 10.4049/jimmunol.167.9.4948. [DOI] [PubMed] [Google Scholar]
- Peserico A., Simone C. Physical and functional HAT/HDAC interplay regulates protein acetylation balance. J. Biomed. Biotechnol. 2011;2011:371832. doi: 10.1155/2011/371832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomerantz J.L., Baltimore D. Two pathways to NF-kappaB. Mol. Cell. 2002;10:693–695. doi: 10.1016/s1097-2765(02)00697-4. [DOI] [PubMed] [Google Scholar]
- Ramakrishnan P., Wang W., Wallach D. Receptor-specific signaling for both the alternative and the canonical NF-kappaB activation pathways by NF-kappaB-inducing kinase. Immunity. 2004;21:477–489. doi: 10.1016/j.immuni.2004.08.009. [DOI] [PubMed] [Google Scholar]
- Ramakrishnan P., Clark P.M., Mason D.E., Peters E.C., Hsieh-Wilson L.C., Baltimore D. Activation of the transcriptional function of the NF-kappaB protein c-Rel by O-GlcNAc glycosylation. Sci. Signal. 2013;6:ra75. doi: 10.1126/scisignal.2004097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramakrishnan P., Yui M.A., Tomalka J.A., Majumdar D., Parameswaran R., Baltimore D. Deficiency of nuclear factor-kappaB c-Rel accelerates the development of autoimmune diabetes in NOD mice. Diabetes. 2016;65:2367–2379. doi: 10.2337/db15-1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao S., Gerondakis S., Woltring D., Shannon M.F. c-Rel is required for chromatin remodeling across the IL-2 gene promoter. J. Immunol. 2003;170:3724–3731. doi: 10.4049/jimmunol.170.7.3724. [DOI] [PubMed] [Google Scholar]
- Rocha S., Martin A.M., Meek D.W., Perkins N.D. p53 represses cyclin D1 transcription through down regulation of Bcl-3 and inducing increased association of the p52 NF-kappaB subunit with histone deacetylase 1. Mol. Cell. Biol. 2003;23:4713–4727. doi: 10.1128/MCB.23.13.4713-4727.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjabi S., Hoffmann A., Liou H.C., Baltimore D., Smale S.T. Selective requirement for c-Rel during IL-12 P40 gene induction in macrophages. Proc. Natl. Acad. Sci. U S A. 2000;97:12705–12710. doi: 10.1073/pnas.230436397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz M.L., Baeuerle P.A. The p65 subunit is responsible for the strong transcription activating potential of NF-kappa B. EMBO J. 1991;10:3805–3817. doi: 10.1002/j.1460-2075.1991.tb04950.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuster S.C., Miller W., Ratan A., Tomsho L.P., Giardine B., Kasson L.R., Harris R.S., Petersen D.C., Zhao F., Qi J. Complete Khoisan and Bantu genomes from southern Africa. Nature. 2010;463:943–947. doi: 10.1038/nature08795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedger L.M., McDermott M.F. TNF and TNF-receptors: from mediators of cell death and inflammation to therapeutic giants - past, present and future. Cytokine Growth Factor Rev. 2014;25:453–472. doi: 10.1016/j.cytogfr.2014.07.016. [DOI] [PubMed] [Google Scholar]
- Sen R., Baltimore D. Multiple nuclear factors interact with the immunoglobulin enhancer sequences. Cell. 1986;46:705–716. doi: 10.1016/0092-8674(86)90346-6. [DOI] [PubMed] [Google Scholar]
- Serhan C.N., Brain S.D., Buckley C.D., Gilroy D.W., Haslett C., O'Neill L.A., Perretti M., Rossi A.G., Wallace J.L. Resolution of inflammation: state of the art, definitions and terms. FASEB J. 2007;21:325–332. doi: 10.1096/fj.06-7227rev. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siebenlist U., Franzoso G., Brown K. Structure, regulation and function of NF-kappa B. Annu. Rev. Cell Biol. 1994;10:405–455. doi: 10.1146/annurev.cb.10.110194.002201. [DOI] [PubMed] [Google Scholar]
- Siggers T., Chang A.B., Teixeira A., Wong D., Williams K.J., Ahmed B., Ragoussis J., Udalova I.A., Smale S.T., Bulyk M.L. Principles of dimer-specific gene regulation revealed by a comprehensive characterization of NF-kappaB family DNA binding. Nat. Immunol. 2011;13:95–102. doi: 10.1038/ni.2151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tourniaire F., Romier-Crouzet B., Lee J.H., Marcotorchino J., Gouranton E., Salles J., Malezet C., Astier J., Darmon P., Blouin E. Chemokine expression in inflamed adipose tissue is mainly mediated by NF-kappaB. PLoS One. 2013;8:e66515. doi: 10.1371/journal.pone.0066515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tumang J.R., Owyang A., Andjelic S., Jin Z., Hardy R.R., Liou M.L., Liou H.C. c-Rel is essential for B lymphocyte survival and cell cycle progression. Eur. J. Immunol. 1998;28:4299–4312. doi: 10.1002/(SICI)1521-4141(199812)28:12<4299::AID-IMMU4299>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- Urban M.B., Schreck R., Baeuerle P.A. NF-kappa B contacts DNA by a heterodimer of the p50 and p65 subunit. EMBO J. 1991;10:1817–1825. doi: 10.1002/j.1460-2075.1991.tb07707.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Bogaert T., Vandevyver S., Dejager L., Van Hauwermeiren F., Pinheiro I., Petta I., Engblom D., Kleyman A., Schutz G., Tuckermann J. Tumor necrosis factor inhibits glucocorticoid receptor function in mice: a strong signal toward lethal shock. J. Biol. Chem. 2011;286:26555–26567. doi: 10.1074/jbc.M110.212365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wajant H., Scheurich P. TNFR1-induced activation of the classical NF-kappaB pathway. FEBS J. 2011;278:862–876. doi: 10.1111/j.1742-4658.2011.08015.x. [DOI] [PubMed] [Google Scholar]
- Walker W.H., Stein B., Ganchi P.A., Hoffman J.A., Kaufman P.A., Ballard D.W., Hannink M., Greene W.C. The v-rel oncogene: insights into the mechanism of transcriptional activation, repression, and transformation. J. Virol. 1992;66:5018–5029. doi: 10.1128/jvi.66.8.5018-5029.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z., Zang C., Cui K., Schones D.E., Barski A., Peng W., Zhao K. Genome-wide mapping of HATs and HDACs reveals distinct functions in active and inactive genes. Cell. 2009;138:1019–1031. doi: 10.1016/j.cell.2009.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All the data and methods necessary to reproduce this study are included in the manuscript and Supplemental Information. Reagent request will be readily fulfilled following the materials transfer policies of Case Western Reserve University.