

Abstract
Gut microbiome community structure is associated with Crohn’s disease (CD) development and response to therapy. Bile acids (BAs) play a central role in modulating intestinal immune responses, and changes in gut bacterial communities can profoundly alter the intestinal BA pool. The liver synthesizes and conjugates primary bile acids (priBAs) that are then deconjugated, epimerized, and dehydroxylated by gut bacteria to produce secondary bile acids (secBAs). We investigated the relationship between the gut microbiome and the fecal BA pool in stool samples obtained from a well-characterized cohort of pediatric CD patients undergoing nutritional therapy to induce disease remission. We found that fecal BA composition was altered in a sub-group of CD patients who did not sustain remission. The microbial community structures associated with priBA and secBA-dominant profiles were distinct. In addition, the fecal BA concentrations were correlated with the abundance of distinct bacterial taxonomic groups. Finally, priBA dominant samples were associated with community-level decreases in enzymes for dehydroxylation but not deconjugation.
Subject terms: Crohn's disease, Metagenomics, Metabolomics
Introduction
Crohn’s disease (CD), a major form of inflammatory bowel disease (IBD), is a relapsing and remitting condition characterized by chronic inflammation throughout the gastrointestinal (GI) tract. The etiology of CD is complex and multifactorial, but convergent data indicate that disease is strongly associated with disturbances to the gut microbiota, i.e., dysbiosis [1–4]. General features of CD-associated dysbiosis include decreases in overall taxonomic diversity and abundance of bacteria within the phyla Firmicutes and Bacteroidetes, which include important ‘anti-inflammatory’ taxa [5, 6]. These changes often correspond with an expansion of members of the Proteobacteria phylum, which include potentially pathogenic bacteria [5].
Changes in the gut microbiome correspond with disturbed microbial metabolic functions in CD/IBD patients, including pathological changes in the metabolism of bile acids (BAs) within the colon [7]. BAs are amphipathic molecules that function mainly to emulsify and solubilize fats for absorption. The human liver synthesizes two primary bile acids (priBAs) from cholesterol—cholic acid (CA) and chenodeoxycholic acid (CDCA)—which are conjugated to glycine or taurine before being secreted into the small intestine. While most priBAs are actively absorbed in the distal small intestine, ~5% are extensively biotransformed by gut bacteria [8, 9]. CA and CDCA are deconjugated from glycine or taurine by bacterial bile salt hydrolase (BSH), which is enriched in the human gut microbiome [10]. Deconjugated CA and CDCA are then biotransformed by 7α-dehydroxylation into the predominant secondary bile acids (secBAs) deoxycholic acid (DCA) and lithocholic acid (LCA), respectively. DCA and LCA are then reabsorbed or excreted in stool, where they normally comprise 80–90% of total fecal BA [11]. 7α-dehydroxylation is carried out by a relatively limited set of bacteria that possess the bile acid inducible (bai) operon [12]. Recent estimates indicate that only 0.0001% of colonic bacteria are capable of performing this reaction, and these bacteria belong mainly to the Clostridium genus [8, 12, 13]. In addition to 7α-dehydroxylation, CDCA can also undergo 7α-dehydrogenation and epimerization to form ursodeoxycholic acid (UDCA), a minor secBA typically present in low concentrations (<2% total fecal BA) [11, 14].
BAs play an important role modulating both the inflammatory responses and bacterial community composition within the gut [15]. BAs have antimicrobial effects that are strain-dependent [16]. Deconjugated BAs and secBAs are more readily absorbed by intestinal epithelial cells and can interact with nuclear receptors such as farnesoid X receptor (FXR), which has been shown to have protective functions in CD/IBD by promoting anti-inflammatory immune responses and maintaining epithelial barrier integrity [17]. Disrupted BA metabolism has been repeatedly observed in IBD patients, with fecal BA pools skewed toward decreased secBAs and increased priBAs relative to healthy controls [18–22]. Together these observations indicate that microbial capacity for BA modification could play an important role in the pathophysiology of IBD.
In this context, we hypothesized that disrupted microbial BA metabolism could help explain the role of the microbiome in outcomes of nutritional therapy in a cohort of well-characterized pediatric CD patients. Exclusive enteral nutrition (EEN) is a liquid formula diet accepted internationally as first-line induction therapy for pediatric CD [23]. Lack of response and/or inability to sustain remission following EEN has been linked to certain microbiome features of CD patient cohorts, including reduced diversity and a persistently increased abundance of Proteobacteria ([3, 24, 25]. However, the mechanisms through which the microbiome influences clinical responses to EEN are unclear [25]. In this study, we used 16S rRNA amplicon data and metagenomic sequence data to examine the relationship between microbial community structure and fecal BA profiles in longitudinal stool samples from pediatric CD patients who were treated with EEN to induce remission.
Materials and methods
Study design, subjects, and sampling
Seventeen pediatric CD patients participated in a longitudinal observational study of microbiome changes associated with EEN therapy (Metagenomic Approach to Remission using Exclusive Enteral Nutrition—MAREEN —2014–2017). All study patients had a confirmed CD diagnosis based on established clinical, endoscopic, histological, and/or radiological criteria. Information on patient demographics, disease location, medications, and results of laboratory tests were collected from medical records. All patients were administered EEN via nasogastric tube. Patients who achieved the primary endpoint of clinical remission by 12 weeks after starting EEN (determined by weighted Pediatric Crohn’s Disease Activity Index score <12.5 [26]) were classified as ‘remission’ and patients with wPCDAI score ≥12.5 were classified as ‘no remission’. All patients received an immunomodulator (azathioprine or methotrexate) for maintenance therapy. Patients who achieved and remained in remission by week 24 were sub-classified as ‘sustained remission’ (SR) whereas patients who experienced a relapse requiring escalation of medical therapy (e.g., oral corticosteroids, biologic therapy, or surgery) by 24 weeks were sub-classified as ‘non-sustained remission’ (NSR). Patients who did not achieve the primary endpoint via EEN maintained the ‘no remission’ classification.
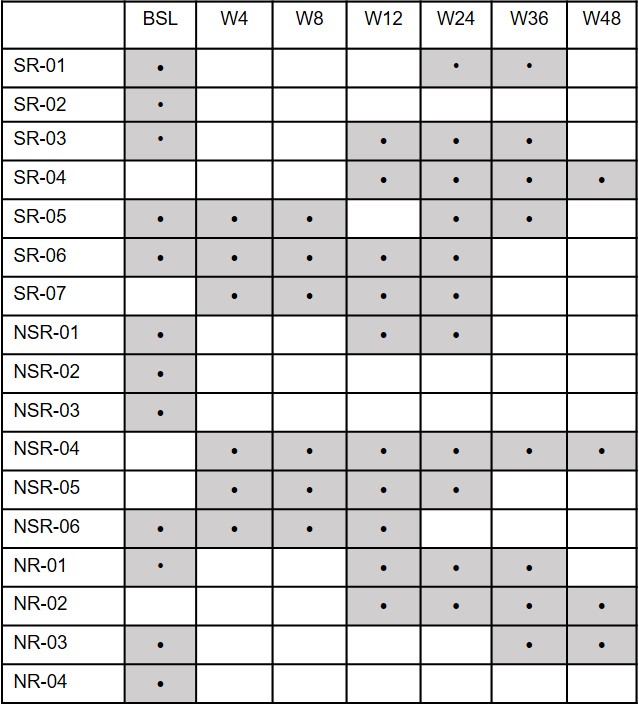
Stool samples were collected prior to starting EEN induction (baseline), at weeks 4, 8, and 12 during EEN treatment and at 12-week intervals during the course of maintenance treatment (Supplementary Fig. 1). Samples were stored at −20 °C for transport and then frozen at −80 °C until analysis. In total, 57 stool samples were analyzed for both fecal bile acid quantification and 16S rRNA and metagenomic shotgun sequencing (see Supplemental methods for sequencing and processing details). Fecal calprotectin levels in stool samples were quantified using the EliA Calprotectin 2 automated immunoassay on the Phadia 250 platform (Thermo Fisher Scientific, Uppsala, Sweden) following manufacturer protocol.
Fecal BA quantification
BA quantification was performed by liquid chromatography tandem mass spectrometry (LC-MS-MS) using the Biocrates Life Sciences Bile Acids Kit (Biocrates, Innsbruck, Austria) according to the manufacturer’s instructions. The analysis was performed at the Sick Kids Analytical Facility for Molecules (AFBM) (Toronto, Canada) using an Agilent UHPLC 1290 LC system coupled to an ABSciex QTRAP 5500 in negative ESI MRM mode. The samples were lyophilized and reconstituted with 75% ethanol, then homogenates were centrifuged for 10 min at 20,000 g. BA quantification was performed on the supernatant. Absolute (micromolar) concentrations of BA were calculated from area ratios using calibration curves generated by internal standards.
Modeling microbiome community structure and building predictive models
A hierarchical Bayesian framework (Bayesian Inference of Microbial Community Structure (BioMiCo)) for characterizing bacterial community structure was used to model the microbial community in sample groups [27]. BioMiCo implements a two-phase modeling procedure that allows the user to build and assess a predictive model based on microbial community structures, as inferred from taxonomic marker genes. The initial training phase learns how operational taxonomy units (OTUs) contribute to assemblages, and how assemblages form communities, and relates this structure to patient clinical metadata. The second test phase applies the trained model to classify independent unknown samples based on maximum posterior probability from the mixture weights learned from the training data [27]. Leave-one-out cross-validation was used to test model prediction on these samples. In the training phase of BioMiCo, 25 communities were set as the maximum number of assemblages and 50,000 iterations were performed after 2000 iterations of burn-in.
Statistical analysis
Continuous variables were expressed as the mean ± SEM or as median and interquartile range [IQR] when not normally distributed. Differences between groups were calculated by t-test (for parametric data) and Mann–Whitney U test (for non-parametric unpaired data). Comparisons of more than two groups were performed using ANOVA or Kruskal–Wallis tests, where appropriate. Categorical variables were compared using Fisher’s exact or chi-squared tests, where appropriate. Statistical analyses were performed using GraphPad Prism version 5.0 for Windows (GraphPad Software).
Alpha diversity analyses were performed using observed OTUs (Chao-1 and Shannon index) and beta diversity analyses were performed using weighted and unweighted UniFrac and Bray-Curtis, implemented in QIIME [28] and R (vegan) [29]. Spearman correlation between BA concentrations and OTU counts was performed using R. To exclude effects of outliers only OTUs found in greater than 5% of samples were used. A p-value < 0.05 was considered statistically significant, and p-values were corrected for false discovery rate (FDR) using Benjamini-Hochberg (BH) correction [30] to accommodate for multiple testing where necessary.
Results
Patient characteristics and response to EEN induction therapy
Demographic and clinical characteristics of the 17 patients that participated in this study are presented in Table 1. Thirteen patients achieved remission following 12 weeks of EEN induction therapy as demonstrated by wPCDAI scores <12.5 (Fig. 1a) and significant decreases in erythrocyte sedimentation rate (ESR), fecal calprotectin, and platelet count values (Table 1). Of the 13 patients who achieved remission with EEN, six subsequently relapsed by week 24 (non-sustained remission, NSR) requiring re-induction with steroids, treatment escalation, or surgery (Fig. 1b). The remaining seven patients maintained stable remission for the remainder of the study period. Although baseline disease activity scores were comparable across groups, NSR patients had significantly higher levels of fecal calprotectin (FCP) than SR patients prior to EEN (5274 ± 1845 versus 2666 ± 537 μg/g, respectively p = 0.027), indicating a greater degree of active inflammation in the lower GI tract. Disease location within the GI tract was similar across groups, with all patients having ileal involvement and the majority (13/17) having ileocolonic disease (L3) (Table 1). Extent of ileal involvement was <10 cm for the majority of patients with available data.
Table 1.
Demographic and clinical characteristics of pediatric Crohn’s disease (CD) patients included in the study
| Patient ID | Age (y), Gender | Disease Location (ParisL) | Ileal disease extent (cm) | Concomitant therapy | FCP (μg/g) | ESR (mm/h) | PLT (×109/L) | Relapse by WK24 | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
| BSL | WK12 | BSL | WK12 | BSL | WK12 | ||||||
| Achieved remission by week 12 of EEN | |||||||||||
| SR-01 | 12, M | L3 | <10 | IMM | 2018 | 211 | 1 | 10 | 305 | 291 | No |
| SR-02 | 9, M | L3 | – | – | 4407 | 704 | 20 | 19 | 440 | 316 | No |
| SR-03 | 11, M | L1 | 10–20 | – | 2292 | 4560 | 18 | 28 | 346 | 300 | No |
| SR-04 | 11, M | L3 | <10 | – | 3644 | 2720 | 27 | 7 | 232 | 184 | No |
| SR-05 | 10, F | L3 | – | – | 2691 | 173 | 5 | 9 | 483 | 412 | No |
| SR-06 | 8, M | L3 | <10 | – | 746 | 1357 | 47 | 43 | 306 | 276 | No |
| SR-07 | 10, F | L3 | <10 | – | – | 4755 | 39 | 19 | 525 | 400 | No |
| NSR-01 | 15, M | L3 | – | – | 6555 | 5221 | 36 | 30 | 488 | 336 | Yes |
| NSR-02 | 12, M | L3 | >20 | ABXa | 6000 | 58 | 40 | 4 | 567 | 367 | Yes |
| NSR-03 | 14, M | L3 | – | ABXa | 6000 | 3904 | 33 | 14 | 571 | 339 | Yes |
| NSR-04 | 10, F | L1 | 10–20 | – | – | 3574 | 43 | 32 | 445 | 331 | Yes |
| NSR-05 | 12, M | L1 | <10 | – | 2012 | 178 | 36 | 16 | 250 | 202 | Yes |
| NSR-06 | 11, M | L3 | <10 | – | 5801 | 1280 | 66 | – | 537 | 444 | Yes |
| p value Week 12 vs BSLb | 0.0151 | 0.0380 | 0.0002 | ||||||||
| Did not achieve remission by week 12 of EEN | |||||||||||
| NR-01 | 10, M | L3 | 10–20 | – | 3781 | 6976 | 17 | 52 | 350 | 371 | – |
| NR-02 | 10, M | L3 | <10 | – | 2745 | 4707 | 46 | 40 | 278 | 258 | – |
| NR-03 | 10, M | L3 | <10 | 5-ASA | 3906 | – | 9 | 16 | 289 | 247 | – |
| NR-04 | 13, M | L3 | <10 | – | 2874 | – | 26 | 46 | 359 | 324 | – |
| p value Week 12 vs BSLb | n/a | 0.2093 | 0.2706 | ||||||||
5-ASA 5-aminosalicylic acid, ABX antibiotics, BSL baseline, ESR erythrocyte sedimentation rate, FCP fecal calprotectin, IMM immunomodulatory, NSR non-sustained remission, NR no remission, PLT platelets, SR sustained remission, wPCDA weighted Pediatric Crohn’s Disease Activity Index
aAntibiotics started on same day or after EEN initiated
bPaired t-test comparing week 12 versus baseline values, where available
Fig. 1.

Analysis of fecal bile acid concentrations in from CD patient samples grouped according to clinical response to EEN induction therapy. a Weighted PCDAI (wPCDAI) scores at baseline and at week 12 of EEN in 13 patients who achieved remission (wPCDAI 12.5 and 4 patients who did not, b Survival graph of percent relapse among 13 patients who achieved remission with EEN. c Percent contribution of each priBA and secBA species (shown in legend) in stools sample from SR, NSR, and NR patients, d Absolute concentrations (uM) of total BA in SR, NSR, and NR patient stool samples, e Percent of priBA in SR, NSR, and NR patient samples. ***p < 0.001
Fecal BA composition is altered in a sub-group of CD patients who do not sustain remission by EEN induction
We analyzed the fecal BA composition of 57 stool samples from 17 CD patients described in Table 1 in relation to outcomes following EEN induction. The percent contribution of priBA and secBA species in each sample identified a distinct subgroup samples belonging to NSR and NR patients wherein priBA comprised >70% of the total fecal BA pool (Fig. 1c). Two samples (1 NSR and 1 NR) were composed nearly entirely of conjugated priBA (Fig. 1c). Notably, a priBA-dominant profile was observed in 11/19 samples (57.9%) from five out of six NSR patients but was not observed in any samples from SR patients (0/26) (p < 0.0001, Fisher’s exact test priBA-dominant profiles in NSR vs SR samples). Total fecal BA concentrations were not significantly different between SR, NSR, and NR samples (Fig. 1d). All available NSR patient samples collected prior to EEN had a priBA-dominant profile with significantly higher % priBA than SR patients (p < 0.001), indicating that a skewed fecal BA profile exists prior to starting EEN therapy in patients who subsequently relapse (Fig. 1e). No priBA-dominant samples were observed in SR patients before and during, or after EEN (Fig. 1e).
PriBA-dominant samples are associated with decreased taxonomic diversity
We analyzed bacterial sequence data from 11 priBA-dominant and 44 secBA-dominant samples (55 of 57 total, 2 samples containing exclusively conjugated BA were excluded due to low bacterial sequence counts) using both 16S rRNA amplicon data (V4V5) and selected taxonomic marker genes within the metagenome assessed with MetaPhlAn2 [31]. 16S rRNA-based analysis identified 1445 (mean 227 ± 85) OTUs and metagenomic sequence analysis identified 209 (mean 37 ± 13) OTUs. Global signatures of priBA-dominant and secBA-dominant communities showed distinct features: priBA-dominant samples displayed decreased alpha diversity (Chao-1 p < 0.001 and Shannon index p < 0.01) (Fig. 2a) and clustered together in beta diversity analysis plots (Fig. 2b).
Fig. 2.

a Alpha diversity plots (Chao 1 and Shannon index) and b beta diversity (PCA) plot of OTUs identified in priBA- vs secBA-dominant samples, c Box plots showing relative abundance of taxa in phyla Bacteroidetes, Firmicutes, and Proteobacteria in priBA- and secBA-dominant samples, by non-parametric Mann–Whitney U test (FDR-adjusted), d Community-level association network of the posterior distributions of assemblages and OTUs, identified in the BioMico analysis. Predominate OTUs identified in assemblages are connected by dashed lines. Diameter of circles represents the posterior probability (PP) of the OTU in the assemblage. *p < 0.05, **p < 0.01, ***p < 0.001
Distinct microbial community structures are associated with priBA and secBA-dominant fecal BA profiles
At the phylum level, we found significantly greater abundance of Proteobacteria in priBA-dominant samples (p < 0.05, Mann–Whitney U test, FDR < 0.05) (Fig. 2c).
To better understand the differences in bacterial community structures associated with priBA- versus secBA-dominant samples, we applied the BioMiCo Bayesian modeling framework to analyze co-occurrence relationships among taxa. Using 16S data, BioMiCo identified three assemblages (A1, A2, A3)—one (A1) associated with priBA-dominant samples and two (A2 and A3) associated with secBA-dominant samples (Fig. 2d). Predominant taxa within the priBA-dominant assemblages were Bacteroides (including B. plebeius), Enterobacteriaceae, Roseburia, Ruminococcus gnavus and Megamonas whereas the predominant taxa found in secBA-dominant assemblage were different groups of Bacteroides (including B. uniformis), Ruminococcaceae, Erysipelotrichaceae, Rikenellaceae, and Lachnospiraceae (Fig. 2d).
BioMiCo analysis of metagenomic taxa data, which compromised considerably fewer (<20%) OTUs than 16S rRNA analysis, identified 13 assemblages. Of these, two were associated with priBA-dominant samples with posterior probabilities (PP) of >0.60 and >0.15. These priBA-dominant assemblages contained multiple taxa belonging to Bacteroides (including B. plebeius, B. ovatus, B. dorei, and B. thetaiotaomicron), as well as Ruminococcus gnavus, Escherichia coli, Clostridium bolteae, and several species of Veillonella (Supplementary Fig. 2). Nine assemblages were associated with secBA-dominant samples. Each had low PP (<0.1) but together defined roughly 0.75 of the distribution and contained different Bacteroides taxa including B. vulgatus, and B uniformis, as well as Faecalibacterium, Subdoligranulum, and several species of Alistipes (Supplementary Fig. 2).
We further compared the relative abundance of taxa in priBA- and secBA-dominant samples using the Kruskal–Wallis test and correcting for multiple testing (Benjimini–Hochberg). Using 16S-based OTU assignments at FDR threshold <0.05, we identified 9 OTUs with significantly higher relative abundance in priBA-dominant samples, and nine OTUs higher in secBA-dominant samples (Supplementary table 1). Parallel analysis based on metagenomic taxonomic assignments found that 24 taxa (FDR < 0.05) had significantly higher relative abundance in priBA-dominant samples and 16 taxa had significantly higher relative abundance in secBA-dominant samples (Supplementary table 2). These analyses identified many of the same taxa as in the 16S analysis in the priBA-dominant assemblage, including B. plebeius and several strains of Megamonas (Veillonellaceae), and in the secBA-dominant assemblages, including Lachnospiraceae, Rikenellaceae, and Ruminococcoceae.
Fecal BA concentrations correlate with abundance of distinct bacterial taxonomic groups
Spearman correlations were used to explore the relationship between the fecal concentrations of priBA and secBA metabolites, and the abundance of bacterial taxonomic groups. Using 16S OTU data we found a total of 195 FDR-significant (q < 0.05) associations. The major priBAs CA and CDCA were highly correlated with taxa belonging to Proteobacteria and in particular Enterobacteriaceae (Fig. 3a). In contrast, the major secBAs LCA and DCA were correlated with OTUs mostly belonging to Firmicutes (Ruminococcaceae and Lachnospiraceae) (Fig. 3a). Interestingly, several minor secBAs, including UDCA and two BAs typically non-detectable in human stool –hyodeoxycholic acid (HDCA) and β−muricholic acid (β−MCA)−correlated with numerous taxa associated with priBA. Specifically, UDCA and HDCA were positively associated with Veillonella dispar, Bifidobacterium, and B. plebeius, as well as OTUs from Streptococcus, Ruminococcaceae, Lachnospiraceae, Enterobacteriaceae, and Clostridiales (Fig. 3a).
Fig. 3.

Heatmaps showing Spearman correlation between fecal BA metabolites and microbial taxa identified by a 16S- or b metagenome sequence-based analysis that were present in more than 5% of samples. Benjimini Hochberg was used to correct for multiple tests to determine significance. c Counts of OTUs from 16S rRNA data and taxa from metagenome data correlated with BA metabolites identified in the phyla Actinobacteria, Bacteroidetes, Firmicutes, and Proteobacteria
For the metagenomic data, we found 43 FDR-significant (q < 0.05) associations. Similar to the 16S rRNA data, CA and CDCA were associated with taxa from Proteobacteria (Escherichia and Klebsiella) and HDCA and UDCA were mainly correlated with Veillonella parvula, Megamonas hypermegale, Megamonas rupellensis, Streptococcus mitis, Clostridium innocuum, Bifodobacterium bifidum, and B. plebeius (Fig. 3b). Both LCA and DCA were correlated with Ruminococcus torques, Erysipelotrichaceae sp., and Subdoligranulum sp. (Fig. 3b).
PriBA-dominant samples are associated with community-level decrease in enzyme gene sequences for BA 7α-dehydroxylation but not deconjugation
PriBAs produced in the liver are conjugated to taurine or glycine and must undergo deconjugation by bacterial BSH in order to be further metabolized to secBA in the colon [8]. The ratio of unconjugated (unconj) to conjugated (conj) BA in priBA-dominant samples was comparable to secBA-dominant samples, indicating that deconjugation was not impaired (Fig. 4a). Consistent with this, examination of metagenomic sequence data for the gene required for deconjugating BA, bsh (K01442), indeed found that the relative abundance of bsh sequences was not significantly different between priBA- and secBA-dominant samples (Fig. 4b). In total, 36 different taxa belonging to the phyla Actinobacteria [1], Bacteroidetes [8], and Firmicutes [26] as well as an unclassified taxonomic group [1] contributed bsh across 53 samples (10 priBA- and 43 secBA-dominant). In two samples, one each from the priBA-and secBA-dominant groups, we did not detect any bsh.
Fig. 4.

a Ratio of unconjugated to conjugated BA metabolites in priBA- versus secBA-dominant samples, b Relative abundance of bile salt hydrolase gene (bsh) sequences in priBA- versus secBA-dominant samples, c Percentage of samples containing bsh from the identified taxa in priBA- versus secBA-dominant samples
There were notable differences in the taxa contributing bsh between sample groups. SecBA-dominant samples contained two-fold more taxa contributing bsh than priBA-dominant samples (35 versus 16, respectively). Using the Mann–Whitney test we found that Alistipes (A. finegoldii p = 0.0027, A. onderdonkii p = 0.011, and A. shahii p = 0.015) in the Bacteroidetes phyla, and Ruminococcus obeum (p = 0.004) and Faecalibacterium prausnitzii (p = 0.003) within the Firmicutes phyla made a significantly greater contribution of bsh in secBA-dominant versus priBA-dominant samples. F. prausnitzii contributed bsh to 32/43 secBA-dominant samples and only 1/10 priBA-dominant samples. In contrast, R. gnavus (Firmicutes) was the most common contributor of bsh in priBA-dominant samples (10/10 samples), with significantly greater abundance compared to secBA-dominant samples (p = 0.013). Figure 4c shows the percentage of samples from each group that contained bsh from the identified taxa.
Metagenomic sequences were then examined for the presence of genes involved in the conversion of unconjugated priBA to secBA, including the gene for 7α-HSDH (hdhA, K00076), and seven genes on the bai operon: baiB (K15868), baiA (K15869), baiCD (K15870), baiF (K15871), baiE (K15872), baiH (K15873), and baiI (K15874).
Sequences for hdhA were identified in 7/11 (63%) of priBA-dominant samples and in 13/44 (~30%) of secBA-dominant samples. E. coli was the most common source of hdhA (19/20 samples), with Campylobacter identified in one sample. Of the bai operon genes, only 4 were detected (baiB, baiCD, baiE, and baiF) across all samples. The majority of these were detected in secBA-dominant samples, with 11/44 containing one or more of these genes from Firmicutes Clostridiales members Lachnospiraceae sp 5_1_57FAA and Clostridium hylemonae, and Actinobacteria Eggerthella sp. (Eggerthella_sp_1_3_56FAA and Eggerthella lenta). Within priBA-dominant samples, only baiB from Eggerthella lenta was detected in 2/11 samples.
Discussion
This study is the first to investigate fecal BA profiles and gut microbiota community structure in relation to outcomes including sustained remission and relapse following nutritional therapy in pediatric CD patients. We found that a fecal BA profile dominated by priBAs occurred among a subset of patients who did not sustain remission after EEN treatment (NSR). Notably, this priBA-dominant profile was most striking in samples collected at baseline prior to EEN therapy. In contrast, patients who achieved and sustained remission (SR) with EEN had exclusively secBA-dominant samples.
Reduced fecal secBA concentrations have been previously associated with IBD, as well as cirrhosis and non-alcoholic fatty liver disease [19, 32, 33]. Since secBA formation is mediated exclusively by gut microbiota via a distributed pathway of deconjugation and dehydroxylation, dysbiosis can profoundly impact conversion of priBA to secBA in the intestine [34]. Gut microbe communities deficient in BSH enzyme, which catalyzes the ‘gateway’ deconjugation reaction, are reported to have low potential for secBA formation [21]. Indeed, previous work has shown that reduced BSH gene abundance and activity is associated with low fecal secBA and high levels of conjBA in IBD patients [19]. Our analysis indicates that the microbiome associated with priBA-dominant samples had BA deconjugation capabilities comparable to secBA-dominant samples. However, priBA-dominant samples had notably fewer taxa contributing bsh genes detected, which is likely a reflection of overall decreased community diversity. While BSH is a conserved function that promotes favorable environments for bacterial growth by detoxifying conjBA, it remains to be seen whether reduced diversity observed within the priBA-dominant community could degrade such robustness of function and predispose patients to exacerbated BA dysmetabolism.
Our analysis indicates that the primary defect in secBA synthesis in priBA-dominant samples was in 7α-dehydroxylation activity. 7α-dehydroxylation of CA and CDCA to form DCA and LCA, respectively, is performed mostly by a restricted group of bacteria in the order of Clostridiales that express bai genes, including Lachnospiraceae, Ruminococcaceae and Blautia [8]. The abundance of Blautia, Ruminococcaceae, and Lachnospiraceae has been correlated with 7α -dehydroxylation capabilities in patients with liver cirrhosis compared to controls, especially in patients with advanced cirrhosis [35]. In a study of fecal microbiome transplant (FMT) recipients and donors, Brown et al. also found Coprococcus, Faecalibacterium, Ruminococcus, and Ruminococcaeae among genera that correlate positively with fecal levels of secBA [36]. Consistent with these previous reports, the concentration of secBAs DCA and LCA showed a significant positive correlation with these taxa within our samples. However, sequence-based analysis identified bai genes in <25% of samples. Lack of bai gene recovery could be due to incomplete annotation of genes in taxa found to encode these genes within our cohort (several Clostridium, Eggerthella, and several Lachnospiraceae). Eggerthella is a member of Actinobacteria previously shown to express genes for 7α-dehydroxylation and other BA modifications, including iso epimers of DCA and LCA [21, 37, 38]. We note that a survey of curated metagenomic data of the human microbiome found similar low-levels of recovery of bai genes to what was found here [39].
Interestingly, priBA-dominant samples had increased concentrations of minor fecal secBA that are normally present at low levels, including UDCA and HDCA. Indeed, HDCA is typically considered a non-human BA, detected in human stool only in trace amounts [15, 40]. UDCA is reported to have protective actions in the colon, including inhibiting release of proinflammatory cytokines by colonic epithelial cells and protecting against the development of colonic inflammation in experimental colitis [41, 42]. UDCA is formed from CDCA by successive reactions involving 7α- and 7β-HSDH enzymes. Our metagenomics analysis identified the gene encoding 7α-HSDH (hdhA) in a greater portion of priBA-dominant samples than secBA-dominant samples, suggesting a direct link with UDCA production. The gene for 7β-HSDH, which is poorly annotated, was not detected. The hdhA gene in priBA-dominant samples was derived mainly from R. gnavus. Ruminococcus species are reported to exhibit both 7α- and 7β-HSDH activity required for UDCA production [43–46]. UDCA can be further metabolized to LCA by bacterial 7α-dehydroxylation. Notably, LCA has been shown to exert more potent anti-inflammatory activity in the colon than UDCA and to also dampen T helper 1 responses, suggesting that LCA derived from microbial metabolism of UDCA contributes to its protective effect [42, 47]. Correlation analysis showed strong positive associations between UDCA and Veillonella, Streptococcus, and Enterobacteriaceae, which are also enriched in patients treated with oral UDCA for primary biliary cholangitis (PBC), suggesting UDCA itself may act on the microbial community to induce characteristic changes [32].
Using Bayesian inference analysis of the structures of the gut communities as well as Kruskal–Wallis testing, we identified the taxa differentially associated with priBA- and secBA-dominant samples. Within priBA-dominant samples, predominant OTUs were from the families Veillonellaceae (Veillonella and Megamonas) and Enterobacteriaceae. Enrichment of Enterobacteriaceae and Veillonellaceae has also been observed in the microbiota of cirrhotic patients with decreased conversion of priBA to secBA [35]. Enterobacteriaceae are highly tolerant of BAs and the expansion of potentially pathogenic symbionts or ‘pathobionts,’ including Enterobacteriaceae, has been repeatedly observed in CD/IBD and is hypothesized to initiate or exacerbate the overactive immune response and the ensuing tissue damage characteristic of disease [2, 48, 49]. Similarly, R. gnavus, the major source of BSH and 7α-HSDH genes in the priBA-dominant samples, is implicated as a pathobiont enriched in IBD patients and especially in those patients with increased disease activity [50]. SecBAs are reported to have more potent bactericidal activity than priBAs [16], which may contribute to pathobiont expansion in priBA-dominant samples. We were unable to measure absolute bacteria counts in the samples in this study so it is currently unclear whether priBA-dominant samples were also associated with bacterial overgrowth.
Given that disrupted BA metabolism has proposed pathophysiological consequences on intestinal immune function and resistance to pathobiont expansion in the GI tract, an imbalance of fecal priBA and secBA may play a mechanistic role in relapse after EEN therapy. PriBA-dominant profiles were detected in multiple NSR patients prior to starting EEN, suggesting that a lack of stable secBA synthesis could contribute to disease exacerbation once normal dietary triggers are reintroduced. An important limitation of the current study is that stool samples were not available for all patients at all timepoints of EEN treatment and follow-up. Further studies are required to better understand whether distinct abilities of the gut microbiome to metabolize BAs can predict responses to EEN. In our sample cohort, microbial communities differed such that we could reliably distinguish the BA phenotypes according to each patient’s microbial signature. Microbiome profiling offers interesting new research avenues to improve patient selection for nutritional therapy in pediatric CD and develop new microbiome-targeted interventions to help patients sustain remission.
Supplementary information
{kind=link}
{kind=link}
Acknowledgements
The authors would like to thank all participating children and their families, as well as the pediatric dietitians (Jennifer Haskett, Lisa Parkinson-McGraw) and the IWK GI-Research Team (Brad MacIntyre). JVL was supported by a Canadian Institutes of Health Research (CIHR)-Canadian Association of Gastroenterology-Crohn’s Colitis Canada New Investigator Award (2015–2019), a Canada Research Chair Tier 2 in Translational Microbiomics (2018–2019) and a Canadian Foundation of Innovation John R. Evans Leadership fund (awards #35235 and #36764), a Nova Scotia Health Research Foundation (NSHRF) establishment award (2015–2017), an IWK Health Centre Research Associateship (for JC), an IWK Health Centre Project grant (Cat. B—2017), a American Gastroenterology Association Pfizer Young Investigator Pilot Research Award in Inflammatory Bowel Disease (2018), a donation from the MacLeod family and by a CIHR-SPOR-Chronic Diseases grant (Inflammation, Microbiome, and Alimentation: Gastro-Intestinal and Neuropsychiatric Effects: the IMAGINE-SPOR chronic disease network). JVL reports consulting, travel and/or speaker fees and research support from AbbVie, Janssen, Nestlé Health Science, Merck, P&G, GSK, Illumina, Otsuka.
Data availability
The 16S rRNA gene and metagenomic sequencing data used in this study will be made available under accession PRJEB 35587 at the European Nucleotide Archive.
Compliance with ethical standards
Conflict of interest
JVL reports consulting, travel and/or speaker fees and research support from AbbVie, Janssen, Nestlé Health Science, Merck, P&G, GSK, Illumina, Otsuka. The other authors have no conflicts to report.
Footnotes
These authors jointly supervised: Jessica Connors, Katherine Dunn
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version of this article (10.1038/s41396-019-0560-3) contains supplementary material, which is available to authorized users.
References
- 1.Morgan XC, Tickle TL, Sokol H, Gevers D, Devaney KL, Ward DV, et al. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol. 2012;13:R79. doi: 10.1186/gb-2012-13-9-r79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gevers D, Kugathasan S, Denson LA, Vázquez-Baeza Y, Van Treuren W, Ren B, et al. The treatment-naive microbiome in new-onset Crohn’s disease. Cell Host Microbe. 2014;15:382–92. doi: 10.1016/j.chom.2014.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lewis JD, Chen EZ, Baldassano RN, Otley AR, Griffiths AM, Lee D, et al. Inflammation, antibiotics, and diet as environmental stressors of the gut microbiome in pediatric Crohn’s disease. Cell Host Microbe. 2015;18:489–500. doi: 10.1016/j.chom.2015.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ni J, Wu GD, Albenberg L, Tomov VT. Gut microbiota and IBD: causation or correlation? Nat Rev Gastroenterol Hepatol. 2017;14:573. doi: 10.1038/nrgastro.2017.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Packey CD, Sartor RB. Commensal bacteria, traditional and opportunistic pathogens, dysbiosis and bacterial killing in inflammatory bowel diseases. Curr Opin Infect Dis. 2009;22:292–301. doi: 10.1097/QCO.0b013e32832a8a5d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sokol H., Pigneur B., Watterlot L., Lakhdari O., Bermudez-Humaran L. G., Gratadoux J.-J., Blugeon S., Bridonneau C., Furet J.-P., Corthier G., Grangette C., Vasquez N., Pochart P., Trugnan G., Thomas G., Blottiere H. M., Dore J., Marteau P., Seksik P., Langella P. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proceedings of the National Academy of Sciences. 2008;105(43):16731–16736. doi: 10.1073/pnas.0804812105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Holmes E, Li JV, Athanasiou T, Ashrafian H, Nicholson JK. Understanding the role of gut microbiome–host metabolic signal disruption in health and disease. Trends Microbiol. 2011;19:349–59. doi: 10.1016/j.tim.2011.05.006. [DOI] [PubMed] [Google Scholar]
- 8.Ridlon JM, Kang DJ, Hylemon PB. Bile salt biotransformations by human intestinal bacteria. J Lipid Res. 2006;47:241–59. doi: 10.1194/jlr.R500013-JLR200. [DOI] [PubMed] [Google Scholar]
- 9.Chiang JY. Bile acids: regulation of synthesis. J Lipid Res. 2009;50:1955–66. doi: 10.1194/jlr.R900010-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jones BV, Begley M, Hill C, Gahan CG, Marchesi JR. Functional and comparative metagenomic analysis of bile salt hydrolase activity in the human gut microbiome. Proc Natl Acad Sci USA. 2008;105:13580–5. doi: 10.1073/pnas.0804437105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Humbert L, Maubert MA, Wolf C, Duboc H, Mahé M, Farabos D, et al. Bile acid profiling in human biological samples: comparison of extraction procedures and application to normal and cholestatic patients. J Chromatogr B. 2012;899:135–45. doi: 10.1016/j.jchromb.2012.05.015. [DOI] [PubMed] [Google Scholar]
- 12.Wells JE, Berr F, Thomas LA, Dowling RH, Hylemon PB. Isolation and characterization of cholic acid 7α-dehydroxylating fecal bacteria from cholesterol gallstone patients. J Hepatol. 2000;32:4–10. doi: 10.1016/s0168-8278(00)80183-x. [DOI] [PubMed] [Google Scholar]
- 13.Wells JE, Williams KB, Whitehead TR, Heuman DM, Hylemon PB. Development and application of a polymerase chain reaction assay for the detection and enumeration of bile acid 7α-dehydroxylating bacteria in human feces. Clin Chim Acta. 2003;331:127–34. doi: 10.1016/s0009-8981(03)00115-3. [DOI] [PubMed] [Google Scholar]
- 14.Begley M, Gahan CG, Hill C. The interaction between bacteria and bile. FEMS Microbiol Rev. 2005;29:625–51. doi: 10.1016/j.femsre.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 15.Wahlström A, Sayin SI, Marschall H, Bäckhed F. Intestinal crosstalk between bile acids and microbiota and its impact on host metabolism. Cell Metab. 2016;24:41–50. doi: 10.1016/j.cmet.2016.05.005. [DOI] [PubMed] [Google Scholar]
- 16.Islam KS, Fukiya S, Hagio M, Fujii N, Ishizuka S, Ooka T, et al. Bile acid is a host factor that regulates the composition of the cecal microbiota in rats. Gastroenterology. 2011;141:1773–81. doi: 10.1053/j.gastro.2011.07.046. [DOI] [PubMed] [Google Scholar]
- 17.Gadaleta RM, van Erpecum KJ, Oldenburg B, Willemsen EC, Renooij W, Murzilli S, et al. Farnesoid X receptor activation inhibits inflammation and preserves the intestinal barrier in inflammatory bowel disease. Gut. 2011;60:463–72. doi: 10.1136/gut.2010.212159. [DOI] [PubMed] [Google Scholar]
- 18.Jansson J, Willing B, Lucio M, Fekete A, Dicksved J, Halfvarson J, et al. Metabolomics reveals metabolic biomarkers of Crohn’s disease. PloS ONE. 2009;4:e6386. doi: 10.1371/journal.pone.0006386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Duboc H, Rajca S, Rainteau D, Benarous D, Maubert MA, Quervain E, et al. Connecting dysbiosis, bile-acid dysmetabolism and gut inflammation in inflammatory bowel diseases. Gut. 2013;62:531–9. doi: 10.1136/gutjnl-2012-302578. [DOI] [PubMed] [Google Scholar]
- 20.Labbé A, Ganopolsky JG, Martoni CJ, Prakash S, Jones ML. Bacterial bile metabolising gene abundance in Crohn’s, ulcerative colitis and type 2 diabetes metagenomes. PloS ONE. 2014;9:e115175. doi: 10.1371/journal.pone.0115175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Heinken A, Ravcheev DA, Baldini F, Heirendt L, Fleming RM, Thiele I. Personalized modeling of the human gut microbiome reveals distinct bile acid deconjugation and biotransformation potential in healthy and IBD individuals. BioRxiv. 2017:229138.
- 22.Franzosa EA, Sirota-Madi A, Avila-Pacheco J, Fornelos N, Haiser HJ, Reinker S, et al. Gut microbiome structure and metabolic activity in inflammatory bowel disease. Nat Microbiol. 2019;4:293. doi: 10.1038/s41564-018-0306-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ruemmele F, Veres G, Kolho K, Griffiths A, Levine A, Escher J, et al. Consensus guidelines of ECCO/ESPGHAN on the medical management of pediatric Crohn’s disease. J Crohn’s Colitis. 2014;8:1179–207. doi: 10.1016/j.crohns.2014.04.005. [DOI] [PubMed] [Google Scholar]
- 24.Dunn KA, Moore-Connors J, MacIntyre B, Stadnyk AW, Thomas NA, Noble A, et al. Early changes in microbial community structure are associated with sustained remission after nutritional treatment of pediatric Crohn’s disease. Inflamm Bowel Dis. 2016;22:2853–62. doi: 10.1097/MIB.0000000000000956. [DOI] [PubMed] [Google Scholar]
- 25.MacLellan A, Connors J, Grant S, Cahill L, Langille M, Van Limbergen J. The impact of exclusive enteral nutrition (EEN) on the gut microbiome in Crohn’s disease: a review. Nutrients. 2017;9:447. doi: 10.3390/nu9050447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Turner D, Griffiths AM, Walters TD, Seah T, Markowitz J, Pfefferkorn M, et al. Mathematical weighting of the pediatric Crohn’s disease activity index (PCDAI) and comparison with its other short versions. Inflamm Bowel Dis. 2011;18:55–62. doi: 10.1002/ibd.21649. [DOI] [PubMed] [Google Scholar]
- 27.Shafiei M, Dunn KA, Boon E, MacDonald SM, Walsh DA, Gu H, et al. BioMiCo: a supervised Bayesian model for inference of microbial community structure. Microbiome. 2015;3:8. doi: 10.1186/s40168-015-0073-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7:335. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Oksanen J, Blanchet FG, Kindt R, Legendre P, Minchin P, O’hara R, et al. vegan: Community Ecology Package.R package version 2.3–0.2015. 2017.
- 30.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc: Ser B. 1995;57:289–300. [Google Scholar]
- 31.Truong DT, Franzosa EA, Tickle TL, Scholz M, Weingart G, Pasolli E, et al. MetaPhlAn2 for enhanced metagenomic taxonomic profiling. Nat Methods. 2015;12:902. doi: 10.1038/nmeth.3589. [DOI] [PubMed] [Google Scholar]
- 32.Tang R, Wei Y, Li Y, Chen W, Chen H, Wang Q, et al. Gut microbial profile is altered in primary biliary cholangitis and partially restored after UDCA therapy. Gut. 2018;67:534–41. doi: 10.1136/gutjnl-2016-313332. [DOI] [PubMed] [Google Scholar]
- 33.Mouzaki M, Comelli EM, Arendt BM, Bonengel J, Fung SK, Fischer SE, et al. Intestinal microbiota in patients with nonalcoholic fatty liver disease. Hepatology. 2013;58:120–7. doi: 10.1002/hep.26319. [DOI] [PubMed] [Google Scholar]
- 34.Narushima S, Itoh K, Miyamoto Y, Park S, Nagata K, Kuruma K, et al. Deoxycholic acid formation in gnotobiotic mice associated with human intestinal bacteria. Lipids. 2006;41:835–43. doi: 10.1007/s11745-006-5038-1. [DOI] [PubMed] [Google Scholar]
- 35.Kakiyama G, Pandak WM, Gillevet PM, Hylemon PB, Heuman DM, Daita K, et al. Modulation of the fecal bile acid profile by gut microbiota in cirrhosis. J Hepatol. 2013;58:949–55. doi: 10.1016/j.jhep.2013.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brown JR, Flemer B, Joyce SA, Zulquernain A, Sheehan D, Shanahan F, et al. Changes in microbiota composition, bile and fatty acid metabolism, in successful faecal microbiota transplantation for Clostridioides difficile infection. BMC Gastroenterol. 2018;18:131. doi: 10.1186/s12876-018-0860-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Devlin AS, Fischbach MA. A biosynthetic pathway for a prominent class of microbiota-derived bile acids. Nat Chem Biol. 2015;11:685. doi: 10.1038/nchembio.1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Harris SC, Devendran S, Méndez-García C, Mythen SM, Wright CL, Fields CJ, et al. Bile acid oxidation by Eggerthella lenta strains C592 and DSM 2243T. Gut Microbes. 2018;9:523–39. doi: 10.1080/19490976.2018.1458180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pasolli E, Schiffer L, Manghi P, Renson A, Obenchain V, Truong DT, et al. Accessible, curated metagenomic data through ExperimentHub. Nat Methods. 2017;14:1023. doi: 10.1038/nmeth.4468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Martin G, Kolida S, Marchesi J, Want E, Sidaway J, Swann JR. In vitro modeling of bile acid processing by the human fecal microbiota. Front Microbiol. 2018;9:1153. doi: 10.3389/fmicb.2018.01153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Martínez-Moya P, Romero-Calvo I, Requena P, Hernández-Chirlaque C, Aranda CJ, González R, et al. Dose-dependent antiinflammatory effect of ursodeoxycholic acid in experimental colitis. Int Immunopharmacol. 2013;15:372–80. doi: 10.1016/j.intimp.2012.11.017. [DOI] [PubMed] [Google Scholar]
- 42.Ward JB, Lajczak NK, Kelly OB, O’Dwyer AM, Giddam AK, Ní Gabhann J, et al. Ursodeoxycholic acid and lithocholic acid exert anti-inflammatory actions in the colon. Am J Physiol-Gastrointest Liver Physiol. 2017;312:G550–8. doi: 10.1152/ajpgi.00256.2016. [DOI] [PubMed] [Google Scholar]
- 43.Hirano S, Masuda N. Epimerization of the 7-hydroxy group of bile acids by the combination of two kinds of microorganisms with 7 alpha- and 7 beta-hydroxysteroid dehydrogenase activity, respectively. J Lipid Res. 1981;22:1060–8. [PubMed] [Google Scholar]
- 44.Akao T, Akao T, Kobashi K. Purification and characterization of 7β-Hydroxysteroid dehydrogenase from Ruminococus sp. of human intestine. J Biochem. 1987;102:613–9. doi: 10.1093/oxfordjournals.jbchem.a122095. [DOI] [PubMed] [Google Scholar]
- 45.Lee JY, Arai H, Nakamura Y, Fukiya S, Wada M, Yokota A. Contribution of the 7beta-hydroxysteroid dehydrogenase from Ruminococcus gnavus N53 to ursodeoxycholic acid formation in the human colon. J Lipid Res. 2013;54:3062–9. doi: 10.1194/jlr.M039834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lepercq P, Gérard P, Béguet F, Grill J, Relano P, Cayuela C, et al. Isolates from normal human intestinal flora but not lactic acid bacteria exhibit 7α-and 7β-hydroxysteroid dehydrogenase activities. Micro Ecol Health Dis. 2004;16:195–201. [Google Scholar]
- 47.Pols TW, Puchner T, Korkmaz HI, Vos M, Soeters MR, de Vries CJ. Lithocholic acid controls adaptive immune responses by inhibition of Th1 activation through the Vitamin D receptor. PloS ONE. 2017;12:e0176715. doi: 10.1371/journal.pone.0176715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu J, Yan Q, Luo F, Shang D, Wu D, Zhang H, et al. Acute cholecystitis associated with infection of Enterobacteriaceae from gut microbiota. Clin Microbiol Infect. 2015;21:851. e1–9. doi: 10.1016/j.cmi.2015.05.017. [DOI] [PubMed] [Google Scholar]
- 49.Buttó LF, Schaubeck M, Haller D. Mechanisms of microbe–host interaction in Crohn’s disease: dysbiosis vs. pathobiont selection. Front Immunol. 2015;6:555. doi: 10.3389/fimmu.2015.00555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hall AB, Yassour M, Sauk J, Garner A, Jiang X, Arthur T, et al. A novel Ruminococcus gnavus clade enriched in inflammatory bowel disease patients. Genome Med. 2017;9:103. doi: 10.1186/s13073-017-0490-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The 16S rRNA gene and metagenomic sequencing data used in this study will be made available under accession PRJEB 35587 at the European Nucleotide Archive.