

Abstract
Immunotherapy, with an increasing number of therapeutic dimensions, is becoming an important mode of treatment for cancer patients. The inhibition of immune checkpoints, which are the source of immune escape for various cancers, is one such immunotherapeutic dimension. It has mainly been aimed at T cells in the past, but NK cells are a newly emerging target. Simultaneously, the number of checkpoints identified has been increasing in recent times. In addition to the classical NK cell receptors KIRs, LIRs, and NKG2A, several other immune checkpoints have also been shown to cause dysfunction of NK cells in various cancers and chronic infections. These checkpoints include the revolutionized CTLA-4, PD-1, and recently identified B7-H3, as well as LAG-3, TIGIT & CD96, TIM-3, and the most recently acknowledged checkpoint-members of the Siglecs family (Siglec-7/9), CD200 and CD47. An interesting dimension of immune checkpoints is their candidacy for dual-checkpoint inhibition, resulting in therapeutic synergism. Furthermore, the combination of immune checkpoint inhibition with other NK cell cytotoxicity restoration strategies could also strengthen its efficacy as an antitumor therapy. Here, we have undertaken a comprehensive review of the literature to date regarding NK cell-based immune checkpoints.
Keywords: natural killer cell (NK), cancer immunotherapy (CI), immune checkpoint inhibitors (ICI), immune checkpoint, immune therapeutics
Introduction
Human natural killer cells constitute 15% of all circulating lymphocytes (1). Discovered in the 1970s, NK cells have mainly been associated with the killing of microbially infected and malignantly transformed allogeneic and autologous cells (2–4). NK cells demonstrate antitumor cell cytotoxicity without prior sensitization and production of cytokines as well as chemokines that regulate various immune responses (5). Recent advancements suggest that the actual role of natural killer cells is not restricted to the killing of malignantly transformed and virally infected cells but also plays a circumstantial role in affecting the players of the adaptive immune system such as DCs and T cells, either directly or indirectly, expanding on its functional domain (5–7). Furthermore, NK cell subsets exhibit major functional differences in their cytotoxicity, cytokine production, and homing capabilities (6). Based on CD56 density on the cell surface, NK cells are divided into CD56bright and CD56dim, which have different phenotypic properties. CD56bright NK cells have the capacity to produce abundant cytokines, while CD56dim NK cells are more cytotoxic and express more immunoglobulin-like receptors as well as FcγRIII (Fcγ receptor III, also known as CD16) (Figure 1) (1, 6, 8, 9).
Figure 1.

Types and functions of NK cells. (a) CD56bright CD16− and CD56dim CD16+ NK cells, termed immature and mature NK cells, respectively, are identified to have functional differences. CD56bright NK cells produce more cytokines, while CD56dim CD16+ NK cells are more cytotoxic and carry out ADCC (antibody-dependent cell-mediated cytotoxicity). (b) NK cell surface receptors, both activating and inhibitory receptors, carry out NK cell functions through a balance of signals. Inhibitory receptors detect MHC-I ligands on normal cells, and if present, activating signals are terminated, thereby maintaining “self-recognition.” These receptors carry ITIM motifs in their cytoplasmic tail, which recruit SHP1/2 through phosphorylation to carry out its function. Such inhibition is termed “dominant inhibition.” (c) Inhibitory receptors are exploited by cancer through upregulation of ligands, thereby avoiding destruction by NK cells. Hence, antibodies such as lirilumab and monalizumab are developed to block such interaction and enhance NK cell cytotoxicity toward cancer cells. This phenomenon is termed immune checkpoint inhibition. (d) The presence of CD16 receptors on NK cells makes them able to carry out ADCC. Therefore, several antibodies, such as rituximab, elotuzumab, and cetuximab, have been clinically evaluated for synergism with immune checkpoint inhibitors.
Both activating and inhibitory receptors are expressed on the surface of NK cells, contributing to the execution of the functions performed by the NK cell (10–17) (Figure 1). Inhibitory receptors specific for MHC-I (major histocompatibility complex class I) antigens tightly regulate NK cell-mediated cytotoxicity and lymphokine production. The inhibitory signal from the MHC-I specific receptor is essential for hematopoietic target cells to avoid destruction by NK cells. This concept is termed the “missing self” and was originally proposed by Ljunggren and Karre (17–20). Such MHC-I-recognizing inhibitory receptors form three families of NK-cell surface receptors, namely KIRs (killer cell immunoglobulin-like receptors), LIRs (Leukocyte immunoglobin-like receptors), and NKG2A (natural killer group 2 A) (10–14, 21). KIRs, members of the immunoglobulin superfamily, are type I transmembrane molecules that recognize classical human leukocyte antigens A, B, and C (HLA class Ia) (10, 11, 13). LIRs, also known as ILTs (immunoglobin-like transcripts), form the second set of receptors and mainly recognize non-classical HLA-G (class Ib) molecules, in addition to HLA class Ia. LIRs belong to the same Ig superfamily as KIRs. NKG2A, a member of the NKG2 group of seven receptors, namely A, B, C, D, E, F, and H, dimerizes with CD94 to form the NKG2A/CD94 receptor. It belongs to the C-type lectin family of receptors that recognizes non-classical HLA-E class I molecule as its ligand (22).
Destruction by NK cells not only requires the detection of MHC-I molecules on transforming cells by inhibitory receptors but also the activation of the NK cell by activating receptors (10–17). Natural cytotoxicity receptors (NCRs) represent the group of natural killer cell surface activating receptors that includes NKp46, NKp30, and NKp44. These receptors, as well as NKG2D and DNAM-1 (DNAX accessory molecule-1), recognize ligands expressed on the surface of virally infected or malignantly transformed cells (15, 23, 24). A number of coreceptors (2B4, NKp80, NTB-A, and CD59) are also expressed, which function only in combination with other activating receptors (23, 25). CD16 (or FcγRIII), also an activating receptor, is expressed mainly by the CD56dim NK-cell subset and is essential for antibody-dependent cellular cytotoxicity (ADCC) against IgG-coated target cells (1, 26) (Figure 1).
Tumors evade the immune system through the establishment of an immune-suppressive tumor microenvironment (27). Immune evasion involving NK cells encompasses several mechanisms (5, 28–30). Hypoxia and the release of various cytokines and soluble factors by tumor cells or other components of the tumor microenvironment such as transforming growth factor-β (TGFβ), IL-6, IL-10, tryptophan catabolites, prostaglandin E2 (PGE2), dickkopf-related protein 2 (DKK2), idoleamine 2,3-dioxygenase (IDO), soluble HLA-G, soluble NKG2D ligands, and galactin-3 (soluble inhibitory receptor for NKp30) have been reported to decrease the activation of NK cells and their cytotoxic activity, production of IFNγ, as well as expression and activation of its activating receptors (28–36). Furthermore, ligand shedding for activating receptors and upregulation of ligands for inhibitory receptors by tumor cells have also been revealed (25, 37). Hence, various strategies have been developed to restore NK cell functions, including adoptive cell transfer, cytokine therapies, and monoclonal antibodies targeting activating as well as inhibitory receptors and the tumor microenvironment (5, 25, 38–44). The exploitation of the NK cell inhibitory receptors by tumors for immune evasion is one such mechanism, termed immune checkpoint inhibition, and has been proven to be the most effective and appreciated therapeutic target (Box 1). Several such inhibitory immune checkpoints involving NK cells other than the MHC class I-associated inhibitory receptors have been identified, including the classical CTLA-4 and PD-1 receptors and TIGIT & CD96, LAG-3, and TIM-3 (Figure 2). B7-CD28 family members such as B7-H3, VISTA (PD-1H), and B7-H7 have recently been reported as plausible targets for such inhibition in regard to NK cells. More recently identified as NK cell-based immune checkpoint inhibition targets are the Siglec family receptors, namely Siglec-7 and -9, CD200, and CD47. Here, we review the recent advances and updates in NK cell-based immune checkpoint inhibition.
Box 1. Immune checkpoint inhibition.
The immune system consists of innate and adaptive immune components that, upon activation, eliminate infectious agents and cancer cells. Inhibitory pathways exist that normally maintain self-tolerance and counter-balance the activation process in order to avoid excessive damage and limit collateral tissue damage during the anti-microbial or anti-cancer immune response. These inhibitory pathways, consisting of receptors and ligands, are termed “immune checkpoints” and are utilized by the cancer cells to evade immune destruction. Inhibition of these checkpoints by developing monoclonal antibodies, thereby relieving the immune cells from inhibition and enabling them to recognize the cancer cells in order to eliminate them, is termed as “immune checkpoint inhibition,” and the agents are called “immune checkpoint inhibitors” (45). Ipilimumab, a CTLA-4 blocker, became the first approved immune checkpoint blocking anti-cancer drug in 2011 in the United States (46). James P. Allison and Tasuku Honjo were honored with the Nobel Prize in Physiology or Medicine in 2018 for their work and discoveries in basic science that allowed the development of checkpoint inhibitor therapies (47).
Figure 2.

Immune checkpoint inhibition observed in Natural Killer cells. (A) Inhibitory receptor-ligand interaction leading to immune escape of cancer cells is termed immune checkpoint inhibition. Inhibitory receptors expressed on the surface of NK cells are illustrated as blue rods, and ligands for these receptors expressed by tumor cells are illustrated as orange rods. (B) Rectangular boxes represent the intracellular domains of the receptors through which inhibition is carried out. Several of these receptors (KIR, ILT2, NKG2A & CD94, TIGIT & CD96, Siglec-7/9, PD-1, and SIRPα) bear 1-3 ITIMs in their cytoplasmic tail and observe ITIM-based inhibition. In addition, TIGIT and PD-1 cytoplasmic tails contain an ITT-like and ITSM motif, respectively. LAG-3, TIM-3, CD200, and CTLA-4 lack an ITIM motif in their cytoplasmic tails. Instead, they have special intracellular tyrosine motifs such as KEELE, Y265, NPXY, and YVKM and YFIP, respectively, which are implicated in carrying out the inhibition process. Antibodies to these receptors are shown within red-outlined boxes. (C) Moreover, several other immune cells, including T cells, B cells, and myeloid cells, express these receptors on their surfaces, as shown on the left panel for each immune checkpoint receptor.
MHC-I-Specific NK Inhibitors
NK cells avoid the killing of healthy autologous cells through MHC-I specific inhibitory receptors such as KIRs and NKG2A & CD94. Ligation results in a transient abortion of activation signals rather than anergy or apoptosis of natural killer cells (18) (Figure 1). These inhibitory receptors carry an inhibitory motif in their cytoplasmic domain called the immunoreceptor tyrosine-based inhibition motif (ITIM). Tyrosine phosphatases SHP-1/SHP-2 are recruited by ITIMs to carry out their function (48). Inhibitory receptors carrying such an inhibitory motif in their cytoplasmic tail constitute a specific group of receptors with various functions. Such inhibition is termed “dominant inhibition,” as it inhibits signaling at a proximal level (Box 2). Other NK cells surface inhibitory receptors, such as TIGIT, PD-1, and the Siglecs family inhibitory receptors, also possess ITIM containing cytoplasmic tails, while some of the inhibitory receptors lack this cytoplasmic inhibitory motif (Box 2).
Box 2. ITIM-based inhibition and intracellular signaling in NK cells.
Dominant inhibition
Negative regulation exhibited by classical NK-cell inhibitory receptors is distinct from negative feedback, as it prevents the activation from happening by blocking activation signals at an early stage (16, 18, 19). A transient abortion of activation signals is achieved rather than anergy or apoptosis as a result of this negative regulation. This type of inhibitory regulation is called dominant inhibition, as the downstream activation signals are prevented from occurring at all (16).
ITIM-based inhibition
This dominant inhibition by NK-cell inhibitory receptors is carried out through a cytoplasmic immunoreceptor tyrosine-based inhibition motif (ITIM). Such ITIM-bearing receptors are members of several receptor families and are expressed in many cell types, such as leukocytes, mast cells, monocytes, dendritic cells, and macrophages with various cellular functions that include allergy, autoimmunity, graft vs. host disease, phagocytosis of red blood cells, and neuronal plasticity in the brain (18, 49). The ITIM domain, at least one, is present in over 300 integral membrane proteins, both type I and type II, as revealed in bioinformatics analyses of entire genes of the human genome (49). Inositol (SHIP1 and SHIP2) or tyrosine (SHP1 and SHP2) phosphatases that contain Src homology (SH) 2 domain are recruited by ITIM upon receptor-ligand binding. Nonetheless, SHP1 and SHP2 tyrosine phosphatases represent the majority ITIM-bearing receptor binders (16, 50). Functional characterization of inhibitory KIR revealed the precise ITIM sequence as the binding site for SHP1 to be “V/IxYxxL/V” (“x” indicates a non-conserved position, “/” indicates either). A sequence of 25 amino acids separates the two ITIMs in the cytoplasmic tail of the single inhibitory receptor, thereby providing a binding site for the tandem SH2 domains of SHP1 and SHP2 (48, 50).
SHP1 recruitment, leading to Vav1 dephosphorylation and Crk phosphorylation by c-Abl, have been the intracellular signaling pathways involved in the inhibition process observed in NKG2A/CD94 and KIR. KIRs recruit SHP1 and SHP2 for its inhibitory action but not SHIP (16, 18, 19, 51, 52). On the other hand, LIR (leukocyte immunoglobulin-like receptors LIR1-5) cytoplasmic tails contain two to four ITIMs. ILT2, the one mainly involved in NK cell inhibition, contains three ITIMs in its cytoplasmic tail (53). The TIGIT cytoplastic tail contains one immunoreceptor tail tyrosine (ITT)-like motif in addition to ITIM. Grb2 and β-arrestin2 phosphorylation by Fyn and Lck leads to the recruitment of SHIP1 to downregulate PI3K, MAPK, and NF-κB (nuclear factor-κB) signaling, resulting in a robust reduction of NK cell-mediated cytotoxicity, its granule polarization and production of cytokines (54–57). Similar to classical NK-cell inhibitory receptors NKG2A/CD94 and KIRs, inhibitory Siglecs also contain one or more ITIM and ITIM-like motifs in their cytoplasmic tail. Upon ligation, ITIMs are phosphorylated by Src family kinases recruiting and activating SRC homology 2 (SH2)-domain-containing proteins, mainly the tyrosine phosphatases SHP1 and SHP2 or the SOCS3 (suppressor of cytokine signaling 3) protein (58). PD-1 carries one immunoreceptor tyrosine-based switch motif (ITSM) in addition to one ITIM in its cytoplasmic tail. ITSM is required for the inhibitory functions associated with PD-1 triggering, such as PI3K/Akt activation and suppression of interleukine-2 production, upon phosphorylation by SHP1 or SHP2. Particularly, Y248 (PD-1 ITSM) was found to interact with SHP1 and SHP2, and its association with SHP2 is required for the inhibition of PI3K/Akt activation (59–61). SIRPα has three extracellular Ig domains and an intracellular tail with two immuno-receptor tyrosine-based inhibitory motifs (ITIMs) (62). SIRPα-contained ITIM motifs inhibit phagocytosis and cellular attachment through SHP1 upon interaction with CD47, and members of the JAK/STAT family signaling pathway have been identified as potential downstream mediators of SIRPα signaling (63, 64).
ITIM-lacking inhibition
CTLA-4 lacks the classic signaling motif (ITIM) in its cytoplasmic tail, and phosphorylation of CTLA-4 tyrosines (YVKM and YFIP) fails to allow for single or tandem SHP2 SH2 domain binding (65). It directly inhibits the activation of Akt, but not of PI3K, by association with protein phosphatase 2A (PPA2) (59–61, 66). However, a CTLA-4 mutant lacking PP2A-binding sites appeared to show increased inhibitory function. Walker et al. termed CTLA-4 intracellular signaling confusing and unclear due to the contradictory nature of the medical literature and proposed to consider moving away from the signaling view toward a quantitative conception of ligand competition (67). LAG-3 has a unique cytoplasmic tail containing three regions. The first region contains a serine phosphorylation site, while the KIEELE motif that is contained in the second region has been revealed to be essential for the inhibitory function of Lag-3 in effector CD4+ T cells. The third region contains glutamic acid-proline (EP) repeats. These regions are conserved in humans and mice (59, 68). The TIM-3 cytoplasmic tail contains five conserved tyrosine residues, with two tyrosines (residues 256 and 263 in mouse TIM-3) being particularly important (69). In the absence of ligand binding, TIM-3 is associated with HLA-B-associated transcript 3 (Bat3), which protects the cell from TIM-3-mediated inhibition. Ligand binding (Galec-9 and CEACAM-1) leads to the phosphorylation of tyrosine residues (Y256 and Y263 in mouse, and Y265, a corresponding tyrosine residue to Y256, in human) causing disruption of the association between TIM-3 and Bat-3 and thereby allowing TIM-3-mediated inhibition to occur (59, 70). TIM-3 can associate with interleukin-inducible T-cell kinase (ITK), the Src kinases Fyn and Lck, and the p85 phosphatidylinositol 3-kinase (PI3K) adaptor protein to positively or negatively regulate IL-2 production via NF-κB/NFAT signaling pathways (69–71). CD200R also lacks the classical ITIM in its cytoplasmic tail but contains three tyrosine residues (Y286, Y289, and Y297), one of which (Y297) is located in an NPxY motif. Phosphorylation of tyrosine residues (majorly, Y286 and Y297) leads to the recruitment of inhibitory adaptor proteins Dok1 and Dok2 and the subsequent inhibition of Ras/mitogen-activated protein kinase (RasGAP) activation (72, 73).
KIRs
The KIR family (also known as CD158) is a diversified and polymorphic group of NK cell receptors comprising inhibitory and activating KIRs, each recognizing a specific HLA class I allotype (HLA-A, -B, or -C) as ligand (13). The inhibitory KIR2DL1, KIR2DL2, and KIR2DL3 recognize HLA-C as their ligand while HLA-B and HLA-A serve as ligands to other KIRs, including inhibitory KIR3DL1 and KIR3DL2. In addition to NK cells, T cell subsets and iNKT cells (invariant natural killer T cells) also expressed KIR (74). MHC-I molecules (HLA-A, -B, and -C), ligands for KIRs and KIRs, themselves exhibit extensive natural polymorphism (75). The diverse allelic combination of the KIR genes (a total of 17 KIR genes on chromosome 19q13, 14), the polymorphism within each gene, and each NK cell expressing some of the KIRs make this complex KIR repertoire capable of recognizing minute alterations in MHC-I expression (51, 76–81).
IPH2101 and lirilumab (IPH2102/BMS-986015) are IgG4 monoclonal antibodies (mAbs) targeting KIR2DL1/2/3 NK inhibitory receptors that are currently in clinical evaluation and development as monotherapy or in combination with other agents, including a molecular targeted agent (lenalidomide), monoclonal antibodies (elotuzumab & rituximab), and immune checkpoint blockers (ipilimumab & nivolumab) (82–99) (Table 1). IPH2101 is clinically evaluated in various hematologic (AML, CLL, NHL) or solid malignancies (breast and ovarian cancers). A dose of up to 10 mg/kg of IPH2101 was reported to be well-tolerated (82–85). So far, IPH2101 as monotherapy has failed to impress clinically in patients with multiple myeloma (MM). A dose-escalation, phase I trial of IPH2101 in MM (relapsed/refractory) as monotherapy, with the primary objective of assessing the maximum dose tolerable and limiting toxicity, reported acceptable safety and tolerability without any evidence of autoimmunity. However, just 11 (34%) patients achieved the best response of stable disease, as evaluated by the International Myeloma Working Group (IMWG) criteria (82). It was revealed that infusion of IPH2101 in MM patients had reduced NK cell surface expression of the KIR2D receptor and the responsiveness of the NK cell (86). Preclinical evidence has demonstrated functional augmentation of NK cells and ligand upregulation for activation of NK cell surface receptors on MM cells by lenalidomide, which, in combination with IPH2101, have shown potent in vivo rejection of lenalidomide-resistant tumor (87) (Figure 3). IPH2101 and lenalidomide as “dual immunotherapy” for MM patients has been reported to achieve a median progression-free survival of 24 months, five objective responses with acceptable toxicity (five severe AEs), and no autoimmunity. Overall, this combination holds promise and warrants further clinical evaluation in MM patients despite the failure of IPH2101 as a single agent (88, 89). A phase II trial of lirilumab was terminated because of failure to meet the objective response criteria (50% decline in M-protein) set for MM patients, with only one (11%) and six (66%) of a total of nine patients enrolled achieving minimal response and stable disease (90). However, in vitro elotuzumab-mediated cell-killing was enhanced by lirilumab and showed synergism in potentiating anti-tumor efficacy in KIR2DL3-transgenic and RAG-deficient mice (91). In vitro augmentation of elotuzumab-mediated ADCC and in vivo synergism in mediating potent elotuzumab anti-MM activity by lirilumab were also reported by Sola et al., setting the rationale for clinical evaluation of this combination in MM patients (92). A phase I (NCT2252263) study evaluating elotuzumab and lirilumab in combination in multiple myeloma patients is currently in development.
Table 1.
Clinical trials evaluating the safety, tolerability and efficacy of NK cell-based immune checkpoint inhibitors or potential immune checkpoint inhibitors for NK cell-based immunotherapy.
| Receptor | National clinical trial no. | Phase, allocation | Sponsors | Participants | Drugs | Combinations | Status | Disease | First posting date |
|---|---|---|---|---|---|---|---|---|---|
| KIR | NCT00552396 | I, Non-randomized | Innate Pharma | 32 | Anti-KIR (1-7F9) | Single agent | Completed | Multiple myeloma | November 1, 2007 |
| NCT00999830 | II, Randomized | Innate Pharma | 27 | IPH2101 | Single agent | Completed | Patients with multiple myeloma in stable partial response after a first line therapy | October 22, 2009 | |
| NCT01256073 | I, Non-randomized | Innate Pharma | 21 | IPH2101 | Single agent | Completed | Acute myeloid leukemia | December 8, 2010 | |
| NCT01222286 | II, Randomized | Innate Pharma | 30 | IPH2101 | Single agent | Completed | Smoldering multiple myeloma | October 18, 2010 | |
| NCT01217203 | I, Non-randomized | Innate Pharma | 15 | IPH2101, lenalidomide | IPH2101 plus lenalidomide | Completed | Patients with multiple myeloma experiencing a first or second relapse | October 8, 2010 | |
| NCT01687387 | II, Randomized | Innate Pharma | 152 | Lirilumab (IPH2102), placebo | Lirilumab vs. placebo | Completed | Elderly Patients with Acute Myeloid Leukemia (AML) in First Complete Remission | September 18, 2012 | |
| NCT01750580 | I, Non-randomized | Bristol-Myers Squibb | 22 | Lirilumab, ipilimumab | Lirilumab plus ipilimumab | Completed | Selected advanced tumor | December 17, 2012 | |
| NCT02252263 | I, Randomized | Innate Pharma | 44 | Elotuzumab, lirilumab (BMS-986015), urelumab | Elotuzumab plus lirilumab or urelumab | Completed | Multiple myeloma | September 30, 2014 | |
| NCT02481297 | II, Non-randomized | M.D. Anderson Cancer Center, Bristol-Myers Squibb | 8 | Lirilumab, rituximab | Lirilumab plus rituximab | Active | Leukemia, chronic lymphocytic leukemia, lymphocytic leukemia | June 25, 2015 | |
| NCT03203876 | I, Non-randomized | Bristol-Myers Squibb/Ono Pharmaceutical Co., Ltd. | 21 | Lirilumab, nivolumab, ipilimumab | Lirilumab plus nivolumab or lirilumab plus nivolumab and ipilimumab | Active, not recruiting | Advanced and/or metastatic solid tumors | June 29, 2017 | |
| NCT01714739 | I/II, Randomized | Bristol-Myers Squibb | 337 | Lirilumab, nivolumab, ipilimumab | Lirilumab plus nivolumab or lirilumab plus nivolumab and ipilimumab | Active, not recruiting | Advanced refractory solid tumors | October 26, 2012 | |
| NCT02593045 | I, Non-randomized | Innate Pharma | 60 | IPH4102, a humanized anti-KIR3DL2 monoclonal antibody | Single agent | Active, not recruiting | Relapsed/refractory cutaneous T-cell lymphomas (CTCL) | October 30, 2015 | |
| NCT03902184 | II, Non-randomized | Innate Pharma | 250 | IPH4102, gemcitabine + oxaliplatin | IPH4102 alone or in combination with chemotherapy | Recruiting | Lymphoma, T-Cell lymphoma, T-Cell, cutaneous lymphoma, T-cell, peripheral mycosis fungoides/sezary syndrome | April 3, 2019 | |
| NKG2A | NCT02331875 | I/II, Non-randomized | Innate Pharma | 3 | Monalizumab (IPH2201) | Single agent | Terminated (lack of recruitment) | Squamous Cell carcinoma of the oral cavity | January 6, 2015 |
| NCT02459301 | I, Non-randomized | Canadian Cancer Trials Group, Innate Pharma | 59 | Monalizumab | Single agent | Completed (November 13, 2019) | Gynecologic malignancies | June 2, 2015 | |
| NCT02921685 | I, Non-randomized | Institut Paoli-Calmettes, Innate Pharma | 18 | Monalizumab | Single agent | Recruiting | Hematologic malignancies | October 3, 2016 | |
| NCT02557516 | I/II, Non-randomized | Innate Pharma | 22 | Monalizumab, ibrutinib | Monalizumab plus ibrutinib | Active, not recruiting | Relapsed, refractory or previously untreated chronic lymphocytic leukemia | September 23, 2015 | |
| NCT02643550 | I/II, Non-randomized | Innate Pharma, Astrazeneca | 140 | Monalizumab, cetuximab, anti-PD(L)1 | Monalizumab plus cetuximab or monalizumab plus cetuximab plus Anti-PD(L)1 | Recruiting | Human Papillomavirus (HPV) (+) and HPV (-) Recurrent or Metastatic HNSCC | December 31, 2015 | |
| NCT02671435 | I/II, Non-randomized | MedImmune LLC | 501 | Durvalumab (MEDI4736), monalizumab | Durvalumab plus monalizumab | Recruiting | Advanced solid tumors | February 2, 2016 | |
| NCT03088059 | II, Non-randomized | European Organization for Research and Treatment of Cancer—EORTC | 340 | Monalizumab, durvalumab, afatinib, palbociclib, standard of care, niraparib, rogaratinib (BAY1163877) | Monalizumab alone or monalizumab plus durvalumab or SOC | Recruiting | Recurrent/metastatic HNSCC | March 23, 2017 | |
| NCT03822351 | II, Randomized | MedImmune LLC | 300 | Monalizumab, durvalumab (MEDI-4736), oleclumab (MEDI-9447) (anti-CD73 antibody), | Durvalumab plus monalizumab or durvalumab plus oleclumab or durvalumab alone | Recruiting | Locally advanced, unresectable (Stage III) non-small cell lung cancer (COAST) | January 30, 2019 | |
| NCT03833440 | II, Randomized | Assistance Publique Hopitaux De Marseille | 120 | Monalizumab, durvalumab, oleclumab, AZD6738 (ATR inhibitor), DOCETAXEL | Durvalumab plus monalizumab or durvalumab plus oleclumab or durvalumab plus AZD6738 or DOCETAXEL alone | Not yet recruiting | Advanced non-small cell lung cancer patients with PD-1 ICI resistance | February 7, 2019 | |
| TIGIT | NCT02794571 | I, Non-randomized | Genentech, Inc. | 300 | Tiragolumab (MTIG7192A), atezolizumab | Tiragolumab alone or tiragolumab plus atezolizumab | Recruiting | Advanced or metastatic tumors | June 9, 2016 |
| NCT03119428 | I, Non-randomized | OncoMed Pharmaceuticals, Inc. | 33 | Etigilimab (OMP-313M32), nivolumab | Etigilimab alone or etigilimab plus nivolumab | Terminated (sponsor decision) | Locally advanced or metastatic solid tumors | April 18, 2017 | |
| NCT03563716 | II, Randomized | Genentech, Inc. | 135 | Tiragolumab (MTIG7192A), atezolizumab | Atezolizumab plus tiragolumab or atezolizumab plus placebo | Active, not recruiting | Locally advanced or metastatic non-small cell lung cancer | June 20, 2018 | |
| NCT03628677 | I, Non-randomized | Arcus Biosciences, Inc. | 242 | AB154 (anti-TIGIT), AB122 (anti-PD-1) | AB154 alone or AB154 plus AB122 | Recruiting | Advanced solid malignancies | August 14, 2018 | |
| LAG-3 | NCT03489369 | I | Symphogen A/S | 30 | Sym022 (anti-LAG-3) | Single agent | Recruiting | Advanced solid tumor malignancies or lymphomas | April 5, 2018 |
| NCT03311412 | I, Non-randomized | Symphogen A/S | 102 | Sym021 (anti-PD-1), Sym022 (anti-LAG-3), Sym023 (anti-TIM-3) | Sym021 or Sym021 plus Sym022 or Sym021 plus Sym023 | Recruiting | Advanced solid tumor malignancies or lymphomas | October 17, 2017 | |
| NCT01968109 | I/II, Randomized | Bristol-Myers Squibb | 2000 | Relatlimab, nivolumab, BMS-986213 | Relatlimab alone or relatlimab plus nivolumab | Recruiting | Advanced solid tumors | October 23, 2013 | |
| NCT02061761 | I/II, Non-randomized | Bristol-Myers Squibb | 109 | BMS-986016 (relatlimab) (anti-Lag-3), nivolumab | Relatlimab alone or relatlimab plus nivolumab | Active, not recruiting | Relapsed or refractory hematologic malignancies | February 13, 2014 | |
| NCT02658981 | I, Non-randomized | Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins, National Cancer Institute (NCI), Bristol-Myers Squibb | 100 | Relatlimab, nivolumab, urelumab | Relatlimab plus nivolumab or urelumab plus nivolumab | Recruiting | Glioblastoma, gliosarcoma, recurrent brain neoplasm | January 20, 2016 | |
| NCT02966548 | I, Non-randomized | Bristol-Myers Squibb, Ono Pharmaceutical Co. Ltd | 45 | Relatlimab, nivolumab | Relatlimab alone or relatlimab plus nivolumab | Recruiting | Advanced solid tumors | November 17, 2016 | |
| NCT03044613 | I, Non-randomized | Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins, Bristol-Myers Squibb | 25 | Nivolumab, relatlimab, carboplatin, paclitaxel, radiation | Nivolumab or nivolumab/relatlimab prior to concurrent chemoradiation plus nivolumab or nivolumab/relatlimab | Active, not recruiting | Gastric cancer, esophageal cancer, gastroesophageal cancer | February 7, 2017 | |
| NCT03459222 | I/II, Non-randomized | Bristol-Myers Squibb | 230 | Relatlimab, nivolumab, BMS-986205 (IDO1 inhibitor), ipilimumab | Relatlimab plus nivolumab plus IDO1 inhibitor or relatlimab plus nivolumab plus ipilimumab | Recruiting | Advanced cancer | March 8, 2018 | |
| NCT03493932 | I | National Institute of Neurological Disorders and Stroke (NINDS) | 20 | Nivolumab, relatlimab | Nivolumab plus relatlimab | Recruiting | Glioblastoma | April 11, 2018 | |
| NCT03623854 | II | Jonsson Comprehensive Cancer Center, National Cancer Institute (NCI) | 20 | Nivolumab, relatlimab | Nivolumab plus relatlimab | Recruiting | Advanced Chordoma | August 9, 2018 | |
| NCT03743766 | II, Randomized | John Kirkwood Bristol-Myers Squibb | 42 | Relatlimab, nivolumab | Nivolumab or relatlimab or nivolumab plus relatlimab | Recruiting | Metastatic melanoma | November 16, 2018 | |
| NCT02614833 | II, randomized | Immutep S.A. | 241 | Eftilagimod alpha, paclitaxel | Eftilagimod Alpha plus Paclitaxel or placebo plus paclitaxel | Active, not recruiting | Metastatic breast carcinoma | November 25, 2015 | |
| NCT02676869 | I | Immutep Australia Pty. Ltd. | 24 | IMP321 (eftilagimod alpha), pembrolizumab | IMP321 (Eftilagimod alpha) adjuvant to Anti-PD-1 | Active, not recruiting | Unresectable or metastatic melanoma | February 8, 2016 | |
| NCT03252938 | I, Non-randomized | IKF Klinische Krebsforschung GmbH at Krankenhaus Nordwest | 50 | IMP321 (LAG-3Ig fusion protein), avelumab | IMP321 alone or IMP321 plus avelumab | Recruiting | Solid tumor peritoneal carcinomatosis | August 17, 2017 | |
| NCT03625323 | II, Non-randomized | Immutep S.A., Merck Sharp & Dohme Corp. | 109 | Eftilagimod alpha, pembrolizumab | Eftilagimod alpha plus pembrolizumab | Recruiting | NSCLC HNSCC | August 10, 2018 | |
| TIM-3 | NCT03489343 | I, Non-randomized | Symphogen A/S | 24 | Sym023 (anti-TIM-3) | Single agent | Active, not recruiting | Advanced solid tumor malignancies or lymphomas | April 5, 2018 |
| NCT02817633 | I, Non-randomized | Tesaro, Inc. | 873 | Cobolimab (TSR-022, an anti-TIM-3 antibody), TSR-042 (dostarlimab, an anti-PD-1 antibody), TSR-033 (an anti-LAG-3 antibody) | Cobolimab alone or cobolimab plus dostarlimab or cobolimab plus dostarlimab plus TSR-033 | Recruiting | Advanced solid tumors | June 29, 2016 | |
| NCT03680508 | II, Non-randomized | University of Hawaii, Tesaro, Inc. | 42 | Cobolimab, dostarlimab | Cobolimab plus dostarlimab | Recruiting | Liver cancer | September 21, 2018 | |
| NCT04139902 | II, Randomized | Diwakar Davar, Tesaro, Inc. | 56 | Dostarlimab, cobolimab | Dostarlimab alone or cobolimab plus dostarlimab | Not yet recruiting | Resectable stage III or oligometastatic stage IV melanoma | October 25, 2019 | |
| NCT03099109 | I, Non-randomized | Eli Lilly and Company | 196 | LY3321367(an anti-TIM-3 antibody), Lodapolimab (LY3300054, an anti-PD-L1 antibody) | LY3321367 Alone or LY3321367 plus lodapolimab | Recruiting | Advanced relapsed/refractory solid tumors | April 4, 2017 | |
| NCT03744468 | I/II, Non-randomized | BeiGene | 162 | BGB-A425 (an anti-TIM-3 antibody), tislelizumab (BGB-A317, an anti-PD-1 antibody) | BGB-A425 plus tislelizumab | Recruiting | Advanced solid tumors | November 16, 2018 | |
| NCT02608268 | I/II, Non-randomized | Novartis Pharmaceuticals | 250 | Spartalizumab, MBG453 (an anti-TIM-3 antibody) | MBG453 alone or MBG453 plus Spartalizumab | Recruiting | Advanced malignancies. | November 18, 2015 | |
| NCT03066648 | I, Randomized | Novartis Pharmaceuticals | 235 | MBG453 (an anti-TIM-3 antibody), decitabine, spartalizumab, | MBG453 alone or MBG453 plus decitabine or MBG453 plus spartalizumab or MBG453 plus decitabine plus spartalizumab | Recruiting | AML or high risk myelodysplastic syndrome | February 28, 2017 | |
| NCT03961971 | I, Non-randomized | Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins, Novartis Pharmaceuticals | 15 | MBG453 (an anti-TIM-3 antibody), spartalizumab (PDR001) | MBG453 plus spartalizumab | not yet recruiting | Recurrent GBM | May 23, 2019 | |
| CD200 | NCT00648739 | I/II, Non-randomized | Alexion Pharmaceuticals | 26 | Samalizumab (ALXN6000) | Single agent | Terminated | Relapsing or refractory CLL or MM | April 1, 2008 |
| NCT02987504 | I, Non-randomized | Alexion Pharmaceuticals, Quintiles, Inc. | 10 | Samalizumab (ALXN6000) | Single agent | Terminated | Advanced solid tumors | December 9, 2016 | |
| CD47 | NCT02953509 | I/II, Non-randomized | Forty Seven, Inc., The Leukemia and Lymphoma Society | 72 | Magrolimab (Hu5F9-G4), rituximab | Magrolimab plus rituximab | Recruiting | Relapsed/refractory B-cell non-hodgkin's lymphoma | November 2, 2016 |
| NCT03248479 | I, Non-randomized | Forty Seven, Inc., California Institute for Regenerative Medicine (CIRM) | 96 | Magrolimab, azacitidine | Magrolimab alone or magrolimab plus azacitidine | Recruiting | Acute myeloid leukemia myelodysplastic syndromes | August 14, 2017 | |
| NCT02953782 | I/II, non-randomized | Forty Seven Inc., California Institute for Regenerative Medicine (CIRM) | 112 | Magrolimab, cetuximab | Magrolimab plus cetuximab | Recruiting | Solid tumor and advanced colorectal cancer | November 3, 2016 | |
| NCT03527147 | I, Non-randomized | Acerta Pharma BV, AstraZeneca | 88 | Magrolimab, rituximab, acalabrutinib, AZD9150, AZD6738, AZD5153 | Magrolimab or rituximab plus acalabrutinib | Recruiting | Relapsed or refractory aggressive non-hodgkin's lymphoma | May 17, 2018 | |
| NCT03869190 | I/II, Randomized | Hoffmann-La Roche, Forty Seven Inc., Tesaro Inc., Seattle Genetics and Astellas, Sanofi | 305 | Magrolimab, atezolizumab, enfortumab vedotin, niraparib, isatuximab, linagliptin, tocilizumab | Atezolizumab plus magrolimab | Recruiting | Urothelial carcinoma | March 11, 2019 | |
| NCT03922477 | I, Non-randomized | Hoffmann-La Roche | 21 | Magrolimab, atezolizumab (anti-PD-L1 antibody) | Magrolimab plus atezolizumab | Recruiting | Acute myeloid leukemia | April 22, 2019 | |
| B7-H3 | NCT02923180 | II, Non-randomized | Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins, MacroGenics | 33 | Enoblituzumab (MGA271) | Neoadjuvant enoblituzumab | Active, not recruiting | Localized intermediate and high-risk prostate cancer | October 4, 2016 |
| NCT02982941 | I, Non-randomized | MacroGenics | 25 | Enoblituzumab | Enoblituzumab | Completed | Neuroblastoma, rhabdomyosarcoma, osteosarcoma, ewing sarcoma, wilms tumor, desmoplastic small round cell tumor | December 6, 2016 | |
| NCT02381314 | I, Non-randomized | MacroGenics | 24 | Enoblituzumab, ipilimumab | Enoblituzumab plus ipilimumab | Completed | Melanoma, non-small cell lung cancer | March 6, 2015 | |
| NCT02475213 | I, Non-randomized | MacroGenics | 157 | Enoblituzumab, pembrolizumab, MGA012 (anti-PD-1 monoclonal antibody) | Enoblituzumab plus pembrolizumab or enoblituzumab plus MGA012 | Recruiting | Melanoma, head and neck cancer, non-small cell lung cancer, urothelial carcinoma | June 18, 2015 | |
| NCT03406949 | I, Non-randomized | MacroGenics | 139 | Orlotamab (MGD009, a humanized B7-H3 × CD3 DART® protein), MGA012 (anti-PD-1 monoclonal antibody) | Orlotamab plus MGA012 | Recruiting | Advanced solid tumors | January 23, 2018 |
Figure 3.
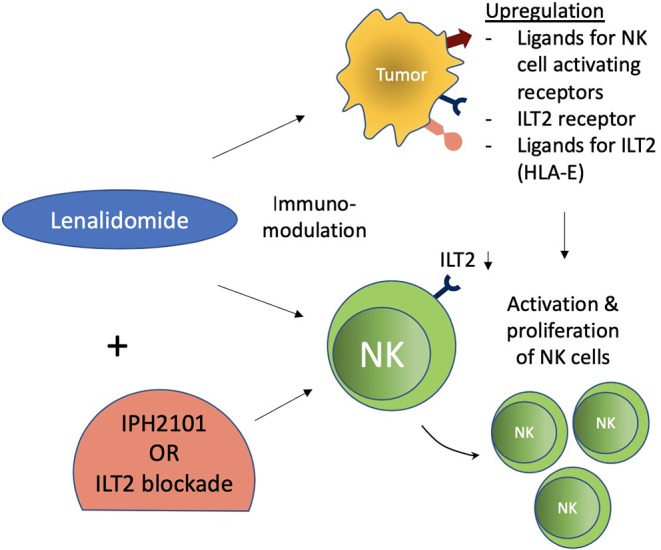
Immunomodulatory effect of lenalidomide on NK cells. Lenalidomide upregulates ligands for NK cell-activating receptors in multiple myeloma and augments NK cell function. Lenalidomide also increases ILT2 expression on CLL and decreases it on NK cells, while its ligand (HLA-E) is reestablished on leukemic cells. These immunomodulatory effects of NK cells were associated with increased NK cell activation and proliferation. ILT2 blockade in this setting further potentiated NK cell functions.
In vivo IPH2101 blockade of KIR resulted in better survival, showing preclinical evidence of efficacy in AML cells (acute myeloid leukemia) (93). Comparatively better clinical efficacy was evident in AML patients, with a median PFS of 7.7 months, RFS of 10.8 months, and OS of 12.7 months. These clinical outcomes were improved with increasing dose, but to a non-significant degree. Only OS showed significant increase with a dose of 1–3 mg/kg dose as compared to the previous dose of 0.3 mg/kg (27.9 vs. 11.8 months, p < 0.034) (84). Safety and tolerability were acceptable, and AEs were mild and transient (83–85). An extension of this study revealed the role of lirilumab as maintenance treatment in elderly patients by prolonging survival and achieving satisfactory safety with repeated administrations (94). However, the results of an effiki trial revealed no significant difference among lirilumab regimens vs. placebo, casting doubts on lirilumab as a single agent in this setting (95). Lirilumab with full doses of azacytidine was well-tolerated in heavily pretreated/relapsed AML patients with high-risk features (96). The effectiveness and tolerability of lirilumab as monotherapy or combined with azacytidine in myelodysplastic syndrome (MDS) patients have been reported in a recent study (97). In a human model, a lirilumab and rituximab combination showed increased NK cytotoxicity in comparison to rituximab alone, in which autologous, EBV-transformed B cells were encountered by the individual's fresh primary NK cells, compared to a cell line model of anti-lymphoma therapy (98). In KIR transgenic and syngenic murine lymphoma models, this combination showed, in vitro and in vivo, an increased cytotoxicity mediated by NK cells and dependent on rituximab (99). These results provide and support a combined therapeutic approach of anti-KIR and anti-CD20 mAbs for clinical evaluation. More recently, a third member of this group of anti-KIR antibodies, IPH4102, a humanized anti-KIR3DL2 monoclonal antibody, has entered the clinical evaluation phase (Table 1). IPH4102, also termed Lacutamab, was declared safe in a phase I clinical evaluation of relapsed/refractory cutaneous T-cell lymphoma, with the most common adverse events including edema, fatigue, and lymphopenia. The clinical activity was encouraging as well, as 16 (36%) of the total of 44 patients achieved global overall response. Patients with relapsed/refractory cutaneous T-cell lymphoma with Sézary syndrome exhibited much better clinical response [15 (43%) of the 35 patients] (100). A phase II clinical trial (NCT03902184) is ongoing investigating IPH4102 as a single agent or in combination with chemotherapy in treating T-cell lymphomas.
LIRs (ILTs)
Leukocyte immunoglobin-like receptor (LIR) or immunoglobin-like transcript (ILT), like KIR, belongs to the Ig superfamily and consists of activating as well as inhibitory receptors (53, 101, 102). Five inhibitory receptors (LIRB1-5) have been identified out of a total of 11 LIR members (103). Numerous immune cells (NK, T, B, and myeloid lineage cells that include macrophages and dendritic cells) variedly express these receptors (53, 101, 102). Among these, LIRB1 (ILT2) and LIRB2 (ILT4) recognize HAL-G as their main ligand, in addition to other ligands, leading to immunogenic tolerance (101, 104). LIRB1 or ILT2 is expressed on natural killer cells (36 ± 18% of normal NK cells), T cells, B cells, monocytes, subsets of DCs, and myeloid-derived suppressive cells (MDSCs), while ILT4 is expressed mainly on myeloid cells (21, 102). Therefore, ILT2 and HLA-G interaction could inhibit NK, T, and B cells' immune functions, thereby representing an immunotherapeutic target (105).
Various primary tumors and metastatic malignancies express HLA-G. It has also been regarded as an indicator of progressive disease and prognosis in various cancers (102, 106). Its expression has been associated with a decrease in NK functions in various cancers such as hepatocellular carcinoma (HCC), ovarian cancer, non-small cell lung carcinoma (NSCLC), glioma, and renal cell carcinoma (RCC) (107–111). Moreover, HLA-G surface expression or soluble HLA-G and/or its interaction with ILT2 has demonstrated inhibition of NK functions including cytotoxicity, cytokine production, and chemokine secretion (82, 102). Cytolysis by decidual NK cells was resisted by HLA-G-expressing target cells (112). HLA-G expression protected target cells against NK cell cytotoxicity in peripheral blood and NK cell lines (113, 114). ILT2 with HLA-G interaction has also shown inhibition of target cell-induced polarized IFN-γ production by NK cells (115). Chemotaxis and secretion of cytokines and chemokines by NK cells (CD56bright and CD56dim) were differentially modulated by soluble HLA-G (sHLA-G). Secretion of CCL2 by both CD56bright and CD56dim NK cells and CCL2, CCL8, and CXCL2-CXCL3 by CD56dim NK cells from peripheral blood were upregulated by sHLA-G (116). HLA-G1/ILT2 interaction was also shown to mitigate MICA/NKG2D activation by inhibiting NK cytotoxicity (117). In gastric cancers, HLA-G/ILT2 interaction led to inhibition of infiltrating NK cell proliferation and cytotoxicity (118). HLA-G plasma expression in B-cell malignancies, however, has been shown to inhibit malignant B cell proliferation due to their expression of ILT2 surface receptors, which is lacking in solid tumor cells (102, 119). This may have been the reason for the failure of LIR-1 blockade in enhancing the cytotoxicity of the NK cells against MM cells (120). However, a dual blockade of LIR-1 and NKG2A was able to increase the cytotoxicity of KIR-negative NK cells (21). These findings suggest that a dual role is played by HLA-G-ILT2 interaction: the role of the classical inhibitory checkpoint in solid malignancies and the role of disease progression in hematological malignancies due to ILT2 expression on the hematological malignant cells. Hence, blocking this checkpoint with antibodies can be considered a possible potential target in solid cancers. Further exploration is needed to confirm its role in hematological cancers. Lenalidomide has also shown immunomodulation of NK cells and leukemic cells in CLL (Chronic Lymphocytic Leukemia). ILT2 expression is increased in CLL on NK cells and decreased on leukemic cells. The immunomodulatory effects of lenalidomide increase ILT2 expression on leukemic cells and partially recover its ligand (HLA-E) expression as well. Moreover, NK cell activation and proliferation were also increased. ILT2 blockade further potentiated NK cell activation and proliferation in this setting (121) (Figure 3).
NKG2A and CD94
NKG2A (also known as CD159) & CD94, a heterodimer inhibitory receptor of the C-type lectin family, recognizes a non-classical MHC-I molecule, HLA-E, as ligand (22). CD94-NKG2A and its HLA-E ligand are non-polymorphic. HLA-E*0101 and HLA-E*0103 represent the only two alleles exhibited by HLA-E in worldwide populations (122, 123). Almost 50% of the NK cells in the peripheral blood express CD94/NKG2A, primarily those that do not express inhibitory KIR. The co-expression of CD94/NKG2A with other inhibitory receptors of different specificity also exists. In addition, γδ and CD 8+ T cells also express CD94/NKG2A (21, 124). Ligation of NKG2A & CD94 to HLA-E expressed on normal cells suppresses signaling activation, thereby avoiding the destruction of normal bystander cells (125).
Tumor cells (hematological as well as solid tumors), in order to avoid killing by NK cells, have shown upregulation of HLA-E expression. In various cancers, poor prognosis has been associated with HLA-E upregulation, including colorectal (126, 127), ovarian (122), gynecologic cancers (123, 128), liver (129), glioblastoma (130, 131), Hodgkin lymphoma (132), chronic lymphocytic leukemia, esophagus, gastric, pancreas, colon, kidney, head and neck, lung and melanoma (124). Blocking of the CD94/NKG2A receptor with an antibody could be used as a therapeutic strategy. Hence, an antibody against CD94/NKG2A (IPH2201-Monalizumab), developed by Innate Pharma, has been employed in various trials (17) (Table 1). In vitro and in vivo findings have suggested the application of humanized anti-NKG2A antibody against hematologic malignancies to be safe and effective (133). Improvement of NK-cell dysfunction by monalizumab in chronic lymphocytic leukemia has been shown in vitro (134). Monalizumab was well-tolerated (IV or SC dosing up to 10 mg/kg) as monotherapy in gynecologic malignancies with no reported DTLs or SAEs. This ongoing trial of heavily pretreated cohorts revealed a stabilized disease in 41% of evaluable patients (128).
A transition from monotherapy to a combined therapeutic approach is on the rise in the field of immune checkpoint inhibitors, mainly because some of these receptors are heavily expressed on several innate and adaptive immune cells simultaneously, as well as due to intercellular interaction and interdependence. Monalizumab is being evaluated in combination with durvalumab, cetuximab, and ibrutinib. Various solid cancers that express HLA-E have infiltrating CD8+ T, NK, and NKG2A+ immune cells (124). These infiltrating NKG2A+ NK cells and CD8+ T cells have demonstrated enhanced NK- and T-cell responses upon receptor blocking (135). It has been reported that PD-1 is coexpressed along with NKG2A in tumor-infiltrating NK cells and CD8+ T cells. In vitro and in vivo blocking of both NKG2A/HLA-E and PD-1/PD-L1 pathways with antibodies have shown complete response rate (124, 135, 136). A combination of monalizumab and durvalumab has shown clinical efficacy and a manageable toxicity profile, with no DTLs, as suggested by preliminary data in patients with heavily pretreated metastatic microsatellite colorectal cancer (137).
In vitro findings have revealed the additive efficacy of anti-NKG2A antibody in combination with other immune-oncology treatments such as anti-EGFR (cetuximab) in an SCCHN cell line and anti-CD20 (obinutuzumab) in cocultures with B cell lines expressing MHC class I (135). The induction of ADCC by cetuximab and the possible inhibition of cetuximab-mediated cytotoxicity by CRC (colorectal cancer)-expressed HLA-E provided the basis for a combined therapeutic approach (135, 138, 139). Preliminary assessment of the safety and efficacy of a monalizumab and cetuximab combination in head and neck squamous cell carcinoma (SCC) that was previously treated, recurrent, and/or metastatic revealed a 27.5% ORR (objective response rate), a 5-month median PFS (progression-free survival), and a 10-month median overall survival (OS). This is an encouraging outcome if compared to historical records of the efficacy of cetuximab alone from previous studies (ORR 12.6%, PFS 2.3 m, OS 5.6 m). The adverse events profile of the combined approach was similar to that of cetuximab alone (140). Recent in vivo analysis has demonstrated that vaccine therapy efficacy is hampered by the induction of NKG2A on CD8+ T cells and that blocking of the NKG2A receptor leads to improved efficacy of vaccine therapy (141). Overall, blocking of NKG2A represents an exciting therapeutic approach, and in particular, its combination with other immune-oncology therapeutic agents is the way forward and permits further exploration.
Tigit and CD96
TIGIT (T cell immunoreceptor with immunoglobulin and ITIM domains) is an immune inhibitory receptor expressed on NK and T cells such as activated NK, T, mT (memory T cells), fTh (follicular T helper cells), and regulatory T cells (Tregs) (56, 142, 143). CD96, a member of the same immunoglobulin superfamily, has a similar inhibitory role but with lower binding affinity for the ligand CD155 as compared to TIGIT. CD226 is an activating receptor that competes in binding to CD155 with TIGIT and CD96 (144–146). CD155 (mainly) and CD112 serve as ligands for TIGIT & CD96 to bind in order to inhibit T cell- and NK cell-mediated immunity (55, 57, 143, 147–151). It certainly is an important receptor mediating innate as well as adaptive immune responses. CD155 is a transmembrane glycoprotein, also known as poliovirus receptor (PVR) as it was first identified as a poliovirus entry receptor. PVR (CD155) is a member of the immunoglobulin superfamily as well as being the fifth member of the nectin-like molecule family and is therefore also known as necl-5 (152). It is barely expressed in normal human tissues, but many tumor cell lines and primary malignancies highly express PVR (54, 153). Of the functions performed by PVR, immunoregulation through its interaction with inhibitory receptors TIGIT and CD96 and activating receptor CD226 is of particular interest. Various cancers have shown upregulation of CD155 with corresponding upregulated NK and T cell expression of TIGIT and CD96 in order to evade anti-tumor immunity by eliciting T cell or NK cell inhibition. Preclinical evidence supports the idea of blockading this checkpoint for the activation of NK cell-mediated antitumor immunity (56, 145, 151, 154). Clinical translation is in its preliminary stages (148).
NK, T (effector and memory), and regulatory T cells express TIGIT. So far, TIGIT blockade has been evaluated, primarily in hematologic tumors, in reference to its expression on T cells, including CD8+, CD3+, and regulatory T cells (154, 155). TIGIT blockade in MM is mostly explored in relation to CD8+ T cells. Multiple myeloma tumor cells show upregulation of CD155 ligand. Immunosurveillance and therapy of multiple myeloma were demonstrated to be dependent on CD226 experessed by NK and T cells (156). TIGIT blockade prevented the T-cell exhaustion mechanism responsible for myeloma escape after stem cell transplantation (157). CD8+ T cells expressing high levels of TIGIT on their surfaces were associated with multiple myeloma progression. Immunity was enhanced against multiple myeloma by its blockade in mice as well as in humans (158). Anti-TIGIT therapy could be tested for its clinical efficacy as monotherapy or combined with other therapeutic agents for the treatment of multiple myeloma patients after the failure of a single checkpoint agent targeting PD-1 receptors (159).
Poor clinical outcomes for patients with acute myeloid leukemia (AML) and CD8+ T cell exhaustion were associated with TIGIT (160). AML cells lysis was significantly augmented by T cells through in vitro blockade of TIGIT and PVR or PVRL2 interaction alone or in combination with the BiTE® antibody construct AMG 330 (161). Patients with higher TIGIT expression in the bone marrow (BM) after alloSCT had a significantly lower incidence of grade II–IV acute graft vs. host disease (aGVHD) (p = 0.048), shorter PFS (p = 0.024), and shorter OS (p = 0.046). Higher TIGIT expression also had lower NK cell counts in the BM after alloSCT, suggesting that TIGIT might play a crucial role in the GVL effect and GVHD to control NK cell activity and proliferation after alloSCT. Based on these observations, it was suggested that TIGIT could be a prognostic predictor after alloSCT and that its blocking could be a potent immunotherapeutic strategy to intensify the graft-vs.-leukemia effect after alloSCT in AML patients (162). Hodgkin and Reed–Sternberg (HRS) cells or Tregs are involved in the inhibition of Th1, CD8+ T cells, and NK cell activity through PD-1 (163). Variable expression of PD-1 and TIGIT was observed on CD3+ T cells in patients with Hodgkin lymphoma, suggesting that TIGIT blockade alone or in combination with other drugs might be used as a potential therapeutic target. However, further evaluation is required (164). As PD-1 or TIM-3 are co-expressed by TIGIT-positive T cells, targeting TIGIT could be an additional mechanism to avert exhaustion of T cells in B-cell Non-Hodgkin Lymphoma (B-NHL) (155). Though HL and NHL express TIGIT on T cells, its expression on NK cells and relevant therapeutic application in this group of patients have not been evaluated so far.
Several tumor mice models have shown TIGIT upregulation on CTL and NK cells during tumor progression. In vivo tumor growth of inoculated B16/F10 was delayed by TIGIT deficiency, while exhaustion of effector cells (CTL and NK cells) targeting tumor was reversed by in vivo TIGIT blocking in combination with in vivo Flt3L overexpression by gene delivery and improved overall survival by significantly suppressing pre-established B16/F10 tumor growth and metastasis (151). CD155 expression was associated with prognosis in human pancreatic cancer. This expression was also inversely correlated with tumor-infiltrating lymphocytes and positively correlated with VEGF expression and angiogenesis (165). However, TIGIT expression was found to be similar between pancreatic cancer patients and healthy controls, while CD226 and CD96 were downregulated (166). There seems to be a dysregulation of the TIGIT/CD96/CD226/CD155 pathway involving NK cells in pancreatic cancer patients, warranting further in vitro and in vivo evaluation.
NK and CD8+ T-cell functions were suppressed by TIGIT-intrinsic expression, thereby helping tumor (colorectal) growth in vivo (167). TIGIT was associated with exhaustion of NK cells in tumor-bearing mice and colonic cancer patients, while this exhaustion was reverted by its blockade, thereby eliciting potent antitumor immunity. The presence of NK cells was important for the therapeutic efficacy of TIGIT and/or PD-L1 blockade or dual blockade of both the checkpoints, as NK cell absence was associated with a lower frequency of IFNy- or TNF-secreting TILs (CD8+) and a higher frequency of PD-1-expressing TILs (CD8+) (167). NK cells constitute 25–50% of liver lymphocytes, which shows their importance for liver immunity. Moreover, the survival and prognosis of HCC patients were positively correlated with NK cell numbers in blood and tumor tissue (168, 169). Tumor progression of the HCC patients was associated with dysfunction of the tumor-infiltrating NK cells, primarily the CD11b-CD27-NK subsets (170). Sun et al. identified exhausted tumor-infiltrating CD96+ NK cells and found their expression to be correlated with poor clinical outcome for HCC patients. NK cell exhaustion was reversed when CD96-CD155 interaction or TGF-β1 was blocked (171).
There are conflicting reports about the effectiveness of TIGIT blockade against metastatic disease. TIGIT is considered not to have any effectiveness against metastasis formation, as the number of lung nodules found was comparable in TIGIT−/− mice and wild-type mice after intravenous injection with B16 melanoma cells (146). In vivo TIGIT signaling blockade improved overall survival by significantly suppressing pre-established B16/F10 tumor growth and metastasis (151). However, this effect was achieved in combination with Flt3L overexpression in vivo by gene delivery. On the other hand, several studies have reported CD96 efficacy against metastatic disease. CD96−/− mice had fewer lung metastases as compared to wild-type mice after intravenous injection with B16 melanoma cells (146). Experimental metastases were inhibited in three different tumor models with monoclonal antibody blockade of CD96. This suppression by CD96 mAb was NK cell-, CD226-, and IFN-γ-dependent but independent of activating Fc receptors. Furthermore, CD96 blockade enhanced metastatic control in the absence of TIGIT (172).
Recently, increased emphasis has been placed on the combination of checkpoint inhibitors in order to achieve a synergistic effect. Improved survival was reported in tumor-bearing mice with dual targeting of PD-1 and TIGIT by enhancing CD8+ T-cell activation (173). Dixon et al. also reported that dual blocking of TIGIT and PD-1 in an MC38 colon carcinoma model leads to a synergistic anti-tumor effect, resulting in complete tumor regression (174). Dual blockade of TIGIT and PD-1 in melanoma patients synergistically increased the proliferation, degranulation, and cytokine secretion of tumor-infiltrating and tumor antigen-specific CD8+ T cells, demonstrating a potential for dual blockage (175). Hong et al. suggested that both PD-1 and TIGIT may serve as potential targets for the treatment of RCC as well (176). In GBM patients, such dual blockade has also shown to enhance anti-cancer immunity as well as survival (177). Though these studies have reflected the efficacy of dual checkpoint blockade in various cancers by exploring the role of T cells, there are some studies that have shown efficacy of dual checkpoint to be dependent on NK cells as well. Anti-TIGIT plus anti-PD-L1 blockade prevented the exhaustion of NK cells in tumor-bearing mice and colon cancer patients (167). On the other hand, a combination of Anti-CD96 with doxorubicin chemotherapy, anti-CTLA-4, or anti-PD-1 has shown more efficacy in inhibiting experimental metastases in three different tumor models (172). Exhausted NK cells from patients with bladder cancer (BC) showed upregulation of TIM-3 and TIGIT in both the periphery and tumor (178). In fact, the role of TIGIT and CD96 in NK cell exhaustion in various cancers is under investigation, and further revelations are needed to establish their potential for targeting either as monotherapy or in combination with other checkpoints.
Siglec-7/9
Sialic acid-binding Immunoglobulin-like lectins (Siglecs) are immunomodulatory sialic acid-binding receptors belonging to the I-type lectin family (179). Siglecs are expressed on various immune cells that include immune cells of both lymphoid and myeloid origin, namely neutrophils, eosinophils, monocytes, macrophages, NK cells, DCs, mast cells, and B and T cells (58, 180–183). Siglecs shows variety in two properties: their expression and their specificity for sialic acid-containing ligands (184). Most of these Siglecs are inhibitory receptors, such as Siglec-2, Siglec-3, Siglec-5, Siglec-6, SIglec-7, Siglec-8, Siglec-9, Siglec-10, and Siglec-11 (58). Of the inhibitory Siglecs, Siglec-7 and Siglec-9 are reported to be expressed on human NK cells (185–187). Similar to classical NK cell inhibitory receptors NKG2A/CD94 and KIRs, inhibitory Siglecs also contain one or more ITIMs and ITIM-like motifs in their cytoplasmic tail. Upon ligation, ITIMs are phosphorylated by Src family kinases, recruiting and activating SRC homology 2 (SH2)-domain-containing proteins, mainly the tyrosine phosphatases SHP1 and SHP2 or the suppressor of cytokine signaling 3 protein (SOCS3) (58).
Changes in sialic acid, a nine-carbon sugar, have been related to cancer (188, 189). Apart from aberrant tumor cell surface expression, sialic acid alterations and the amount or density of sialic acid have been associated with tumorigenesis and cancer progression. These include hypersialylation, xenosialylation (uptake of Neu5Gc), and sialic acid alterations, including C5-hydroxyl modification of sialic acid [which generates 2-keto-3-deoxy-D-glycerol-D-galac- to-nononic acid (KDN)] and O-acetylation of sialic acid (particularly 9-O-acetylation) (189). Hypersilaylation has been associated with several cancers such as oral cancer, RCC (renal cell carcinoma), HNSCC (head and neck squamous cell carcinoma), breast cancer, prostate cancer, and colon cancer (184). KDN changes have been associated with ovarian cancer (190) and carcinoma of the head and neck (191), while O-acetylation of sialic acid has been reported in colorectal cancer (192, 193).
Siglec-sialic acid interactions are involved in the modulation of immune tolerance and can be targeted for eliciting anti-tumor immunity (184). Targeting some of these inhibitory checkpoints (anti-Siglec-2; Inotuzumab ozogamicin and anti-Siglec-3; Gemtuzumab ozogamicin) with antibodies conjugated to a cytotoxic agent have already been tested for their clinical efficacy (182, 183, 194). Human NK cells mostly upregulate Siglec-7 and Siglec-9. Furthermore, in cancer, peripheral NK cells also upregulate Siglec-9, primarily on CD56dim CD16+ NK cells (58, 187). Blocking of Siglec-7 and Siglec-9 with Fab fragments increased the in vitro cytotoxicity of NK cells against tumor cells (K562). In an in vivo model of immunodeficient mice with transferred human NK cells and human tumor cells, killing of tumor cells was mediated by the sialoglycan-dependent NK cell inhibition (187). High and sustainable cytotoxicity against leukemia cells was shown by a Siglec-7(negative) phenotype of developed NK-92MI cell line (195). Recruitment of Siglec-7 was essential for inhibition of human NK cell activation by artificially increased sialylated glycans on cancer cells. The susceptibility to NK cell killing of various remodeled tumor lines of breast, brain, colon, liver, or lymphoid tissue (Siglec-7-abundant tumor cell lines) was increased after sialidase treatment. Siglec-7 engagement provided protection against NK cell killing by inhibiting both antibody-dependent and antibody-independent cytotoxicity. Cancer cells expressing ligands for Siglec-7 can protect themselves from the innate immune response as well as therapeutically relevant ADCC (196). In vitro fusion of sialidase to antibodies targeting HER2 enhanced the HER2+ tumor cell killing by NK cells. This enhanced NK cell-mediated killing was possible through cutting off the sialic acid ligands by sialidase, specifically the ones bound by Siglec-7 and Siglec-9. This demonstrates that glycocalyx-selective desialylation of tumors could make them more susceptible to ADCC with such antibody-sialidase conjugates (197). Siaglec-7 show high preference for engagement with cis sialic acid residues on the surface of human NK cells results in masking of these siaglec-7 (198). Siglec-9 has also shown such self-sialic acid interactions (188). Engineered overexpression of α2,8-linked disialic acids on tumor cells showed a slight reduction in NK cell cytotoxicity, which was intensified after treatment of NK cells with sialidase, which cleaves the cis ligands from the NK cell surface. α2,8-linked disialic acids are overexpressed by the GD3 ganglioside produced at high levels in cancers like melanoma, making them susceptible to such sialidase-treatment-driven NK cell cytotoxicity (199).
In vitro NK cell cytotoxicity was potently enhanced by high-affinity anti-Siglec-9 antibodies through blocking sialic acid expressed on tumor target cells. These antibodies targeting Siglec-9 also had improved anti-tumor response, induced by the NKG2A blockade (188). Siglec-9 is also upregulated on tumor-infiltrated CD8+ T cells in NSCLC, ovarian, and colorectal cancers (181, 188). A subset of intratumoral effector memory CD8+ T cells in melanoma have also shown Siglec-9 upregulation and suppression by its engagement through phosphorylating SHP1 (200). In vitro and in vivo targeting of the sialoglycan-SAMP/Siglec pathway resulted in increased anticancer immunity. Other inhibitory receptors such as PD-1 were also co-expressed by T cells expressing Siglec-9, hinting at a potential for co-inhibition (181). The multi-mode action of Siaglec-9 is apparent from the fact it is expressed on various types of immune cells (188). These data lend support to the idea that anti-Siglec-7 and anti-Siglec-9 blocking antibodies should be developed for cancer immunotherapy, potentially in combination with other immune checkpoint inhibitors.
LAG-3
LAG-3 (Lymphocyte Activation Gene-3) is also a member of the immunoglobulin superfamily receptors with inhibitory properties. LAG-3 was discovered as an upregulated molecule on the surface of activated CD4+ T cells, CD8+ T cells, and NK cells (201). In addition to these cells, Lag-3 is expressed on several other immune cells including TILs, regulatory T cells, iNKT cells, B cells, and DCs (68, 202–207). It recognizes MHC class II molecules as ligands and shares structural similarity with CD4 molecules but binds to MHC-II molecules with greater affinity than CD4 (201, 208, 209). LSECtin, expressed in liver and several other tumors and a member of the DC-SIGN family, has also been described as a potential ligand for LAG-3-expressing immune cells (210). Engagement of LAG-3 inhibits T cell effector function and is involved in T-cell exhaustion (211–213). It also promotes the suppressive activity of regulatory T cells (203, 214). Blocking of LAG-3 has been shown to induce improvement in T-cell functions (212). Relatlimab is an anti-LAG-3 monoclonal antibody being investigated in several ongoing clinical trials, either alone or combined with PD-1 blockade, in various cancers (215). LAG-3 and PD-1 have shown synergism in T-cell functional regulation to promote tumor immune escape (216).
Though LAG-3 is expressed on NK cells, its role in the regulation of NK cells has not been well-established. Knockout of the LAG-3 gene in a mouse model resulted in the inability of NK cells to kill certain tumor targets. However, this deletion had no effect on MHC class I-mismatched cytolytic activity (217). Human NK cells, on the other hand, showed an opposite result. Antibodies blocking the LAG-3 pathway were unable to induce human NK cell cytotoxicity. Soluble Lag-3, capable of binding to MHC-II molecules, also had no effect on human NK cell killing capability. However, in this research, cytokine production was not investigated (218). In patients with HIV, viral control was associated with low LAG-3 expression on NK cells along with other inhibitory molecules (219). Wiskott-Aldrich Syndrome protein (WASp) deficiency is associated with a high susceptibility to cancer, most probably due to impairment of the anti-cancer capacity of NK cells and DCs (220, 221). WASp knockout NK cells showed an association of cellular exhaustion and NK cell memory with enhanced LAG-3 expression (222). There seems to be an evident association; however, the direct impact of LAG-3 on NK cell functions and underlying mechanisms needs further investigation. In contrast to NK cells, its regulation of NKT (Natural Killer T cell) functions has been well-reported. In patients with chronic HIV, exhaustion of iNKT cells and reduction in IFN-γ production were associated with elevated expression of LAG-3 (204). The LAG-3 signaling pathway, through arresting the cell cycle in the S phase, also down-modulated the proliferation of activated CD1d-restricted NKT cells (223).
A soluble recombinant LAG-3-Ig fusion protein, Eftilagimod alpha (IMP321), has been used as an immunological adjuvant for vaccination against various infections and cancer. It has also been applied as monotherapy or combined with chemotherapy in cancer (224). IMP321 was able to induce NK cells to produce cytokines (IFN-γ and/or TNF-α) in healthy individuals (52 of 60 donors) and, to a lower extent, in 21 untreated metastatic cancer patients in an ex vivo short-term experiment (225). In metastatic renal cancer patients, IMP321, in a dose-escalation study (P003), induced NK cell activation as monotherapy (226). IMP321 with standard chemotherapy was associated with enhanced activation of NK cells for several months in breast cancer patients (227). Hence, LAG-3 has the potential to activate T cells as well as NK cells. Hence, it can be further explored as a potential target for checkpoint inhibition. Furthermore, CD56bright, CD16−, and CD62L+ NK cells were identified as the dominant subset of cytokine-induced memory-like (CIML) NK cells, with sustained expression of NKG2A involved in inhibition of killing of the HLA-E-positive target cells in a recent study on CIML NK cells. A minor CIML NK cell subset, KIR+ and NKG2C+, was shown to express LAG-3, suggesting CIML NK cells as a potential target for dual checkpoint inhibition (228).
TIM-3
A co-inhibitory receptor, TIM-3 (T-cell immunoglobulin and mucin domain 3), recognizes galectin-9 as ligand, which is upregulated in various cancers and chronic infections (68, 229–234). In addition, the TIM-3 variable IgV domain has also been reported to bind to other ligands, such as HMGB1 (high mobility group protein B1 proteins), Ceacam-1 (carcinoembryonic antigen cell adhesion molecule 1), and PtdSer (phosphatidylserine) (229, 230). TIM-3 expression is diverse, encompassing several types of immune cells, including CD4+ T cells, CD8+ T cells, regulatory T cells, B cells, NK cells, NKT cells, and myeloid cells (230, 235). TIM-3 engagement with its ligands induces immune tolerance by exhausting T cells as well as NK cells (232, 236–238). This pathway upregulation is associated with exhaustion of T and NK cells in various chronic infections as well as cancers, making TIM-3 a negative regulator of T and NK cell immunity (232, 234). Correspondingly, its blockade has reversed T or NK cell dysfunction (238–242). Co-expression of TIM-3 and PD-1 was involved in mediating the exhaustion of CD8+ T cells in various cancers and chronic viral infections (243–248). Studies have revealed a reversal of T-cell exhaustion and reduction in tumor growth with TIM-3 and/or PD-1 blockade together (245, 247, 249, 250). Antibodies for TIM-3, such as Sym023, Cobolimab, LY3321367, BGB-A425, and MBG453, in combination with several anti-PD-1/PD-L1 antibodies, are under clinical investigation for their efficacy against various cancers (Table 1).
TIM-3 expression on NK cells has several aspects to it. It has been regarded as a maturation, activation, and prognostic marker. TIM-3 is highly expressed in the resting CD56+/CD3+ NK-cell population as compared to the CD56+/CD3+ NKT and CD56−/CD3+ T-cell populations (251). A fraction of the mature CD56dimCD16+ NK cell subset in blood from healthy adults displayed TIM-3 expression, while its expression was heterogeneous in the immature CD56brightCD16− NK-cell subsets. Moreover, several cytokines (IL-12, IL-15, and IL-18) strongly induce TIM-3 expression, primarily in the immature CD56bright NK cells (251, 252). IL-12 and IL-18-induced activation and IL-15-induced maturation of NK cells are the main cause of TIM-3 expression in these cells, identifying TIM-3 expression in NK cells as a marker for the activation and differentiation or both (241, 251). Upregulation of TIM-3 in the peripheral NK cells is observed in several cancers, namely gastric cancer (253), lung adenocarcinoma (238), advanced melanoma (242), and bladder cancer (178) leading to NK cell exhaustion. Increased levels of TIM-3 in NK cells with tumor growth shows TIM-3 expression to be a prognostic biomarker (178, 238, 241, 242, 253). Tumor-infiltrating NK cells in patients with GIST (gastrointestinal stromal tumors) and bladder cancer were also revealed to express TIM-3 (178, 254). Similar to TIM-3 and PD-1 co-expression in T cells, exhausted tumor-infiltrated NK cells have also shown detectable co-expression in MHC-I-deficient tumors (255). However, patients with GIST lacked PD-1 co-expression in TIM-3+ tumor-infiltrating NK cells (254).
TIM-3 has been described as an activation marker and activation limiter of NK cells, since its engagement to its ligand (Gal-9) and blockade have shown contrasting results (256). TIM-3 engagement with Gal-9 was able to induce IFN-y production by NK cells with no effect on cytotoxic ability (251), and anti-Gal-9 antibody blockade of TIM-3 reduced the production of IFN-γ by NK cells from healthy donors in response to primary AML blasts (231). TIM-3 blockade on IL-15-stimulated NK cells also resulted in cytotoxicity reduction for two PCC lines (human pancreatic cancer cell), Mia-Paca-2 and Capan-2, though a PSC line panel (human pancreatic stellate cell) showed no significant difference in cytotoxicity (257). In vitro, Gal-9 downregulated TIM-3 on NK cells and triggered NK cell activation in HIV-1 infection; however, a subset of these NK cells (immature CD56bright NK cells) were not affected by this decrease in TIM-3 expression; rather, an increase in the surface expression of TIM-3 was witnessed with a concomitant increase in Gal-9 expression during late primary HIV-1 infection (258).
TIM-3 upregulation on NK cells and its association with NK cell exhaustion and dysfunction has been reported in a number of studies, defining TIM-3 as a negative regulator. TIM-3 engagement with agonist antibody or Gal-9-expressing target cells revealed significant suppression of NK cell-mediated cytotoxicity in NKL, a human NK-cell line, or primary human peripheral blood NK cells from PBMCs (252). TIM-3 expression on NK cells was also increased in chronic HIV infection associated with a dysfunctional phenotype of NK cells (237). During early pregnancy, an upregulation of TIM-3 in NK cells was observed, leading to inhibition of NK cell cytotoxicity toward trophoblast in a Gal-9 dependent pathway (259). NK cell expression of TIM-3 receptors was also upregulated by chronic hepatitis B infection, causing subsequent suppression of NK cell function, and was rescued by TIM-3 blocking (260). TIM-3 blockade also had increased NK cell cytotoxicity against K562 target cells (261). Blockade of the TIM-3 pathway in patients with several cancers, such as advanced melanoma and lung adenocarcinoma, restored NK cell cytotoxicity. TIM-3 blockade enhanced effector function in peripheral NK cells from bladder cancer patients (178).
It has been suggested that, when the expression of TIM-3 is upregulated, it initially enhances NK cell cytotoxicity, and that chronic activation leads to overexpressed or dysregulated TIM-3 expression, resulting in a subset of dysfunctional or exhausted NK cells (256, 258). TIM-3 unselective binding to its ligands, Gal-9 inhibition of NK cells in a TIM-3-independent manner (236), and Gal-9 downmodulation of TIM-3 in chronic HIV patients (258) may account for some of these shortcomings upon further investigation. The ineffectiveness of TIM-3 blockade in intra-tumor NK cells also reveals the role of tumor-specific factors, and its potentiation after activation with IL-15 suggests that inflammatory cues also have a role to play (178).
CD200R
CD200R is another inhibitory receptor expressed on T, B, NK, and myeloid cells (262–264). It recognizes CD200 as its ligand, which is expressed on various normal tissues such as the central nervous system, retina, hair follicular cells, vascular endothelial cells, and thymocytes, as well as activated T, B, and DCs, in addition to its expression on various tumors (265). CD200 is considered as a marker of tumor progression since it is overexpressed on various cancers of both hematopoietic and non-hematopoietic origin, such as acute myeloid leukemia, multiple myeloma, hairy cell leukemias, B cell chronic leukemias, melanoma, and ovarian, rectal cancer, and bladder cancer, and its expression is associated with the worst prognosis (266–274). Moreover, CD200 expression can also be induced on cancer cells (275, 276). In fact, cancer cell expression of CD200 had no effect on suppression of anti-cancer responses by CD200–CD200R signaling (277). Therefore, CD200 blockade represents a potential therapeutic option not restricted to the treatment of CD200-expressing tumors (265). The inhibitory CD200–CD200R pathway appears to inhibit the effector functions of T cell in an indirect manner via the regulation of macrophages and DCs. Hence, tumor growth could be inhibited by blockade of CD200–CD200R interaction, lending support to the idea that antagonistic CD200 or CD200R antibodies are an option in cancer treatment (278). Samalizumab (a humanized Anti-CD200 antibody) was well-tolerated and demonstrated changes in CD4-positive T cells and CD200-positive B-CLL in a dose-dependent manner as well as inducing a dose-dependent linear increase in serum AUC and modest Th1 cytokine responses (279).
In the case of NK cells, there is evidence of direct involvement of the CD200–CD200R inhibitory pathway resulting in the suppression of NK cells. In AML patients, NK cell anti-tumor responses were suppressed by the overexpression of CD200, thereby increasing risk for relapse in these patients (280). NK cell subpopulations in AML patients expressed CD200R, suggesting NK cell suppression from CD200–CD200R interaction. Moreover, in patients with CD200hi, antibody blocking CD200 restored NK cell activity. These data suggest that CD200–CD200R interaction is directly responsible for NK cell suppression in AML patients (264). These are the only studies directly indicating that target cells expressing CD200 could inhibit the cytotoxic as well as the IFNγ-producing activities of NK cell. Liu et al., while investigating the CD200 signaling part in CD200+ melanoma growth and metastasis restriction, revealed a significantly reduced number of NK cells in livers undergoing metastatic tumor growth that were deficient in CD200. However, how deficiency in CD200 could affect local NK responses in liver remains to be explained (272). It suggests that the CD200–CD200R checkpoint is an appreciable target for checkpoint blockade in hematologic and solid tumors. Since NK cells have been targeted in relation to other checkpoint receptors in AML and multiple myeloma, such as KIR and NKG2A, a combination of checkpoint targets could be validated in this group of patients.
CD47
CD47, also referred to as integrin-associated protein (IAP), is a glycoprotein of the Ig superfamily that is expressed ubiquitously (281). It was first discovered on leukocytes as a membrane protein taking part in signaling mediated by β3 integrin. It is a transmembrane protein that also interacts with thrombospondin-1 (TSP-1) and signal regulatory protein-alpha (SIRPα), in addition to integrins (282–285). Of the functions performed by CD47, its engagement with SIRPα and thrombospondin-1 has established its role as an inhibitory receptor involved in immune evasion by cancers through inhibition of phagocytosis, antigen presentation, and T/NK cell inhibition (286–289). Consequently, targeting these signaling pathways with antibodies has revealed the therapeutic candidacy of this checkpoint (283, 289–292).
CD-47 has been disclosed to play an inhibitory role in NK cell-mediated anti-viral or anti-tumor cytotoxicity. Deficiency in CD47 was shown to impair the immune responses of NK cells to LCMV infection (293). NK cell cytotoxicity was associated with CD47 expression on HNSCC cell lines. High CD47-expressing HNSCC cell lines exhibited NK cytotoxicity at lower levels compared to those with low expression of CD47. Pretreating cells with neutralizing MHC-1 or anti-CD47 antibodies led to an increase in NK cell cytotoxicity against HNSCC cell lines (289). This study did not reveal an associated CD47-receptor ligating partner. Both ligands SIRPα and TSP-1 have been associated with NK cell-mediated cytotoxicity. In immunocompetent syngeneic mice, RCC or melanoma cell tumor formation was markedly suppressed by anti-SIRPα antibody through blockade of CD47 interaction. Selective depletion of NK cells greatly attenuated the antitumor effect of anti-SIRPα Ab, in addition to macrophages and CD8+ T cells (294). However, in vitro killing of the tumor cells by NK cells was not inhibited by the same antibody, suggesting CD47 function in NK cells to be independent of SIRPα. In a similar fashion, thrombospondin-1 was indicated in inhibiting early NK cell proliferation and enhancing late expansion, but the role of CD47 was not identified (295). Therefore, there is still much to be unraveled in this regard. Nonetheless, attacking CD-47 with antibodies is worth exploring, not only in the context of macrophages, dendritic cells, and T cells but also NK cells. Dual blockade of CD47 in combination with PD-L1 has also been explored and has shown to enhance immunotherapy against circulating tumor cells (296). Magrolimab (Hu5F9-G4), an anti-CD47 antibody, is under investigation in several phase I and II clinical trials in various combinations with other agents such as rituximab, cetuximab, azacitidine, acalabrutinib, and atezolizumab (Table 1).
B7-CD28 Family Receptors
The B7-CD28 family of ligands and receptors play important roles in T-cell co-stimulation and co-inhibition. Recognition of their role in relation to NK cells in on the rise, with accumulating evidence. Primarily, CTlA-4 and PD-1 checkpoints, just like T cells, have been implicated in NK cell dysfunction in various cancers. Newer checkpoints from this family, including B7-H3, VISTA (PD-1H), and B7-H7, are being discovered to show inhibition of T and NK cell-mediated cytotoxicity (297).
CTLA-4
CTLA-4 (Cytotoxic T lymphocyte-associated antigen 4), a co-inhibitory receptor, is expressed on several immune cells, such as activated T lymphocytes (CD4+ T cells & CD8+ T cells), regulatory T cells, tumor-infiltrating NK cells, and splenic Kit+ CD11b− NK cells in tumor-bearing mice, and is induced on mouse NK cells upon IL-2 stimulation (298–300). CTLA-4 competes with CD28, a costimulatory receptor, for ligands B7-1 (CD80) and B7-2 (CD86) on cancer cells or antigen-presenting cells. CTLA-4 is well-established as a negative regulator of T-cell activation and controller of peripheral T-cell tolerance and autoreactivity (298, 301–308). Blockade of CTLA-4 with antibody (ipilimumab) has improved T-cell function in various cancers (309).
Previous studies have indicated CTLA-4/CD28/CD80/CD86 pathway involvement in NK cell-mediated cytotoxicity. In vivo, CD28 triggering the proliferation of NK cells, their cytotoxicity, and secretion of cytokines has been described in a number of studies (310, 311). Ligands B7-1 and B7-2 on cancer cells both also appear to improve human NK cell cytotoxicity (312–316). Similarly, CTLA-4 expressed by NK cells inhibited IFN-γ production in response to B7-1 induced by dendritic cells (299). In mice, Kit+ CD11b− NK cells capable of B7-H1-dependent immuno-ablative functions were elicited by IL-18 produced by tumor cells. Kit+ CD11b− NK cells also reported upregulation of CTLA-4; however, its involvement in tumor progression in NK cell-controlled cancers has not been investigated (300). These studies reveal an undeniable role for this pathway in NK cell-mediated toxicity. However, co-stimulation mediated by CD80-CD28/CTLA-4 being absent in human NK cells has also been proposed (317). Co-stimulation of CD28/B7 was also denied to play any significant part in peripheral NK cells in murine CMV infection (317).
CTLA-4+ tumor-infiltrating NK cells embody an anti-CTLA-4 monoclonal antibody-based prospective immunotherapeutic target. Blocking CTLA-4 might relieve the suppressed NK cells in an indirect manner, as shown in Figure 4. The expression of CTLA-4 on Tregs is thought to be essential for their suppressive functions (318). NK cell cytotoxicity-suppression with an increase in CTLA-4 positive Tregs was correlated with poor prognosis in cetuximab-treated head and neck cancer patients (319). Ipilimumab, an anti-CTLA-4 monoclonal antibody, leading to depletion of regulatory T cells, resulted in clinical efficacy in melanoma patients in an Fc-mediated manner, which may be partly due to relieved NK cell cytotoxicity suppressed by Tregs (320, 321). The availability of IL-2 released by activated CD4-positive T cells was also restricted by Tregs, which is involved in NK cell proliferation, accumulation, and activation (322, 323). It has been demonstrated that CTLA-4 engagement on CD4+ T cells inhibits IL-2 production and CTLA-4 blockade, leading to a rise in IL-2-producing effector cells, again showing anti-CTLA-4 blockade affecting NK cell cytotoxicity in an indirect way (324, 325). Increased frequency of a subset of NK cells (mature circulating CD3− CD56dim CD16+ NK cells) with increased TIM-3 expression had also shown a correlation with clinical outcome in melanoma patients during treatment with anti-CTLA-4 blockade (326). A different subset of NK-cells, CD56brightCD16dim, was involved in mediating the regression of pancreatic cancer from ipilimumab (327). These cells reported an upregulation of p46-activating receptors and TRAIL. Ipilimumab was also able to trigger ADCC via the engagement of FcyRIIIA receptors present on primary NK cells as well as NK cells and γδT cells activated by IL-2 by reacting with CTLA-4 present on melanoma cell lines and tissues. Furthermore, Ipilimumab and CTLA-4-positive melanoma cell interaction also led to TNF-α release by NK cells (328). Delay in tumor growth and survival prolongation were witnessed in melanoma with a combination of CTLA-4 blockade and IL-2 immunotherapy, indicating synergism. Tumor infiltration of the immune cells, including CD8-positive T cells and NK cells, was increased with CTLA-4 blockade, while a reduction in the proportion of tumor-infiltrating NK cells that were exhausted and differentiated was observed with IL-2 (329). Ipilimumab demonstrated induction of IL-2R α-chain expression on the phenotype of NK cells, with subsequent enhanced response to IL-2 stimulation and cytotoxicity, and this was associated with better clinical response in advanced melanoma patients (330).
Figure 4.
Indirect effects of CTLA-4 blockade on NK cells. (A) Treg increase was correlated with NK cell suppression and decrease IL-2 availability to NK cells, which may be reversed with CTLA-4 blockade. (B) CTLA-4 blockade with antibodies increased CD4+ T-cell proliferation and IL-2 production. (C) Ipilimumab blockade of CTLA-4 on tumor cells has been reported to be associated with ADCC, TNF-α release by NK cells, and induction of the IL-2Rα chain on NK cells. (D) Ipilimumab was associated with an increased frequency of CD3− CD56dim CD16+ NK cells with increased TIM-3 expression. CD56bright CD16− NK cells with increased expression of p46 receptor and TRAIL have also been reported. (E) A combination of IL-2 plus ipilimumab was reported to be associated with increased NK cell infiltration of tumor, as well as a decrease in exhausted and differentiated NK cells.
PD-1
Programmed cell Death-1 (PD-1) is expressed on various immune cells, including T (CD4+ & CD8+), B cells and myeloid cells, NK cells, NKT cells and other innate lymphoid cells (IL2) (298, 306, 331–334). Ligands for PD-1, PD-L1, and PD-L2 upregulation have been reported on in various cancers, and their interactions have led to T-cell inhibition, resulting in immune escape (298, 306). High PD-1 expression on NK cells is detectable in the peripheral blood of approximately a quarter of healthy individuals (335). However, PD-1 expression on NK cells is upregulated in cancer patients such as in ascites of ovarian-carcinoma patients, peripheral blood of Kaposi sarcoma patients, and renal cell carcinoma and multiple myeloma patients (336–340). It is also upregulated on peripheral and tumor-infiltrating NK cells in digestive cancers such as esophageal, gastric, biliary, liver, and colorectal cancer (341). Chronic infections such as HIV (Human Immunodeficiency Virus), HCV (hepatitis C virus), HCMV (Human cytomegalovirus), and M. tuberculosis have also shown enhanced PD-1 expression on NK cells (342, 343).
PD-1 expression on NK cells is diverse, varying from cancer to cancer. PD-1 expression is generally lacking in CD56bright NK cells. CD56dim NK cells, however, have demonstrated PD-1 expression that is restricted to the NKG2A− KIR+CD57+ phenotype, a fully mature NK cell (335). The NKG2A− KIR+CD57+ phenotype NK cell is considered to have strikingly downregulated activating receptors such as NKp30 and NKp46. Furthermore, a correlation was witnessed between PD-1 expression and NK cell anti-tumor activity impairment, while disrupting PD-1 and PD-L1 interaction by antibody led to partial restoration (344). PTLD pediatric transplant patients have also demonstrated NK cell functional alteration, with increased PD-1 and decreased NKp46 and NKG2D expression (345). On the other hand, CD56bright NK cells were shown to express PD-1 in chronic HCV patients (332). Meanwhile, in patients with digestive cancers, both types of NK cells (CD56bright and CD56dim NK cells) have demonstrated increased PD-1 expression (341). Furthermore, newly identified hepatic resident infiltrated CD3−CD49a+CD56+NK cells in HCC tissues also revealed enormous expression of PD-1 on their surfaces, which was associated with reduced survival in hepatocellular carcinoma patients (341).
NK cell upregulation of PD-1 expression in several cancers suggests a dysfunctional state of the NK cell, probably due to over-stimulation by tumor cells deficient in MHC-I. Comparisons of PD-1+ NK cells and PD-1− NK cells have revealed PD-1+NK cells to be functionally exhausted, with impaired cytotoxicity and cytokine production and reduced proliferative capability (335, 341). Blockade with anti-PD-1 mAbs has been shown to revert NK cell functional capability. Mouse tumor-resident NK cells exhibit PD-1 expression, and antitumor immune response by NK cells was elicited with anti-PD-1 blockade (346). Antibodies against PD-1 were able to reverse impaired degranulation toward an ovarian carcinoma cell line (335). In vitro, NK cell-mediated cytotoxicity toward autologous MM cells was enhanced by anti-PD-1 antibody (339). Killing of mouse glioma stem cells (GL261GSCs) by mouse NK cells was also promoted by PD-1 blockade (347).
In PTLD patients, IFN-γ release, but not cytotoxicity, was enhanced by anti-PD-1 pathway disruption, indicating a partial dependence on the PD-1 pathway (345). Hence, more studies are required to clearly determine the role of PD-1 blockade in the context of NK cells. Blocking PD-L1 has also been shown to improve NK cell-based anti-tumor responses (348). In HN cancer patients, activated PD-1+ NK cell dysfunction was mediated by PD-L1 (349). CD56dim PD+NK cells expressed in Hodgkin lymphoma were efficiently suppressed by PD-L1-expressing myeloid cells, which was reversed by anti-PD-1 blockade in vitro (350). ADCC toward multiple types of carcinoma cells obtained by avelumab, an anti-PD-L1 antibody, was augmented with epigenetic priming of NK cells and tumor (351). In another study, Avelumab triggered NK cells to produce cytokines and mediate the killing of triple-negative breast cancer cells (352). PD-L1-independent killing of CRC cells that were grown in 3D cultures by densely activated primary human NK cells has also been demonstrated (353). Again, it seems that the biology of NK cells in the context of checkpoint inhibition is very complex and needs further exploration.
B7-H3
B7 homolog 3 protein (B7-H3) is a ligand molecule belonging to the B7-CD28 family for which the receptor, probably present on T cells and NK cells, as it appears to inhibit both T- and NK-cell functions, is yet to be discovered (297). B7-H3 is believed to be both co-stimulatory and co-inhibitory in regulating T-cell functions (354–356). B7-H3 co-stimulates T-cell activation by binding to TLT-2, whereas binding to unidentified receptor(s) leads to co-inhibition of T cells. Meanwhile, it is an inhibitor for NK cells and osteoblastic cells by ligating unknown receptor(s) (355).
There is a limited expression of B7-H3 on various normal tissues such as pancreas, liver, small intestine, colon, heart, thymus, spleen, placenta, and testis (357). However, aberrant expression of B7-H3 is seen in various malignancies in which it is associated with poor outcome, including RCC, breast cancer, lung cancer, esophageal squamous cancer, gastric cancer, pancreatic cancer, gallbladder cancer, colorectal cancer, prostate cancer, ovarian cancer, cervical cancer, endometrial cancer, osteosarcoma, and neuroblastoma (358, 359). Circulating serum B7-H3 levels are significantly higher in patients with lung cancer, renal cell carcinoma, hepatocellular carcinoma, colorectal carcinoma, and glioma than in healthy volunteers (360). Suppression of NK cell-mediated cytotoxicity is one of the various mechanisms involved in tumor evasion by B7-H3-expressing cells (297). Glioma malignancy grade and reduced survival were correlated with the expression of B7-H3 in tumor and endothelial cells. Soluble B7-H3 in the supernatant of glioma cells and cell-bound B7-H3 were able to suppress natural killer cell-mediated tumor cell lysis. Susceptibility to killing was confirmed in an in vivo model of B7-H3-silenced glioma cell lines (361). Monoclonal antibody-mediated masking of the 4Ig-B7-H3 molecule, identified as a neuroblastoma-associated molecule, on cell transfectants or on freshly isolated neuroblastoma cells protected it from killing by NK cells (362). Similarly, in a case of neuroblastoma arising in an ovarian cystic teratoma, B7-H3 was expressed in addition to substantial amounts of HLA class-I molecules, suggesting a protective immune-evasive mechanism by neuroblastoma cells against NK cell-mediated lysis. Additionally, receptors such as DNAM-1 (CD226) and CD16 were expressed with lower intensity on NK cells isolated from peritoneal fluid of these patients in comparison to NK cells from the peripheral blood (363). Bispecific killer cell engager (BiKE) treatment, one of the two forms of antibody therapeutics, was demonstrated to significantly inhibit cell growth by inducing natural killer cells when exploring B7-H3 as a potential target in NSCLC (364).
B7-H3 binding Fc-optimized humanized IgG1 mAb, Enoblituzumab, is currently being explored (Table 1). In order to improve functional effectivity, including ADCC, enoblituzumab is Fc-engineered. It has been shown to inhibit tumor growth in renal and bladder carcinoma xenografts positive for B7-H3 (365). MGA271, an Fc-optimized humanized mAb targeting B7-H3, has shown safety and antitumor efficacy in several tumor types that were refractory B7-H3-expressing cancers or where the vasculature of the patients with these cancers were positive for B7-H3, as revealed from an interim analysis of an ongoing dose-escalating phase I study (366). This anti-tumor activity was attributed to an increase in the T-cell repertoire clonality in patients. Further characterization of enoblituzumab including its pharmacological kinetics and dynamics as well as its safety profile, dosage toleration, and antitumor activity against relapsed or refractory solid malignancies with positive expression of B7-H3 receptors in younger patients (children and young adults), is being evaluated in an open-label phase I study (NCT02982941) (367). Orlotamab (MGD009, a humanized B7-H3 x CD3 DART® protein), a bispecific antibody targeting CD3 in addition to B7-H3, developed by MacrGenics is under clinical investigation (NCT03406949) for its safety and efficacy in combination with anti-PD-1 antibody (MGA012) in relapsed or refractory tumors with B7-H3 expression (Table 1). Plenty remains to be unraveled regarding B7-H3, particularly the discovery of its receptors on NK cells and T cells. All in all, B7-H3 presents itself as a potential candidate for checkpoint-based immunotherapy against T cells as well as NK cells.
Conclusions
Natural killer cells represent a distinct group of anti-tumor response agents with functions like MHC-unrestricted cytotoxicity, cytokine production, and immunologic memory, making them a key player in the innate as well as the adaptive immune response system. The development of several cancers has been associated with the presence of dysfunctional NK cells. Hence, restoration of such NK cells could be a potential option for anti-tumor immunotherapy. One approach to such restoration is the inhibition of immune checkpoints, which is the manipulation of inhibitory receptors on the immune cell surface by cancer cells for immune escape. Immune checkpoint inhibition has been successful in the context of T cells. NK cells have recently been focused on for the same purpose. The immune checkpoint inhibitors, such as monalizumab and lirilumab, aimed at these inhibitory receptors present at the surface of NK cells have been assessed as monotherapy and have shown good safety profiles but mild success in terms of prolonging progression-free survival. Therefore, the combinations of immune checkpoint inhibitors, such as CTLA-4 and PD-1 inhibitors, that are being tried for synergistic response targeting T cells could also be tried in the context of NK cells (368), as anti-PD-1 and anti-PD-L1 inhibitors have also been shown to enhance NK cell-mediated cytotoxicity (346, 348, 352). Similarly, NKG2A potentiating CD8 T-cell immunity induced by cancer vaccines also stresses the potential of combination therapy (141). Hence, a combination of the two, an anti-PD-1 or anti-PD-L1 inhibitor and an NK cell-specific checkpoint inhibitor such as anti-KIR or anti-NKG2A inhibitor, could be of value for combined checkpoint inhibition-based immunotherapy. With the addition of newer checkpoints such as B7-H3, CD200R, CD47, and Siglecs 7/9, it seems more logical to combine these checkpoints for synergistic anti-tumor response.
Another important aspect in the context of immune checkpoint inhibition is the unraveling of the importance of cancer profiling based on biomarkers and the identification of NK cell phenotypes for individual patients in order to get a good idea of where an individual is more likely to get benefit from a given immune checkpoint inhibitor or a combination of inhibitors and cytokine therapy or virotherapy. Inhibitory NK cell receptors may function in different subsets of NK cells or in distinct states of differentiation, such as the acquisition of KIR and loss of NKG2A observed during NK-cell differentiation. Hence, lirilumab will not be effective if the KIR-negative NK cells are responsible for tumor growth (369). Similarly, CD56bright NK cells exhibit potent antitumor responses following IL-15 priming (370). Interleukin-7 also selectively enhances natural kill cytotoxicity mediated by the CD56bright natural killer subpopulation (371). Hence, cytokine therapy would work well if the CD56bright NK cells are required for cancer elimination. Furthermore, as the contribution of several other factors in addition to inhibitory receptors leads to an immune escape of NK cells, for example, downregulation of activating receptors, molecular checkpoints of the activation pathways such as Cbl, GSK-3β, DGKζ, and CIS, and tumor environment suppressors, a combination of strategies countering each of these processes could lead to a more complementary and synergistic potent immune response (369). This would certainly lead to a more comprehensive cancer treatment approach composed of immune checkpoint blockade, augmentation of activating receptors or their molecular checkpoints, and cancer environment checkpoint blockade. Yet another way to improve checkpoint blockade efficacy is its integration with other standard cancer therapeutics, such as surgery, chemotherapy, and radiotherapy, in particular (372, 373). These are some of the areas that should be evaluated in order to increase the clinical efficacy of immune checkpoint blockade in the future.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- 1.Cooper MA, Fehniger TA, Caligiuri MA. The biology of human natural killer-cell subsets. Trends Immunol. (2001) 22:633–40. 10.1016/S1471-4906(01)02060-9 [DOI] [PubMed] [Google Scholar]
- 2.Greenberg AH. The origins of the NK cell, or a Canadian in King Ivan's court. Clin Invest Med Med Clin Exp. (1994) 17:626–31 [PubMed] [Google Scholar]
- 3.Spits H, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, et al. Innate lymphoid cells–a proposal for uniform nomenclature. Nat Rev Immunol. (2013) 13:145–9. 10.1038/nri3365 [DOI] [PubMed] [Google Scholar]
- 4.Fuchs A, Colonna M. Natural killer (NK) and NK-like cells at mucosal epithelia: mediators of anti-microbial defense and maintenance of tissue integrity. Eur J Microbiol Immunol. (2011) 1:257–66. 10.1556/EuJMI.1.2011.4.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chiossone L, Dumas PY, Vienne M, Vivier E. Natural killer cells and other innate lymphoid cells in cancer. Nat Rev Immunol. (2018) 18:671–88. 10.1038/s41577-018-0061-z [DOI] [PubMed] [Google Scholar]
- 6.Moretta A, Marcenaro E, Parolini S, Ferlazzo G, Moretta L. NK cells at the interface between innate and adaptive immunity. Cell Death Different. (2008) 15:226–33. 10.1038/sj.cdd.4402170 [DOI] [PubMed] [Google Scholar]
- 7.Pallmer K, Oxenius A. Recognition and regulation of T cells by NK cells. Front Immunol. (2016) 7:251. 10.3389/fimmu.2016.00251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vossen MT, Matmati M, Hertoghs KM, Baars PA, Gent MR, Leclercq G, et al. CD27 defines phenotypically and functionally different human NK cell subsets. J Immunol. (2008) 180:3739–45. 10.4049/jimmunol.180.6.3739 [DOI] [PubMed] [Google Scholar]
- 9.Fu B, Tian Z, Wei H. Subsets of human natural killer cells and their regulatory effects. Immunology. (2014) 141:483–9. 10.1111/imm.12224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wagtmann N, Rajagopalan S, Winter CC, Peruzzi M, Long EO. Killer cell inhibitory receptors specific for HLA-C and HLA-B identified by direct binding and by functional transfer. Immunity. (1995) 3:801–9. 10.1016/1074-7613(95)90069-1 [DOI] [PubMed] [Google Scholar]
- 11.Moretta A, Bottino C, Vitale M, Pende D, Biassoni R, Mingari MC, et al. Receptors for HLA class-I molecules in human natural killer cells. Annu Rev Immunol. (1996) 14:619–48. 10.1146/annurev.immunol.14.1.619 [DOI] [PubMed] [Google Scholar]
- 12.Moretta L, Mingari MC, Pende D, Bottino C, Biassoni R, Moretta A. The molecular basis of natural killer (NK) cell recognition and function. J Clin Immunol. (1996) 16:243–53. 10.1007/BF01541388 [DOI] [PubMed] [Google Scholar]
- 13.Moretta A, Biassoni R, Bottino C, Pende D, Vitale M, Poggi A, et al. Major histocompatibility complex class I-specific receptors on human natural killer and T lymphocytes. Immunol Rev. (1997) 155:105–17. 10.1111/j.1600-065X.1997.tb00943.x [DOI] [PubMed] [Google Scholar]
- 14.Bottino C, Biassoni R, Millo R, Moretta L, Moretta A. The human natural cytotoxicity receptors (NCR) that induce HLA class I-independent NK cell triggering. Hum Immunol. (2000) 61:1–6. 10.1016/S0198-8859(99)00162-7 [DOI] [PubMed] [Google Scholar]
- 15.Moretta A, Bottino C, Vitale M, Pende D, Cantoni C, Mingari MC, et al. Activating receptors and coreceptors involved in human natural killer cell-mediated cytolysis. Annu Rev Immunol. (2001) 19:197–223. 10.1146/annurev.immunol.19.1.197 [DOI] [PubMed] [Google Scholar]
- 16.Long EO, Kim HS, Liu D, Peterson ME, Rajagopalan S. Controlling natural killer cell responses: integration of signals for activation and inhibition. Annu Rev Immunol. (2013) 31:227–58. 10.1146/annurev-immunol-020711-075005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moretta L, Bottino C, Pende D, Vitale M, Mingari MC, Moretta A. Different checkpoints in human NK-cell activation. Trends Immunol. (2004) 25:670–6. 10.1016/j.it.2004.09.008 [DOI] [PubMed] [Google Scholar]
- 18.Long EO. Regulation of immune responses through inhibitory receptors. Annu Rev Immunol. (1999) 17:875–904. 10.1146/annurev.immunol.17.1.875 [DOI] [PubMed] [Google Scholar]
- 19.Long EO. Negative signaling by inhibitory receptors: the NK cell paradigm. Immunol Rev. (2008) 224:70–84. 10.1111/j.1600-065X.2008.00660.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ljunggren HG, Karre K. Host resistance directed selectively against H-2-deficient lymphoma variants. Analysis of the mechanism. J Exp Med. (1985) 162:1745–59. 10.1084/jem.162.6.1745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Godal R, Bachanova V, Gleason M, McCullar V, Yun GH, Cooley S, et al. Natural killer cell killing of acute myelogenous leukemia and acute lymphoblastic leukemia blasts by killer cell immunoglobulin-like receptor-negative natural killer cells after NKG2A and LIR-1 blockade. Biol Blood Marrow Transplant. (2010) 16:612–21. 10.1016/j.bbmt.2010.01.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Borrego F, Kabat J, Kim DK, Lieto L, Maasho K, Pena J, et al. Structure and function of major histocompatibility complex (MHC) class I specific receptors expressed on human natural killer (NK) cells. Mol Immunol. (2002) 38:637–60. 10.1016/S0161-5890(01)00107-9 [DOI] [PubMed] [Google Scholar]
- 23.Moretta L, Montaldo E, Vacca P, Del Zotto G, Moretta F, Merli P, et al. Human natural killer cells: origin, receptors, function, and clinical applications. Int Arch Allergy Immunol. (2014) 164:253–64. 10.1159/000365632 [DOI] [PubMed] [Google Scholar]
- 24.Vitale M, Cantoni C, Della Chiesa M, Ferlazzo G, Carlomagno S, Pende D, et al. An historical overview: the discovery of how NK cells can kill enemies, recruit defense troops, and more. Front Immunol. (2019) 10:1415. 10.3389/fimmu.2019.01415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chiossone L, Vienne M, Kerdiles YM, Vivier E. Natural killer cell immunotherapies against cancer: checkpoint inhibitors and more. Semin Immunol. (2017) 31:55–63. 10.1016/j.smim.2017.08.003 [DOI] [PubMed] [Google Scholar]
- 26.Wang W, Erbe AK, Hank JA, Morris ZS, Sondel PM. NK cell-mediated antibody-dependent cellular cytotoxicity in cancer immunotherapy. Front Immunol. (2015) 6:368. 10.3389/fimmu.2015.00368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. (2011) 144:646–74. 10.1016/j.cell.2011.02.013 [DOI] [PubMed] [Google Scholar]
- 28.Stojanovic A, Correia MP, Cerwenka A. Shaping of NK cell responses by the tumor microenvironment. Cancer Microenviron. (2013) 6:135–46. 10.1007/s12307-012-0125-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vitale M, Cantoni C, Pietra G, Mingari MC, Moretta L. Effect of tumor cells and tumor microenvironment on NK-cell function. Eur J Immunol. (2014) 44:1582–92. 10.1002/eji.201344272 [DOI] [PubMed] [Google Scholar]
- 30.Terrén I, Orrantia A, Vitallé J, Zenarruzabeitia O, Borrego F. NK cell metabolism and tumor microenvironment. Front Immunol. (2019) 10:2278. 10.3389/fimmu.2019.02278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zingoni A, Vulpis E, Nardone I, Soriani A, Fionda C, Cippitelli M, et al. Targeting NKG2D and NKp30 ligands shedding to improve NK cell-based immunotherapy. Crit Rev Immunol. (2016) 36:445–60. 10.1615/CritRevImmunol.2017020166 [DOI] [PubMed] [Google Scholar]
- 32.Krzywinska E, Kantari-Mimoun C, Kerdiles Y, Sobecki M, Isagawa T, Gotthardt D, et al. Loss of HIF-1α in natural killer cells inhibits tumour growth by stimulating non-productive angiogenesis. Nat Commun. (2017) 8:1597. 10.1038/s41467-017-01599-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Konjevic GM, Vuletic AM, Mirjacic Martinovic KM, Larsen AK, Jurisic VB. The role of cytokines in the regulation of NK cells in the tumor environment. Cytokine. (2019) 117:30–40. 10.1016/j.cyto.2019.02.001 [DOI] [PubMed] [Google Scholar]
- 34.Lee JC, Lee KM, Kim DW, Heo DS. Elevated TGF-beta1 secretion and down-modulation of NKG2D underlies impaired NK cytotoxicity in cancer patients. J Immunol. (2004) 172:7335–40. 10.4049/jimmunol.172.12.7335 [DOI] [PubMed] [Google Scholar]
- 35.Park A, Lee Y, Kim MS, Kang YJ, Park YJ, Jung H, et al. Prostaglandin E2 secreted by thyroid cancer cells contributes to immune escape through the suppression of natural killer (NK) Cell cytotoxicity and NK cell differentiation. Front Immunol. (2018) 9:1859 10.3389/fimmu.2018.01859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xiao Q, Wu J, Wang W-J, Chen S, Zheng Y, Yu X, et al. DKK2 imparts tumor immunity evasion through β-catenin-independent suppression of cytotoxic immune-cell activation. Nat Med. (2018) 24:262–70. 10.1038/nm.4496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang W, Guo H, Geng J, Zheng X, Wei H, Sun R, et al. Tumor-released Galectin-3, a soluble inhibitory ligand of human NKp30, plays an important role in tumor escape from NK cell attack. J Biol Chem. (2014) 289:33311–9. 10.1074/jbc.M114.603464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Suen WC, Lee WY, Leung KT, Pan XH, Li G. Natural killer cell-based cancer immunotherapy: a review on 10 years completed clinical trials. Cancer Invest. (2018) 36:431–57. 10.1080/07357907.2018.1515315 [DOI] [PubMed] [Google Scholar]
- 39.Davis ZB, Vallera DA, Miller JS, Felices M. Natural killer cells unleashed: checkpoint receptor blockade and BiKE/TriKE utilization in NK-mediated anti-tumor immunotherapy. Semin Immunol. (2017) 31:64–75. 10.1016/j.smim.2017.07.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Assal A, Kaner J, Pendurti G, Zang X. Emerging targets in cancer immunotherapy: beyond CTLA-4 and PD-1. Immunotherapy. (2015) 7:1169–86. 10.2217/imt.15.78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Burugu S, Dancsok AR, Nielsen TO. Emerging targets in cancer immunotherapy. Semin Cancer Biol. (2018) 52(Pt 2):39–52. 10.1016/j.semcancer.2017.10.001 [DOI] [PubMed] [Google Scholar]
- 42.Grossenbacher SK, Aguilar EG, Murphy WJ. Leveraging natural killer cells for cancer immunotherapy. Immunotherapy. (2017) 9:487–97. 10.2217/imt-2017-0013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Guillerey C, Huntington ND, Smyth MJ. Targeting natural killer cells in cancer immunotherapy. Nat Immunol. (2016) 17:1025–36. 10.1038/ni.3518 [DOI] [PubMed] [Google Scholar]
- 44.Rezvani K, Rouce R, Liu E, Shpall E. Engineering natural killer cells for cancer immunotherapy. Mol Therapy. (2017) 25:1769–81. 10.1016/j.ymthe.2017.06.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell. (2015) 27:450–61. 10.1016/j.ccell.2015.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cameron F, Whiteside G, Perry C. Ipilimumab: first global approval. Drugs. (2011) 71:1093–104. 10.2165/11594010-000000000-00000 [DOI] [PubMed] [Google Scholar]
- 47.Li J, Dong C. Nobel goes to immune checkpoint-innovative cancer treatment by immunotherapy. Sci China Life Sci. (2018) 61:1445–50. 10.1007/s11427-018-9409-7 [DOI] [PubMed] [Google Scholar]
- 48.Burshtyn DN, Yang W, Yi T, Long EO. A novel phosphotyrosine motif with a critical amino acid at position−2 for the SH2 domain-mediated activation of the tyrosine phosphatase SHP-1. J Biol Chem. (1997) 272:13066–72. 10.1074/jbc.272.20.13066 [DOI] [PubMed] [Google Scholar]
- 49.Daeron M, Jaeger S, Du Pasquier L, Vivier E. Immunoreceptor tyrosine-based inhibition motifs: a quest in the past and future. Immunol Rev. (2008) 224:11–43. 10.1111/j.1600-065X.2008.00666.x [DOI] [PubMed] [Google Scholar]
- 50.Burshtyn DN, Scharenberg AM, Wagtmann N, Rajagopalan S, Berrada K, Yi T, et al. Recruitment of tyrosine phosphatase HCP by the killer cell inhibitor receptor. Immunity. (1996) 4:77–85. 10.1016/S1074-7613(00)80300-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Purdy AK, Campbell KS. Natural killer cells and cancer: regulation by the killer cell Ig-like receptors (KIR). Cancer Biol Therapy. (2009) 8:2211–20. 10.4161/cbt.8.23.10455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Thielens A, Vivier E, Romagne F. NK cell MHC class I specific receptors (KIR): from biology to clinical intervention. Curr Opin Immunol. (2012) 24:239–45. 10.1016/j.coi.2012.01.001 [DOI] [PubMed] [Google Scholar]
- 53.Kang X, Kim J, Deng M, John S, Chen H, Wu G, et al. Inhibitory leukocyte immunoglobulin-like receptors: immune checkpoint proteins and tumor sustaining factors. Cell Cycle. (2016) 15:25–40. 10.1080/15384101.2015.1121324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gao J, Zheng Q, Xin N, Wang W, Zhao C. CD155, an onco-immunologic molecule in human tumors. Cancer Sci. (2017) 108:1934–8. 10.1111/cas.13324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liu S, Zhang H, Li M, Hu D, Li C, Ge B, et al. Recruitment of Grb2 and SHIP1 by the ITT-like motif of TIGIT suppresses granule polarization and cytotoxicity of NK cells. Cell Death Different. (2013) 20:456–64. 10.1038/cdd.2012.141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Anderson AC, Joller N, Kuchroo VK. Lag-3, TIM-3, and TIGIT: co-inhibitory receptors with specialized functions in immune regulation. Immunity. (2016) 44:989–1004. 10.1016/j.immuni.2016.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li M, Xia P, Du Y, Liu S, Huang G, Chen J, et al. T-cell immunoglobulin and ITIM domain (TIGIT) receptor/poliovirus receptor (PVR) ligand engagement suppresses interferon-gamma production of natural killer cells via beta-arrestin 2-mediated negative signaling. J Biol Chem. (2014) 289:17647–57. 10.1074/jbc.M114.572420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Crocker PR, Paulson JC, Varki A. Siglecs and their roles in the immune system. Nat Rev Immunol. (2007) 7:255–66. 10.1038/nri2056 [DOI] [PubMed] [Google Scholar]
- 59.Nirschl CJ, Drake CG. Molecular pathways: coexpression of immune checkpoint molecules: signaling pathways and implications for cancer immunotherapy. Clin Cancer Res. (2013) 19:4917–24. 10.1158/1078-0432.CCR-12-1972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Parry RV, Chemnitz JM, Frauwirth KA, Lanfranco AR, Braunstein I, Kobayashi SV, et al. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol Cell Biol. (2005) 25:9543–53. 10.1128/MCB.25.21.9543-9553.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Boussiotis VA. Molecular and biochemical aspects of the PD-1 checkpoint pathway. N Engl J Med. (2016) 375:1767–78. 10.1056/NEJMra1514296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kharitonenkov A, Chen Z, Sures I, Wang H, Schilling J, Ullrich A. A family of proteins that inhibit signalling through tyrosine kinase receptors. Nature. (1997) 386:181–6. 10.1038/386181a0 [DOI] [PubMed] [Google Scholar]
- 63.Tsai RK, Discher DE. Inhibition of “self” engulfment through deactivation of myosin-II at the phagocytic synapse between human cells. J Cell Biol. (2008) 180:989–1003. 10.1083/jcb.200708043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Alblas J, Honing H, de Lavalette CR, Brown MH, Dijkstra CD, van den Berg TK. Signal regulatory protein alpha ligation induces macrophage nitric oxide production through JAK/STAT- and phosphatidylinositol 3-kinase/Rac1/NAPDH oxidase/H2O2-dependent pathways. Mol Cell Biol. (2005) 25:7181–92. 10.1128/MCB.25.16.7181-7192.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Schneider H, Rudd CE. Tyrosine phosphatase SHP-2 binding to CTLA-4: absence of direct YVKM/YFIP motif recognition. Biochem Biophys Res Commun. (2000) 269:279–83. 10.1006/bbrc.2000.2234 [DOI] [PubMed] [Google Scholar]
- 66.Chuang E, Fisher TS, Morgan RW, Robbins MD, Duerr JM, Vander Heiden MG, et al. The CD28 and CTLA-4 receptors associate with the serine/threonine phosphatase PP2A. Immunity. (2000) 13:313–22. 10.1016/S1074-7613(00)00031-5 [DOI] [PubMed] [Google Scholar]
- 67.Walker LS, Sansom DM. Confusing signals: recent progress in CTLA-4 biology. Trends Immunol. (2015) 36:63–70. 10.1016/j.it.2014.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Workman CJ, Dugger KJ, Vignali DA. Cutting edge: molecular analysis of the negative regulatory function of lymphocyte activation gene-3. J Immunol. (2002) 169:5392–5. 10.4049/jimmunol.169.10.5392 [DOI] [PubMed] [Google Scholar]
- 69.Lee J, Su EW, Zhu C, Hainline S, Phuah J, Moroco JA, et al. Phosphotyrosine-dependent coupling of TIM-3 to T-cell receptor signaling pathways. Mol Cell Biol. (2011) 31:3963–74. 10.1128/MCB.05297-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.van de Weyer PS, Muehlfeit M, Klose C, Bonventre JV, Walz G, Kuehn EW. A highly conserved tyrosine of TIM-3 is phosphorylated upon stimulation by its ligand galectin-9. Biochem Biophys Res Commun. (2006) 351:571–6. 10.1016/j.bbrc.2006.10.079 [DOI] [PubMed] [Google Scholar]
- 71.Tomkowicz B, Walsh E, Cotty A, Verona R, Sabins N, Kaplan F, et al. TIM-3 suppresses anti-CD3/CD28-induced TCR activation and IL-2 expression through the NFAT signaling pathway. PLoS ONE. (2015) 10:e0140694. 10.1371/journal.pone.0140694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhang S, Phillips JH. Identification of tyrosine residues crucial for CD200R-mediated inhibition of mast cell activation. J Leukoc Biol. (2006) 79:363–8. 10.1189/jlb.0705398 [DOI] [PubMed] [Google Scholar]
- 73.Mihrshahi R, Barclay AN, Brown MH. Essential roles for Dok2 and RasGAP in CD200 receptor-mediated regulation of human myeloid cells. J Immunol. (2009) 183:4879–86. 10.4049/jimmunol.0901531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Uhrberg M, Valiante NM, Young NT, Lanier LL, Phillips JH, Parham P. The repertoire of killer cell Ig-like receptor and CD94:NKG2A receptors in T cells: clones sharing identical alpha beta TCR rearrangement express highly diverse killer cell Ig-like receptor patterns. J Immunol. (2001) 166:3923–32. 10.4049/jimmunol.166.6.3923 [DOI] [PubMed] [Google Scholar]
- 75.Campbell KS, Purdy AK. Structure/function of human killer cell immunoglobulin-like receptors: lessons from polymorphisms, evolution, crystal structures and mutations. Immunology. (2011) 132:315–25. 10.1111/j.1365-2567.2010.03398.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Uhrberg M, Valiante NM, Shum BP, Shilling HG, Lienert-Weidenbach K, Corliss B, et al. Human diversity in killer cell inhibitory receptor genes. Immunity. (1997) 7:753–63. 10.1016/S1074-7613(00)80394-5 [DOI] [PubMed] [Google Scholar]
- 77.Chan HW, Kurago ZB, Stewart CA, Wilson MJ, Martin MP, Mace BE, et al. DNA methylation maintains allele-specific KIR gene expression in human natural killer cells. J Exp Med. (2003) 197:245–55. 10.1084/jem.20021127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Parham P. Killer cell immunoglobulin-like receptor diversity: balancing signals in the natural killer cell response. Immunol Lett. (2004) 92:11–3. 10.1016/j.imlet.2003.11.016 [DOI] [PubMed] [Google Scholar]
- 79.Parham P. MHC class I molecules and KIRs in human history, health and survival. Nat Rev Immunol. (2005) 5:201–14. 10.1038/nri1570 [DOI] [PubMed] [Google Scholar]
- 80.Parham P. The genetic and evolutionary balances in human NK cell receptor diversity. Semin Immunol. (2008) 20:311–6. 10.1016/j.smim.2008.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yawata M, Yawata N, Draghi M, Little AM, Partheniou F, Parham P. Roles for HLA and KIR polymorphisms in natural killer cell repertoire selection and modulation of effector function. J Exp Med. (2006) 203:633–45. 10.1084/jem.20051884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Benson DM, Jr, Hofmeister CC, Padmanabhan S, Suvannasankha A, Jagannath S, Abonour R, et al. A phase 1 trial of the anti-KIR antibody IPH2101 in patients with relapsed/refractory multiple myeloma. Blood. (2012) 120:4324–33. 10.1182/blood-2012-06-438028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Vey N, Bourhis J-H, Dombret H, Bordessoule D, Prébet T, Charbonnier A, et al. A phase I study of the anti-natural killer inhibitory receptor (KIR) monoclonal antibody (1-7F9, IPH2101) in elderly patients with acute myeloid leukemia (AML): clinical and immunological effects of a single dose followed by repeated dosing. Blood. (2009) 114:632 10.1182/blood.V114.22.632.632 [DOI] [Google Scholar]
- 84.Vey N, Bourhis JH, Boissel N, Bordessoule D, Prebet T, Charbonnier A, et al. A phase 1 trial of the anti-inhibitory KIR mAb IPH2101 for AML in complete remission. Blood. (2012) 120:4317–23. 10.1182/blood-2012-06-437558 [DOI] [PubMed] [Google Scholar]
- 85.Korde N, Carlsten M, Lee MJ, Minter A, Tan E, Kwok M, et al. A phase II trial of pan-KIR2D blockade with IPH2101 in smoldering multiple myeloma. Haematologica. (2014) 99:e81–3. 10.3324/haematol.2013.103085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Carlsten M, Korde N, Kotecha R, Reger R, Bor S, Kazandjian D, et al. Checkpoint inhibition of KIR2D with the monoclonal antibody IPH2101 induces contraction and hyporesponsiveness of NK cells in patients with myeloma. Clin Cancer Res. (2016) 22:5211–22. 10.1158/1078-0432.CCR-16-1108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Benson DM, Jr, Bakan CE, Zhang S, Collins SM, Liang J, Srivastava S, et al. IPH2101, a novel anti-inhibitory KIR antibody, and lenalidomide combine to enhance the natural killer cell versus multiple myeloma effect. Blood. (2011) 118:6387–91. 10.1182/blood-2011-06-360255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Cohen AD, Hofmeister CC, Nikhil MC, Jagannath S, Efebera YA, Spitzer G, et al. A phase I trial of anti-KIR monoclonal antibody IPH2101 and lenalidomide for multiple myeloma. Blood. (2013) 122:3181 10.1182/blood.V122.21.3181.3181 [DOI] [Google Scholar]
- 89.Benson DM, Jr, Cohen AD, Jagannath S, Munshi NC, Spitzer G, Hofmeister CC, et al. A phase I trial of the anti-KIR antibody IPH2101 and lenalidomide in patients with relapsed/refractory multiple myeloma. Clin Cancer Res. (2015) 21:4055–61. 10.1158/1078-0432.CCR-15-0304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Vey N, Goncalves A, Karlin L, Lebouvier-Sadot S, Broussais F, Marie D, et al. A phase 1 dose-escalation study of IPH2102 (lirilumab, BMS-986015, LIRI), a fully human anti KIR monoclonal antibody (mAb) in patients (pts) with various hematologic (HEM) or solid malignancies (SOL). J Clin Oncol. (2015) 33(15_suppl):3065 10.1200/jco.2015.33.15_suppl.306526304881 [DOI] [Google Scholar]
- 91.Robbins M, Jure-Kunkel M, Dito G, Andre P, Zhang H-f, Bezman N, et al. Effects of IL-21, KIR blockade, and CD137 agonism on the non-clinical activity of elotuzumab. Blood. (2014) 124:4717 10.1182/blood.V124.21.4717.4717 [DOI] [Google Scholar]
- 92.Sola C, Blery M, Bonnafous C, Bonnet E, Fuseri N, Graziano RF, et al. Lirilumab Enhances anti-tumor efficacy of elotuzumab. Blood. (2014) 124:4711 10.1182/blood.V124.21.4711.4711 [DOI] [Google Scholar]
- 93.Romagne F, Andre P, Spee P, Zahn S, Anfossi N, Gauthier L, et al. Preclinical characterization of 1-7F9, a novel human anti-KIR receptor therapeutic antibody that augments natural killer-mediated killing of tumor cells. Blood. (2009) 114:2667–77. 10.1182/blood-2009-02-206532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bourhis J-H, Recher C, Etienne A, Charbonnier A, Andre P, Rey J, et al. Repeated dosing of anti-KIR (IPH2101) as maintenance therapy in ederly patients with acute myeloid leukemia. Blood. (2013) 122:2696 10.1182/blood.V122.21.2696.2696 [DOI] [Google Scholar]
- 95.Vey N, Dumas P-Y, Recher C, Gastaud L, Lioure B, Bulabois C-E, et al. Randomized phase 2 trial of lirilumab (anti-KIR monoclonal antibody, mAb) as maintenance treatment in elderly patients (pts) with acute myeloid leukemia (AML): results of the effikir trial. Blood. (2017) 130(Suppl. 1):889 10.1182/blood.V130.Suppl_1.889.889 [DOI] [Google Scholar]
- 96.Daver N, Garcia-Manero G, Basu S, Cortes JE, Ravandi F, Jabbour EJ, et al. Phase IB/II study of lirilumab in combination with azacytidine (AZA) in patients (pts) with relapsed acute myeloid leukemia (AML). Blood. (2016) 128:1641 10.1182/blood.V128.22.1641.1641 [DOI] [Google Scholar]
- 97.Yalniz FF, Daver N, Rezvani K, Kornblau S, Ohanian M, Borthakur G, et al. A pilot trial of lirilumab with or without azacitidine for patients with myelodysplastic syndrome. Clin Lymphoma Myeloma Leukemia. (2018) 18:658–63.e2. 10.1016/j.clml.2018.06.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Binyamin L, Alpaugh RK, Hughes TL, Lutz CT, Campbell KS, Weiner LM. Blocking NK cell inhibitory self-recognition promotes antibody-dependent cellular cytotoxicity in a model of anti-lymphoma therapy. J Immunol. (2008) 180:6392–401. 10.4049/jimmunol.180.9.6392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kohrt HE, Thielens A, Marabelle A, Sagiv-Barfi I, Sola C, Chanuc F, et al. Anti-KIR antibody enhancement of anti-lymphoma activity of natural killer cells as monotherapy and in combination with anti-CD20 antibodies. Blood. (2014) 123:678–86. 10.1182/blood-2013-08-519199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Bagot M, Porcu P, Marie-Cardine A, Battistella M, William B, Vermeer MH, et al. IPH4102, a first-in-class anti-KIR3DL2 monoclonal antibody, in patients with relapsed or refractory cutaneous T-cell lymphoma: an international, first-in-human, open-label, phase 1 trial. Lancet Oncol. (2019) 20:1160–70. 10.1016/S1470-2045(19)30320-1 [DOI] [PubMed] [Google Scholar]
- 101.Zhang F, Zheng J, Kang X, Deng M, Lu Z, Kim J, et al. Inhibitory leukocyte immunoglobulin-like receptors in cancer development. Sci China Life Sci. (2015) 58:1216–25. 10.1007/s11427-015-4925-1 [DOI] [PubMed] [Google Scholar]
- 102.Rouas-Freiss N, Moreau P, LeMaoult J, Carosella ED. The dual role of HLA-G in cancer. J Immunol Res. (2014) 2014:359748. 10.1155/2014/359748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hogan L, Bhuju S, Jones DC, Laing K, Trowsdale J, Butcher P, et al. Characterisation of bovine leukocyte Ig-like receptors. PLoS One. (2012) 7:e34291. 10.1371/journal.pone.0034291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Navarro F, Llano M, Bellon T, Colonna M, Geraghty DE, Lopez-Botet M. The ILT2(LIR1) and CD94/NKG2A NK cell receptors respectively recognize HLA-G1 and HLA-E molecules co-expressed on target cells. Eur J Immunol. (1999) 29:277–83. 10.1002/(SICI)1521-4141(199901)29:01<277::AID-IMMU277>3.0.CO;2-4 [DOI] [PubMed] [Google Scholar]
- 105.Lin A, Yan W-H. Heterogeneity of HLA-G expression in cancers: facing the challenges. Front Immunol. (2018) 9:2164. 10.3389/fimmu.2018.02164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Carosella ED, Moreau P, Lemaoult J, Rouas-Freiss N. HLA-G: from biology to clinical benefits. Trends Immunol. (2008) 29:125–32. 10.1016/j.it.2007.11.005 [DOI] [PubMed] [Google Scholar]
- 107.Lin A, Yan WH, Xu HH, Gan MF, Cai JF, Zhu M, et al. HLA-G expression in human ovarian carcinoma counteracts NK cell function. Ann Oncol. (2007) 18:1804–9. 10.1093/annonc/mdm356 [DOI] [PubMed] [Google Scholar]
- 108.Lin A, Zhu CC, Chen HX, Chen BF, Zhang X, Zhang JG, et al. Clinical relevance and functional implications for human leucocyte antigen-g expression in non-small-cell lung cancer. J Cell Mol Med. (2010) 14:2318–29. 10.1111/j.1582-4934.2009.00858.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Lin A, Chen HX, Zhu CC, Zhang X, Xu HH, Zhang JG, et al. Aberrant human leucocyte antigen-G expression and its clinical relevance in hepatocellular carcinoma. J Cell Mol Med. (2010) 14:2162–71. 10.1111/j.1582-4934.2009.00917.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Wiendl H, Mitsdoerffer M, Hofmeister V, Wischhusen J, Bornemann A, Meyermann R, et al. A functional role of HLA-G expression in human gliomas: an alternative strategy of immune escape. J Immunol. (2002) 168:4772. 10.4049/jimmunol.168.9.4772 [DOI] [PubMed] [Google Scholar]
- 111.Bukur J, Rebmann V, Grosse-Wilde H, Luboldt H, Ruebben H, Drexler I, et al. Functional role of human leukocyte antigen-G upregulation in renal cell carcinoma. Cancer Res. (2003) 63:4107–11. [PubMed] [Google Scholar]
- 112.Chumbley G, King A, Robertson K, Holmes N, Loke YW. Resistance of HLA-G and HLA-A2 transfectants to lysis by decidual NK cells. Cell Immunol. (1994) 155:312–22. 10.1006/cimm.1994.1125 [DOI] [PubMed] [Google Scholar]
- 113.Pazmany L, Mandelboim O, Vales-Gomez M, Davis DM, Reyburn HT, Strominger JL. Protection from natural killer cell-mediated lysis by HLA-G expression on target cells. Science. (1996) 274:792–5. 10.1126/science.274.5288.792 [DOI] [PubMed] [Google Scholar]
- 114.Rouas-Freiss N, Marchal RE, Kirszenbaum M, Dausset J, Carosella ED. The alpha1 domain of HLA-G1 and HLA-G2 inhibits cytotoxicity induced by natural killer cells: is HLA-G the public ligand for natural killer cell inhibitory receptors? Proc Natl Acad Sci USA. (1997) 94:5249–54. 10.1073/pnas.94.10.5249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Favier B, Lemaoult J, Lesport E, Carosella ED. ILT2/HLA-G interaction impairs NK-cell functions through the inhibition of the late but not the early events of the NK-cell activating synapse. FASEB J. (2010) 24:689–99. 10.1096/fj.09-135194 [DOI] [PubMed] [Google Scholar]
- 116.Morandi F, Ferretti E, Castriconi R, Dondero A, Petretto A, Bottino C, et al. Soluble HLA-G dampens CD94/NKG2A expression and function and differentially modulates chemotaxis and cytokine and chemokine secretion in CD56bright and CD56dim NK cells. Blood. (2011) 118:5840. 10.1182/blood-2011-05-352393 [DOI] [PubMed] [Google Scholar]
- 117.Menier C, Riteau B, Carosella ED, Rouas-Freiss N. MICA triggering signal for NK cell tumor lysis is counteracted by HLA-G1-mediated inhibitory signal. Int J Cancer. (2002) 100:63–70. 10.1002/ijc.10460 [DOI] [PubMed] [Google Scholar]
- 118.Wan R, Wang ZW, Li H, Peng XD, Liu GY, Ou JM, et al. Human leukocyte antigen-G inhibits the anti-tumor effect of natural killer cells via immunoglobulin-like transcript 2 in gastric cancer. Cell Physiol Biochem. (2017) 44:1828–41. 10.1159/000485819 [DOI] [PubMed] [Google Scholar]
- 119.Naji A, Menier C, Maki G, Carosella ED, Rouas-Freiss N. Neoplastic B-cell growth is impaired by HLA-G/ILT2 interaction. Leukemia. (2012) 26:1889. 10.1038/leu.2012.62 [DOI] [PubMed] [Google Scholar]
- 120.Heidenreich S, Zu Eulenburg C, Hildebrandt Y, Stubig T, Sierich H, Badbaran A, et al. Impact of the NK cell receptor LIR-1 (ILT-2/CD85j/LILRB1) on cytotoxicity against multiple myeloma. Clin Dev Immunol. (2012) 2012:652130. 10.1155/2012/652130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Villa-Álvarez M, Sordo-Bahamonde C, Lorenzo-Herrero S, Gonzalez-Rodriguez AP, Payer AR, Gonzalez-Garcia E, et al. Ig-like transcript 2 (ILT2) blockade and lenalidomide restore NK cell function in chronic lymphocytic leukemia. Front Immunol. (2018) 9:2917. 10.3389/fimmu.2018.02917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Zheng H, Lu R, Xie S, Wen X, Wang H, Gao X, et al. Human leukocyte antigen-E alleles and expression in patients with serous ovarian cancer. Cancer Sci. (2015) 106:522–8. 10.1111/cas.12641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Gooden M, Lampen M, Jordanova ES, Leffers N, Trimbos JB, van der Burg SH, et al. HLA-E expression by gynecological cancers restrains tumor-infiltrating CD8(+) T lymphocytes. Proc Natl Acad Sci USA. (2011) 108:10656–61. 10.1073/pnas.1100354108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Soulas C, Remark R, Brezar V, Lopez J, Bonnet E, Caraguel F, et al. Abstract 2714: combination of monalizumab and durvalumab as a potent immunotherapy treatment for solid human cancers. Cancer Res. (2018) 78(13 Suppl.):2714 10.1158/1538-7445.AM2018-2714 [DOI] [Google Scholar]
- 125.Borrego F, Masilamani M, Kabat J, Sanni TB, Coligan JE. The cell biology of the human natural killer cell CD94/NKG2A inhibitory receptor. Mol Immunol. (2005) 42:485–8. 10.1016/j.molimm.2004.07.031 [DOI] [PubMed] [Google Scholar]
- 126.Levy EM, Bianchini M, Von Euw EM, Barrio MM, Bravo AI, Furman D, et al. Human leukocyte antigen-E protein is overexpressed in primary human colorectal cancer. Int J Oncol. (2008) 32:633–41. 10.3892/ijo.32.3.633 [DOI] [PubMed] [Google Scholar]
- 127.Bossard C, Bezieau S, Matysiak-Budnik T, Volteau C, Laboisse CL, Jotereau F, et al. HLA-E/beta2 microglobulin overexpression in colorectal cancer is associated with recruitment of inhibitory immune cells and tumor progression. Int J Cancer. (2012) 131:855–63. 10.1002/ijc.26453 [DOI] [PubMed] [Google Scholar]
- 128.Tinker A, Hirte H, Provencher D, Butler M, Ritter H, Tu D, et al. Dose ranging study of monalizumab (IPH2201) in patients with gynecologic malignancies: a trial of the Canadian Cancer Trials Group (CCTG): IND221. Eur J Cancer. (2016) 69:S97–S8. 10.1016/S0959-8049(16)32889-1 [DOI] [PubMed] [Google Scholar]
- 129.Sun C, Xu J, Huang Q, Huang M, Wen H, Zhang C, et al. High NKG2A expression contributes to NK cell exhaustion and predicts a poor prognosis of patients with liver cancer. Oncoimmunology. (2017) 6:e1264562. 10.1080/2162402X.2016.1264562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Kren L, Slaby O, Muckova K, Lzicarova E, Sova M, Vybihal V, et al. Expression of immune-modulatory molecules HLA-G and HLA-E by tumor cells in glioblastomas: an unexpected prognostic significance? Neuropathology. (2011) 31:129–34. 10.1111/j.1440-1789.2010.01149.x [DOI] [PubMed] [Google Scholar]
- 131.Wolpert F, Roth P, Lamszus K, Tabatabai G, Weller M, Eisele G. HLA-E contributes to an immune-inhibitory phenotype of glioblastoma stem-like cells. J Neuroimmunol. (2012) 250:27–34. 10.1016/j.jneuroim.2012.05.010 [DOI] [PubMed] [Google Scholar]
- 132.Kren L, Fabian P, Slaby O, Janikova A, Soucek O, Sterba J, et al. Multifunctional immune-modulatory protein HLA-E identified in classical Hodgkin lymphoma: possible implications. Pathol Res Pract. (2012) 208:45–9. 10.1016/j.prp.2011.11.004 [DOI] [PubMed] [Google Scholar]
- 133.Ruggeri L, Urbani E, Andre P, Mancusi A, Tosti A, Topini F, et al. Effects of anti-NKG2A antibody administration on leukemia and normal hematopoietic cells. Haematologica. (2016) 101:626–33. 10.3324/haematol.2015.135301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.McWilliams EM, Mele JM, Cheney C, Timmerman EA, Fiazuddin F, Strattan EJ, et al. Therapeutic CD94/NKG2A blockade improves natural killer cell dysfunction in chronic lymphocytic leukemia. Oncoimmunology. (2016) 5:e1226720. 10.1080/2162402X.2016.1226720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Andre P, Denis C, Soulas C, Bourbon-Caillet C, Lopez J, Arnoux T, et al. Anti-NKG2A mAb is a checkpoint inhibitor that promotes anti-tumor immunity by unleashing both T and NK cells. Cell. (2018) 175:1731–43.e13. 10.1016/j.cell.2018.10.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Ager C, Reilley M, Nicholas C, Bartkowiak T, Jaiswal A, Curran M, et al. 31st annual meeting and associated programs of the society for immunotherapy of cancer (SITC 2016): part two: National Harbor, MD, USA. 9–13 November 2016. J Immunother Cancer. (2016) 4(Suppl. 1):107–221. 10.1186/s40425-016-0173-6 [DOI] [Google Scholar]
- 137.Segal NH, Naidoo J, Curigliano G, Patel S, Sahebjam S, Papadopoulos KP, et al. First-in-human dose escalation of monalizumab plus durvalumab, with expansion in patients with metastatic microsatellite-stable colorectal cancer. J Clin Oncol. (2018) 36(15_suppl):3540 10.1200/JCO.2018.36.15_suppl.3540 [DOI] [Google Scholar]
- 138.Levy EM, Sycz G, Arriaga JM, Barrio MM, von Euw EM, Morales SB, et al. Cetuximab-mediated cellular cytotoxicity is inhibited by HLA-E membrane expression in colon cancer cells. Innate Immun. (2009) 15:91–100. 10.1177/1753425908101404 [DOI] [PubMed] [Google Scholar]
- 139.Ferris RL, Lenz H-J, Trotta AM, García-Foncillas J, Schulten J, Audhuy F, et al. Rationale for combination of therapeutic antibodies targeting tumor cells and immune checkpoint receptors: harnessing innate and adaptive immunity through IgG1 isotype immune effector stimulation. Cancer Treat Rev. (2018) 63:48–60. 10.1016/j.ctrv.2017.11.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Cohen R, Fayette J, Posner M, Lefebvre G, Bauman J, Salas S, et al. Abstract CT158: Phase II study of monalizumab, a first-in-class NKG2A monoclonal antibody, in combination with cetuximab in previously treated recurrent or metastatic squamous cell carcinoma of the head and neck (R/M SCCHN): preliminary assessment of safety and efficacy. Cancer Res. (2018) 78(13 Suppl.):CT158 10.1158/1538-7445.AM2018-CT158 [DOI] [Google Scholar]
- 141.Van Montfoort N, Borst L, Korrer MJ, Sluijter M, Marijt KA, Santegoets SJ, et al. NKG2A blockade potentiates CD8 T cell immunity induced by cancer vaccines. Cell. (2018) 175:1744–55.e15. 10.1016/j.cell.2018.10.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Blake SJ, Dougall WC, Miles JJ, Teng MW, Smyth MJ. Molecular pathways: targeting CD96 and TIGIT for cancer immunotherapy. Clin Cancer Res. (2016) 22:5183–8. 10.1158/1078-0432.CCR-16-0933 [DOI] [PubMed] [Google Scholar]
- 143.Zhou XM, Li WQ, Wu YH, Han L, Cao XG, Yang XM, et al. Intrinsic expression of immune checkpoint molecule TIGIT could help tumor growth in vivo by suppressing the function of NK and CD8(+) T cells. Front Immunol. (2018) 9:2821. 10.3389/fimmu.2018.02821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Deuss FA, Watson GM, Fu Z, Rossjohn J, Berry R. Structural basis for CD96 immune receptor recognition of nectin-like protein-5, CD155. Structure. (2019) 27:219–228. 10.1016/j.str.2018.10.023 [DOI] [PubMed] [Google Scholar]
- 145.Dougall WC, Kurtulus S, Smyth MJ, Anderson AC. TIGIT and CD96: new checkpoint receptor targets for cancer immunotherapy. Immunol Rev. (2017) 276:112–20. 10.1111/imr.12518 [DOI] [PubMed] [Google Scholar]
- 146.Chan CJ, Martinet L, Gilfillan S, Souza-Fonseca-Guimaraes F, Chow MT, Town L, et al. The receptors CD96 and CD226 oppose each other in the regulation of natural killer cell functions. Nat Immunol. (2014) 15:431–8. 10.1038/ni.2850 [DOI] [PubMed] [Google Scholar]
- 147.Deuss FA, Gully BS, Rossjohn J, Berry R. Recognition of nectin-2 by the natural killer cell receptor T cell immunoglobulin and ITIM domain (TIGIT). J Biol Chem. (2017) 292:11413–22. 10.1074/jbc.M117.786483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Solomon BL, Garrido-Laguna I. TIGIT: a novel immunotherapy target moving from bench to bedside. Cancer Immunol Immunother. (2018) 67:1659–67. 10.1007/s00262-018-2246-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Joller N, Hafler JP, Brynedal B, Kassam N, Spoerl S, Levin SD, et al. Cutting edge: TIGIT has T cell-intrinsic inhibitory functions. J Immunol. (2011) 186:1338–42. 10.4049/jimmunol.1003081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Stanietsky N, Simic H, Arapovic J, Toporik A, Levy O, Novik A, et al. The interaction of TIGIT with PVR and PVRL2 inhibits human NK cell cytotoxicity. Proc Natl Acad Sci USA. (2009) 106:17858. 10.1073/pnas.0903474106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Tian Z. TIGIT blockade prevents CTL and NK cell exhaustion and leads to tumor rejection in mice. J Immunol. (2018) 200(1 Suppl.):57.50. [Google Scholar]
- 152.Bowers JR, Readler JM, Sharma P, Excoffon K. Poliovirus receptor: more than a simple viral receptor. Virus Res. (2017) 242:1–6. 10.1016/j.virusres.2017.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Kucan Brlic P, Lenac Rovis T, Cinamon G, Tsukerman P, Mandelboim O, Jonjic S. Targeting PVR (CD155) and its receptors in anti-tumor therapy. Cell Mol Immunol. (2019) 16:51–63. 10.1038/s41423-018-0168-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Liu XG, Hou M, Liu Y. TIGIT, a novel therapeutic target for tumor immunotherapy. Immunol Invest. (2017) 46:172–82. 10.1080/08820139.2016.1237524 [DOI] [PubMed] [Google Scholar]
- 155.Yang Z-Z. Expression and function of tigit in B-cell non-hodgkin lymphoma. Blood. (2016) 128:4138 10.1182/blood.V128.22.4138.4138 [DOI] [Google Scholar]
- 156.Guillerey C, Ferrari de Andrade L, Vuckovic S, Miles K, Ngiow SF, Yong MC, et al. Immunosurveillance and therapy of multiple myeloma are CD226 dependent. J Clin Invest. (2015) 125:2077–89. 10.1172/JCI77181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Minnie SA, Kuns RD, Gartlan KH, Zhang P, Wilkinson AN, Samson L, et al. Myeloma escape after stem cell transplantation is a consequence of T-cell exhaustion and is prevented by TIGIT blockade. Blood. (2018) 132:1675–88. 10.1182/blood-2018-01-825240 [DOI] [PubMed] [Google Scholar]
- 158.Guillerey C, Harjunpaa H, Carrie N, Kassem S, Teo T, Miles K, et al. TIGIT immune checkpoint blockade restores CD8(+) T-cell immunity against multiple myeloma. Blood. (2018) 132:1689–94. 10.1182/blood-2018-01-825265 [DOI] [PubMed] [Google Scholar]
- 159.Lesokhin AM, Ansell SM, Armand P, Scott EC, Halwani A, Gutierrez M, et al. Nivolumab in patients with relapsed or refractory hematologic malignancy: preliminary results of a phase Ib study. J Clin Oncol. (2016) 34:2698–704. 10.1200/JCO.2015.65.9789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Kong Y, Zhu L, Schell TD, Zhang J, Claxton DF, Ehmann WC, et al. T-cell immunoglobulin and ITIM Domain (TIGIT) associates with CD8+ T-cell exhaustion and poor clinical outcome in AML patients. Clin Cancer Res. (2016) 22:3057–66. 10.1158/1078-0432.CCR-15-2626 [DOI] [PubMed] [Google Scholar]
- 161.Stamm H, Wellbrock J, Fiedler W. Interaction of PVR/PVRL2 with TIGIT/DNAM-1 as a novel immune checkpoint axis and therapeutic target in cancer. Mammalian Genome. (2018) 29:694–702. 10.1007/s00335-018-9770-7 [DOI] [PubMed] [Google Scholar]
- 162.Hattori N, Kawaguchi Y, Sasaki Y, Shimada S, Murai S, Abe M, et al. Monitoring TIGIT/DNAM-1 and PVR/PVRL2 immune checkpoint's expression levels in allogeneic stem cell transplantation for acute myeloid leukemia. Biol Blood Marrow Transplant. (2019) 25:861–7. 10.1016/j.bbmt.2019.01.013 [DOI] [PubMed] [Google Scholar]
- 163.Aldinucci D, Gloghini A, Pinto A, De Filippi R, Carbone A. The classical Hodgkin's lymphoma microenvironment and its role in promoting tumour growth and immune escape. J Pathol. (2010) 221:248–63. 10.1002/path.2711 [DOI] [PubMed] [Google Scholar]
- 164.Li W, Blessin NC, Simon R, Kluth M, Fischer K, Hube-Magg C, et al. Expression of the immune checkpoint receptor TIGIT in Hodgkin's lymphoma. BMC Cancer. (2018) 18:1209. 10.1186/s12885-018-5111-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Nishiwada S, Sho M, Yasuda S, Shimada K, Yamato I, Akahori T, et al. Clinical significance of CD155 expression in human pancreatic cancer. Anti Cancer Res. (2015) 35:2287–97. [PubMed] [Google Scholar]
- 166.Peng YP, Xi CH, Zhu Y, Yin LD, Wei JS, Zhang JJ, et al. Altered expression of CD226 and CD96 on natural killer cells in patients with pancreatic cancer. Oncotarget. (2016) 7:66586–94. 10.18632/oncotarget.11953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Zhang Q, Bi J, Zheng X, Chen Y, Wang H, Wu W, et al. Blockade of the checkpoint receptor TIGIT prevents NK cell exhaustion and elicits potent anti-tumor immunity. Nat Immunol. (2018) 19:723–32. 10.1038/s41590-018-0132-0 [DOI] [PubMed] [Google Scholar]
- 168.Chew V, Chen J, Lee D, Loh E, Lee J, Lim KH, et al. Chemokine-driven lymphocyte infiltration: an early intratumoural event determining long-term survival in resectable hepatocellular carcinoma. Gut. (2012) 61:427–38. 10.1136/gutjnl-2011-300509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Sun C, Sun HY, Xiao WH, Zhang C, Tian ZG. Natural killer cell dysfunction in hepatocellular carcinoma and NK cell-based immunotherapy. Acta pharmacologica Sinica. (2015) 36:1191–9. 10.1038/aps.2015.41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Zhang QF, Yin WW, Xia Y, Yi YY, He QF, Wang X, et al. Liver-infiltrating CD11b(-)CD27(-) NK subsets account for NK-cell dysfunction in patients with hepatocellular carcinoma and are associated with tumor progression. Cell Mol Immunol. (2017) 14:819–29. 10.1038/cmi.2016.28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Sun H, Huang Q, Huang M, Wen H, Lin R, Zheng M, et al. Human CD96 correlates to natural killer cell exhaustion and predicts the prognosis of human hepatocellular carcinoma. Hepatology. (2019) 70:168–83. 10.1002/hep.30347 [DOI] [PubMed] [Google Scholar]
- 172.Blake SJ, Stannard K, Liu J, Allen S, Yong MC, Mittal D, et al. Suppression of metastases using a new lymphocyte checkpoint target for cancer immunotherapy. Cancer Discov. (2016) 6:446–59. 10.1158/2159-8290.CD-15-0944 [DOI] [PubMed] [Google Scholar]
- 173.He W, Zhang H, Han F, Chen X, Lin R, Wang W, et al. CD155T/TIGIT signaling regulates CD8(+) T-cell metabolism and promotes tumor progression in human gastric cancer. Cancer Res. (2017) 77:6375–88. 10.1158/0008-5472.CAN-17-0381 [DOI] [PubMed] [Google Scholar]
- 174.Dixon KO, Schorer M, Nevin J, Etminan Y, Amoozgar Z, Kondo T, et al. Functional anti-TIGIT antibodies regulate development of autoimmunity and antitumor immunity. J Immunol. (2018) 200:3000–7. 10.4049/jimmunol.1700407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Chauvin JM, Pagliano O, Fourcade J, Sun Z, Wang H, Sander C, et al. TIGIT and PD-1 impair tumor antigen-specific CD8(+) T cells in melanoma patients. J Clin Invest. (2015) 125:2046–58. 10.1172/JCI80445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Hong X, Wang X, Wang T, Zhang X. Correlation of T cell immunoglobulin and ITIM Domain (TIGIT) and programmed death 1 (PD-1) with clinicopathological characteristics of renal cell carcinoma may indicate potential targets for treatment. Med Sci Monit. (2018) 24:6861–72. 10.12659/MSM.910388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Hung AL, Maxwell R, Theodros D, Belcaid Z, Mathios D, Luksik AS, et al. TIGIT and PD-1 dual checkpoint blockade enhances antitumor immunity and survival in GBM. Oncoimmunology. (2018) 7:e1466769. 10.1080/2162402X.2018.1466769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Farkas AM, Audenet F, Anastos H, Galsky M, Sfakianos J, Bhardwaj N. TIM-3 and TIGIT mark Natural Killer cells susceptible to effector dysfunction in human bladder cancer. J Immunol. (2018) 200(1 Suppl.):124.14 10.1158/1538-7445.AM2018-4745 [DOI] [Google Scholar]
- 179.Macauley MS, Crocker PR, Paulson JC. Siglec-mediated regulation of immune cell function in disease. Nat Rev Immunol. (2014) 14:653–66. 10.1038/nri3737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Beatson R, Tajadura-Ortega V, Achkova D, Picco G, Tsourouktsoglou TD, Klausing S, et al. The mucin MUC1 modulates the tumor immunological microenvironment through engagement of the lectin Siglec-9. Nat Immunol. (2016) 17:1273–81. 10.1038/ni.3552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Stanczak MA, Siddiqui SS, Trefny MP, Thommen DS, Boligan KF, von Gunten S, et al. Self-associated molecular patterns mediate cancer immune evasion by engaging Siglecs on T cells. J Clin Invest. (2018) 128:4912–23. 10.1172/JCI120612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Kantarjian HM, DeAngelo DJ, Stelljes M, Martinelli G, Liedtke M, Stock W, et al. Inotuzumab ozogamicin versus standard therapy for acute lymphoblastic leukemia. N Engl J Med. (2016) 375:740–53. 10.1056/NEJMoa1509277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Amadori S, Suciu S, Selleslag D, Aversa F, Gaidano G, Musso M, et al. Gemtuzumab ozogamicin versus best supportive care in older patients with newly diagnosed acute myeloid leukemia unsuitable for intensive chemotherapy: results of the randomized phase III EORTC-GIMEMA AML-19 trial. J Clin Oncol. (2016) 34:972–9. 10.1200/JCO.2015.64.0060 [DOI] [PubMed] [Google Scholar]
- 184.Lubbers J, Rodriguez E, van Kooyk Y. Modulation of immune tolerance via siglec-sialic acid interactions. Front Immunol. (2018) 9:2807. 10.3389/fimmu.2018.02807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Nicoll G, Ni J, Liu D, Klenerman P, Munday J, Dubock S, et al. Identification and characterization of a novel siglec, siglec-7, expressed by human natural killer cells and monocytes. J Biol Chem. (1999) 274:34089–95. 10.1074/jbc.274.48.34089 [DOI] [PubMed] [Google Scholar]
- 186.Falco M, Biassoni R, Bottino C, Vitale M, Sivori S, Augugliaro R, et al. Identification and molecular cloning of p75/AIRM1, a novel member of the sialoadhesin family that functions as an inhibitory receptor in human natural killer cells. J Exp Med. (1999) 190:793–802. 10.1084/jem.190.6.793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Jandus C, Boligan KF, Chijioke O, Liu H, Dahlhaus M, Demoulins T, et al. Interactions between Siglec-7/9 receptors and ligands influence NK cell-dependent tumor immunosurveillance. J Clin Invest. (2014) 124:1810–20. 10.1172/JCI65899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Bénac O, Gaudin M, Ors M, Roy AL, Blanc HR, Soulas C, et al. Abstract 2713: Preclinical development of first-in-class antibodies targeting Siglec-9 immune checkpoint for cancer immunotherapy. Cancer Res. (2018) 78(13 Suppl.):2713 10.1158/1538-7445.AM2018-2713 [DOI] [Google Scholar]
- 189.Adams OJ, Stanczak MA, von Gunten S, Laubli H. Targeting sialic acid-Siglec interactions to reverse immune suppression in cancer. Glycobiology. (2018) 28:640–7. 10.1093/glycob/cwx108 [DOI] [PubMed] [Google Scholar]
- 190.Inoue S, Lin SL, Chang T, Wu SH, Yao CW, Chu TY, et al. Identification of free deaminated sialic acid (2-keto-3-deoxy-D-glycero-D-galacto-nononic acid) in human red blood cells and its elevated expression in fetal cord red blood cells and ovarian cancer cells. J Biol Chem. (1998) 273:27199–204. 10.1074/jbc.273.42.27199 [DOI] [PubMed] [Google Scholar]
- 191.Wang F, Xie B, Wang B, Troy FA, 2nd. LC-MS/MS glycomic analyses of free and conjugated forms of the sialic acids, Neu5Ac, Neu5Gc and KDN in human throat cancers. Glycobiology. (2015) 25:1362–74 10.1093/glycob/cwv051 [DOI] [PubMed] [Google Scholar]
- 192.Corfield AP, Myerscough N, Warren BF, Durdey P, Paraskeva C, Schauer R. Reduction of sialic acid O-acetylation in human colonic mucins in the adenoma-carcinoma sequence. Glycoconj J. (1999) 16:307–17. 10.1023/A:1007026314792 [DOI] [PubMed] [Google Scholar]
- 193.Shen Y, Kohla G, Lrhorfi AL, Sipos B, Kalthoff H, Gerwig GJ, et al. O-acetylation and de-O-acetylation of sialic acids in human colorectal carcinoma. Eur J Biochem. (2004) 271:281–90. 10.1046/j.1432-1033.2003.03927.x [DOI] [PubMed] [Google Scholar]
- 194.Castaigne S, Pautas C, Terre C, Raffoux E, Bordessoule D, Bastie JN, et al. Effect of gemtuzumab ozogamicin on survival of adult patients with de-novo acute myeloid leukaemia (ALFA-0701): a randomised, open-label, phase 3 study. Lancet. (2012) 379:1508–16. 10.1016/S0140-6736(12)60485-1 [DOI] [PubMed] [Google Scholar]
- 195.Huang CH, Liao YJ, Fan TH, Chiou TJ, Lin YH, Twu YC. A developed NK-92MI cell line with Siglec-7(neg) phenotype exhibits high and sustainable cytotoxicity against leukemia cells. Int J Mol Sci. (2018) 19:1073 10.3390/ijms19041073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Hudak JE, Canham SM, Bertozzi CR. Glycocalyx engineering reveals a Siglec-based mechanism for NK cell immunoevasion. Nat Chem Biol. (2014) 10:69–75. 10.1038/nchembio.1388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Xiao H, Woods EC, Vukojicic P, Bertozzi CR. Precision glycocalyx editing as a strategy for cancer immunotherapy. Proc Natl Acad Sci USA. (2016) 113:10304–9. 10.1073/pnas.1608069113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Avril T, North SJ, Haslam SM, Willison HJ, Crocker PR. Probing the cis interactions of the inhibitory receptor Siglec-7 with alpha2,8-disialylated ligands on natural killer cells and other leukocytes using glycan-specific antibodies and by analysis of alpha2,8-sialyltransferase gene expression. J Leukoc Biol. (2006) 80:787–96. 10.1189/jlb.1005559 [DOI] [PubMed] [Google Scholar]
- 199.Nicoll G, Avril T, Lock K, Furukawa K, Bovin N, Crocker PR. Ganglioside GD3 expression on target cells can modulate NK cell cytotoxicity via siglec-7-dependent and -independent mechanisms. Eur J Immunol. (2003) 33:1642–8. 10.1002/eji.200323693 [DOI] [PubMed] [Google Scholar]
- 200.Haas Q, Boligan KF, Jandus C, Schneider C, Simillion C, Stanczak MA, et al. Siglec-9 regulates an effector memory CD8+ T-cell subset that congregates in the melanoma tumor microenvironment. Cancer Immunol Res. (2019) 7:707–18. 10.1158/2326-6066.CIR-18-0505 [DOI] [PubMed] [Google Scholar]
- 201.Triebel F, Jitsukawa S, Baixeras E, Roman-Roman S, Genevee C, Viegas-Pequignot E, et al. LAG-3, a novel lymphocyte activation gene closely related to CD4. J Exp Med. (1990) 171:1393–405. 10.1084/jem.171.5.1393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Demeure CE, Wolfers J, Martin-Garcia N, Gaulard P, Triebel F. T Lymphocytes infiltrating various tumour types express the MHC class II ligand lymphocyte activation gene-3 (LAG-3): role of LAG-3/MHC class II interactions in cell–cell contacts. Eur J Cancer. (2001) 37:1709–18. 10.1016/S0959-8049(01)00184-8 [DOI] [PubMed] [Google Scholar]
- 203.Huang CT, Workman CJ, Flies D, Pan X, Marson AL, Zhou G, et al. Role of LAG-3 in regulatory T cells. Immunity. (2004) 21:503–13. 10.1016/j.immuni.2004.08.010 [DOI] [PubMed] [Google Scholar]
- 204.Juno JA, Stalker AT, Waruk JL, Oyugi J, Kimani M, Plummer FA, et al. Elevated expression of LAG-3, but not PD-1, is associated with impaired iNKT cytokine production during chronic HIV-1 infection and treatment. Retrovirology. (2015) 12:17. 10.1186/s12977-015-0142-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Kisielow M, Kisielow J, Capoferri-Sollami G, Karjalainen K. Expression of lymphocyte activation gene 3 (LAG-3) on B cells is induced by T cells. Eur J Immunol. (2005) 35:2081–8. 10.1002/eji.200526090 [DOI] [PubMed] [Google Scholar]
- 206.Andreae S, Buisson S, Triebel F. MHC class II signal transduction in human dendritic cells induced by a natural ligand, the LAG-3 protein (CD223). Blood. (2003) 102:2130–7. 10.1182/blood-2003-01-0273 [DOI] [PubMed] [Google Scholar]
- 207.Workman CJ, Wang Y, El Kasmi KC, Pardoll DM, Murray PJ, Drake CG, et al. LAG-3 regulates plasmacytoid dendritic cell homeostasis. J Immunol. (2009) 182:1885–91. 10.4049/jimmunol.0800185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Baixeras E, Huard B, Miossec C, Jitsukawa S, Martin M, Hercend T, et al. Characterization of the lymphocyte activation gene 3-encoded protein. A new ligand for human leukocyte antigen class II antigens. J Exp Med. (1992) 176:327–37. 10.1084/jem.176.2.327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Huard B, Prigent P, Tournier M, Bruniquel D, Triebel F. CD4/major histocompatibility complex class II interaction analyzed with CD4- and lymphocyte activation gene-3 (LAG-3)-Ig fusion proteins. Eur J Immunol. (1995) 25:2718–21. 10.1002/eji.1830250949 [DOI] [PubMed] [Google Scholar]
- 210.Xu F, Liu J, Liu D, Liu B, Wang M, Hu Z, et al. LSECtin Expressed on melanoma cells promotes tumor progression by inhibiting antitumor T-cell responses. Cancer Res. (2014) 74:3418. 10.1158/0008-5472.CAN-13-2690 [DOI] [PubMed] [Google Scholar]
- 211.Workman CJ, Vignali DAA. Negative regulation of T cell homeostasis by lymphocyte activation gene-3 (CD223). J Immunol. (2005) 174:688. 10.4049/jimmunol.174.2.688 [DOI] [PubMed] [Google Scholar]
- 212.Maçon-Lemaître L, Triebel F. The negative regulatory function of the lymphocyte-activation gene-3 co-receptor (CD223) on human T cells. Immunology. (2005) 115:170–8. 10.1111/j.1365-2567.2005.02145.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Blackburn SD, Shin H, Haining WN, Zou T, Workman CJ, Polley A, et al. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat Immunol. (2009) 10:29–37. 10.1038/ni.1679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Camisaschi C, Casati C, Rini F, Perego M, De Filippo A, Triebel F, et al. LAG-3 expression defines a subset of CD4(+)CD25(high)Foxp3(+) regulatory T cells that are expanded at tumor sites. J Immunol. (2010) 184:6545–51. 10.4049/jimmunol.0903879 [DOI] [PubMed] [Google Scholar]
- 215.He Y, Rivard CJ, Rozeboom L, Yu H, Ellison K, Kowalewski A, et al. Lymphocyte-activation gene-3, an important immune checkpoint in cancer. Cancer Sci. (2016) 107:1193–7. 10.1111/cas.12986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Woo SR, Turnis ME, Goldberg MV, Bankoti J, Selby M, Nirschl CJ, et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res Cancer Res. (2012) 72:917–27. 10.1158/0008-5472.CAN-11-1620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Miyazaki T, Dierich A, Benoist C, Mathis D. Independent modes of natural killing distinguished in mice lacking Lag3. Science. (1996) 272:405–8. 10.1126/science.272.5260.405 [DOI] [PubMed] [Google Scholar]
- 218.Huard B, Tournier M, Triebel F. LAG-3 does not define a specific mode of natural killing in human. Immunol Lett. (1998) 61:109–12. 10.1016/S0165-2478(97)00170-3 [DOI] [PubMed] [Google Scholar]
- 219.Taborda NA, Hernandez JC, Lajoie J, Juno JA, Kimani J, Rugeles MT, et al. Short communication: low expression of activation and inhibitory molecules on NK cells and CD4(+) T cells is associated with viral control. AIDS Res Hum Retroviruses. (2015) 31:636–40. 10.1089/aid.2014.0325 [DOI] [PubMed] [Google Scholar]
- 220.Romagnani C, Babic M. NK/DC crosstalk in immunosurveillance: a broken relationship caused by WASP-deficiency. Eur J Immunol. (2014) 44:958–61. 10.1002/eji.201444514 [DOI] [PubMed] [Google Scholar]
- 221.Catucci M, Zanoni I, Draghici E, Bosticardo M, Castiello MC, Venturini M, et al. Wiskott-Aldrich syndrome protein deficiency in natural killer and dendritic cells affects antitumor immunity. Eur J Immunol. (2014) 44:1039–45. 10.1002/eji.201343935 [DOI] [PubMed] [Google Scholar]
- 222.Kritikou JS, Dahlberg CI, Baptista MA, Wagner AK, Banerjee PP, Gwalani LA, et al. IL-2 in the tumor microenvironment is necessary for Wiskott-Aldrich syndrome protein deficient NK cells to respond to tumors in vivo. Sci Rep. (2016) 6:30636. 10.1038/srep30636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Byun HJ, Jung WW, Lee DS, Kim S, Kim SJ, Park CG, et al. Proliferation of activated CD1d-restricted NKT cells is down-modulated by lymphocyte activation gene-3 signaling via cell cycle arrest in S phase. Cell Biol Int. (2007) 31:257–62. 10.1016/j.cellbi.2006.11.002 [DOI] [PubMed] [Google Scholar]
- 224.Sierro S, Romero P, Speiser DE. The CD4-like molecule LAG-3, biology and therapeutic applications. Expert Opin Therap Targets. (2011) 15:91–101. 10.1517/14712598.2011.540563 [DOI] [PubMed] [Google Scholar]
- 225.Brignone C, Grygar C, Marcu M, Schakel K, Triebel F. A soluble form of lymphocyte activation gene-3 (IMP321) induces activation of a large range of human effector cytotoxic cells. J Immunol. (2007) 179:4202–11. 10.4049/jimmunol.179.6.4202 [DOI] [PubMed] [Google Scholar]
- 226.Brignone C, Escudier B, Grygar C, Marcu M, Triebel F. A phase I pharmacokinetic and biological correlative study of IMP321, a novel MHC class II agonist, in patients with advanced renal cell carcinoma. Clin Cancer Res. (2009) 15:6225–31. 10.1158/1078-0432.CCR-09-0068 [DOI] [PubMed] [Google Scholar]
- 227.Brignone C, Gutierrez M, Mefti F, Brain E, Jarcau R, Cvitkovic F, et al. First-line chemoimmunotherapy in metastatic breast carcinoma: combination of paclitaxel and IMP321 (LAG-3Ig) enhances immune responses and antitumor activity. J Transl Med. (2010) 8:71. 10.1186/1479-5876-8-71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Jin J, Ahn Y-O, Kim TM, Keam B, Kim D-W, Heo DS. The CD56bright CD62L+ NKG2A+ immature cell subset is dominantly expanded in human cytokine-induced memory-like NK cells. bioRxiv. (2018) 2018:405134 10.1101/405134 [DOI] [Google Scholar]
- 229.Du W, Yang M, Turner A, Xu C, Ferris RL, Huang J, et al. TIM-3 as a target for cancer immunotherapy and mechanisms of action. Int J Mol Sci. (2017) 18:E645. 10.3390/ijms18030645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.He Y, Cao J, Zhao C, Li X, Zhou C, Hirsch FR. TIM-3, a promising target for cancer immunotherapy. Onco Targets Ther. (2018) 11:7005–9. 10.2147/OTT.S170385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Folgiero V, Cifaldi L, Pira GL, Goffredo BM, Vinti L, Locatelli F. TIM-3/Gal-9 interaction induces IFNγ-dependent IDO1 expression in acute myeloid leukemia blast cells. J Hematol Oncol. (2015) 8:36. 10.1186/s13045-015-0134-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Li H, Wu K, Tao K, Chen L, Zheng Q, Lu X, et al. TIM-3/galectin-9 signaling pathway mediates T-cell dysfunction and predicts poor prognosis in patients with hepatitis B virus-associated hepatocellular carcinoma. Hepatology. (2012) 56:1342–51. 10.1002/hep.25777 [DOI] [PubMed] [Google Scholar]
- 233.Ji P, Chen D, Bian J, Xia R, Song X, Wen W, et al. Upregulation of TIM-3 on CD4+ tumor infiltrating lymphocytes predicts poor prognosis in human non-small-cell lung cancer. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi. (2015) 31:808–11. [PubMed] [Google Scholar]
- 234.Nebbia G, Peppa D, Schurich A, Khanna P, Singh HD, Cheng Y, et al. Upregulation of the TIM-3/galectin-9 pathway of T cell exhaustion in chronic hepatitis B virus infection. PLoS ONE. (2012) 7:e47648. 10.1371/journal.pone.0047648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Gao X, Zhu Y, Li G, Huang H, Zhang G, Wang F, et al. TIM-3 expression characterizes regulatory T cells in tumor tissues and is associated with lung cancer progression. PLoS ONE. (2012) 7:e30676. 10.1371/journal.pone.0030676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Golden-Mason L, McMahan RH, Strong M, Reisdorph R, Mahaffey S, Palmer BE, et al. Galectin-9 functionally impairs natural killer cells in humans and mice. J Virol. (2013) 87:4835. 10.1128/JVI.01085-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Finney CA, Ayi K, Wasmuth JD, Sheth PM, Kaul R, Loutfy M, et al. HIV infection deregulates TIM-3 expression on innate cells: combination antiretroviral therapy results in partial restoration. J Acquir Immune Defic Syndr. (1999). (2013) 63:161–7. 10.1097/QAI.0b013e318285cf13 [DOI] [PubMed] [Google Scholar]
- 238.Xu L, Huang Y, Tan L, Yu W, Chen D, Lu C, et al. Increased TIM-3 expression in peripheral NK cells predicts a poorer prognosis and TIM-3 blockade improves NK cell-mediated cytotoxicity in human lung adenocarcinoma. Int Immunopharmacol. (2015) 29:635–41. 10.1016/j.intimp.2015.09.017 [DOI] [PubMed] [Google Scholar]
- 239.Ngiow SF, von Scheidt B, Akiba H, Yagita H, Teng MW, Smyth MJ. Anti-TIM3 antibody promotes T cell IFN-gamma-mediated antitumor immunity and suppresses established tumors. Cancer Res. (2011) 71:3540–51. 10.1158/0008-5472.CAN-11-0096 [DOI] [PubMed] [Google Scholar]
- 240.Wu W, Shi Y, Li S, Zhang Y, Liu Y, Wu Y, et al. Blockade of TIM-3 signaling restores the virus-specific CD8(+) T-cell response in patients with chronic hepatitis B. Eur J Immunol. (2012) 42:1180–91. 10.1002/eji.201141852 [DOI] [PubMed] [Google Scholar]
- 241.Gallois A, Silva I, Osman I, Bhardwaj N. Reversal of natural killer cell exhaustion by TIM-3 blockade. Oncoimmunology. (2014) 3:e946365. 10.4161/21624011.2014.946365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.da Silva IP, Gallois A, Jimenez-Baranda S, Khan S, Anderson AC, Kuchroo VK, et al. Reversal of NK-cell exhaustion in advanced melanoma by TIM-3 blockade. Cancer Immunol Res. (2014) 2:410–22. 10.1158/2326-6066.CIR-13-0171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Jin HT, Anderson AC, Tan WG, West EE, Ha SJ, Araki K, et al. Cooperation of TIM-3 and PD-1 in CD8 T-cell exhaustion during chronic viral infection. Proc Natl Acad Sci USA. (2010) 107:14733–8. 10.1073/pnas.1009731107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Zhou Q, Munger ME, Veenstra RG, Weigel BJ, Hirashima M, Munn DH, et al. Coexpression of TIM-3 and PD-1 identifies a CD8+ T-cell exhaustion phenotype in mice with disseminated acute myelogenous leukemia. Blood. (2011) 117:4501–10. 10.1182/blood-2010-10-310425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Fourcade J, Sun Z, Benallaoua M, Guillaume P, Luescher IF, Sander C, et al. Upregulation of TIM-3 and PD-1 expression is associated with tumor antigen-specific CD8+ T cell dysfunction in melanoma patients. J Exp Med. (2010) 207:2175–86. 10.1084/jem.20100637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Linedale R, Schmidt C, King BT, Ganko AG, Simpson F, Panizza BJ, et al. Elevated frequencies of CD8 T cells expressing PD-1, CTLA-4 and TIM-3 within tumour from perineural squamous cell carcinoma patients. PLoS ONE. (2017) 12:e0175755. 10.1371/journal.pone.0175755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Liu F, Zeng G, Zhou S, He X, Sun N, Zhu X, et al. Blocking TIM-3 or/and PD-1 reverses dysfunction of tumor-infiltrating lymphocytes in HBV-related hepatocellular carcinoma. Bull Cancer. (2018) 105:493–501. 10.1016/j.bulcan.2018.01.018 [DOI] [PubMed] [Google Scholar]
- 248.Shen H, Sheng H, Lu JJ, Feng C, Yao M, Pan H, et al. Expression and distribution of programmed death receptor 1 and T cell immunoglobulin mucin 3 in breast cancer microenvironment and its relationship with clinicopathological features. Zhonghua Yi Xue Za Zhi. (2018) 98:1352–7. 10.3760/cma.j.issn.0376-2491.2018.17.014 [DOI] [PubMed] [Google Scholar]
- 249.Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, Anderson AC. Targeting TIM-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J Exp Med. (2010) 207:2187–94. 10.1084/jem.20100643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Liu J, Zhang S, Hu Y, Yang Z, Li J, Liu X, et al. Targeting PD-1 and TIM-3 pathways to reverse CD8 T-cell exhaustion and enhance ex vivo T-cell responses to autologous dendritic/tumor vaccines. J Immunother. (2016) 39:171–80. 10.1097/CJI.0000000000000122 [DOI] [PubMed] [Google Scholar]
- 251.Gleason MK, Lenvik TR, McCullar V, Felices M, O'Brien MS, Cooley SA, et al. TIM-3 is an inducible human natural killer cell receptor that enhances interferon gamma production in response to galectin-9. Blood. (2012) 119:3064–72. 10.1182/blood-2011-06-360321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Ndhlovu LC, Lopez-Vergès S, Barbour JD, Jones RB, Jha AR, Long BR, et al. TIM-3 marks human natural killer cell maturation and suppresses cell-mediated cytotoxicity. Blood. (2012) 119:3734–43. 10.1182/blood-2011-11-392951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Wang Z, Zhu J, Gu H, Yuan Y, Zhang B, Zhu D, et al. The clinical significance of abnormal TIM-3 expression on NK cells from patients with gastric cancer. Immunol Invest. (2015) 44:578–89. 10.3109/08820139.2015.1052145 [DOI] [PubMed] [Google Scholar]
- 254.Komita H, Koido S, Hayashi K, Kan S, Ito M, Kamata Y, et al. Expression of immune checkpoint molecules of T cell immunoglobulin and mucin protein 3/galectin-9 for NK cell suppression in human gastrointestinal stromal tumors. Oncol Rep. (2015) 34:2099–105. 10.3892/or.2015.4149 [DOI] [PubMed] [Google Scholar]
- 255.Seo H, Jeon I, Kim BS, Park M, Bae EA, Song B, et al. IL-21-mediated reversal of NK cell exhaustion facilitates anti-tumour immunity in MHC class I-deficient tumours. Nat Commun. (2017) 8:15776. 10.1038/ncomms15776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Han G, Chen G, Shen B, Li Y. TIM-3: an activation marker and activation limiter of innate immune cells. Front Immunol. (2013) 4:449. 10.3389/fimmu.2013.00449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Van Audenaerde JRM, De Waele J, Marcq E, Van Loenhout J, Lion E, Van den Bergh JMJ, et al. Interleukin-15 stimulates natural killer cell-mediated killing of both human pancreatic cancer and stellate cells. Oncotarget. (2017) 8:56968–79. 10.18632/oncotarget.18185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Jost S, Moreno-Nieves UY, Garcia-Beltran WF, Rands K, Reardon J, Toth I, et al. Dysregulated TIM-3 expression on natural killer cells is associated with increased Galectin-9 levels in HIV-1 infection. Retrovirology. (2013) 10:74. 10.1186/1742-4690-10-74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Sun J, Yang M, Ban Y, Gao W, Song B, Wang Y, et al. TIM-3 is upregulated in NK cells during early pregnancy and inhibits NK cytotoxicity toward trophoblast in galectin-9 dependent pathway. PLoS ONE. (2016) 11:e0147186. 10.1371/journal.pone.0147186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Ju Y, Hou N, Meng J, Wang X, Zhang X, Zhao D, et al. T cell immunoglobulin- and mucin-domain-containing molecule-3 (TIM-3) mediates natural killer cell suppression in chronic hepatitis B. J Hepatol. (2010) 52:322–9. 10.1016/j.jhep.2009.12.005 [DOI] [PubMed] [Google Scholar]
- 261.Hou H, Liu W, Wu S, Lu Y, Peng J, Zhu Y, et al. TIM-3 negatively mediates natural killer cell function in LPS-induced endotoxic shock. PLoS ONE. (2014) 9:e110585. 10.1371/journal.pone.0110585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Wright GJ, Cherwinski H, Foster-Cuevas M, Brooke G, Puklavec MJ, Bigler M, et al. Characterization of the CD200 receptor family in mice and humans and their interactions with CD200. J Immunol. (2003) 171:3034–46. 10.4049/jimmunol.171.6.3034 [DOI] [PubMed] [Google Scholar]
- 263.Rijkers ES, de Ruiter T, Baridi A, Veninga H, Hoek RM, Meyaard L. The inhibitory CD200R is differentially expressed on human and mouse T and B lymphocytes. Mol Immunol. (2008) 45:1126–35. 10.1016/j.molimm.2007.07.013 [DOI] [PubMed] [Google Scholar]
- 264.Coles SJ, Wang EC, Man S, Hills RK, Burnett AK, Tonks A, et al. CD200 expression suppresses natural killer cell function and directly inhibits patient anti-tumor response in acute myeloid leukemia. Leukemia. (2011) 25:792–9. 10.1038/leu.2011.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Rygiel TP, Meyaard L. CD200R signaling in tumor tolerance and inflammation: a tricky balance. Curr Opin Immunol. (2012) 24:233–8. 10.1016/j.coi.2012.01.002 [DOI] [PubMed] [Google Scholar]
- 266.Moreaux J, Hose D, Reme T, Jourdan E, Hundemer M, Legouffe E, et al. CD200 is a new prognostic factor in multiple myeloma. Blood. (2006) 108:4194–7. 10.1182/blood-2006-06-029355 [DOI] [PubMed] [Google Scholar]
- 267.Tonks A, Hills R, White P, Rosie B, Mills KI, Burnett AK, et al. CD200 as a prognostic factor in acute myeloid leukaemia. Leukemia. (2007) 21:566–8. 10.1038/sj.leu.2404559 [DOI] [PubMed] [Google Scholar]
- 268.McWhirter JR, Kretz-Rommel A, Saven A, Maruyama T, Potter KN, Mockridge CI, et al. Antibodies selected from combinatorial libraries block a tumor antigen that plays a key role in immunomodulation. Proc Natl Acad Sci USA. (2006) 103:1041–6. 10.1073/pnas.0510081103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Brunetti L, Di Noto R, Abate G, Gorrese M, Gravetti A, Raia M, et al. CD200/OX2, a cell surface molecule with immuno-regulatory function, is consistently expressed on hairy cell leukaemia neoplastic cells. Br J Haematol. (2009) 145:665–7. 10.1111/j.1365-2141.2009.07644.x [DOI] [PubMed] [Google Scholar]
- 270.Siva A, Xin H, Qin F, Oltean D, Bowdish KS, Kretz-Rommel A. Immune modulation by melanoma and ovarian tumor cells through expression of the immunosuppressive molecule CD200. Cancer Immunol Immunother. (2008) 57:987–96. 10.1007/s00262-007-0429-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Moreaux J, Veyrune JL, Reme T, De Vos J, Klein B. CD200: a putative therapeutic target in cancer. Biochem Biophys Res Commun. (2008) 366:117–22. 10.1016/j.bbrc.2007.11.103 [DOI] [PubMed] [Google Scholar]
- 272.Liu JQ, Talebian F, Wu L, Liu Z, Li MS, Wu L, et al. A critical role for CD200R signaling in limiting the growth and metastasis of CD200+ melanoma. J Immunol. (2016) 197:1489–97. 10.4049/jimmunol.1600052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Bisgin A, Meng WJ, Adell G, Sun XF. Interaction of CD200 overexpression on tumor cells with CD200R1 overexpression on stromal cells: an escape from the host immune response in rectal cancer patients. J Oncol. (2019) 2019:5689464. 10.1155/2019/5689464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Rexin P, Tauchert A, Hanze J, Heers H, Schmidt A, Hofmann R, et al. The immune checkpoint molecule CD200 is associated with tumor grading and metastasis in bladder cancer. Anti Cancer Res. (2018) 38:2749–54. 10.21873/anticanres.12517 [DOI] [PubMed] [Google Scholar]
- 275.Stumpfova M, Ratner D, Desciak EB, Eliezri YD, Owens DM. The immunosuppressive surface ligand CD200 augments the metastatic capacity of squamous cell carcinoma. Cancer Res. (2010) 70:2962–72 10.1158/0008-5472.CAN-09-4380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Gorczynski RM, Chen Z, Diao J, Khatri I, Wong K, Yu K, et al. Breast cancer cell CD200 expression regulates immune response to EMT6 tumor cells in mice. Breast Cancer Res Treat. (2010) 123:405–15. 10.1007/s10549-009-0667-8 [DOI] [PubMed] [Google Scholar]
- 277.Rygiel TP, Karnam G, Goverse G, van der Marel AP, Greuter MJ, van Schaarenburg RA, et al. CD200-CD200R signaling suppresses anti-tumor responses independently of CD200 expression on the tumor. Oncogene. (2012) 31:2979–88. 10.1038/onc.2011.477 [DOI] [PubMed] [Google Scholar]
- 278.Pilch Z, Tonecka K, Skorzynski M, Sas Z, Braniewska A, Kryczka T, et al. The pro-tumor effect of CD200 expression is not mimicked by agonistic CD200R antibodies. PLoS ONE. (2019) 14:e0210796 10.1371/journal.pone.0210796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Mahadevan D, Lanasa MC, Whelden M, Faas SJ, Ulery TL, Kukreja A, et al. First-in-human phase I dose escalation study of a humanized anti-CD200 antibody (samalizumab) in patients with advanced stage B cell chronic lymphocytic leukemia (B-CLL) or multiple myeloma (MM). Blood. (2010) 116:2465 10.1182/blood.V116.21.2465.2465 [DOI] [Google Scholar]
- 280.Atfy M, Ebian H, Mostafa S, Atteia HH. CD200 suppresses the natural killer cells and decreased its activity in acute myeloid leukemia patients. J Leukemia. (2015) 3:190 10.4172/2329-6917.1000190 [DOI] [Google Scholar]
- 281.Oldenborg P-A. CD47: a cell surface glycoprotein which regulates multiple functions of hematopoietic cells in health and disease. ISRN Hematol. (2013) 2013:614619. 10.1155/2013/614619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Brown E, Hooper L, Ho T, Gresham H. Integrin-associated protein: a 50-kD plasma membrane antigen physically and functionally associated with integrins. J Cell Biol. (1990) 111(6 Pt 1):2785–94. 10.1083/jcb.111.6.2785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Barclay AN, Van den Berg TK. The interaction between signal regulatory protein alpha (SIRPalpha) and CD47: structure, function, and therapeutic target. Annu Rev Immunol. (2014) 32:25–50. 10.1146/annurev-immunol-032713-120142 [DOI] [PubMed] [Google Scholar]
- 284.Kaur S, Soto-Pantoja DR, Stein EV, Liu C, Elkahloun AG, Pendrak ML, et al. Thrombospondin-1 signaling through CD47 inhibits self-renewal by regulating c-Myc and other stem cell transcription factors. Sci Rep. (2013) 3:1673. 10.1038/srep01673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Gao Q, Chen K, Gao L, Zheng Y, Yang Y-G. Thrombospondin-1 signaling through CD47 inhibits cell cycle progression and induces senescence in endothelial cells. Cell Death Dis. (2016) 7:e2368. 10.1038/cddis.2016.155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Matlung HL, Szilagyi K, Barclay NA, van den Berg TK. The CD47-SIRPalpha signaling axis as an innate immune checkpoint in cancer. Immunol Rev. (2017) 276:145–64. 10.1111/imr.12527 [DOI] [PubMed] [Google Scholar]
- 287.Kaur S, Chang T, Singh SP, Lim L, Mannan P, Garfield SH, et al. CD47 signaling regulates the immunosuppressive activity of VEGF in T cells. J Immunol. (2014) 193:3914–24. 10.4049/jimmunol.1303116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Soto-Pantoja DR, Terabe M, Ghosh A, Ridnour LA, DeGraff WG, Wink DA, et al. CD47 in the tumor microenvironment limits cooperation between antitumor T-cell immunity and radiotherapy. Cancer Res. (2014) 74:6771–83. 10.1158/0008-5472.CAN-14-0037-T [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Kim MJ, Lee JC, Lee JJ, Kim S, Lee SG, Park SW, et al. Association of CD47 with natural killer cell-mediated cytotoxicity of head-and-neck squamous cell carcinoma lines. Tumour Biol. (2008) 29:28–34. 10.1159/000132568 [DOI] [PubMed] [Google Scholar]
- 290.Liu J, Wang L, Zhao F, Tseng S, Narayanan C, Shura L, et al. Pre-Clinical development of a humanized anti-CD47 antibody with anti-cancer therapeutic potential. PLoS ONE. (2015) 10:e0137345. 10.1371/journal.pone.0137345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Weng TY, Huang SS, Yen MC, Lin CC, Chen YL, Lin CM, et al. A novel cancer therapeutic using thrombospondin 1 in dendritic cells. Mol Ther. (2014) 22:292–302. 10.1038/mt.2013.236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Liu X, Pu Y, Cron K, Deng L, Kline J, Frazier WA, et al. CD47 blockade triggers T cell-mediated destruction of immunogenic tumors. Nat Med. (2015) 21:1209–15. 10.1038/nm.3931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Nath PR, Gangaplara A, Pal-Nath D, Mandal A, Maric D, Sipes JM, et al. CD47 Expression in natural killer cells regulates homeostasis and modulates immune response to lymphocytic choriomeningitis virus. Front Immunol. (2018) 9:2985. 10.3389/fimmu.2018.02985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Yanagita T, Murata Y, Tanaka D, Motegi SI, Arai E, Daniwijaya EW, et al. Anti-SIRPalpha antibodies as a potential new tool for cancer immunotherapy. JCI Insight. (2017) 2:e89140. 10.1172/jci.insight.89140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Pierson BA, Gupta K, Hu WS, Miller JS. Human natural killer cell expansion is regulated by thrombospondin-mediated activation of transforming growth factor-beta 1 and independent accessory cell-derived contact and soluble factors. Blood. (1996) 87:180–9. 10.1182/blood.V87.1.180.bloodjournal871180 [DOI] [PubMed] [Google Scholar]
- 296.Lian S, Xie R, Ye Y, Lu Y, Cheng Y, Xie X, et al. Dual blockage of both PD-L1 and CD47 enhances immunotherapy against circulating tumor cells. Sci Rep. (2019) 9:4532. 10.1038/s41598-019-40241-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Janakiram M, Shah UA, Liu W, Zhao A, Schoenberg MP, Zang X. The third group of the B7-CD28 immune checkpoint family: HHLA2, TMIGD2, B7x, and B7-H3. Immunol Rev. (2017) 276:26–39. 10.1111/imr.12521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Buchbinder EI, Desai A. CTLA-4 and PD-1 pathways: similarities, differences, and implications of their inhibition. Am J Clin Oncol. (2016) 39:98–106. 10.1097/COC.0000000000000239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Stojanovic A, Fiegler N, Brunner-Weinzierl M, Cerwenka A. CTLA-4 is expressed by activated mouse NK cells and inhibits NK Cell IFN-gamma production in response to mature dendritic cells. J Immunol. (2014) 192:4184–91. 10.4049/jimmunol.1302091 [DOI] [PubMed] [Google Scholar]
- 300.Terme M, Ullrich E, Aymeric L, Meinhardt K, Coudert JD, Desbois M, et al. Cancer-induced immunosuppression: IL-18-elicited immunoablative NK cells. Cancer Res. (2012) 72:2757–67. 10.1158/0008-5472.CAN-11-3379 [DOI] [PubMed] [Google Scholar]
- 301.Tivol EA, Borriello F, Schweitzer AN, Lynch WP, Bluestone JA, Sharpe AH. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity. (1995) 3:541–7. 10.1016/1074-7613(95)90125-6 [DOI] [PubMed] [Google Scholar]
- 302.Khattri R, Auger JA, Griffin MD, Sharpe AH, Bluestone JA. Lymphoproliferative disorder in CTLA-4 knockout mice is characterized by CD28-regulated activation of Th2 responses. J Immunol. (1999) 162:5784–91. [PubMed] [Google Scholar]
- 303.Kosmaczewska A, Ciszak L, Bocko D, Frydecka I. Expression and functional significance of CTLA-4, a negative regulator of T cell activation. Arch Immunol Ther Exp. (2001) 49:39–46. [PubMed] [Google Scholar]
- 304.Bluestone JA. Is CTLA-4 a master switch for peripheral T cell tolerance? J Immunol. (1997) 158:1989–93. [PubMed] [Google Scholar]
- 305.Scheipers P, Reiser H. Role of the CTLA-4 receptor in t cell activation and immunity. Immunol Res. (1998) 18:103–15. 10.1007/BF02788753 [DOI] [PubMed] [Google Scholar]
- 306.Fife BT, Bluestone JA. Control of peripheral T-cell tolerance and autoimmunity via the CTLA-4 and PD-1 pathways. Immunol Rev. (2008) 224:166–82. 10.1111/j.1600-065X.2008.00662.x [DOI] [PubMed] [Google Scholar]
- 307.Bour-Jordan H, Esensten JH, Martinez-Llordella M, Penaranda C, Stumpf M, Bluestone JA. Intrinsic and extrinsic control of peripheral T-cell tolerance by costimulatory molecules of the CD28/ B7 family. Immunol Rev. (2011) 241:180–205. 10.1111/j.1600-065X.2011.01011.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Yi JS, Cox MA, Zajac AJ. T-cell exhaustion: characteristics, causes and conversion. Immunology. (2010) 129:474–81. 10.1111/j.1365-2567.2010.03255.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Seidel JA, Otsuka A, Kabashima K. Anti-PD-1 and anti-CTLA-4 therapies in cancer: mechanisms of action, efficacy, and limitations. Front Oncol. (2018) 8:86. 10.3389/fonc.2018.00086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Nandi D, Gross JA, Allison JP. CD28-mediated costimulation is necessary for optimal proliferation of murine NK cells. J Immunol. (1994) 152:3361–9. [PubMed] [Google Scholar]
- 311.Geldhof AB, Moser M, Lespagnard L, Thielemans K, De Baetselier P. Interleukin-12–activated natural killer cells recognize B7 costimulatory molecules on tumor cells and autologous dendritic cells. Blood. (1998) 91:196. 10.1182/blood.V91.1.196 [DOI] [PubMed] [Google Scholar]
- 312.Yeh KY, Pulaski BA, Woods ML, McAdam AJ, Gaspari AA, Frelinger JG, et al. B7–1 enhances natural killer cell-mediated cytotoxicity and inhibits tumor growth of a poorly immunogenic murine carcinoma. Cell Immunol. (1995) 165:217–24. 10.1006/cimm.1995.1208 [DOI] [PubMed] [Google Scholar]
- 313.Chambers BJ, Salcedo M, Ljunggren HG. Triggering of natural killer cells by the costimulatory molecule CD80 (B7–1). Immunity. (1996) 5:311–7. 10.1016/S1074-7613(00)80257-5 [DOI] [PubMed] [Google Scholar]
- 314.Wilson JL, Charo J, Martin-Fontecha A, Dellabona P, Casorati G, Chambers BJ, et al. NK cell triggering by the human costimulatory molecules CD80 and CD86. J Immunol. (1999) 163:4207–12. [PubMed] [Google Scholar]
- 315.Luque I, Reyburn H, Strominger JL. Expression of the CD80 and CD86 molecules enhances cytotoxicity by human natural killer cells. Hum Immunol. (2000) 61:721–8. 10.1016/S0198-8859(00)00136-1 [DOI] [PubMed] [Google Scholar]
- 316.Kelly JM, Takeda K, Darcy PK, Yagita H, Smyth MJ. A role for IFN-γ in Primary and secondary immunity generated by NK cell-sensitive tumor-expressing CD80 in vivo. J Immunol. (2002) 168:4472. 10.4049/jimmunol.168.9.4472 [DOI] [PubMed] [Google Scholar]
- 317.Lang S, Vujanovic NL, Wollenberg B, Whiteside TL. Absence of B7.1-CD28/CTLA-4-mediated co-stimulation in human NK cells. Eur J Immunol. (1998) 28:780–6. 10.1002/(SICI)1521-4141(199803)28:03<780::AID-IMMU780>3.0.CO;2-8 [DOI] [PubMed] [Google Scholar]
- 318.Takahashi T, Tagami T, Yamazaki S, Uede T, Shimizu J, Sakaguchi N, et al. Immunologic self-tolerance maintained by CD25(+)CD4(+) regulatory T cells constitutively expressing cytotoxic T lymphocyte-associated antigen 4. J Exp Med. (2000) 192:303–10. 10.1084/jem.192.2.303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Jie HB, Schuler PJ, Lee SC, Srivastava RM, Argiris A, Ferrone S, et al. CTLA-4(+) regulatory T cells increased in cetuximab-treated head and neck cancer patients suppress NK cell cytotoxicity and correlate with poor prognosis. Cancer Res. (2015) 75:2200–10. 10.1158/0008-5472.CAN-14-2788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Simpson TR, Li F, Montalvo-Ortiz W, Sepulveda MA, Bergerhoff K, Arce F, et al. Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J Exp Med. (2013) 210:1695–710. 10.1084/jem.20130579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Romano E, Kusio-Kobialka M, Foukas PG, Baumgaertner P, Meyer C, Ballabeni P, et al. Ipilimumab-dependent cell-mediated cytotoxicity of regulatory T cells ex vivo by nonclassical monocytes in melanoma patients. Proc Natl Acad Sci USA. (2015) 112:6140–5. 10.1073/pnas.1417320112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Lotze MT, Matory YL, Ettinghausen SE, Rayner AA, Sharrow SO, Seipp CA, et al. In vivo administration of purified human interleukin 2. II. Half life, immunologic effects, and expansion of peripheral lymphoid cells in vivo with recombinant IL 2. J Immunol. (1985) 135:2865–75. [PubMed] [Google Scholar]
- 323.Hannani D, Vetizou M, Enot D, Rusakiewicz S, Chaput N, Klatzmann D, et al. Anticancer immunotherapy by CTLA-4 blockade: obligatory contribution of IL-2 receptors and negative prognostic impact of soluble CD25. Cell Res. (2015) 25:208–24. 10.1038/cr.2015.3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Krummel MF, Allison JP. CTLA-4 engagement inhibits IL-2 accumulation and cell cycle progression upon activation of resting T cells. J Exp Med. (1996) 183:2533–40. 10.1084/jem.183.6.2533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325.Kerdiles Y, Ugolini S, Vivier E. T cell regulation of natural killer cells. J Exp Med. (2013) 210:1065–8. 10.1084/jem.20130960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.Tallerico R, Cristiani CM, Staaf E, Garofalo C, Sottile R, Capone M, et al. IL-15, TIM-3 and NK cells subsets predict responsiveness to anti-CTLA-4 treatment in melanoma patients. Oncoimmunology. (2017) 6:e1261242. 10.1080/2162402X.2016.1261242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Frankel TL, Burns W, Royal RE. Regression of pancreatic cancer from ipilimumab (anti-CTLA-4) mediated by an NK-cell subset (CD56brightCD16dim). J Am Coll Surg. (2009) 209:S120 10.1016/j.jamcollsurg.2009.06.299 [DOI] [Google Scholar]
- 328.Laurent S, Queirolo P, Boero S, Salvi S, Piccioli P, Boccardo S, et al. The engagement of CTLA-4 on primary melanoma cell lines induces antibody-dependent cellular cytotoxicity and TNF-α production. J Transl Med. (2013) 11:108. 10.1186/1479-5876-11-108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329.Kohlhapp FJ, Broucek JR, Hughes T, Huelsmann EJ, Lusciks J, Zayas JP, et al. NK cells and CD8+ T cells cooperate to improve therapeutic responses in melanoma treated with interleukin-2 (IL-2) and CTLA-4 blockade. J Immunother Cancer. (2015) 3:18. 10.1186/s40425-015-0063-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.Pires Da Silva IED, Gallois A, Lui KP, Shapiro RL, Pavlick AC, Bhardwaj N, et al. The effect of ipilimumab on natural killer cells identifies the subset of advanced melanoma patients with clinical response. J Clin Oncol. (2015) 33(15_suppl:9065 10.1200/jco.2015.33.15_suppl.9065 [DOI] [Google Scholar]
- 331.Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol. (2008) 26:677–704. 10.1146/annurev.immunol.26.021607.090331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Golden-Mason L, Klarquist J, Wahed AS, Rosen HR. Cutting edge: programmed death-1 expression is increased on immunocytes in chronic hepatitis C virus and predicts failure of response to antiviral therapy: race-dependent differences. J Immunol. (2008) 180:3637–41. 10.4049/jimmunol.180.6.3637 [DOI] [PubMed] [Google Scholar]
- 333.Chang WS, Kim JY, Kim YJ, Kim YS, Lee JM, Azuma M, et al. Cutting edge: programmed death-1/programmed death ligand 1 interaction regulates the induction and maintenance of invariant NKT cell anergy. J Immunol. (2008) 181:6707–10. 10.4049/jimmunol.181.10.6707 [DOI] [PubMed] [Google Scholar]
- 334.Taylor S, Huang Y, Mallett G, Stathopoulou C, Felizardo TC, Sun MA, et al. PD-1 regulates KLRG1(+) group 2 innate lymphoid cells. J Exp Med. (2017) 214:1663–78. 10.1084/jem.20161653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.Pesce S, Greppi M, Tabellini G, Rampinelli F, Parolini S, Olive D, et al. Identification of a subset of human natural killer cells expressing high levels of programmed death 1: a phenotypic and functional characterization. J Allergy Clin Immunol. (2017) 139:335–46.e3. 10.1016/j.jaci.2016.04.025 [DOI] [PubMed] [Google Scholar]
- 336.Mariotti FR, Petrini S, Ingegnere T, Tumino N, Besi F, Scordamaglia F, et al. PD-1 in human NK cells: evidence of cytoplasmic mRNA and protein expression. Oncoimmunology. (2019) 8:1557030. 10.1080/2162402X.2018.1557030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337.Beldi-Ferchiou A, Lambert M, Dogniaux S, Vely F, Vivier E, Olive D, et al. PD-1 mediates functional exhaustion of activated NK cells in patients with Kaposi sarcoma. Oncotarget. (2016) 7:72961–77. 10.18632/oncotarget.12150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338.MacFarlane AWT, Jillab M, Plimack ER, Hudes GR, Uzzo RG, Litwin S, et al. PD-1 expression on peripheral blood cells increases with stage in renal cell carcinoma patients and is rapidly reduced after surgical tumor resection. Cancer Immunol Res. (2014) 2:320–31. 10.1158/2326-6066.CIR-13-0133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339.Benson DM, Jr, Bakan CE, Mishra A, Hofmeister CC, Efebera Y, Becknell B, et al. The PD-1/PD-L1 axis modulates the natural killer cell versus multiple myeloma effect: a therapeutic target for CT-011, a novel monoclonal anti-PD-1 antibody. Blood. (2010) 116:2286–94. 10.1182/blood-2010-02-271874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 340.Giuliani M, Janji B, Berchem G. Activation of NK cells and disruption of PD-L1/PD-1 axis: two different ways for lenalidomide to block myeloma progression. Oncotarget. (2017) 8:24031–44. 10.18632/oncotarget.15234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341.Liu Y, Cheng Y, Xu Y, Wang Z, Du X, Li C, et al. Increased expression of programmed cell death protein 1 on NK cells inhibits NK-cell-mediated anti-tumor function and indicates poor prognosis in digestive cancers. Oncogene. (2017) 36:6143–53. 10.1038/onc.2017.209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342.Alvarez IB, Pasquinelli V, Jurado JO, Abbate E, Musella RM, de la Barrera SS, et al. Role played by the programmed death-1-programmed death ligand pathway during innate immunity against Mycobacterium tuberculosis. J Infect Dis. (2010) 202:524–32. 10.1086/654932 [DOI] [PubMed] [Google Scholar]
- 343.Norris S, Coleman A, Kuri-Cervantes L, Bower M, Nelson M, Goodier MR. PD-1 expression on natural killer cells and CD8(+) T cells during chronic HIV-1 infection. Viral Immunol. (2012) 25:329–32. 10.1089/vim.2011.0096 [DOI] [PubMed] [Google Scholar]
- 344.Della Chiesa M, Pesce S, Muccio L, Carlomagno S, Sivori S, Moretta A, et al. Features of memory-like and PD-1(+) human NK cell subsets. Front Immunol. (2016) 7:351. 10.3389/fimmu.2016.00351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345.Wiesmayr S, Webber SA, Macedo C, Popescu I, Smith L, Luce J, et al. Decreased NKp46 and NKG2D and elevated PD-1 are associated with altered NK-cell function in pediatric transplant patients with PTLD. Eur J Immunol. (2012) 42:541–50. 10.1002/eji.201141832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346.Hsu J, Hodgins JJ, Marathe M, Nicolai CJ, Bourgeois-Daigneault MC, Trevino TN, et al. Contribution of NK cells to immunotherapy mediated by PD-1/PD-L1 blockade. J Clin Invest. (2018) 128:4654–68. 10.1172/JCI99317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347.Huang BY, Zhan YP, Zong WJ, Yu CJ, Li JF, Qu YM, et al. The PD-1/B7-H1 pathway modulates the natural killer cells versus mouse glioma stem cells. PLoS ONE. (2015) 10:e0134715. 10.1371/journal.pone.0134715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 348.Oyer JL, Gitto SB, Altomare DA, Copik AJ. PD-L1 blockade enhances anti-tumor efficacy of NK cells. Oncoimmunology. (2018) 7:e1509819. 10.1080/2162402X.2018.1509819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349.Concha-Benavente F, Kansy B, Moskovitz J, Moy J, Chandran U, Ferris RL. PD-L1 mediates dysfunction in activated PD-1+ NK cells in head and neck cancer patients. Cancer Immunol Res. (2018) 6:1548. 10.1158/2326-6066.CIR-18-0062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 350.Vari F, Arpon D, Keane C, Hertzberg MS, Talaulikar D, Jain S, et al. Immune evasion via PD-1/PD-L1 on NK cells and monocyte/macrophages is more prominent in Hodgkin lymphoma than DLBCL. Blood. (2018) 131:1809. 10.1182/blood-2017-07-796342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351.Hicks KC, Fantini M, Donahue RN, Schwab A, Knudson KM, Tritsch SR, et al. Epigenetic priming of both tumor and NK cells augments antibody-dependent cellular cytotoxicity elicited by the anti-PD-L1 antibody avelumab against multiple carcinoma cell types. Oncoimmunology. (2018) 7:e1466018. 10.1080/2162402X.2018.1466018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352.Juliá EP, Amante A, Pampena MB, Mordoh J, Levy EM. Avelumab, an IgG1 anti-PD-L1 immune checkpoint inhibitor, triggers NK cell-mediated cytotoxicity and cytokine production against triple negative breast cancer cells. Front Immunol. (2018) 9:2140. 10.3389/fimmu.2018.02140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 353.Lanuza PM, Vigueras A, Olivan S, Prats AC, Costas S, Llamazares G, et al. Activated human primary NK cells efficiently kill colorectal cancer cells in 3D spheroid cultures irrespectively of the level of PD-L1 expression. Oncoimmunology. (2018) 7:e1395123. 10.1080/2162402X.2017.1395123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354.Chapoval AI, Ni J, Lau JS, Wilcox RA, Flies DB, Liu D, et al. B7-H3: a costimulatory molecule for T cell activation and IFN-gamma production. Nat Immunol. (2001) 2:269–74. 10.1038/85339 [DOI] [PubMed] [Google Scholar]
- 355.Hofmeyer KA, Ray A, Zang X. The contrasting role of B7-H3. Proc Natl Acad Sci USA. (2008) 105:10277–8. 10.1073/pnas.0805458105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 356.Suh WK, Gajewska BU, Okada H, Gronski MA, Bertram EM, Dawicki W, et al. The B7 family member B7-H3 preferentially down-regulates T helper type 1-mediated immune responses. Nat Immunol. (2003) 4:899–906. 10.1038/ni967 [DOI] [PubMed] [Google Scholar]
- 357.Nygren MK, Tekle C, Ingebrigtsen VA, Fodstad O. B7-H3 and its relevance in cancer; immunological and non-immunological perspectives. Front Biosci. (2011) 3:989–93. 10.2741/e304 [DOI] [PubMed] [Google Scholar]
- 358.Picarda E, Ohaegbulam KC, Zang X. Molecular pathways: targeting B7-H3 (CD276) for human cancer immunotherapy. Clin Cancer Res. (2016) 22:3425–31. 10.1158/1078-0432.CCR-15-2428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 359.Ye Z, Zheng Z, Li X, Zhu Y, Zhong Z, Peng L, et al. B7-H3 Overexpression predicts poor survival of cancer patients: a meta-analysis. Cell Physiol Biochem. (2016) 39:1568–80. 10.1159/000447859 [DOI] [PubMed] [Google Scholar]
- 360.Ni L, Dong C. New B7 family checkpoints in human cancers. Mol Cancer Ther. (2017) 16:1203–11. 10.1158/1535-7163.MCT-16-0761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 361.Lemke D, Pfenning PN, Sahm F, Klein AC, Kempf T, Warnken U, et al. Costimulatory protein 4IgB7H3 drives the malignant phenotype of glioblastoma by mediating immune escape and invasiveness. Clin Cancer Res. (2012) 18:105–17. 10.1158/1078-0432.CCR-11-0880 [DOI] [PubMed] [Google Scholar]
- 362.Castriconi R, Dondero A, Augugliaro R, Cantoni C, Carnemolla B, Sementa AR, et al. Identification of 4Ig-B7-H3 as a neuroblastoma-associated molecule that exerts a protective role from an NK cell-mediated lysis. Proc Natl Acad Sci USA. (2004) 101:12640–5. 10.1073/pnas.0405025101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 363.Tabellini G, Benassi M, Marcenaro E, Coltrini D, Patrizi O, Ricotta D, et al. Primitive neuroectodermal tumor in an ovarian cystic teratoma: natural killer and neuroblastoma cell analysis. Case Rep Oncol. (2014) 7:70–8. 10.1159/000357802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 364.Zhao Q, Liu J, Yang S. Therapeutically targeting B7-H3 via chimeric antigen receptors and bispecific killer cell engagers in non-small cell lung cancer. J Immunol. (2018) 200(1 Suppl.):179.13. [Google Scholar]
- 365.Loo D, Alderson RF, Chen FZ, Huang L, Zhang W, Gorlatov S, et al. Development of an Fc-enhanced anti-B7-H3 monoclonal antibody with potent antitumor activity. Clin Cancer Res. (2012) 18:3834–45. 10.1158/1078-0432.CCR-12-0715 [DOI] [PubMed] [Google Scholar]
- 366.Powderly J, Cote G, Flaherty K, Szmulewitz RZ, Ribas A, Weber J, et al. Interim results of an ongoing Phase I, dose escalation study of MGA271 (Fc-optimized humanized anti-B7-H3 monoclonal antibody) in patients with refractory B7-H3-expressing neoplasms or neoplasms whose vasculature expresses B7-H3. J Immunother Cancer. (2015) 3(Suppl. 2):O8 10.1186/2051-1426-3-S2-O8 [DOI] [Google Scholar]
- 367.Desantes K, Maris JM, McDowell K, Mackall C, Shankar S, Vasselli J, et al. A phase 1, open-label, dose escalation study of enoblituzumab (MGA271) in pediatric patients with B7-H3-expressing relapsed or refractory solid tumors. J Clin Oncol. (2017) 35(15_suppl):TPS2596-TPS 10.1200/JCO.2017.35.15_suppl.TPS2596 [DOI] [Google Scholar]
- 368.Helmink BA, Gaudreau PO, Wargo JA. Immune checkpoint blockade across the cancer care continuum. Immunity. (2018) 48:1077–80. 10.1016/j.immuni.2018.06.003 [DOI] [PubMed] [Google Scholar]
- 369.Li Y, Sun R. Tumor immunotherapy: new aspects of natural killer cells. Chin J Cancer Res. (2018) 30:173–96. 10.21147/j.issn.1000-9604.2018.02.02 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 370.Wagner JA, Rosario M, Romee R, Berrien-Elliott MM, Schneider SE, Leong JW, et al. CD56bright NK cells exhibit potent antitumor responses following IL-15 priming. J Clin Invest. (2017) 127:4042–58. 10.1172/JCI90387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 371.Dadmarz R, Bockstoce DC, Golub SH. Interleukin-7 selectively enhances natural kill cytotoxicity mediated by the CD56bright natural killer subpopulation. Lymphokine Cytokine Res. (1994) 13:349–57. [PubMed] [Google Scholar]
- 372.Kareva I. A combination of immune checkpoint inhibition with metronomic chemotherapy as a way of targeting therapy-resistant cancer cells. Int J Mol Sci. (2017) 18:2134. 10.3390/ijms18102134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 373.Patel SA, Minn AJ. Combination cancer therapy with immune checkpoint blockade: mechanisms and strategies. Immunity. (2018) 48:417–33. 10.1016/j.immuni.2018.03.007 [DOI] [PMC free article] [PubMed] [Google Scholar]