

Abstract
Background
Atypical chronic myeloid leukemia (aCML) is a hematologic disorder characterized by leukocytosis with increased dysplastic neutrophils and their precursors. In CSF3R gene, the activation mutation including T618I is frequently reported in aCML but is rarely accompanied by truncation mutations. Herein, we report a unique aCML patient with two CSF3R mutations (T618I and Y779*) in the same DNA strand.
Methods
High‐coverage next‐generation sequencing for 40 genes related with myeloid leukemia was performed. Sanger sequencing was performed to confirm CSF3R mutations. To confirm whether two CSF3R mutations are in cis or not, TA cloning was used. Clinical information and bone marrow pathology were reviewed by two hematopathologists.
Results
In the patient diagnosed with aCML in bone marrow study, two CSF3R mutations, (T618I and Y779*) a SETBP1 mutation (G870S) and an U2AF1 mutation (Q157P), were identified by high‐coverage next‐generation sequencing. The two CSF3R mutations were confirmed to be located in the same DNA strand by TA cloning, indicating that the two mutations are harbored in one malignant clone. The SETBP1 mutation is known to be related with poor prognosis in aCML. Likewise, the patient was refractory to hydroxyurea and showed disease progression. Additionally, we discussed the potential therapeutic targets by reviewing the molecular profile of the patient.
Conclusion
We believe that the accurate diagnosis and maximum therapeutic chance could be achieved by profiling the mutations and their characteristics.
Keywords: atypical chronic myeloid leukemia, CSF3R, next generation sequencing, SETBP1, U2AF1
1. INTRODUCTION
Atypical chronic myeloid leukemia (aCML) is a rare disease belonging to the myelodysplastic/myeloproliferative neoplasms (MDS/MPN) group according to the 2016 World Health Organization (WHO) classification of hematologic malignancies. While aCML shows sharing hematologic findings with CML with BCR‐ABL1, the major differential diagnostic points include the absence of BCR‐ABL1 rearrangement and presence of prominent dysgranulopoiesis. Clinically, aCML is an aggressive disease with a poor prognosis.1 For these reasons, there is an unmet need for discovering molecular markers to expand treatment options and to monitor the disease. Recently, deep sequencing using next‐generation sequencing (NGS) is applied in a variety of diseases and can help solve the unmet need. Using NGS, multiple cancer genes are tested with a high mutation detection sensitivity. Driver and key mutations found by NGS could be useful therapeutic targets.2, 3
In aCML, poor prognosis is known to be related with female gender, older age (>65 years), leukocytosis >50 × 109/L, and the presence of circulating precursors.4 In molecular level, although mutations from several genes including KRAS, NRAS, SETBP1, CSF3R, ASXL1, and ETNK1 are frequently observed in aCML,5 mutations from SETBP1 and ASXL1 are considered to be associated with poor prognosis till now.5, 6, 7 In therapeutic aspect, mutations in genes related with JAK‐STAT, MAPK, ROCK, and SRC family‐TNK2 kinase signaling are gaining attention as therapeutic targets recently.5, 8, 9
Herein, we describe a case of aCML with a unique molecular profile revealed by NGS. The patient had double mutations of Y779* (first case in atypical CML) and T618I (known mutation) in the CSF3R gene. By using TA cloning, we also found that the two mutations are located in cis, suggesting that the two mutations are on the same RNA transcript of one malignant clone. In addition, two gain‐of‐function mutations in the SETBP1 and U2AF1 genes were found with high variant allele frequencies (VAFs). We discussed the biological, diagnostic, and therapeutic significance of these mutations.
2. MATERIALS AND RESULTS
2.1. Patient
The patient was a 75‐year‐old woman transferred from an outside hospital because of abnormal CBC with leukocytosis. She experienced anorexia, sweating, and weight loss. Physical examination revealed mild splenomegaly with right upper quadrant abdominal discomfort. CBC showed Hb 9.2 g/dL, white blood cells (WBC) 72 K/µL with blasts 2% and granulocytic precursors 29%, and platelets 359 K/µL (Figure 1). Peripheral blood smear demonstrated dysplastic features in the granulocytic series (Figure 1). Bone marrow (BM) study was performed and revealed granulocytic proliferation with dysplasia (Figure 1), increased megakaryocytes, and focal fibrosis. Chromosome analysis showed 47,XX,+14[19]/46,XX[1]. Molecular genetic studies were performed to screen BCR/ABL1, PDGFRA, PDGFRB, and FGFR1 rearrangements, and point mutations of JAK2, CALR, and MPL, and the results were all negative. Collectively, the patient was diagnosed as having aCML based on the 2016 WHO diagnostic criteria.10 After patient started hydroxyurea therapy, leukocytosis and blast count were decreased initially (Figure 1C). However, leukocytosis and blast count started to be increased after 3 months of hydroxyurea therapy, suggesting the refractoriness to the medication.
Figure 1.
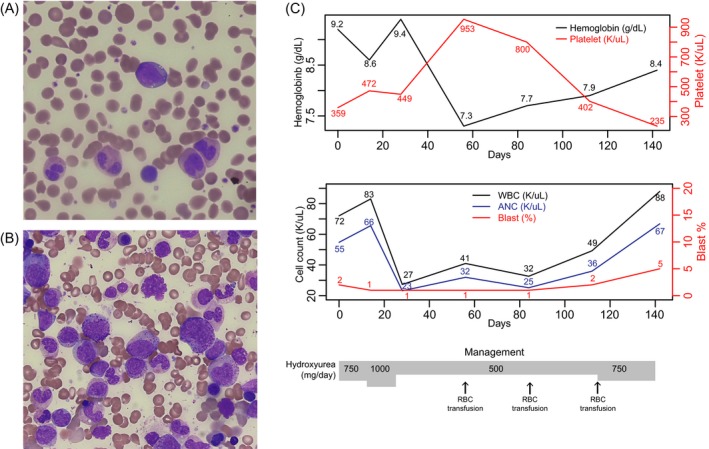
Peripheral blood smear (A) and bone marrow aspirate (B) in Wright and Giemsa stains. A blast and dysplastic granulocytes were shown (A) and granulocytic proliferation and granulocytic dysplasia in bone marrow (B). C, Laboratory findings including hemoglobin, platelet, white blood cell count, and blast count during the 140 days of hydroxyurea therapy
2.2. Next‐generation sequencing and gene mutations
Next‐generation sequencing was performed to detect gene mutations other than the aforementioned major driver mutations in MPN. DNA was extracted using the Promega DNA Extraction Kit (Promega) following the manufacturer's instructions. Isolation of DNA was performed using the RecoverAll Total Nucleic Acid Isolation Kit (Thermo Fisher Scientific). The library preparation was conducted using the Ion Chef System (Thermo Fisher Scientific, San Francisco, CA, USA) per the manufacturer's instructions. Sequencing was conducted on the Ion S5 XL Sequencer using the Ion 530 Chip and Ion 530 kit‐Chef (Thermo Fisher Scientific). Alignment and base calling were performed using the Ion Reporter (Version 5.6.0) with Oncomine™ Myeloid Research‐530‐w2.3.1 workflow based on the reference genome hg19. Variant detection and annotation were performed using Ion Reporter software (Version 5.6). The DNA panel from OncomineTM myeloid research assay was designed to test the following genes: ABL1, ASXL1, BCOR, BRAF, CALR, CBL, CEBPA, CSF3R, DNMT3A, ETV6, EZH2, FLT3, GATA2, HRAS, IDH1, IDH2, IKZF1, JAK2, KIT, KRAS, MPL, MYD88, NF1, NPM1, NRAS, PHF6, PRPF8, PTPN11, RB1, RUNX1, SETBP1, SF3B1, SH2B3, SRSF2, STAG2, TET2, TP53, U2AF1L5, WT1, and ZRSR2. As a result, we detected two pathogenic variants in the CSF3R gene, one variant each in SETBP1 and in U2AF1 (Table 1). The two CSF3R mutations were a truncation mutation c.2337T>G (Y779*) in receptor cytoplasmic domain and the well‐known activating mutation c.1853C>T (T618I) affecting the extracellular domain. The mutation in SETBP1 was c.2608G>A (G870S), and the mutation in U2AF1 was c.470A>C (Q157P). The variant allele frequencies (VAFs) of CSF3R Y779*, CSF3R T618I, SETBP1 G870S, and U2AF1 Q157P were 48.4%, 45.9%, 48.9%, and 47.2%, respectively (Table 1). All mutations were confirmed by Sanger sequencing.
Table 1.
Mutations revealed by next‐generation sequencing
| Gene | cDNA change | AA change | VAF (%) | Coverage |
|---|---|---|---|---|
| CSF3R | c.2337T>G | p.Tyr779Ter | 48.4 | 1989 |
| CSF3R | c.1853C>T | p.Thr618Ile | 45.9 | 1998 |
| SETBP1 | c.2608G>A | p.Gly870Ser | 48.9 | 1999 |
| U2AF1 | c.470A>C | p.Gln157Pro | 47.2 | 1998 |
Abbreviation: VAF, variant allele frequencie.
2.3. TA cloning for CSF3R double mutations
We additionally performed TA cloning to determine whether the 2 mutations of CSF3R were on the same mutant allele. PCR was performed by using SimpliAmp thermal cycler (Life Technologies) (primer sequence and reaction conditions are available upon request), and the PCR products were ligated into a pGEM‐T easy vector system (Promega). Ten subcloned DNA was sequenced (Cosmo Genetech Inc, Seoul, Korea), and the sequence data were analyzed using DNASTAR™ Lasergene software (Thermo Fisher). As a result, the sequences from colonies 1, 3, 5, 6, and 10 harbored both T618I and Y779* mutations, while those from the remaining colonies had no mutations. These results demonstrated that the two mutations were on the same allele (Figure 2B).
Figure 2.
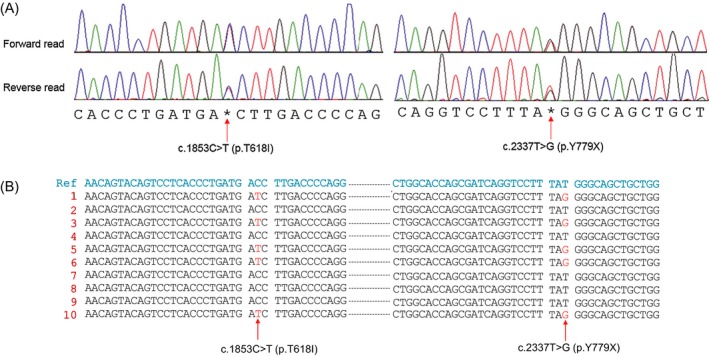
Sanger sequencing confirmation for two CSF3R mutations (A). TA cloning to identify whether the two CSF3R mutations were in the same DNA strand or not (B). In the lanes of 1, 3, 5, 6, and 10, c.1853C>T and c.2337T>G were found simultaneously, while no CSF3R mutations were found in other lanes. These findings suggest that the two CSF3R mutations are located in the same DNA strand, finally indicating that the two mutations are harbored in the same leukemic clone
3. DISCUSSION
In this report, we described a patient with aCML having a unique genetic profile: double mutations in CSF3R, T618I, and T779*, along with SETBP1 G870S, U2AF1 Q157P, and trisomy 14. Unlike CML with BCR‐ABL1, aCML has is no molecular hallmark. The detection of multiple gene mutations was possible through NGS covering a panel of recurrently mutated genes in myeloid neoplasms, and we could detect those mutations.
The CSF3R gene encodes the receptor for colony‐stimulating factor 3, a cytokine that controls the production and differentiation of granulocytes. The pathogenic variant in CSF3R is commonly found in hematologic malignancies including acute myeloid leukemia, chronic neutrophilic leukemia, and atypical CML.11 Double mutations in CSF3R, one in the domain and the other in the C‐terminal as in our patient, are previously reported in a small proportion of aCML. However, the T779* mutation of CSF3R has been reported in congenital neutropenia or acute myeloid leukemia as a sole mutation.12, 13 Thus, the combination of T618I and Y779* mutations is novel, and our patient with aCML is the first report. Y779* co‐existing with T618I is novel in aCML and seemed to be associated with delayed receptor internalization based on the location of the mutation.14 The two mutations were suggested to be on the same allele based on the VAF information, and TA cloning results confirmed the status. First, the malignant clone harbors both mutations simultaneously in the same DNA strand. Hence, this finding helps in establishing the strategy for a molecular target therapy. The T618I is known to be related with cell growth via JAK‐STAT signaling and the JAK‐STAT signaling could be inhibited by JAK inhibitors in this intractable disease (Figure 3). Additionally, the truncation mutation, Y779*, could be related with cell growth signaling via SFK‐TNK2 and this could be inhibited by SFK‐TNK2 kinase inhibitors. Finally, the malignant clone has two potential therapeutic targets. Second, it can be inferred that the two mutations, T618I and Y779*, play a synchronistic or complementary role in forming a tumor clone via activation and delayed receptor internalization of CSF3R.
Figure 3.
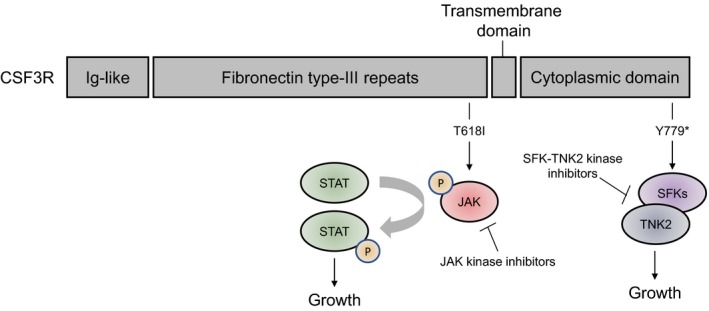
Location of two mutations in CSF3R. The T618I is related with JAK signaling, and the signal could be inhibited by JAK kinase inhibitor. The CSF3R truncation mutation (Y779*) is related with proliferation signaling via SFK‐TNK2, and this proliferation signaling is potentially inhibited by SFK‐TNK2 kinase inhibitor
The SETBP1 gene encodes SET binding protein 1, which could bind the SET nuclear oncogene which is involved in DNA replication. The gain‐of‐function mutation of SETBP1 in our patient, G870S, is located in the SKI homologous region and has been reported mostly in chronic myelomonocytic leukemia, myelodysplastic syndrome, and acute myeloid leukemia based on the COSMIC database.15 Furthermore, G870S is known to be associated with higher WBC counts and worse prognosis.6 Likewise, our patient was refractory to hydroxyurea treatment and showed disease progression with high leukocyte count and increasing blast count (Figure 1C). The U2AF1 gene encodes a U2 small nuclear RNA auxiliary factor 1, which plays a critical role in RNA splicing. The gain‐of‐function mutation of U2AF1 detected in our patient was Q157P. The Q157 residue is located in the second zinc finger domain,16 and Q157P is a known pathogenic mutation that alters splicing of many important genes in myeloid disorders,17 while the prognostic significance has not been fully investigated in aCML. Interestingly, several molecular features in the patient are considered to be related with dysplasia, which is a typical bone marrow finding in aCML SETBP1 mutation is reported to be related with dysplastic morphology in MDS/MPN.18 The mutation in aCML having dysplasia as one of diagnostic hallmark is frequently found with 24%.6 The gain‐of‐function mutation in U2AF1 is thought to contribute dysplastic hematopoiesis. In the report, the authors described that hundreds of exons were differentially spliced with association of U2AF1 mutation and that the phenomenon could be related with dysplasia and tumorigenesis.17 In addition, sole trisomy 14 was found in chromosome analysis. The chromosomal aberration is also reported to be related with dysplastic blood cancer, while further investigation is needed for its biology and clinical significance.19
In the aspect of treatment, there was additional chance of a clinical trial with several therapeutic targets, although the patient was lost to follow‐up with disease progression, unfortunately. For T618I of CSF3R, ruxolitinib showed efficacy according to several case reports.9, 20, 21 The G870S of SETBP1 is reported to imply ruxolitinib unresponsiveness.22 On the other hand, the SETBP1 mutation was suggested as a novel target of fingolimod (FTY720) in an in vitro study.23
For differential diagnosis of aCML with CSF3R mutation, chronic neutrophilic leukemia (CNL) is an important disease in that CSF3R mutations are more frequent in CNL. The CSF3R mutation frequencies are reported to be 43% and less than 10% in CNL and aCML, respectively.10, 24 In the diagnostic criteria, the main difference between the two diseases is that increased neutrophil precursors more than 10% of WBC with dysplasia were observed in aCML while not in CNL.10 In molecular aspect, the genetic drivers seemed to be more heterogenous in aCML than CNL.7 Atypical CML is associated with the presence of SETBP1 and/or ENTK1 mutations while CNL is associated the presence of CSF3R mutations.10 In an analysis of 14 CNL and 58 aCML patients, Meggendorfer et al reported that CSF3R mutations are statistically more frequent in CNL.24 In the study, the mutation frequencies of ASXL1, SETBP1, SRSF2, and TET2 are not rare in both aCML and CNL, although their frequencies are seemed to be higher in aCML. As in our case, morphological findings such as the proportion of neutrophil precursors and dysplasia are critical in differential diagnosis between CNL and aCML with mutated CSF3R till now. Molecular markers discriminating the two diseases need to be further investigated.
In summary, we detected double mutations of CSF3R in cis with gain‐of‐function mutations of SETBP1 and U2AF1 in our patient with aCML through deep sequencing and TA cloning. We believe that the accurate diagnosis and maximum therapeutic chance could be achieved by profiling the mutations and their characteristics.
CONFLICT OF INTEREST
The authors declare that they have no competing interests.
Yun JW, Yoon J, Jung CW, et al. Next‐generation sequencing reveals unique combination of mutations in cis of CSF3R in atypical chronic myeloid leukemia. J Clin Lab Anal. 2020;34:e23064 10.1002/jcla.23064
DATA AVAILABILITY STATEMENT
All the information about the case report is available from the corresponding author on reasonable request.
REFERENCES
- 1. Dhakal P, Gundabolu K, Amador C, Rayamajhi S, Bhatt VR. Atypical chronic myeloid leukemia: a rare entity with management challenges. Future Oncol. 2018;14(2):177‐185. [DOI] [PubMed] [Google Scholar]
- 2. Griffith M, Spies NC, Krysiak K, et al. CIViC is a community knowledgebase for expert crowdsourcing the clinical interpretation of variants in cancer. Nat Genet. 2017;49(2):170‐174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ommen HB. Monitoring minimal residual disease in acute myeloid leukaemia: a review of the current evolving strategies. Ther Adv Hematol. 2016;7(1):3‐16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Breccia M, Biondo F, Latagliata R, Carmosino I, Mandelli F, Alimena G. Identification of risk factors in atypical chronic myeloid leukemia. Haematologica. 2006;91(11):1566‐1568. [PubMed] [Google Scholar]
- 5. Schwartz LC, Mascarenhas J. Current and evolving understanding of atypical chronic myeloid leukemia. Blood Rev. 2019;33:74‐81. [DOI] [PubMed] [Google Scholar]
- 6. Piazza R, Valletta S, Winkelmann N, et al. Recurrent SETBP1 mutations in atypical chronic myeloid leukemia. Nat Genet. 2013;45(1):18‐24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dao KH, Tyner JW. What's different about atypical CML and chronic neutrophilic leukemia? Hematology Am Soc Hematol Educ Program. 2015;2015:264‐271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rocca S, Carra G, Poggio P, Morotti A, Brancaccio M. Targeting few to help hundreds: JAK, MAPK and ROCK pathways as druggable targets in atypical chronic myeloid leukemia. Mol Cancer. 2018;17(1):40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Maxson JE, Gotlib J, Pollyea DA, et al. Oncogenic CSF3R mutations in chronic neutrophilic leukemia and atypical CML. N Engl J Med. 2013;368(19):1781‐1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391‐2405. [DOI] [PubMed] [Google Scholar]
- 11. Liongue C, Ward AC. Granulocyte colony‐stimulating factor receptor mutations in myeloid malignancy. Front Oncol. 2014;4:93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Germeshausen M, Kratz CP, Ballmaier M, Welte K. RAS and CSF3R mutations in severe congenital neutropenia. Blood. 2009;114(16):3504‐3505. [DOI] [PubMed] [Google Scholar]
- 13. Skokowa J, Steinemann D, Katsman‐Kuipers JE, et al. Cooperativity of RUNX1 and CSF3R mutations in severe congenital neutropenia: a unique pathway in myeloid leukemogenesis. Blood. 2014;123(14):2229‐2237. [DOI] [PubMed] [Google Scholar]
- 14. Zhang H, Reister Schultz A, Luty S, et al. Characterization of the leukemogenic potential of distal cytoplasmic CSF3R truncation and missense mutations. Leukemia. 2017;31(12):2752‐2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tate JG, Bamford S, Jubb HC, et al. COSMIC: the Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 2019;47(D1):D941‐D947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Clark WD, Bailey BJ, Clegg TJ. Epiglottitis and laryngotracheobronchitis. Am Fam Physician. 1983;28(4):189‐194. [PubMed] [Google Scholar]
- 17. Ilagan JO, Ramakrishnan A, Hayes B, et al. U2AF1 mutations alter splice site recognition in hematological malignancies. Genome Res. 2015;25(1):14‐26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kanagal‐Shamanna R, Luthra R, Yin CC, et al. Myeloid neoplasms with isolated isochromosome 17q demonstrate a high frequency of mutations in SETBP1, SRSF2, ASXL1 and NRAS. Oncotarget. 2016;7(12):14251‐14258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cui W, Bueso‐Ramos CE, Yin CC, et al. Trisomy 14 as a sole chromosome abnormality is associated with older age, a heterogenous group of myeloid neoplasms with dysplasia, and a wide spectrum of disease progression. J Biomed Biotechnol. 2010;2010:365318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dao KH, Solti MB, Maxson JE, et al. Significant clinical response to JAK1/2 inhibition in a patient with CSF3R‐T618I‐positive atypical chronic myeloid leukemia. Leuk Res Rep. 2014;3(2):67‐69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Freedman JL, Desai AV, Bailey LC, et al. Atypical chronic myeloid leukemia in two pediatric patients. Pediatr Blood Cancer. 2016;63(1):156‐159. [DOI] [PubMed] [Google Scholar]
- 22. Ammatuna E, Eefting M, van Lom K, Kavelaars FG, Valk PJ, Touw IP. Atypical chronic myeloid leukemia with concomitant CSF3R T618I and SETBP1 mutations unresponsive to the JAK inhibitor ruxolitinib. Ann Hematol. 2015;94(5):879‐880. [DOI] [PubMed] [Google Scholar]
- 23. Cristobal I, Garcia‐Orti L, Cirauqui C, Alonso MM, Calasanz MJ, Odero MD. PP2A impaired activity is a common event in acute myeloid leukemia and its activation by forskolin has a potent anti‐leukemic effect. Leukemia. 2011;25(4):606‐614. [DOI] [PubMed] [Google Scholar]
- 24. Meggendorfer M, Haferlach T, Alpermann T, et al. Specific molecular mutation patterns delineate chronic neutrophilic leukemia, atypical chronic myeloid leukemia, and chronic myelomonocytic leukemia. Haematologica. 2014;99(12):e244‐e246. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All the information about the case report is available from the corresponding author on reasonable request.