

Abstract
Neural crest (NC) cells are a multipotent stem cell population that give rise to a diverse array of cell types in the body, including peripheral neurons, Schwann cells (SC), craniofacial cartilage and bone, smooth muscle cells, and melanocytes. NC formation and differentiation into specific lineages takes place in response to a set of highly regulated signaling and transcriptional events within the neural plate border. Premigratory NC cells initially are contained within the dorsal neural tube from which they subsequently emigrate, migrating to often distant sites in the periphery. Following their migration and differentiation, some NC‐like cells persist in adult tissues in a nascent multipotent state, making them potential candidates for autologous cell therapy. This review discusses the gene regulatory network responsible for NC development and maintenance of multipotency. We summarize the genes and signaling pathways that have been implicated in the differentiation of a postmigratory NC into mature myelinating SC. We elaborate on the signals and transcription factors involved in the acquisition of immature SC fate, axonal sorting of unmyelinated neuronal axons, and finally the path toward mature myelinating SC, which envelope axons within myelin sheaths, facilitating electrical signal propagation. The gene regulatory events guiding development of SC in vivo provides insights into means for differentiating NC‐like cells from adult human tissues into functional SC, which have the potential to provide autologous cell sources for the treatment of demyelinating and neurodegenerative disorders.
Keywords: demyelinating disorders, gene regulatory network, neural crest, Schwann cells
Significance statement.
Neural crest (NC) cells have attracted attention for their multipotent nature and ease of isolation from adult tissues. This concise review reports the advantages of using NCs for the treatment of demyelinating disorders and spinal cord injury (SCI), over other cell sources such as induced pluripotent stem cells and embryonic stem cells. Adult tissue‐derived NCs are easy to expand in vitro and can be derived from autologous sources. Moreover, differentiation of NCs to Schwann cells (SC) can be easily achieved without genetic mutation, making them safe for translation from a laboratory to a clinical setting. Adult NC‐derived SC are functional and can myelinate neurons in vitro and spinal cord in vivo in mice. Hence, NCs derived from adult tissue are a promising cell source for the treatment of demyelinating disorders and SCI. Furthermore, this technology can also be used for disease modeling and drug testing, making way for personalized therapeutics for neurological disorders.
1. INTRODUCTION TO THE NEURAL CREST CELLS
Often referred to as the “fourth germ layer,” the neural crest (NC) is a multipotent and migratory stem cell population that contributes to a wide array of organs and tissues in the vertebrate embryo, including autonomic ganglia, sensory neurons, adrenal and thyroid glands, cartilage and bone of the face, smooth muscle cells of some major arteries, and melanocytes in the skin.1, 2 NC formation is first observed at stage 9 of human embryogenesis and extends till stage 20 as per the Carnegie staging system.3 Neural crest stem cells (NCSC) were first identified in rodents by Stemple and Anderson and isolated using cell sorting for NC‐specific cell surface protein p75NTR (neurotrophin receptor [NTR]). These p75NTR+ cells could self‐renew and generate neurons and glia of the PNS as well as myofibroblasts.4 The high degree of self‐renewal and regenerative capacity of NC makes it a very attractive source for stem cell‐based therapies.
During embryonic development, formation of NC stem cells originates in response to a set of signaling events between neural and non‐neural ectoderm, a region termed as the neural plate border.5 Induction is initiated at the plate border by signals including fibroblast growth factor (FGF), bone morphogenetic protein (BMP), and Wnt.5, 6 On establishment of the neural plate border territory, new signaling events are established for specification of bona fide NCSCs. This process results in expression of NC specifiers (Snail2, Sox8, Sox9, Sox10, FoxD3, c‐Myc, and Id family members) that in turn mediate changes in shape, motility, and adhesive properties leading to delamination of NCSCs from the neuroepithelium and initiating cell lineage decisions.6 NC precursor cells within the dorsal neural tube then undergo epithelial to mesenchymal transition and migrate along precise pathways,7 eventually settling and differentiating into specialized cell types, according to their axial origin as well as environmental cues encountered during the process.8 Cranial NCSCs form craniofacial structures of the head including cartilage and bone tissue of the skull and face, as well as cranial neurons, glia, and connective tissue of the face. Trunk NCSCs differentiate into dorsal root ganglia (DRG), containing sensory neurons and satellite glial cells, endocrine cells of the adrenal glands, and Schwann cells (SC) along the spinal nerves. Some of these NCSCs also differentiate into melanocytes in the skin. Finally, vagal NCSCs populate the enteric nervous system (ENS) along the length of the gut and contribute to the connective tissues of the arteries, septation of the outflow tract and cardiac ganglion.1
2. SOURCES OF NC‐LIKE STEM CELLS IN ADULT TISSUES
2.1. NC characteristics are retained postembryonically in adult tissues
Although NC cells are often referred to as stem or stem‐like cells, they are a transient population in the embryo that becomes progressively restricted in developmental potential during the course of development and rapidly lose their multipotency. Nevertheless, recent evidence suggests that many NC derivatives, including skin, cornea, gut, and peripheral nerves, contain stem or precursor cells with the ability to give rise to multiple NC derivatives.1, 9 These tissues provide potential sources for obtaining multipotent neural crest‐like stem cells (NClSCs) for the purpose of regenerative medicine and cell therapy, and consequently, they are under study in several laboratories. Whether NC‐like cells from the adult tissue come from embryonic NCs and remain dormant in a multipotent state or they attain a specialized fate and dedifferentiate to their NC progeny in specific culture conditions has not been established yet. The answers to these questions can only be attained by lineage‐tracing experiments tracking migration and differentiation of NCs starting from embryo development and throughout adulthood.
NClSCs have been isolated from mouse bone marrow and DRG. Nagoshi et al used double‐transgenic mouse strains P0 and Wnt1‐Cre/Floxed‐EGFP to map and isolate NClSCs from bone marrow, DRG, and whisker pad using cell‐sorting. P0 and Wnt1 are marker genes expressed in NC cells in mouse embryos.10 Interestingly, DRG‐derived NClSCs generated more primary and secondary spheres compared with bone marrow and whisker pad–derived cells and showed increased expression of NC markers p75NTR, Sox10, Nestin, and Musashi1. Additionally, 75% of DRG‐derived spheres showed tri‐lineage differentiation to neurons, glia, and myofibroblasts, compared with only 7.3% of spheres derived from whisker pad and 3.3% in spheres derived from bone marrow.10 These results highlight the tissue‐dependent differences in the abundance and self‐renewal capacity of adult NClSCs, which may ultimately determine the suitability for cellular therapies. Furthermore, bone marrow–derived NCs could be differentiated into SC that showed myelination around DRG neuronal axons in vitro, indicating their potential for use in peripheral nerve regeneration.11
During development, NCs also migrate to the cornea, where their derivatives remain throughout adulthood. The cornea is a transparent avascular structure that covers the front of the eye and helps to focus light on the retina. The cornea consists of three major cellular components: a stratified epithelium, a collagenous stroma containing keratocytes, and a layer of endothelium.1 Experiments with chick‐quail chimeras demonstrated that cranial NC cells form the corneal endothelium and keratocytes.12, 13, 14, 15 Lineage tracing experiments demonstrated that upon transplantation from late into early chick embryos quail NC‐derived keratocytes followed normal migratory routes giving rise to smooth muscle cells, myofibrils, keratocytes, and endothelial cells but failed to differentiate into neurons or cartilage, suggesting restricted plasticity of these NC‐derived progenitors.16 Another study however demonstrated differentiation of corneal progenitor cells into keratocytes, fibroblasts, myofibroblasts, adipocytes, chondrocytes, and neural cells, indicating similar differentiation potential to NCSC.17
Stem cell derivatives of NC origin persist in the adult rodent gut from which they can be isolated based on CD49b expression. Sorted cells showed expression of NC marker genes, such as p75NTR, Sox10, and Nestin, along with markers specific to enteric glia such as S100B and glial fibrillary acidic protein.18 Multipotent progenitors from fetal gut exhibited more plasticity and degree of self‐renewal compared with those derived from adult gut. In addition, upon transplantation into chick embryos, fetal progenitors gave rise primarily to neurons, whereas postnatal gut progenitors turned predominantly into glia.19 In vivo grafting experiments suggest therapeutic potential of these progenitors for the treatment of ENS disorders.20, 21
Cardiac NC cells play a role in septation of cardiac outflow tract into pulmonary and aortic branches.1 In vitro clonal expansion of NCISCs isolated from cardiac tissue suggested that only a small fraction of them were capable of self‐renewal and generation of NC derivatives.22 Stem cells from neonatal rodent heart demonstrate a “NC‐like” behavior, and when grown as cardiospheres, they could differentiate into PNS neurons, glia, smooth muscle cells, and cardiomyocytes, as well as migrate to tissues characteristic for NC derivatives in ovo.23 Interestingly, cardiac resident Nestin+ progenitors migrated to areas damaged by infarction and contributed to reparative vascularization, indicating their potential for the treatment of various heart diseases.24
Although the presence of NClSCs in developed tissues provides evidence of an alternative cell source for cell therapy, most studies of this type have been limited to rodents, due to inaccessibility of human NCs in organs such as gut, heart, DRG and spine, as shown in Figure S1. Interestingly though, NCISCs have also been isolated from adult human tissues such as skin and dental pulp. The isolation of NClSCs from skin tissue enhances their therapeutic potential, mostly because of the accessibility and size of skin tissue that can provide autologous cells for cell therapies.25, 26, 27, 28 Fernandes et al showed that endogenous adult dermal precursors residing in the papillae of hair and whisker follicles could give rise to multipotent NClSCs.25 Similarly, Sieber‐Blum et al reported the presence of multipotent NCISCs in the adult mammalian hair follicle, which could give rise to neurons, melanocytes, smooth muscle cells, SC, and chondrocytes in vitro. These multipotent cells were termed epidermal neural‐crest cells,26 and when grafted in a mouse model of SCI, they integrated into the host spinal tissue yielding improvements in touch perception and sensory connectivity.29
2.2. Multipotent NClSCs from interfollicular epidermis
Recently, in our laboratory, Bajpai et al devised a method to reprogram postnatal human epidermal keratinocytes (KCs) to NClSC (termed KC‐NC) by mimicking signaling events that occur at the neural plate border. Transcriptomic analysis confirmed that epidermally derived NCISCs were similar to those generated from human embryonic stem cells (ESCs) and maintained the multilineage differentiation potential into melanocytes, neurons, SC, and mesenchymal cells in vitro and in ovo.30 In a subsequent study, we identified the factors that promote expansion of KC‐NC and maintain the NC phenotype. Specifically, we showed that FGF2 was necessary and sufficient for expression of Sox10, but both FGF2 and IGF1 worked synergistically to upregulate FoxD3. In addition, inhibition of TGF‐β1 further enhanced Sox10 expression.31 We also demonstrated that the same signaling factors can be used to obtain multipotent and functional NClSCs from cultures of human inter‐follicular KC isolated from elderly donors.32 Interestingly, NClSC from older donors exhibited significantly younger epigenetic age than epidermal KC, perhaps indicating greater potential for cell therapies. Given the accessibility, high proliferative capacity, and ease of reprogramming without genetic modification, KC‐NC represent an abundant, autologous source of functional therapeutic cells for regenerative medicine. They can also provide an excellent culture system for studying human disease, similar to induced pluripotent stem cells (iPSCs) but without the need for genetic modification or reprogramming to the pluripotent state.
2.3. Schwann cell precursors contribute to NC derivatives
Recent evidence suggests that NC cells that become associated with peripheral nerves acquire a partial glial phenotype, assuming the characteristics of a “Schwann cell precursor” (SCP). Intriguingly, it has been shown that late embryonic stages, many melanocytes originate from nerve‐associated SCPs.33 This occurs well after NC cells destined to form melanocytes have emigrated from the neural tube. Similarly, lineage analysis in mice has shown that cranial parasympathetic ganglia as well as a subpopulation of enteric neurons arise from this cell population.34, 35, 36 This is consistent with the possibility that later NC derivatives may arise from an SCP population that represents a nascent stem cell population associated with peripheral nerves. These SCPs express characteristic marker genes that are different from those expressed by migratory NCSC but similar to markers expressed by immature SC. For example, SCPs express genes encoding myelin basic protein (MBP), peripheral myelin protein 22 (PMP22), desert hedgehog, Cadherin 19, Connexin 29, GAP43, BFABP, and other Schwann cell markers, many of which are also associated with differentiation into myelinating SC.37, 38, 39, 40, 41 However, rather than being restricted to differentiate into SC, SCPs remain multipotent and appear to have the ability to contribute to numerous lineages at much later times in embryogenesis than normally associated with NC migration. In fact, recent studies also suggest that SCPs may give rise to chromaffin cells of the adrenal medulla, as evidenced by single cell RNA sequencing (scRNA‐seq) of the developing adrenomedullary cells in mice.42 This raises the intriguing possibility that these may represent true stem cells with remarkable multipotency and regenerative ability.
The transition from migratory NC cell to SCPs likely occurs when NC cells approach and/or become associated with peripheral nerves emanating from sensory and autonomic ganglia. At this point, the NC‐derived cells upregulate genes typically associated with SC while downregulating more typical NC markers, thus assuming a more glial like state.38, 43 Although these cells may be biased toward glial lineages, they remain multipotent and we speculate that perhaps they “dedifferentiate” similar and give rise to other NC fates similar to radial glia which are neuronal progenitors in the central nervous system. Whether SCPs maintain their multipotency into adulthood and the full range of cell types into which they can differentiate remain open questions.
3. GENE REGULATION IN NC CELLS: FATE ACQUISITION AND MULTIPOTENCY MAINTENANCE
3.1. An NC gene regulatory network controls lineage diversification into neuronal, melanocytic, and glial, including Schwann cell lineages
The NC is an excellent model system for studying questions of stem cell biology due to its multipotency, motility, and ability to form a broad array of derivatives. These are as diverse as neurons and SC of the peripheral nervous system, craniofacial cartilage, and bone, as well as skin melanocytes. These inherent stem cell properties have potentially important implications in regenerative medicine to treat disorders like familial dysautonomia, cleft palate, and NC‐related heart conditions such as Persistent Truncus Arteriosus, as well as to understand anomalies in differentiation that lead to cancers such as melanomas and Schwannomas.
The recent expansion of molecular biological techniques including single‐cell RNA‐seq, ChIP‐seq, and ATAC‐seq have facilitated the dissection of the genetic program controlling NC development. These genomic approaches have provided important insights into gene regulatory mechanisms and uncovered new regulatory factors involved in control of NC formation and diversification.44, 45 It is now clear that an intricate array of transcription factors and signaling molecules act in concert to imbue NC cells with its broad multipotency and migratory ability. These factors have been proposed to act via a multistep NC gene regulatory network (GRN) that integrates transcriptional inputs and diverse environmental signals.2, 5, 46, 47 The NC GRN consists of a series of hierarchically arranged regulatory steps, including induction of the prospective NC at the neural plate border, specification of multipotent NC cells within the dorsal neural tube, control of their delamination via an epithelial to mesenchymal transition to produce a migratory population, and finally, diversification into distinct cell lineages.
The NC GRN posits that the process of NC formation is comprised of a logical series of distinct regulatory steps that flow seamlessly from one to the other. First, signaling molecules, including Wnts, FGFs, and BMPs, in the gastrula stage embryo initiate the process of NC induction by inducing transcription factors like Msx, Pax3/7, Zic1, and Dlx3/5 at the border between the neural plate and nonneural ectoderm. The region where these genes are coexpressed defines the neural plate border—a domain primed to form bona fide NC cells. These in turn function in combination with signaling molecules to regulate “NC specifier genes” like Snail/Slug, AP‐2, FoxD3, Twist, Id, cMyc, and Sox8/9/10. The NC GRN itself can be envisioned as a series of sequential binary decisions leading to differentiation into derivative fates. Recently Twist1 has been reported to play a critical role as a regulator of NC fate decision, and it biases NC commitment toward a mesenchymal fate.45 Conversely, FoxD3 represses the mesenchymal program of delaminating NCs.48 The postmigratory program begins following the downregulation of transcription factors associated with the neural tube program such as Zic3 and Pax8.
Expression of the Sox family genes initiates within the dorsal neural tube and defines cells with the potential to emigrate from the neural tube and form migratory NC cells. In particular, the SoxE transcription factors (Sox8/9/10) are critical regulators of most NC lineages.5, 49, 50 Specifically, Sox8 and Sox9 are expressed early in the newly induced NC, preceding Sox10, which serves as a nearly pan migratory NC marker. At later developmental stages, Sox9 and Sox10 persist in specific NC subpopulations. Whereas Sox9 is maintained in NC‐derived chondrocytes, Sox10 persists in neuronal, glial, and melanocyte lineages and controls their specification in combination with different cofactors in each lineage.
Sox10 regulates differentiation of sensory and autonomic lineages by regulating expression of achaete‐scute homolog 1 and the paired homeodomain (Phox2b) transcription factors that are essential for neurogenesis in the autonomic lineage.51 In the DRG, transient expression of Sox10 regulates expression of neurogenin.52 Sox10 also binds to endothelin receptor‐B (EDNRB) to regulate development of the ENS, and disruption in this binding has been shown to cause Hirschsprung disease.53 Sox10 also is critical for emergence of the glial lineage by regulating Oct6 and Krox20 (Egr2) transcription factors, which are critical for myelination.54, 55, 56 Its expression persists through subsequent stages of terminal differentiation,49 regulating expression of myelin proteins, including protein zero (P0)57, MBP, PMP22, and the gap junction protein connexin 32.58
3.2. Diverse signaling pathways govern NC cell fate during embryogenesis
Many studies have focused on discovering the regulatory pathways that control NC fate acquisition in order to better understand cell fate specification during embryonic development in vivo and develop bioinspired strategies to differentiate NC into different lineages in vitro.
After formation of the neural tube, NC cells separate from the neuroepithelium and migrate to distant anatomic locations, while making cell lineage decisions in response to multiple morphogenetic signals.59, 60 However, some NCs remain unspecified and retain their stemness and multipotency.9, 61 In general, there is significant heterogeneity with respect to the differentiation capacity of NCs, with some giving rise to multiple derivatives while others differentiating into a subset of cell types,62, 63 depending on anatomic location and the presence of fate specifying signals. NCs from all axial levels give rise to neurons, glia, and pigment cells, but mesenchymal cell specification depends on axial position. Cranial NCs give rise to skeletal mesenchyme; trunk NCs give rise to dorsal fin mesenchyme in amphibians and fish; and vagal NCs contribute to smooth muscle cells of the cardiac outflow tract.64, 65
One of the earliest NC markers includes the family of receptor tyrosine kinases (RTK), which plays an important role in cell migration and survival. The RTK family are critical for development of NC derivatives in vivo.66 At migratory stages, the neurotrophin factor NT‐3 binds to the trkC receptor and induces sensory neurogenesis.67 GDNF binds to the RET receptor during ENS development.66 Neuregulin binds to receptor ErbB3 and is required for glia formation in the peripheral nervous system.68 Binding of steel factor to the c‐kit receptor promotes formation of melanocyte precursors,60 whereas cartilage precursors express platelet‐derived growth factor receptor α.69
The timing of NC migration during development also affects the fate they attain as cell specific genes are expressed at different times during migration. Early‐migrating cells express transcription factors Brn3 or neurogenin‐2 and commit to sensory neuron lineage.70, 71 Late‐migrating cells express melanocytic‐specific markers in the migration staging area after departure of the early‐migrating NCs.72 These signals contribute to the molecular heterogeneity of the premigratory NCs.
Three main classes of signals, namely Wnts, BMP2/4, and TGFβ1/2/3, influence NC cell fate during and following migration (Figure 1).73, 74 Their interplay, timing, and relative intensity determine the proportion of various derivatives generated during development. Wnts and the downstream β‐catenin pathway have been found to promote pigment cell formation in zebrafish embryo, possibly through activation of Nacre (Mitfa), a gene necessary and sufficient for pigment cell formation; conversely, inhibition of Wnt signaling leads to generation of neurons at the expense of pigment cells.75, 76 Conditional deletion of β‐catenin in mice prevented sensory ganglion formation, whereas constitutive activation led to formation of sensory neurons at the expense of other NC derivatives.77, 78 The second class of signaling molecules, BMP2/4 are known to induce autonomic neurogenesis via expression of Ascl1 (MASH1), a basic helix‐loop‐helix transcription factor expressed in autonomic neuron precursors prior to differentiation. Interestingly, continuous BMP signaling is required for commitment of NCs to neuronal fate.79 Although BMP2/4 have been identified as factors promoting specification of NCs to an autonomic lineage, it does not prevent sensory neurogenesis in NCs prespecified to a sensory fate in vivo.70
Figure 1.
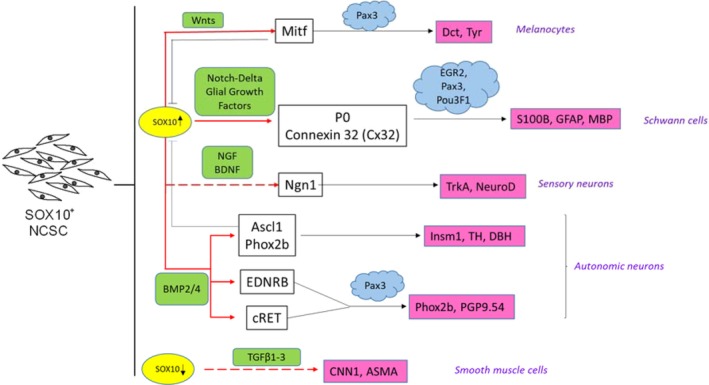
Role of Sox10 in differentiation to neural crest (NC)‐specific lineages. Sox10 is required for differentiation of NCs to neurons, melanocytes, and Schwann cells, but downregulated for smooth muscle cell differentiation. Red arrows represent direct binding sites of Sox10 to lineage specific transcription factors. Dashed red lines indicate no evidence for direct binding of Sox10. Green boxes represent extrinsic signals during differentiation. Factors that act synergistically with Sox10 are depicted in blue clouds. Downstream targets of gene regulation by Sox10 are depicted in pink boxes
The members of TGFβ superfamily—1/2/3 are also able to specify NC cell fates, through a different mechanism than BMP2/4, since they signal through a separate receptor complex and have different downstream effectors.75 TGFβ1/2/3 were shown to promote cardiac smooth muscle specification of rat NCs, as evidenced by the expression of smooth muscle actin.74 TGFβ1/2/3 are also expressed in the developing heart and thought to be active during the induction of cardiomyogenesis in NCs, as well as at final stages of cardiac cushion tissue formation.80
The development of SC from NCs results from exposure of NCs to neuregulin‐1 (NRG1, also known as Glial Growth Factor), an inductive signal of the neuregulin family. NRG1 has been found to suppress neuronal differentiation and specify NCs to a Schwann cell fate.81 The molecular mechanisms governing differentiation of NCs to SC are elaborated later in this review. The interplay of signaling pathways and their effect on different transcription factors are summarized in Figure 1.
4. MOLECULAR MECHANISMS UNDERLYING DIFFERENTIATION OF NC CELLS TO SC
During development, NCSC) delaminate from the neural tube and translocate to various regions of the embryo. Some localize adjacent to the developing nerves, where they differentiate into SCPs, which in turn give rise to a variety of peripheral glial cells that ultimately perform a plethora of functions linked to myelination, neuronal support, regulation of synaptic connectivity and sensory function. ErbB3/Nrg1 interactions and the transcription factor Zeb2 are involved in lineage specification and myelination82, 83, 84; however, the precise mechanisms dictating the fate acquisition from NC to SCP and mature myelinating or nonmyelinating SC, as well as the role of metabolic pathways in SC development and myelination, are not yet completely understood.
4.1. The role of Sox10, Pax3, and HDAC1 in NC lineage specification to SCP
The transcription factors Sox10 and Pax3 are responsible for maintenance of NC multipotency and for differentiation into SC and melanocytes. Although Sox10 is an important NC specifier,85 it is not the sole transcription factor setting the lineage specification and differentiation mechanisms in motion. Peripheral glial lineage specification is abolished when HDAC1/2 knockout mice (flanked by LoxP)86 are crossed with mice expressing Cre recombinase (Cre) under the control of Wnt1 promoter (Wnt1‐Cre).87, 88 Furthermore, HDAC1/2 has been shown to interact with Sox10 and activate two promoters,89, 90 the early lineage marker Myelin Protein Zero (MPZ or P0)61 and Pax3. In turn, a positive feedback loop is set in motion, where Pax3 and Sox10 activate the Sox10 MCS4 enhancer, to maintain high levels of Sox10 but also upregulate expression of Fabp7, another early SCP marker.90, 91 Interestingly, Sox10 expression precedes neuronal differentiation92, 93 but is downregulated during neurogenesis. In addition to Sox10 and Pax3, another key transcription factor FoxD3 plays a key role in glial fate acquisition, by biasing migrating NC against alternative melanocytic fate.94, 95, 96 Considering that Sox10 is also necessary for melanocytic differentiation and survival, the combined action of Sox10 and FoxD3 is necessary for commitment of NC to peripheral glial fate acquisition.
4.2. Path to myelination—Signaling during radial axonal sorting dictates lineage specification
Following HDAC1/2‐Sox10‐Pax3‐related signaling events, SCPs become immature SC, where the process of radial sorting takes place. Radial sorting refers to the process by which SC choose which axons to myelinate during development.97 It relies on the establishment of Schwann cell polarity and cytoskeletal remodeling through Schwann cell‐axon interactions, ultimately resulting in deposition of extracellular matrix (ECM), terminal Schwann cell differentiation, and myelin production. As expected, a variety of signals are necessary for these processes. ECM components like Laminin 211 and 411, Collagen XV and associated integrins α6β1, α7β1, as well as dystroglycan glycosylation enzymes play a pivotal role in attachment and sorting.98, 99, 100, 101, 102, 103, 104, 105 Intracellular signaling regulates cytoskeletal remodeling and radial sorting through molecules such as ILK, FAK, RhoGTPases, Rac1, Cdc42, Profilin, Merlin/NF2, and N‐WASp106, 107, 108, 109, 110, 111, 112, as summarized in Figure S2.
In addition, cell‐cell communications between neurons and SC regulate axonal myelination. Such interactions are mediated through Neuregulin 1 (Nrg1) type III on the surface of axons and ErbB2/3 on the surface of SC.98 Indeed inhibition of the ErbB pathway impaired axonal sorting in zebrafish nerves,113 strongly suggesting that ErbB2/3‐Nrg1 is necessary for Schwann cell radial sorting.114, 115, 116 Furthermore, the Erk/Akt signaling pathways downstream of Nrg1 were implicated in formation of abnormal Remak bundles containing unsorted large caliber axons, possibly through the inactivation of Gab1 (Grb2 associated binder 1), which in turn indirectly decreases Erk but not Akt phosphorylation.117 Finally, the Wnt/β‐catenin signaling has also been implicated in axonal radial sorting,118 as conditional inactivation of β‐catenin resulted in mild radial sorting defects and impaired lamellipodia formation.119
4.3. Metabolic signaling pathway networks in Schwann cell development and myelination
Recent studies revealed an increasingly important role for metabolism in neuronal and glial biology. Accumulating evidence supports the role of key metabolic pathways in SCP development120, 121; the importance of metabolic crosstalk between SC and axons122, 123, 124; and the contributions of metabolic abnormalities to the etiology of axonal degradation and myelin related disorders.125, 126, 127, 128, 129 Specifically, mTORC1, a multiprotein complex and major integrator of several growth factor (e.g., IGF‐1) signaling pathways such as PI3K/AKT and MAPK,130, 131 drives Schwann cell proliferation or myelin production, in a context‐specific manner that depends on developmental stage. On the one hand, mTORC1 impeded myelin production by promoting proliferation of immature SC and inhibiting their terminal differentiation and nerve development.132, 133, 134 On the other hand, myelin production was arrested in mTOR core kinase knockout mice and mTORC1 was identified as the cause of the deficiency due to its prime role in lipid formation and protein biosynthesis.135 In addition, LKB1, a serine threonine kinase upstream of AMPK, a key regulator of cellular energetics,136, 137, 138 plays a pivotal role in myelin production and axonal sorting in SC, with significant repercussions for PNS myelination. Collectively, these studies shed light on the etiology of myelopathies by providing links between metabolism and signaling during development, thereby providing valuable insights for development of therapeutic strategies that may involve the use of NC derived SC for cellular therapies.139, 140, 141
5. NCSCs: A CELL SOURCE FOR THE TREATMENT OF DEMYELINATING DISORDERS
Demyelinating disorders are generally defined as diseases resulting in loss of myelin from neuronal cells with relative preservation of axons. These disorders are a result of damage to myelin sheaths or to the cells that produce them. Demyelinated axons tend to degenerate, resulting in decrease or loss of neurological function. Demyelinating disorders affect both the CNS and PNS and can be caused by auto‐immunity, certain infectious agents, or genetic factors.142 Demyelinating diseases affecting the CNS include in multiple sclerosis (MS), acute‐disseminated encephalomyelitis, acute haemorrhagic leucoencephalitis, and progressive multifocal leukoencephalopathy, with MS being the most common disabling neurological illness affecting young and middle‐aged adults in North America and Europe.143 Demyelinating diseases affecting the PNS include the Guillain‐Barré syndrome, Charcot‐Marie‐Tooth disease, progressive inflammatory neuropathy, and copper deficiency. These diseases often result from loss of function of cells responsible for myelination—oligodendrocytes in the CNS or SC in the PNS. Although the causes and pathology of each of these diseases have been extensively studied, the likelihood of reoccurrence is high and treatment options are limited. Peripheral nerve injuries are also known to result in loss of myelin in and around the site of injury. The incidence of peripheral nerve injuries is fairly high, with a conservative estimates placing it between 13 and 23 per 100,000 people per year.144 Although nerve grafts have dominated the field of experimental treatment for peripheral nerve injuries, their use is often limited by technical difficulties, invasiveness, and mediocre outcomes.145
The last few decades have seen attempts to use SC expanded in culture for neural repair.146 SC play a central role in nerve repair—they become activated after a nerve injury and assume a primitive phenotype, upregulating genes encoding for the production of neurotropic factors and stimulating axonal regeneration. Additionally, once activated, SC produce ECM molecules such as collagen and laminin creating guided tunnels for axonal growth and regeneration.145 Various stem cell sources have been examined for the purpose of generating SC for the treatment of neurological diseases and peripheral nerve injury. Here, we provide a brief review of the therapeutic potential of these stem cells and discuss NCISC as an alternate cell source for Schwann cell engineering.
5.1. Stem cells for generation of autologous SC
Multipotent stem cells isolated from various adult sources may serve as an autologous source of SC. Multipotent mesenchymal stem cells (MSCs) have dominated this field, given their abundance and accessibility via minimally invasive procedures. Adipose tissue has been shown to be a rich source of MSCs that could be coaxed to generate SC in vitro and myelinate axons in the spinal cord in vivo.147, 148, 149, 150, 151, 152 However, these cells have limited in vitro expansion capacity, which hinders their clinical usefulness. Adult neural stem cells isolated from the brain have been shown to differentiate to S100/p75NTR positive SC and improve axonal regeneration in mouse models of peripheral nerve injury.153, 154 However, neural tissue may not be an ideal source of stem cells due to its limited accessibility, invasive procurement, and frequent contamination with fibroblasts that may overpopulate the cultures upon cell expansion in vitro.155 Moreover, the potency and regenerative potential of neural stem cells may be affected by the neurodegenerative disease affecting the patient, calling the effectiveness of this cell source into question.
Since the discovery of the reprograming factors by Yamanaka et al,156 the focus of cell engineering has drastically shifted to reprogramming patient‐specific somatic cells to pluripotent stem cells). These iPSCs can then be coaxed to differentiate to any cell type by exposing them to signals that mimic embryonic development. In this regard, iPSCs have been considered as a potentially unlimited source of SC,157, 158, 159 which have been shown to induce axonal regrowth and facilitate myelination in mouse models of SCI.160, 161 ESCs derived from blastocyst stage embryos can also be used to produce SC, but significant ethical concerns are plaguing their clinical use. Clinical cases with patients undergoing cell‐based therapies for SCI have been evaluated in detail by Harrop et al.162 Though there has been considerable progress in the treatment of SCI and other neurodegenerative diseases, there clearly is a need of more accessible and easily expandable adult stem cell sources for derivation of myelinating SC.
5.2. Adult tissue–derived NCISCs for Schwann cell therapy
NC cells give rise to SC in vivo during development, thus making them one of the most suitable candidates as an autologous cell source of SC. ESCs and iPSCs go through an intermediate NC fate when coaxed to differentiate into SC, and some of these studies were discussed previously.157 Although most studies establish differentiation of pluripotent cells to mature SC, as evidenced by expression of S100β and MBP, few have demonstrated Schwann cell function in vivo.
As discussed above, several adult human tissues can also serve as potential sources of NCISCs. In one of the first studies, adult skin‐derived precursor cells (SKPs) were shown to have the potential to generate SC. SKPs are multipotent cells that are present in the skin dermis and express NC markers including Pax3, Snail, Slug, and NGFR.28 However, SKPs do not express the key NC transcription factors, SOX10 and FOXD3, and they have not been reported to give rise to melanocytes, chondrocytes, or osteocytes, all cells known to be derived from NCs. More recently, lineage tracing studies showed that murine SKPs are of mesodermal origin, in contrast to NCSC that originate from the ectoderm. Nevertheless, mesodermal SKPs gave rise to myelinating SC in the presence of signaling cues such as heregulin β, FGF, and forskolin, suggesting plasticity of developmentally defined lineage boundaries163 (Table 1 ). Furthermore, SKP‐derived SC were shown to myelinate axons as efficiently as CNS‐derived precursors, thus establishing them as a potent cell source for the treatment of SCI.169 Similarly, rat‐derived SKP‐SCs aided sensory,171 motor,175, 176 and behavioral recovery,172 and enhanced peripheral nerve regeneration compared with acellular nerve grafts.174, 177 Finally, SC generated from SKPs were used successfully for the treatment of incomplete cervical SCI.170, 175, 178
Table 1.
Studies describing differentiation of adult‐tissue derived neural crest (NC) cells to Schwann cells (SC)
| Source | Culture conditions | NC markers expressed | SC markers | Functionality | References |
|---|---|---|---|---|---|
| Human epidermal neural crest cells isolated from hair follicles | Alpha‐modified MEM containing retinoic acid (RA) (35 ng/mL), SB431542 (10 μM), rhFGF2 (10 ng/mL), PDGF‐BB (5 ng/mL), forskolin (5 μM) and neuregulin‐1 (200 ng/mL) | SOX10, p75 | SOX10, KROX20, p75NTR, MBP and S100B | Coculture with primary dorsal root ganglion (DRG) neurons | Sakaue and Sieber‐Blum164 |
| Human‐derived hair follicle cells | MesenPRO medium containing neuregulin‐1 (20 ng/mL), transfection with miR‐21 agonist agomir‐21 | p75 | S100β, GFAP | Ni et al165 | |
| Human neonatal foreskin | EBM2 basal medium, FBS (2%), ciliary neurotrophic factor (100 ng/mL), NRG1 (100 ng/mL), FGF2 (4 ng/mL), ascorbic acid (200 mg/mL), Glutamax (0.5X), SB431542 (10 mM) | SOX10, FOXD3, PAX3/7, SNAI2, TFAP2A, MSX1/2, cMYC, and SOX9 | S100β, GFAP, MPZ, PLP1 | NC migration toward DRG in chicken embryo | Bajpai et al30 |
| Human‐derived dental pulp cells | αMEM supplemented with forskolin (5 μM), bFGF (10 ng/mL), PDGF (5 ng/mL), recombinant human neuregulin‐β1 (200 ng/mL) | CD271, SOX10, nestin | S100β | Al‐Zer et al166 | |
| Human bone marrow | DMEM/F12 supplemented with N2 supplement, CNTF (10 ng/mL), l bFGF (10 ng/m), dbcAMP (1 mM), neuregulin‐1β (20 ng/mL) | Nestin, Sox9, TWIST, SLUG, p75, SNAIL1, Brn3a, MSI1 | S100β, MBP, P0 | NC migration toward DRG in chicken embryo | Coste et al167 |
| Human skin mesenchymal stem cells | Alpha‐MEM containing FBS (10%), RA (35 ng/mL), forskolin (5 μM), rh‐FGF‐2 (10 ng/mL), rhPDGF‐AA (5 ng/mL) and neuregulin‐1β (200 ng/mL) | p75NTR, Sox10, Notch1, integrin‐4α, Ap2α and Pax6 | S100β, MBP | BDNF secretion by ELISA | Saulite et al.168 |
| Postnatal rat bone marrow | DMEM/F12 supplemented with FBS (5%), all‐trans‐retinoic acid (tRA) (35 ng/mL), forskolin (5 mM), βheregulin‐1 (HRG‐1b) (200 ng/mL), FGF2 (10 ng/mL), PDGF‐AA (10 ng/mL), and N2 (1%) | p75, nestin | S100β, GFAP, CNPase p75 | Coculture with primary dorsal root ganglion (DRG) neurons | Shi et al11 |
| Human skin–derived precursors | Neurobasal medium containing 1% FBS plus 1% N2 supplement, 4 μM forskolin, and 10 ng/mL heregulin β | Pax3, snail, slug and NGFR | S100β, p75 | — | Toma et al28 |
| Mice skin–derived precursors | — | Nestin | MBP, GFAP | Myelination of spinal cord in mice | Mozafari et al169 |
| Neonatal SKPs | — | — | — | Myelination of spinal cord in rats | Vasudeva et al170 |
| Rodent/human skin mesenchymal precursors | DMEM/F12 at 3:1, N2 supplement (2%), neuregulin‐1β (10 ng/mL), and forskolin (4 μM) | — | GFAP, S100β, P0, p75NTR, and NECL4 | Alignment of MBP expressing Schwann cells in vivo | Krause et al.163 |
| Rat SKPs | — | — | p75 | Sensory (thermal) recovery, axonal regeneration | Shakhbazau et al171 |
| Rat SKPs | DMEM/F12, Neuregulin (50 ng/mL), forskolin (5 μM), N2 supplement (1%) | — | p75, nestin, MBP | Integration and ensheathment of regenerating axons and myelination, behavioral recovery | Khuong et al172 |
| Mouse SKPs | Serum‐free DMEM/F12 supplemented with dibutyryl cyclic AMP (10 mM), bFGF (10 ng/mL), and neuregulin (20 ng/mL) | — | GFAP, S100β | — | Kanget al173 |
| Rat SKPs | DMEM/F12 media with forksolin (4 μM), neuregulin‐1 (10 ng/mL), N2 supplement (1%) | — | S100β, p75 | Secretion of bioactive neurotrophins, axonal regeneration | Walsh et al174 |
| Rat SKPs | DMEM/F‐12 (3:1) containing forskolin (5 μM), neuregulin‐1 (50 ng/mL), N2 supplement (1%) | — | S100β, p75, P0 | Axonal growth and myelination | Biernaskie et al175 |
Abbreviations: BDNF, brain‐derived neurotrophic factor; DMEM, Dulbecco's Modified Eagle Medium; ELISA, enzyme‐linked immunosorbent assay; FGF, fibroblast growth factor; GFAP, glial fibrillary acidic protein; MBP, myelin basic protein; NRG1, neuregulin‐1; SKP, skin‐derived precursor cell.
Recently, NCISCs were isolated from the bulge of hair follicles and coaxed to differentiate into SC, a process that required NRG1 and micro‐RNA miR‐21.165 These SC expressed SOX10, p75NTR (NGFR), KROX20, MBP, and S100β and interacted with axons in a DRG coculture model, thus providing evidence of their functionality. Dental pulp stem cells derived from human wisdom teeth have also been shown to resemble NCSC and were capable of differentiating into S100β‐expressing SC.166 Finally, human and rat bone marrow–derived NC‐like cells were also capable of differentiating into SC in vitro.11, 167
Recently, our laboratory showed that cultures of KC from interfollicular epidermis could be the source of NClSCs (termed KC‐NC), after induction with specific chemical cues.30 KC‐derived NCISCs could be coaxed to differentiate into functional neurons, SC, melanocytes, osteocytes, chondrocytes, adipocytes, and smooth muscle cells in vitro. Most notably, upon transplantation into chick embryos, KC‐NCs migrated along stereotypical pathways and gave rise to multiple NC derivatives, providing strong support of their NC‐like phenotype. Specifically, these cells gave rise to BLBP+ glial cells in ovo and were localized around the axon bundles. Interestingly, KC‐NC from aged donors maintained the same differentiation potential in vitro and in ovo,32 indicating the potential of adult epidermis as a source of KC‐NC for the treatment of neurodegenerative diseases. Current studies in our laboratory employ the hypomyelinating Shiverer/Rag2−/− mouse—a model of congenital hypomyelinating disease that has become the gold standard for the assessment of myelinating cell preparations—to examine whether KC‐NC or SC derived from them can be used to myelinate axons and rescue the shiverer phenotype. Table 1 depicts studies describing differentiation of adult tissue–derived NClSCs to SC.
6. CONCLUSION AND FUTURE PERSPECTIVES
NC cells have attracted great interest due to their ability to differentiate into multiple cell types.156 Although NC stem cells are a transient cell population in developing embryos, multiple investigators have isolated cells with NC‐like characteristics, including transcriptional profile and differentiation potential, from a variety of adult tissues like DRG, bone marrow, skin, carotid body, whisker pad, heart, gut, and cranial tissues like cornea, iris, hard palate, dental pulp, and oral mucosa.1 Regardless of source, NC‐like cells have been coaxed to differentiate into SC, neurons, chondrocytes, smooth muscle cells, and even cardiomyocytes,2, 179, 180 providing a multipotent stem cell source for the treatment of demyelinating and other neurodegenerative disorders. Patient‐derived NCISCs harboring mutations for neurogenic diseases may also be used to study disease pathogenesis as well as provide a platform for drug screening further increasing their clinical potential.1
The use of SC derived from adult tissue–derived NClSCs for the treatment of SCI has gained momentum in recent years, after demonstration of successful myelination of axons in vitro and in vivo. Though these results are very promising, most studies use cells derived from mice or rats, which are implanted in a mouse contusion model of SCI. However, the potential of these cells for the treatment of demyelinating disease in large animal models or humans has yet to be established. Furthermore, most studies have focused on myelination after SCI and not treatment of demyelinating disorders of the CNS or PNS, leaving the field fairly unexplored in this domain. Finally, isolation of NCISCs from patients suffering from neurodegenerative disorders and their differentiation to SC and neurons may help identify abnormalities in myelin production and nerve conduction, aiding in better disease diagnosis and development of patient‐specific therapies.
CONFLICT OF INTEREST
The authors indicated no potential conflicts of interest.
AUTHOR CONTRIBUTIONS
P.M. wrote the manuscript, specifically parts on introduction to the topic, sources of neural crest and their genetic regulation, their application for treatment of demyelinating disorders and made the manuscript figures. G.T. wrote the manuscript, specifically mechanisms underlying differentiation of neural crest cells to Schwann cells and made the manuscript figures. M.E.B. wrote the manuscript, specifically content on how Schwann cell precursors contribute to neural crest derivatives, and gene regulatory networks controlling lineage diversification. S.T.A. conceived overall review content, writing, and editing the manuscript and project supervision.
Supporting information
Figure S1: Adult sources for in vitro expansion of multipotent stem cells exhibiting an NC‐like phenotype. From the above mentioned sources, only cells from dental pulp and hair follicles have been successfully isolated and studied in humans
Figure S2: A Neural Crest Stem Cell's journey to myelination. Delaminating, post migratory NC follow Wnt and FGF signals and upregulate key transcription factors like Sox10, FoxD3 and Pax3. By recruiting HDAC1/2, myelin protein 0 is upregulated leading to a Schwann cell precursor phenotype. During this immature Schwann cell stage, Schwann cells initially approach multiple axons and after undergoing significant cytoskeletal reorganization, each one myelinates a single axon, a process referred to as axonal sorting. Finally, the mature Schwann cell envelopes along the neuronal axon with myelin sheath to facilitate saltatory electrical impulse conduction.
ACKNOWLEDGMENTS
This work was supported by grants from the National Institutes of Health R01 EB023114 (S.T.A.) and the New York Stem Cell Science NYSTEM (Contract #C30290GG, S.T.A.).
Mehrotra P, Tseropoulos G, Bronner ME, Andreadis ST. Adult tissue–derived neural crest‐like stem cells: Sources, regulatory networks, and translational potential. STEM CELLS Transl Med. 2020;9:328–341. 10.1002/sctm.19-0173
Contributor Information
Pihu Mehrotra, Email: pihumehr@buffalo.edu.
Stelios T. Andreadis, Email: sandread@buffalo.edu.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Liu JA, Cheung M. Neural crest stem cells and their potential therapeutic applications. Dev Biol. 2016;419:199‐216. [DOI] [PubMed] [Google Scholar]
- 2. Martik ML, Bronner ME. Regulatory logic underlying diversification of the neural crest. Trends Genet. 2017;33:715‐727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. O'Rahilly R, Müller F. The development of the neural crest in the human. J Anat. 2007;211:335‐351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Stemple DL, Anderson DJ. Isolation of a stem cell for neurons and glia from the mammalian neural crest. Cell. 1992;71:973‐985. [DOI] [PubMed] [Google Scholar]
- 5. Betancur P, Bronner‐Fraser M, Sauka‐Spengler T. Assembling neural crest regulatory circuits into a gene regulatory network. Annu Rev Cell Dev Biol. 2010;26:581‐603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sauka‐Spengler T, Bronner‐Fraser M. A gene regulatory network orchestrates neural crest formation. Nat Rev Mol Cell Biol. 2008;9:557‐568. [DOI] [PubMed] [Google Scholar]
- 7. Thiery JP, Sleeman JP. Complex networks orchestrate epithelial–mesenchymal transitions. Nat Rev Mol Cell Biol. 2006;7:131‐142. [DOI] [PubMed] [Google Scholar]
- 8. Le Douarin N, Kalcheim C. The Neural Crest. Cambridge, UK: Cambridge University Press; 1999. [Google Scholar]
- 9. Morrison SJ, White PM, Zock C, Anderson DJ. Prospective identification, isolation by flow cytometry, and in vivo self‐renewal of multipotent mammalian neural crest stem cells. Cell. 1999;96:737‐749. [DOI] [PubMed] [Google Scholar]
- 10. Nagoshi N, Shibata S, Kubota Y, et al. Ontogeny and multipotency of neural crest‐derived stem cells in mouse bone marrow, dorsal root ganglia, and whisker pad. Cell Stem Cell. 2008;2:392‐403. [DOI] [PubMed] [Google Scholar]
- 11. Shi H, Gong Y, Qiang L, et al. Derivation of Schwann cell precursors from neural crest cells resident in bone marrow for cell therapy to improve peripheral nerve regeneration. Biomaterials. 2016;89:25‐37. [DOI] [PubMed] [Google Scholar]
- 12. Hassell JR, Birk DE. The molecular basis of corneal transparency. Exp Eye Res. 2010;91:326‐335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gage PJ, Rhoades W, Prucka SK, Hjalt T. Fate maps of neural crest and mesoderm in the mammalian eye. Invest Ophthalmol Vis Sci. 2005;46:4200‐4208. [DOI] [PubMed] [Google Scholar]
- 14. Whikehart, D. , Corneal Endothelium: Overview. 2010.
- 15. Williams AL, Bohnsack BL. Neural crest derivatives in ocular development: discerning the eye of the storm. Birth Defects Res C Embryo Today. 2015;105:87‐95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lwigale PY, Cressy PA, Bronner‐Fraser M. Corneal keratocytes retain neural crest progenitor cell properties. Dev Biol. 2005;288:284‐293. [DOI] [PubMed] [Google Scholar]
- 17. Du Y, et al. Multipotent stem cells in human corneal stroma. Stem Cells. 2005;23:1266‐1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Joseph NM, He S, Quintana E, Kim YG, Núñez G, Morrison SJ. Enteric glia are multipotent in culture but primarily form glia in the adult rodent gut. J Clin Invest. 2011;121:3398‐3411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kruger GM, Mosher JT, Bixby S, Joseph N, Iwashita T, Morrison SJ. Neural crest stem cells persist in the adult gut but undergo changes in self‐renewal, neuronal subtype potential, and factor responsiveness. Neuron. 2002;35:657‐669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dettmann HM, Zhang Y, Wronna N, et al. Isolation, expansion and transplantation of postnatal murine progenitor cells of the enteric nervous system. PLoS One. 2014;9:e97792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hotta R, Stamp LA, Foong JPP, et al. Transplanted progenitors generate functional enteric neurons in the postnatal colon. J Clin Invest. 2013;123:1182‐1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ito K, Sieber‐Blum M. In vitro clonal analysis of quail cardiac neural crest development. Dev Biol. 1991;148:95‐106. [DOI] [PubMed] [Google Scholar]
- 23. Tomita Y, Matsumura K, Wakamatsu Y, et al. Cardiac neural crest cells contribute to the dormant multipotent stem cell in the mammalian heart. J Cell Biol. 2005;170:1135‐1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. El‐Helou V, et al. Cardiac resident nestin+ cells participate in reparative vascularisation. J Cell Physiol. 2013;228:1844‐1853. [DOI] [PubMed] [Google Scholar]
- 25. Fernandes KJ, et al. A dermal niche for multipotent adult skin‐derived precursor cells. Nat Cell Biol. 2004;6:1082‐1093. [DOI] [PubMed] [Google Scholar]
- 26. Sieber‐Blum M, Grim M, Hu YF, Szeder V. Pluripotent neural crest stem cells in the adult hair follicle. Dev Dyn. 2004;231:258‐269. [DOI] [PubMed] [Google Scholar]
- 27. Toma JG, Akhavan M, Fernandes KJL, et al. Isolation of multipotent adult stem cells from the dermis of mammalian skin. Nat Cell Biol. 2001;3:778‐784. [DOI] [PubMed] [Google Scholar]
- 28. Toma JG, McKenzie IA, Bagli D, Miller FD. Isolation and characterization of multipotent skin‐derived precursors from human skin. Stem Cells. 2005;23:727‐737. [DOI] [PubMed] [Google Scholar]
- 29. Sieber‐Blum M, Schnell L, Grim M, Hu YF, Schneider R, Schwab ME. Characterization of epidermal neural crest stem cell (EPI‐NCSC) grafts in the lesioned spinal cord. Mol Cell Neurosci. 2006;32:67‐81. [DOI] [PubMed] [Google Scholar]
- 30. Bajpai VK, Kerosuo L, Tseropoulos G, et al. Reprogramming postnatal human epidermal keratinocytes toward functional neural crest fates. Stem Cells. 2017;35:1402‐1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tseropoulos G, Moghadasi Boroujeni S, Bajpai VK, Lei P, Andreadis ST. Derivation of neural crest stem cells from human epidermal keratinocytes requires FGF‐2, IGF‐1, and inhibition of TGF‐β1. Bioeng Transl Med. 2018;3:256‐264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Boroujeni SM, et al. Neural crest stem cells from human epidermis of aged donors maintain their multipotency in vitro and in vivo. Sci Rep. 2019;9:9750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Adameyko I, Lallemend F, Aquino JB, et al. Schwann cell precursors from nerve innervation are a cellular origin of melanocytes in skin. Cell. 2009;139:366‐379. [DOI] [PubMed] [Google Scholar]
- 34. Dyachuk V, Furlan A, Shahidi MK, et al. Parasympathetic neurons originate from nerve‐associated peripheral glial progenitors. Science. 2014;345:82‐87. [DOI] [PubMed] [Google Scholar]
- 35. Espinosa‐Medina I, Jevans B, Boismoreau F, et al. Dual origin of enteric neurons in vagal Schwann cell precursors and the sympathetic neural crest. Proc Natl Acad Sci. 2017;114:11980‐11985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Uesaka T, Nagashimada M, Enomoto H. Neuronal differentiation in Schwann cell lineage underlies postnatal neurogenesis in the enteric nervous system. J Neurosci. 2015;35:9879‐9888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jaegle M, Mandemakers W, Broos L, et al. The POU factor Oct‐6 and Schwann cell differentiation. Science. 1996;273:507‐510. [DOI] [PubMed] [Google Scholar]
- 38. Buchstaller J, Sommer L, Bodmer M, Hoffmann R, Suter U, Mantei N. Efficient isolation and gene expression profiling of small numbers of neural crest stem cells and developing Schwann cells. J Neurosci. 2004;24:2357‐2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Takahashi M, Osumi N. Identification of a novel type II classical cadherin: rat cadherin19 is expressed in the cranial ganglia and Schwann cell precursors during development. Dev Dyn. 2005;232:200‐208. [DOI] [PubMed] [Google Scholar]
- 40. D'antonio M, et al. Gene profiling and bioinformatic analysis of Schwann cell embryonic development and myelination. Glia. 2006;53:501‐515. [DOI] [PubMed] [Google Scholar]
- 41. Li J, Habbes HW, Eiberger J, Willecke K, Dermietzel R, Meier C. Analysis of connexin expression during mouse Schwann cell development identifies connexin29 as a novel marker for the transition of neural crest to precursor cells. Glia. 2007;55:93‐103. [DOI] [PubMed] [Google Scholar]
- 42. Furlan A, et al. Multipotent peripheral glial cells generate neuroendocrine cells of the adrenal medulla. Science. 2017;357:eaal3753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Jessen KR, Mirsky R. Schwann cell precursors; multipotent glial cells in embryonic nerves. Front Mol Neurosci. 2019;12:69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rada‐Iglesias A, Bajpai R, Prescott S, Brugmann SA, Swigut T, Wysocka J. Epigenomic annotation of enhancers predicts transcriptional regulators of human neural crest. Cell Stem Cell. 2012;11:633‐648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Soldatov R, et al. Spatiotemporal structure of cell fate decisions in murine neural crest. Science. 2019;364(6444):eaas9536. [DOI] [PubMed] [Google Scholar]
- 46. Meulemans D, Bronner‐Fraser M. Gene‐regulatory interactions in neural crest evolution and development. Dev Cell. 2004;7:291‐299. [DOI] [PubMed] [Google Scholar]
- 47. Simões‐Costa M, Bronner ME. Establishing neural crest identity: a gene regulatory recipe. Development. 2015;142:242‐257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Stewart RA, Arduini BL, Berghmans S, et al. Zebrafish foxd3 is selectively required for neural crest specification, migration and survival. Dev Biol. 2006;292:174‐188. [DOI] [PubMed] [Google Scholar]
- 49. Kelsh RN. Sorting out Sox10 functions in neural crest development. Bioessays. 2006;28:788‐798. [DOI] [PubMed] [Google Scholar]
- 50. Sauka‐Spengler T, Bronner‐Fraser M. Insights from a sea lamprey into the evolution of neural crest gene regulatory network. Biol Bull. 2008;214:303‐314. [DOI] [PubMed] [Google Scholar]
- 51. Kim J, Lo L, Dormand E, Anderson DJ. SOX10 maintains multipotency and inhibits neuronal differentiation of neural crest stem cells. Neuron. 2003;38:17‐31. [DOI] [PubMed] [Google Scholar]
- 52. Carney TJ, Dutton KA, Greenhill E, et al. A direct role for Sox10 in specification of neural crest‐derived sensory neurons. Development. 2006;133:4619‐4630. [DOI] [PubMed] [Google Scholar]
- 53. Zhu L, Lee HO, Jordan CRS, Cantrell VA, Southard‐Smith EM, Shin MK. Spatiotemporal regulation of endothelin receptor‐B by SOX10 in neural crest–derived enteric neuron precursors. Nat Genet. 2004;36:732‐737. [DOI] [PubMed] [Google Scholar]
- 54. Schreiner S, Cossais F, Fischer K, et al. Hypomorphic Sox10 alleles reveal novel protein functions and unravel developmental differences in glial lineages. Development. 2007;134:3271‐3281. [DOI] [PubMed] [Google Scholar]
- 55. Svaren J, Meijer D. The molecular machinery of myelin gene transcription in Schwann cells. Glia. 2008;56:1541‐1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Finzsch M, Schreiner S, Kichko T, et al. Sox10 is required for Schwann cell identity and progression beyond the immature Schwann cell stage. J Cell Biol. 2010;189:701‐712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Peirano RI, Goerich DE, Riethmacher D, Wegner M. Protein zero gene expression is regulated by the glial transcription factor Sox10. Mol Cell Biol. 2000;20:3198‐3209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Bondurand N, Girard M, Pingault V, Lemort N, Dubourg O, Goossens M. Human Connexin 32, a gap junction protein altered in the X‐linked form of Charcot–Marie–Tooth disease, is directly regulated by the transcription factor SOX10. Hum Mol Genet. 2001;10:2783‐2795. [DOI] [PubMed] [Google Scholar]
- 59. Weston JA. 6 sequential segregation and fate of developmentally restricted intermediate cell populations in the neural crest lineage. Curr Top Dev Biol. 1991;25:133‐153. [DOI] [PubMed] [Google Scholar]
- 60. Wehrle‐Haller B, Weston JA. Receptor tyrosine kinase‐dependent neural crest migration in response to differentially localized growth factors. Bioessays. 1997;19:337‐345. [DOI] [PubMed] [Google Scholar]
- 61. Hagedorn L, Suter U, Sommer L. P0 and PMP22 mark a multipotent neural crest‐derived cell type that displays community effects in response to TGF‐beta family factors. Development. 1999;126:3781‐3794. [DOI] [PubMed] [Google Scholar]
- 62. Sieber‐Blum M, Ito K, Richardson MK, Langtimm CJ, Duff RS. Distribution of pluripotent neural crest cells in the embryo and the role of brain‐derived neurotrophic factor in the commitment to the primary sensory neuron lineage. Dev Neurobiol. 1993;24:173‐184. [DOI] [PubMed] [Google Scholar]
- 63. Le Douarin NM, Dupin E. Cell lineage analysis in neural crest ontogeny. Dev Neurobiol. 1993;24:146‐161. [DOI] [PubMed] [Google Scholar]
- 64. Beall AC, Rosenquist TH. Smooth muscle cells of neural crest origin form the aorticopulmonary septum in the avian embryo. Anat Rec. 1990;226:360‐366. [DOI] [PubMed] [Google Scholar]
- 65. Phillips MT, Kirby ML, Forbes G. Analysis of cranial neural crest distribution in the developing heart using quail‐chick chimeras. Circ Res. 1987;60:27‐30. [DOI] [PubMed] [Google Scholar]
- 66. Fantauzzo KA, Soriano P. Receptor tyrosine kinase signaling: regulating neural crest development one phosphate at a time. Curr Top Dev Biol. 2015;111:135‐182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Henion PD, Garner AS, Large TH, Weston JA. trkC‐mediated NT‐3 signaling is required for the early development of a subpopulation of neurogenic neural crest cells. Dev Biol. 1995;172:602‐613. [DOI] [PubMed] [Google Scholar]
- 68. Meyer D, Birchmeier C. Multiple essential functions of neuregulin in development. Nature. 1995;378:386‐390. [DOI] [PubMed] [Google Scholar]
- 69. Schatteman GC, Morrison‐Graham K, van Koppen A, Weston JA, Bowen‐Pope DF. Regulation and role of PDGF receptor alpha‐subunit expression during embryogenesis. Development. 1992;115:123‐131. [DOI] [PubMed] [Google Scholar]
- 70. Greenwood AL, Turner EE, Anderson DJ. Identification of dividing, determined sensory neuron precursors in the mammalian neural crest. Development. 1999;126:3545‐3559. [DOI] [PubMed] [Google Scholar]
- 71. Ma Q, Fode C, Guillemot F, Anderson DJ. Neurogenin1 and neurogenin2 control two distinct waves of neurogenesis in developing dorsal root ganglia. Genes Dev. 1999;13:1717‐1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Reedy MV, Faraco CD, Erickson CA. The delayed entry of thoracic neural crest cells into the dorsolateral path is a consequence of the late emigration of melanogenic neural crest cells from the neural tube. Dev Biol. 1998;200:234‐246. [DOI] [PubMed] [Google Scholar]
- 73. LaBonne C, Bronner‐Fraser M. Neural crest induction in Xenopus: evidence for a two‐signal model. Development. 1998;125:2403‐2414. [DOI] [PubMed] [Google Scholar]
- 74. Shah NM, Groves AK, Anderson DJ. Alternative neural crest cell fates are instructively promoted by TGFβ superfamily members. Cell. 1996;85:331‐343. [DOI] [PubMed] [Google Scholar]
- 75. Dorsky RI, Moon RT, Raible DW. Environmental signals and cell fate specification in premigratory neural crest. Bioessays. 2000;22:708‐716. [DOI] [PubMed] [Google Scholar]
- 76. Dorsky RI, Moon RT, Raible DW. Control of neural crest cell fate by the Wnt signalling pathway. Nature. 1998;396:370‐373. [DOI] [PubMed] [Google Scholar]
- 77. Hari L, Brault V, Kléber M, et al. Lineage‐specific requirements of β‐catenin in neural crest development. J Cell Biol. 2002;159:867‐880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Lee H‐Y, Kléber M, Hari L, et al. Instructive role of Wnt/ß‐catenin in sensory fate specification in neural crest stem cells. Science. 2004;303:1020‐1023. [DOI] [PubMed] [Google Scholar]
- 79. Lo L, Sommer L, Anderson DJ. MASH1 maintains competence for BMP2‐induced neuronal differentiation in post‐migratory neural crest cells. Curr Biol. 1997;7:440‐450. [DOI] [PubMed] [Google Scholar]
- 80. Dickson MC, Slager HG, Duffie E, Mummery CL, Akhurst RJ. RNA and protein localisations of TGF beta 2 in the early mouse embryo suggest an involvement in cardiac development. Development. 1993;117:625‐639. [DOI] [PubMed] [Google Scholar]
- 81. Shah NM, Marchionni MA, Isaacs I, Stroobant P, Anderson DJ. Glial growth factor restricts mammalian neural crest stem cells to a glial fate. Cell. 1994;77:349‐360. [DOI] [PubMed] [Google Scholar]
- 82. Aquino JB, Sierra R. Schwann cell precursors in health and disease. Glia. 2018;66:465‐476. [DOI] [PubMed] [Google Scholar]
- 83. Luca Franco Castelnovo VB, Melfi S, Ballabio M, Colleoni D, Magnaghi V. Schwann cell development, maturation and regeneration: a focus on classic and emerging intracellular signaling pathways. Neural Regen Res. 2017;12:1013‐1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Bronner ME, Simões‐Costa M. The neural crest migrating into the twenty‐first century. Curr Top Dev Biol. 2016;116:115‐134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Weider M, Wegner M. SoxE factors: transcriptional regulators of neural differentiation and nervous system development. Semin Cell Dev Biol. 2017;63:35‐42. [DOI] [PubMed] [Google Scholar]
- 86. Jacob C, Lotscher P, Engler S, et al. HDAC1 and HDAC2 control the specification of neural crest cells into peripheral glia. J Neurosci. 2014;34:6112‐6122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Auerbach R. Analysis of the developmental effects of a lethal mutation in the house mouse. J Exp Zool. 1954;127:305‐329. [Google Scholar]
- 88. Franz T. Defective ensheathment of motoric nerves in the Splotch mutant mouse. Cells Tissues Organs. 1990;138:246‐253. [DOI] [PubMed] [Google Scholar]
- 89. Antonellis A, Huynh JL, Lee‐Lin SQ, et al. Identification of neural crest and glial enhancers at the mouse Sox10 locus through transgenesis in zebrafish. PLoS Genet. 2008;4:e1000174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Wahlbuhl M, Reiprich S, Vogl MR, Bösl MR, Wegner M. Transcription factor Sox10 orchestrates activity of a neural crest‐specific enhancer in the vicinity of its gene. Nucleic Acids Res. 2011;40:88‐101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Kurtz A, Zimmer A, Schnütgen F, Brüning G, Spener F, Müller T. The expression pattern of a novel gene encoding brain‐fatty acid binding protein correlates with neuronal and glial cell development. Development. 1994;120:2637‐2649. [DOI] [PubMed] [Google Scholar]
- 92. Britsch S, Goerich DE, Riethmacher D, et al. The transcription factor Sox10 is a key regulator of peripheral glial development. Genes Dev. 2001;15:66‐78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Kuhlbrodt K, Herbarth B, Sock E, Hermans‐Borgmeyer I, Wegner M. Sox10, a novel transcriptional modulator in glial cells. J Neurosci. 1998;18:237‐250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Nitzan E, Krispin S, Pfaltzgraff ER, Klar A, Labosky PA, Kalcheim C. A dynamic code of dorsal neural tube genes regulates the segregation between neurogenic and melanogenic neural crest cells. Development. 2013;140:2269‐2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Nitzan E, Pfaltzgraff ER, Labosky PA, Kalcheim C. Neural crest and Schwann cell progenitor‐derived melanocytes are two spatially segregated populations similarly regulated by Foxd3. Proc Natl Acad Sci USA. 2013;110:12709‐12714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Thomas AJ, Erickson CA. FOXD3 regulates the lineage switch between neural crest‐derived glial cells and pigment cells by repressing MITF through a non‐canonical mechanism. Development. 2009;136:1849‐1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Feltri ML, Poitelon Y, Previtali SC. How Schwann cells sort axons: new concepts. Neuroscientist. 2016;22:252‐265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Berti C, Bartesaghi L, Ghidinelli M, et al. Non‐redundant function of dystroglycan and β1 integrins in radial sorting of axons. Development. 2011;138:4025‐4037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Feltri ML, Porta DG, Previtali SC, et al. Conditional disruption of β1 integrin in Schwann cells impedes interactions with axons. J Cell Biol. 2002;156:199‐210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Occhi S, Zambroni D, del Carro U, et al. Both laminin and Schwann cell dystroglycan are necessary for proper clustering of sodium channels at nodes of Ranvier. J Neurosci. 2005;25:9418‐9427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Patton BL, Wang B, Tarumi YS, Seburn KL, Burgess RW. A single point mutation in the LN domain of LAMA2 causes muscular dystrophy and peripheral amyelination. J Cell Sci. 2008;121:1593‐1604. [DOI] [PubMed] [Google Scholar]
- 102. Pellegatta M, et al. α6β1 and α7β1 integrins are required in Schwann cells to sort axons. J Neurosci. 2013;33:17995‐18007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Saito F, Masaki T, Saito Y, et al. Defective peripheral nerve myelination and neuromuscular junction formation in fukutin‐deficient chimeric mice. J Neurochem. 2007;101:1712‐1722. [DOI] [PubMed] [Google Scholar]
- 104. Wallquist W, Plantman S, Thams S, et al. Impeded interaction between Schwann cells and axons in the absence of laminin α4. J Neurosci. 2005;25:3692‐3700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Yang D, Bierman J, Tarumi YS, et al. Coordinate control of axon defasciculation and myelination by laminin‐2 and‐8. J Cell Biol. 2005;168:655‐666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Benninger Y, Thurnherr T, Pereira JA, et al. Essential and distinct roles for cdc42 and rac1 in the regulation of Schwann cell biology during peripheral nervous system development. J Cell Biol. 2007;177:1051‐1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Grove M, Komiyama NH, Nave KA, Grant SG, Sherman DL, Brophy PJ. FAK is required for axonal sorting by Schwann cells. J Cell Biol. 2007;176:277‐282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Guo L, Moon C, Niehaus K, Zheng Y, Ratner N. Rac1 controls Schwann cell myelination through cAMP and NF2/merlin. J Neurosci. 2012;32:17251‐17261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Nodari A, Zambroni D, Quattrini A, et al. β1 integrin activates Rac1 in Schwann cells to generate radial lamellae during axonal sorting and myelination. J Cell Biol. 2007;177:1063‐1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Novak N, Bar V, Sabanay H, et al. N‐WASP is required for membrane wrapping and myelination by Schwann cells. J Cell Biol. 2011;192:243‐250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Pereira JA, Benninger Y, Baumann R, et al. Integrin‐linked kinase is required for radial sorting of axons and Schwann cell remyelination in the peripheral nervous system. J Cell Biol. 2009;185:147‐161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Jin F, Dong B, Georgiou J, et al. N‐WASp is required for Schwann cell cytoskeletal dynamics, normal myelin gene expression and peripheral nerve myelination. Development. 2011;138:1329‐1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Raphael AR, Lyons DA, Talbot WS. ErbB signaling has a role in radial sorting independent of Schwann cell number. Glia. 2011;59:1047‐1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Nave K‐A, Salzer JL. Axonal regulation of myelination by neuregulin 1. Curr Opin Neurobiol. 2006;16:492‐500. [DOI] [PubMed] [Google Scholar]
- 115. Newbern J, Birchmeier C. Nrg1/ErbB signaling networks in Schwann cell development and myelination. Semin Cell Dev Biol. 2010;21(9):922‐928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Taveggia C, Zanazzi G, Petrylak A, et al. Neuregulin‐1 type III determines the ensheathment fate of axons. Neuron. 2005;47:681‐694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Shin YK, Jang SY, Park SY, et al. Grb2‐associated binder‐1 is required for neuregulin‐1‐induced peripheral nerve myelination. J Neurosci. 2014;34:7657‐7662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Grigoryan T, et al. Wnt/Rspondin/β‐catenin signals control axonal sorting and lineage progression in Schwann cell development. Proc Natl Acad Sci. 2013;110(45):18174‐18179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Lewallen KA, Shen YAA, de la Torre AR, Ng BK, Meijer D, Chan JR. Assessing the role of the cadherin/catenin complex at the Schwann cell–axon interface and in the initiation of myelination. J Neurosci. 2011;31:3032‐3043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Beirowski B. The LKB1‐AMPK and mTORC1 metabolic signaling networks in schwann cells control axon integrity and myelination: assembling and upholding nerves by metabolic signaling in Schwann cells. Bioessays. 2019;41:e1800075. [DOI] [PubMed] [Google Scholar]
- 121. Grigoryan T, Birchmeier W. Molecular signaling mechanisms of axon–glia communication in the peripheral nervous system. Bioessays. 2015;37:502‐513. [DOI] [PubMed] [Google Scholar]
- 122. Morrison BM, Lee Y, Rothstein JD. Oligodendroglia: metabolic supporters of axons. Trends Cell Biol. 2013;23:644‐651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Baltan S. Can lactate serve as an energy substrate for axons in good times and in bad, in sickness and in health? Metab Brain Dis. 2015;30:25‐30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Saab AS, Tzvetanova ID, Nave KA. The role of myelin and oligodendrocytes in axonal energy metabolism. Curr Opin Neurobiol. 2013;23:1065‐1072. [DOI] [PubMed] [Google Scholar]
- 125. Eckersley L. Role of the Schwann cell in diabetic neuropathy. Int Rev Neurobiol. 2002;50:293‐321. [DOI] [PubMed] [Google Scholar]
- 126. Chowdhury SK, Smith DR, Fernyhough P. The role of aberrant mitochondrial bioenergetics in diabetic neuropathy. Neurobiol Dis. 2013;51:56‐65. [DOI] [PubMed] [Google Scholar]
- 127. Kim ES, Isoda F, Kurland I, Mobbs CV. Glucose‐induced metabolic memory in Schwann cells: prevention by PPAR agonists. Endocrinology. 2013;154:3054‐3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Zenker J, Ziegler D, Chrast R. Novel pathogenic pathways in diabetic neuropathy. Trends Neurosci. 2013;36:439‐449. [DOI] [PubMed] [Google Scholar]
- 129. Goncalves NP, et al. Schwann cell interactions with axons and microvessels in diabetic neuropathy. Nat Rev Neurol. 2017;13:135‐147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Dibble CC, Manning BD. Signal integration by mTORC1 coordinates nutrient input with biosynthetic output. Nat Cell Biol. 2013;15:555‐564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Laplante M, Sabatini DM. Regulation of mTORC1 and its impact on gene expression at a glance. J Cell Sci. 2013;126:1713‐1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Wong KM, Babetto E, Beirowski B. Axon degeneration: make the Schwann cell great again. Neural Regen Res. 2017;12:518‐524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Beirowski B, Wong KM. mTORC1 promotes Schwann cell cycling and myelinogenesis. Cell Cycle. 2017;16:1637‐1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Wong KM, Beirowski B. Multiple lines of inhibitory feedback on AKT kinase in Schwann cells lacking TSC1/2 hint at distinct functions of mTORC1 and AKT in nerve development. Commun Integr Biol. 2018;11:e1433441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Norrmen C, et al. mTORC1 controls PNS myelination along the mTORC1‐RXRgamma‐SREBP‐lipid biosynthesis axis in Schwann cells. Cell Rep. 2014;9:646‐660. [DOI] [PubMed] [Google Scholar]
- 136. Maillet V, Boussetta N, Leclerc J, et al. LKB1 as a gatekeeper of hepatocyte proliferation and genomic integrity during liver regeneration. Cell Rep. 2018;22:1994‐2005. [DOI] [PubMed] [Google Scholar]
- 137. Bays JL, Campbell HK, Heidema C, Sebbagh M, DeMali KA. Linking E‐cadherin mechanotransduction to cell metabolism through force‐mediated activation of AMPK. Nat Cell Biol. 2017;19:724‐731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Jansen M, ten Klooster JP, Offerhaus GJ, Clevers H. LKB1 and AMPK family signaling: the intimate link between cell polarity and energy metabolism. Physiol Rev. 2009;89:777‐798. [DOI] [PubMed] [Google Scholar]
- 139. Beirowski B, Babetto E, Golden JP, et al. Metabolic regulator LKB1 is crucial for Schwann cell‐mediated axon maintenance. Nat Neurosci. 2014;17:1351‐1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Pooya S, Liu X, Kumar VBS, et al. The tumour suppressor LKB1 regulates myelination through mitochondrial metabolism. Nat Commun. 2014;5:4993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Tzvetanova ID, Nave KA. Axons hooked to Schwann cell metabolism. Nat Neurosci. 2014;17:1293‐1295. [DOI] [PubMed] [Google Scholar]
- 142. Love S. Demyelinating diseases. J Clin Pathol. 2006;59:1151‐1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Kaufman DM, Geyer HL, Milstein MJ. Kaufman's Clinical Neurology for Psychiatrists. Amsterdam, Netherlands: Elsevier Health Sciences; 2016. [Google Scholar]
- 144. Muheremu A, Ao Q. Past, present, and future of nerve conduits in the treatment of peripheral nerve injury. Biomed Res Int. 2015;2015:1‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Rodrigues MCO, Rodrigues AA, Glover LE, Voltarelli J, Borlongan CV. Peripheral nerve repair with cultured schwann cells: getting closer to the clinics. Scientific World Journal. 2012;2012:1‐10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Duncan I, et al. Transplantation of rat Schwann cells grown in tissue culture into the mouse spinal cord. J Neurol Sci. 1981;49:241‐252. [DOI] [PubMed] [Google Scholar]
- 147. Lopatina T, Kalinina N, Karagyaur M, et al. Adipose‐derived stem cells stimulate regeneration of peripheral nerves: BDNF secreted by these cells promotes nerve healing and axon growth de novo. PLoS One. 2011;6:e17899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Kingham PJ, Kalbermatten DF, Mahay D, Armstrong SJ, Wiberg M, Terenghi G. Adipose‐derived stem cells differentiate into a Schwann cell phenotype and promote neurite outgrowth in vitro. Exp Neurol. 2007;207:267‐274. [DOI] [PubMed] [Google Scholar]
- 149. di Summa PG, Kingham PJ, Raffoul W, Wiberg M, Terenghi G, Kalbermatten DF. Adipose‐derived stem cells enhance peripheral nerve regeneration. J Plast Reconstr Aesthet Surg. 2010;63:1544‐1552. [DOI] [PubMed] [Google Scholar]
- 150. Xu Y, Liu L, Li Y, et al. Myelin‐forming ability of Schwann cell‐like cells induced from rat adipose‐derived stem cells in vitro. Brain Res. 2008;1239:49‐55. [DOI] [PubMed] [Google Scholar]
- 151. Xu Y, Liu Z, Liu L, et al. Neurospheres from rat adipose‐derived stem cells could be induced into functional Schwann cell‐like cells in vitro. BMC Neurosci. 2008;9:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Jiang L, Zhu JK, Liu XL, Xiang P, Hu J, Yu WH. Differentiation of rat adipose tissue‐derived stem cells into Schwann‐like cells in vitro. Neuroreport. 2008;19:1015‐1019. [DOI] [PubMed] [Google Scholar]
- 153. Heine W, et al. Transplanted neural stem cells promote axonal regeneration through chronically denervated peripheral nerves. Exp Neurol. 2004;189:231‐240. [DOI] [PubMed] [Google Scholar]
- 154. Murakami T, Fujimoto Y, Yasunaga Y, et al. Transplanted neuronal progenitor cells in a peripheral nerve gap promote nerve repair. Brain Res. 2003;974:17‐24. [DOI] [PubMed] [Google Scholar]
- 155. Niapour A, Karamali F, Karbalaie K, et al. Novel method to obtain highly enriched cultures of adult rat Schwann cells. Biotechnol Lett. 2010;32:781‐786. [DOI] [PubMed] [Google Scholar]
- 156. Takahashi K, Tanabe K, Ohnuki M, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861‐872. [DOI] [PubMed] [Google Scholar]
- 157. Ma M‐S, Boddeke E, Copray S. Pluripotent stem cells for Schwann cell engineering. Stem Cell Rev Report. 2015;11:205‐218. [DOI] [PubMed] [Google Scholar]
- 158. Kim H‐S, Lee J, Lee DY, et al. Schwann cell precursors from human pluripotent stem cells as a potential therapeutic target for myelin repair. Stem Cell Rep. 2017;8:1714‐1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Lee G, Chambers SM, Tomishima MJ, Studer L. Derivation of neural crest cells from human pluripotent stem cells. Nat Protoc. 2010;5:688‐701. [DOI] [PubMed] [Google Scholar]
- 160. Tsuji O, Miura K, Okada Y, et al. Therapeutic potential of appropriately evaluated safe‐induced pluripotent stem cells for spinal cord injury. Proc Natl Acad Sci. 2010;107:12704‐12709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Wang A, Tang Z, Park IH, et al. Induced pluripotent stem cells for neural tissue engineering. Biomaterials. 2011;32:5023‐5032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Harrop JS, Hashimoto R, Norvell D, et al. Evaluation of clinical experience using cell‐based therapies in patients with spinal cord injury: a systematic review. J Neurosurg Spine. 2012;17(Suppl1):230‐246. [DOI] [PubMed] [Google Scholar]
- 163. Krause MP, Dworski S, Feinberg K, et al. Direct genesis of functional rodent and human Schwann cells from skin mesenchymal precursors. Stem Cell Rep. 2014;3:85‐100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Sakaue M, Sieber‐Blum M. Human epidermal neural crest stem cells as a source of Schwann cells. Development. 2015;142:3188‐3197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Ni Y, Zhang K, Liu X, et al. miR‐21 promotes the differentiation of hair follicle‐derived neural crest stem cells into Schwann cells. Neural Regen Res. 2014;9:828‐836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Al‐Zer H, et al. Enrichment and schwann cell differentiation of neural crest‐derived dental pulp stem cells. In Vivo. 2015;29:319‐326. [PubMed] [Google Scholar]
- 167. Coste C, Neirinckx V, Sharma A, et al. Human bone marrow harbors cells with neural crest‐associated characteristics like human adipose and dermis tissues. PLoS One. 2017;12:e0177962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Saulite L, Vavers E, Zvejniece L, Dambrova M, Riekstina U. The differentiation of skin mesenchymal stem cells towards a schwann cell phenotype: impact of sigma‐1 receptor activation. Mol Neurobiol. 2018;55:2840‐2850. [DOI] [PubMed] [Google Scholar]
- 169. Mozafari S, Laterza C, Roussel D, et al. Skin‐derived neural precursors competitively generate functional myelin in adult demyelinated mice. J Clin Invest. 2015;125:3642‐3656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Vasudeva VS, Abd‐El‐Barr MM, Chi JH. Implantation of neonatal skin–derived precursor schwann cells improves outcomes after incomplete cervical spinal cord injury in rats. Neurosurgery. 2015;77:N15‐N17. [DOI] [PubMed] [Google Scholar]
- 171. Shakhbazau A, Mohanty C, Kumar R, Midha R. Sensory recovery after cell therapy in peripheral nerve repair: effects of naive and skin precursor‐derived Schwann cells. J Neurosurg. 2014;121:423‐431. [DOI] [PubMed] [Google Scholar]
- 172. Khuong HT, Kumar R, Senjaya F, et al. Skin derived precursor Schwann cells improve behavioral recovery for acute and delayed nerve repair. Exp Neurol. 2014;254:168‐179. [DOI] [PubMed] [Google Scholar]
- 173. Kang HK, Min SK, Jung SY, et al. The potential of mouse skin‐derived precursors to differentiate into mesenchymal and neural lineages and their application to osteogenic induction in vivo. Int J Mol Med. 2011;28:1001‐1011. [DOI] [PubMed] [Google Scholar]
- 174. Walsh S, Biernaskie J, Kemp SWP, Midha R. Supplementation of acellular nerve grafts with skin derived precursor cells promotes peripheral nerve regeneration. Neuroscience. 2009;164:1097‐1107. [DOI] [PubMed] [Google Scholar]
- 175. Biernaskie J, Sparling JS, Liu J, et al. Skin‐derived precursors generate myelinating Schwann cells that promote remyelination and functional recovery after contusion spinal cord injury. J Neurosci. 2007;27:9545‐9559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Grochmal J, Dhaliwal S, Stys PK, van Minnen J, Midha R. Skin‐derived precursor schwann cell myelination capacity in focal tibial demyelination. Muscle Nerve. 2014;50:262‐272. [DOI] [PubMed] [Google Scholar]
- 177. Zhu C, Huang J, Xue C, et al. Skin derived precursor Schwann cell‐generated acellular matrix modified chitosan/silk scaffolds for bridging rat sciatic nerve gap. Neurosci Res. 2018;135:21‐31. [DOI] [PubMed] [Google Scholar]
- 178. Sparling JS, Bretzner F, Biernaskie J, et al. Schwann cells generated from neonatal skin‐derived precursors or neonatal peripheral nerve improve functional recovery after acute transplantation into the partially injured cervical spinal cord of the rat. J Neurosci. 2015;35:6714‐6730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Menendez L, Kulik MJ, Page AT, et al. Directed differentiation of human pluripotent cells to neural crest stem cells. Nat Protoc. 2013;8:203‐212. [DOI] [PubMed] [Google Scholar]
- 180. Tamura Y, Matsumura K, Sano M, et al. Neural crest–derived stem cells migrate and differentiate into cardiomyocytes after myocardial infarction. Arterioscler Thromb Vasc Biol. 2011;31:582‐589. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: Adult sources for in vitro expansion of multipotent stem cells exhibiting an NC‐like phenotype. From the above mentioned sources, only cells from dental pulp and hair follicles have been successfully isolated and studied in humans
Figure S2: A Neural Crest Stem Cell's journey to myelination. Delaminating, post migratory NC follow Wnt and FGF signals and upregulate key transcription factors like Sox10, FoxD3 and Pax3. By recruiting HDAC1/2, myelin protein 0 is upregulated leading to a Schwann cell precursor phenotype. During this immature Schwann cell stage, Schwann cells initially approach multiple axons and after undergoing significant cytoskeletal reorganization, each one myelinates a single axon, a process referred to as axonal sorting. Finally, the mature Schwann cell envelopes along the neuronal axon with myelin sheath to facilitate saltatory electrical impulse conduction.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.