

Abstract
Background
Emerging evidence suggests that endothelial-to-mesenchymal transition (EndMT) in endothelial dysfunction due to persistent inflammation is a key component and emerging concept in the pathogenesis of vascular diseases. Ginsenoside Rg3 (Rg3), an active compound from red ginseng, has been known to be important for vascular homeostasis. However, the effect of Rg3 on inflammation-induced EndMT has never been reported. Here, we hypothesize that Rg3 might reverse the inflammation-induced EndMT and serve as a novel therapeutic strategy for vascular diseases.
Methods
EndMT was examined under an inflammatory condition mediated by the NOD1 agonist, γ-d-glutamyl-meso-diaminopimelic acid (iE-DAP), treatment in human umbilical vein endothelial cells. The expression of EndMT markers was determined by Western blot analysis, real-time polymerase chain reaction, and immunocytochemistry. The underlying mechanisms of Rg3-mediated EndMT regulation were investigated by modulating the microRNA expression.
Results
The NOD1 agonist, iE-DAP, led to a fibroblast-like morphology change with a decrease in the expression of endothelial markers and an increase in the expression of the mesenchymal marker, namely EndMT. On the other hand, Rg3 markedly attenuated the iE-DAP–induced EndMT and preserved the endothelial phenotype. Mechanically, miR-139 was downregulated in cells with iE-DAP–induced EndMT and partly reversed in response to Rg3 via the regulation of NF-κB signaling, suggesting that the Rg3–miR-139-5p-NF-κB axis is a key mediator in iE-DAP-induced EndMT.
Conclusion
These results suggest, for the first time, that Rg3 can be used to inhibit inflammation-induced EndMT and may be a novel therapeutic option against EndMT-associated vascular diseases.
Keywords: endothelial-to-mesenchymal transition, ginsenoside Rg3, iE-DAP, inflammation, microRNA
1. Introduction
Endothelial cells (ECs) line the inner surface of the blood vessels and play a key role in maintaining homeostasis of the vascular system during exposure to various stimuli [1], [2], [3]. However, ECs can be dysfunctional under sustained pathological conditions, such as chronic inflammation, hypertension, obesity, and diabetes [4], [5], and this endothelial dysfunction can lead to endothelial-to-mesenchymal transition (EndMT), which is a newly recognized, key feature in the pathogenesis of a variety of diseases including, fibrosis [6], tumor progression [7], and pulmonary arterial hypertension (PAH) [8]. The features of EndMT are similar to those of the well-established epithelial-to-mesenchymal transition (EMT) [9]. EndMT is a process of losing endothelial markers such as platelet and endothelial cell adhesion molecule 1 (PECAM1/CD31) and vascular endothelial (VE)–cadherin and acquiring mesenchymal markers such as alpha-smooth muscle actin, smooth muscle protein 22 alpha (SM22α), vimentin, type I collagen, fibronectin, fibroblast-specific protein 1, and N-cadherin [1], [10], [11], [12]. The EndMT process can be regulated by various pathological stimuli, microRNAs (miRNAs), and many signaling pathways [1], [3]. In particular, miRNAs are emerging as key mediators during the EndMT process because of the capacity to regulate multiple mRNAs and have an advantage in the regulation of phenotypic plasticity in ECs. However, the underlying mechanism of the EndMT process and how EndMT contributes to the progression of a variety of diseases are less well understood.
Nucleotide-binding oligomerization domain–like receptors (NLRs) are members of the intracellular innate immune receptor family [13], [14]. NLRs sense pathogens and play a key role in the host defense mechanism [13], [14]. Nucleotide-binding oligomerization domain-containing proteins 1 and 2 (NOD1 and NOD2), members of the NLR family, are involved in inflammatory cytokine production and several diseases such as cardiac fibrosis and Crohn's disease [13], [14], [15], [16]. NOD1 recognizes γ-d-glutamyl-meso-diaminopimelic acid (iE-DAP), and its activation triggers nuclear factor-kB (NF-κB) signaling [15], [17]. To date, most studies on NOD1 have focused on the role of immune cells. However, a recent study showed the important role of NOD1 in ECs in which activation of NOD1 can lead to endothelial dysfunction, and in turn, contribute to pathological angiogenesis [18]. In addition, several studies have shown that various bacterial endotoxins and viruses promote endothelial dysfunction and EndMT, implying the strong involvement of NOD1 in these responses [19], [20], [21], [22], [23]. Furthermore, whether activation of NOD1 contributes to EndMT has not been studied.
Ginsenoside Rg3 (Rg3), a steroidal saponin extracted from ginseng, has various pharmacological effects [24]. It has been well studied for its antitumor effect, which induces tumor cell apoptosis and inhibits viability, migration, metastasis, and proliferation of tumor cells [24], [25], [26], [27], [28]. It also has suppressive effects on tumor angiogenesis by inhibiting the proliferation of tumor ECs [29], [30]. Various studies have shown that Rg3 inhibits the inflammatory response induced by proinflammatory mediators such as lipopolysaccharide (LPS) and interleukin-1 beta (IL-1β) and ischemia/reperfusion [31], [32], [33], [34], [35]. A recent study demonstrated that 20(R)-Rg3 plays a liver protective role in mice by suppressing inflammation mediated by the PI3K/Akt pathway [36]. Rg3 can also regulate vascular tone. Previous studies have shown that Rg3-enriched Korean Red Ginseng stabilized blood pressure in hypertensive rats and that Rg3 modulated vascular contraction through endothelial nitric oxide production [37], [38], [39]. Although the protective effects of Rg3 under the conditions of an inflammatory response have been shown, whether the role of Rg3 is beneficial in EndMT induced by proinflammatory conditions such as NOD1 activation is not known.
Therefore, in this study, we sought to define the role of Rg3 in the NOD1-mediated EndMT process and its underlying mechanism mediated by miRNA. Here, we provide evidence for a beneficial role of Rg3 in inflammation-induced EndMT and define a novel mechanism by which Rg3-mediated inhibition of EndMT is regulated via the suppression of AKT/NF-κB signaling by miR-139-5p upregulation.
2. Methods
2.1. Cell culture and transfection
Human umbilical vein endothelial cells (HUVECs; Lonza, Basel, Switzerland and Yale VBT Core, New Haven, USA) were cultured at 37°C in a 5% CO2 incubator with EBM-2 basal medium supplemented with EGM-2 (Lonza) and 1% penicillin–streptomycin (Welgene, Daegu, Korea). For experimental treatments, HUVECs (passages 3–7) were grown to 70% to 90% confluence. HEK293T cells were cultured in Dulbecco's modified Eagle's medium (Hyclone, Logan, USA) supplemented with 10% fetal bovine serum (Gibco, Grand Island, USA) and 1% penicillin–streptomycin (Welgene). small interfering RNA (siRNA) (Stealth siRNA; Invitrogen, Grand Island, USA) and microRNA (miRNA) mimics (miRVanaTM; Ambion, MA, USA) were transfected with Lipofectamine RNAiMAX (Invitrogen) as per the manufacturer's instructions.
2.2. Reagents
Rg3 was purchased from (Sigma, St. Louis, USA) and dissolved in dimethyl sulfoxide. Ginsenoside Rb1 was obtained from the Korea Ginseng and Tobacco Research Institute (Daejeon, Korea) and dissolved in 100% ethanol. ML130 was purchased from Selleckchem (Houston, USA) and iE-DAP was purchased from InvivoGen (San Diego, USA). Rg3, Rb1, iE-DAP, and ML130 were used at the indicated doses and time points.
2.3. Real-time polymerase chain reaction
Total RNA was isolated with an miRNeasy RNA isolation kit (Qiagen, Hilden, Germany). Purified RNA was reverse transcribed using the TaqMan MicroRNA Reverse Transcription Kit (Life Technologies, Carlsbad, USA). miRNA quantitative reverse transcriptase polymerase chain reaction was performed with the TaqMan Universal Master Mix II, no UNG (Life Technologies), and miR-139-5p was detected with Taqman probes. Data were normalized to those of the internal control small RNA RNU6B. For the mRNA, purified RNA was reverse transcribed using the qPCRBIO cDNA Synthesis Kit (PCR Biosystems, London, UK). Quantitative reverse transcriptase polymerase chain reaction was performed with the qPCRBIO SyGreen Blue Mix Lo-ROX (PCR Biosystems) as per the manufacturer's instructions. Ribosomal 18S RNA was used as the internal control.
2.4. Western blotting
HUVECs were lysed with radioimmunoprecipitation assay (RIPA) buffer (GenDEPOT, Barker, TX, USA) containing a protease and phosphatase inhibitor cocktail (Roche Diagnostics, Risch-Rotkreuz, Switzerland). Thereafter, centrifugation was performed at 13,000 rpm and 4°C for 15 min. Protein concentrations were determined using the Pierce BCA protein assay kit (Thermo Fisher Scientific, Waltham, USA). Equal amounts of total proteins were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis and transferred onto polyvinyl difluoride membranes (Millipore, Burlington, USA). Immunoblotting was performed with primary antibodies specific to CD31 (1:2000, Cell Signaling, Danvers, USA), VE-cadherin (1:2000, Santa Cruz, Dallas, USA), fibronectin (1:3000, Santa Cruz), N-cadherin (1:3000, BD Biosciences, Franklin Lakes, USA), SM22α (1:3000, Abcam, Cambridge, UK), NF-κB p65 (1:3000, Cell Signaling), lamin B1 (1:2000, Santa Cruz), IκBα (1:2000, Cell Signaling), p IκBα (1:2000, Cell Signaling), importin-α3 (1:3000, Abcam), TAK1 (1:2000, Cell Signaling), Akt (1:2000, Cell Signaling), phosphoAkt (ser-473) (1:2000, Cell Signaling), and GAPDH (1:5000, Cell Signaling). Immunodetection was accomplished using horseradish peroxidase (HRP)-conjugated mouse (1:4000, Thermo Fisher Scientific) and rabbit (1:4000, Cell Signaling) secondary antibodies. The enhanced chemiluminescence detection method was used for development (Thermo Fisher Scientific). Nuclear and cytoplasmic extractions were performed with the EpiQuik Nuclear Extraction Kit I (EpiGentek, Farmingdale, USA).
2.6. Immunofluorescence
HUVECs were fixed with 4% paraformaldehyde for 5 min and washed with phosphate-buffered saline (PBS). Triton X-100 (0.1%) was used for permeabilization for 5 min (permeabilization was not performed for VE-cadherin staining). After blocking with 1% bovine serum albumin (BSA) for 1 h, the primary antibodies were applied overnight at 4°C. Rhodamine phalloidin (1:1000, Sigma), VE-cadherin (1:300, Cell Signaling), fibronectin (1:100, Santa Cruz), N-cadherin (1:200, BD Biosciences), SM22α (1:500, Abcam), and NF-κB p65 (1:400, Cell Signaling) were used. The secondary antibodies were donkey antigoat Alexa Fluor 488, donkey antimouse Alexa Fluor 568, and goat antirabbit Alexa Fluor 647. Immunostaining images were obtained using a confocal microscope (Zeiss LSM-700, Carl Zeiss, Oberkochen, Germany).
2.7. Lentivirus production
For miR-139-5p overexpression in HUVECs, a lentivirus bearing miR-139-5p was obtained from System Biosciences. The Lenti-X HTX packaging system (Clontech, Mountain View, USA) with Lenti-X concentrator was used to generate the lentivirus particles for in vitro cellular transduction.
2.8. Cell viability assay
Cell viability was measured using the WST-1 assay kit (Daeil LabService, Seoul, Korea) as per the manufacturer's instructions. HUVECs (5 × 103 cells per well) were plated to a 96-well plate and treated with various concentrations of Rg3 or Rb1 for 24 hr, followed by 1 hr incubation with WST-1 at 37°C and 5% CO2. The absorbance was measured at 450 nm using an enzyme-linked immunosorbent assay (ELISA) plate reader (Bio-Rad, Model 550, Hercules, CA, USA). The cell viability was calculated as relative absorbance compared with the control.
2.9. Statistical analysis
All experiments were performed at least three times, and analyses were performed with GraphPad Prism 5.0 software. When two groups were compared, statistical differences were assessed with unpaired two-tailed Student t test. A p value < 0.05 was considered statistically significant.
3. Results
3.1. iE-DAP induces EndMT in HUVECs
To identify whether NOD1 activation can induce EndMT, we first examined the effect of iE-DAP (a NOD1 ligand) on the EndMT process. Because filamentous actin is a characteristic of EndMT [40], [41], we analyzed morphology change and cytoskeleton reorganization by rhodamine–phalloidin staining. We found that treatment with iE-DAP (20 μg/mL) led to a fibroblast-like cell morphology and actin stress fiber development in HUVECs (Fig. 1A). We then investigated the mRNA and protein expression of EndMT markers on the 2nd, 4th, and 6th d after iE-DAP treatment. We found that iE-DAP significantly increased the mRNA levels of mesenchymal markers, fibronectin, N-cadherin, and SM22α (Fig. 1B). Western blotting showed that fibronectin, N-cadherin, and SM22α protein expression markedly increased in iE-DAP–treated ECs whereas that of the endothelial markers, CD31 and VE-cadherin, significantly decreased (Fig. 1C). These data indicate that NOD1 activation by iE-DAP contributes to EndMT. On the basis of these findings, we investigated whether inhibition of NOD1 activity suppresses EndMT. We found that pretreatment with a NOD1 inhibitor (ML130, 10 μM) for 2 h reversed the iE-DAP–induced expression of EndMT markers (Fig. 1D). Thus, we demonstrated that NOD1 activation by iE-DAP leads to EndMT in HUVECs.
Fig. 1.

iE-DAP induces EndMT in HUVECs. (A) Rhodamine–phalloidin staining images of HUVECs treated with iE-DAP (20 μg/mL) in 2% FBS medium for 2 d. Scale bar = 50 μm. (B) mRNA expression of mesenchymal markers, fibronectin (FN), N-cadherin (N-Cad), and smooth muscle protein 22 alpha (SM22α) in response to iE-DAP treatment (20 μg/mL) for 2, 4, and 6 d. (C) Protein expression of endothelial and mesenchymal markers after iE-DAP treatment for 2, 4, and 6 d. VE-cadherin (VE-Cad), FN, N-Cad, and SM22α. (D) Protein expression of endothelial markers and mesenchymal markers induced by iE-DAP (20 μg/mL) changes with or without pretreatment with ML130. *P < 0.05, **P < 0.01, and ***P < 0.001 compared with the controls as determined by the unpaired two-tailed Student t test. Error bars, s.e.m. N = 3 experiments per condition.
EndMT, endothelial-to-mesenchymal transition; FBS, fetal bovine serum; HUVEC, human umbilical vein endothelial cell; iE-DAP, γ-d-glutamyl-meso-diaminopimelic acid; s.e.m., standard error of the mean; VE, vascular endothelial.
3.2. Rg3 ameliorates iE-DAP–induced EndMT in HUVECs
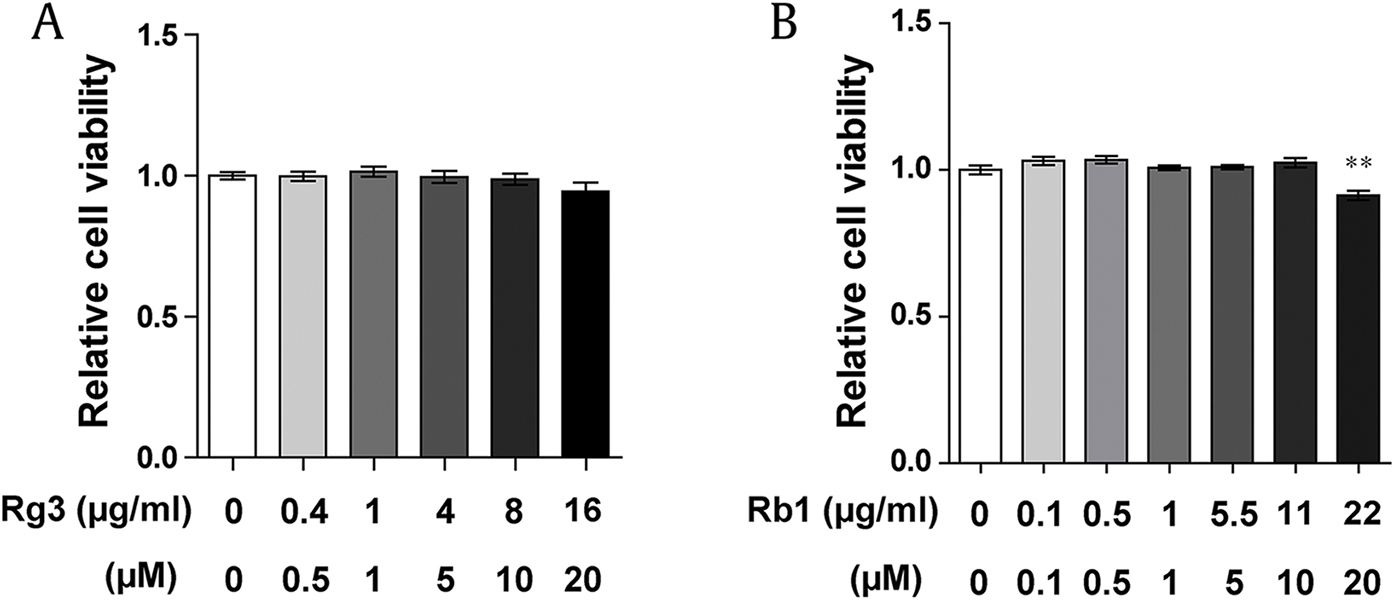
Previous studies have shown that Rg3 and Rb1 protect vascular ECs [38], [39], [42]. The specific roles of Rg3 and Rb1 in EndMT remain unclear. Before examining the effect of Rg3 and Rb1 on the EndMT process, the effect of Rg3 and Rb1 on HUVEC viability was examined. We found that Rg3 (0.4, 0.8, 4, 8, and 16 μg/mL) and Rb1 treatment (0.1, 0.5, 1, 5.5, and 11 μg/mL) did not affect the viability of HUVECs, whereas 22 μg/mL of Rb1 significantly decreased the viability of HUVECs (Supplementary Fig. 1A and 1B). Therefore, we selected 10 μg/mL of Rg3 and Rb1 for the subsequent experiments because this dosage level did not influence HUVEC viability.
To investigate whether these ginsenosides have beneficial effects on iE-DAP–induced EndMT, we examined the protein expression of EndMT markers. We found that Rg3 substantially inhibited the iE-DAP–induced EndMT and preserved the EC phenotype (Fig. 2A) whereas treatment with ginsenoside Rb1 had no significant effect on the iE-DAP–induced EndMT (data not shown). We also analyzed EndMT markers by immunocytochemistry and found that changes in the expression of the iE-DAP–induced EndMT markers were consistent with the results of the protein expression analysis (Fig. 2B). These findings indicate that Rg3 inhibited the iE-DAP–induced EndMT in HUVECs and may be a potential therapeutic option against many EndMT-mediated diseases.
Fig. 2.

Rg3 ameliorates iE-DAP–induced EndMT in HUVECs. (A) Protein expressions of endothelial and mesenchymal markers. (B) Immunostaining images of VE-cadherin (VE-Cad), fibronectin (FN), N-cadherin (N-Cad), and smooth muscle protein 22 alpha (SM22α) after treatment with iE-DAP (20 μg/mL) in conjunction with or without Rg3 treatment (10 μg/mL) for 2, 4, and 6 d. The medium added with iE-DAP (20 μg/mL) and Rg3 (10 μg/mL) was changed every other day. Scale bar = 50 μm. *P < 0.05, **P < 0.01, and ***P < 0.001 compared with the controls, as determined by the unpaired two-tailed Student t test. Error bars, s.e.m. N = 3 experiments per condition.
EndMT, endothelial-to-mesenchymal transition; HUVEC, human umbilical vein endothelial cell; iE-DAP, γ-d-glutamyl-meso-diaminopimelic acid; Rg3, ginsenoside Rg3; s.e.m., standard error of the mean; VE, vascular endothelial.
3.3. Upregulation of miR-139-5p expression on Rg3 treatment inhibits iE-DAP–induced EndMT
Several molecular mechanisms have been shown to regulate EndMT. In particular, miRNAs are key emerging mediators in EndMT because of their capacity to regulate multiple targets and are therefore advantageous in regulating phenotypic plasticity in ECs. In addition, several studies have shown the association between ginseng and miRNAs in different contexts [30], [43], [44], [45], [46]. In a previous study, it was demonstrated that miR-139-5p is highly expressed in ECs and plays a key role in maintaining vascular homeostasis [47]. Furthermore, several studies have shown that miR-139-5p plays a critical role in the regulation of EMT [48], [49]. Thus, to determine the molecular mechanism underlying the effect of Rg3 on iE-DAP–induced EndMT, we first assessed the effect of iE-DAP on miR-139-5p expression. We found that iE-DAP treatment led to a significant decrease in miR-139-5p expression after 2, 4, and 6 d (Fig. 3A), whereas Rg3 treatment significantly upregulated miR-139-5p expression in the basal state (Fig. 3B). To further investigate the relationship between Rg3 and miR-139-5p in iE-DAP–induced EndMT, we tested whether downregulation of miR-139-5p expression in response to iE-DAP is restored by Rg3 treatment. As shown in Fig. 3C, the miR-139-5p level was restored on treatment with Rg3 in conjunction with iE-DAP on the 2nd, 4th, and 6th d. Furthermore, western blotting and immunohistochemistry showed that the iE-DAP–induced expression of the mesenchymal markers, fibronectin and N-cadherin, was significantly inhibited by miR-139-5p overexpression (Fig. 3D and E). Taken together, these findings support that miR-139-5p may play a critical role in the beneficial effect of Rg3 on iE-DAP–induced EndMT.
Fig. 3.

iE-DAP–induced EndMT is regulated by miR-139-5p. (A) Mature miR-139-5p expression in response to iE-DAP treatment (20 μg/mL) for 2, 4, and 6 d. (B) Mature miR-139-5p expression in response to Rg3 treatment (10 μg/mL) for 2 d. (C) Mature miR-139-5p expression in response to iE-DAP (20 μg/mL) with or without concurrent treatment with Rg3 (10 μg/mL) for 2, 4, and 6 d. (D) Protein expression. (E) Immunostaining images of fibronectin (FN) and N-cadherin (N-Cad) in response to iE-DAP treatment (20 μg/mL) with or without miR-139-5p overexpression (12 nM) for 2 d. Scale bar = 50 μm. *P < 0.05, **P < 0.01, and ***P < 0.001 compared with the controls, as determined by the unpaired two-tailed Student t test. Error bars, s.e.m. N = 3 experiments per condition.
EndMT, endothelial-to-mesenchymal transition; iE-DAP, γ-d-glutamyl-meso-diaminopimelic acid; Rg3, ginsenoside Rg3; s.e.m., standard error of the mean.
3.4. The Rg3–miR-139-5p axis suppresses iE-DAP–induced Akt/NF-κB signaling in HUVECs
We further investigated downstream signaling of the Rg3–miR-139-5p axis in this context. Several studies have shown that Rg3 inhibits NF-κB signaling in cancer cells and ECs [50], [51]. In addition, activation of NF-κB signaling is closely associated with the process of EndMT induction [52], [53]. Therefore, to determine whether Rg3 affects NF-κB activation in ECs, we first examined the effect of Rg3 on NF-κB nuclear accumulation by determining the p65 protein levels in the nuclear fraction. We found that iE-DAP treatment led to a robust increase in p65 protein levels, which was abrogated by cotreatment with Rg3 (Fig. 4A). Given that Rg3 can regulate the expression of miR-139-5p, we then determined the detailed downstream signaling of miR-139-5p regulated by Rg3. We tested the effect of miR-139-5p on this response and found that iE-DAP treatment resulted in a robust increase in p65 protein levels, which was abrogated with concurrent overexpression of miR-139-5p in the nuclear fraction (Fig. 4B). Translocation of NF-κB from the cytoplasm to the nucleus via importin proteins requires the phosphorylation of IκB-α and its subsequent degradation, thus allowing the activation of NF-κB target genes. In this canonical NF-κB signaling pathway, importin proteins play an essential role in the translocation of NF-κB to the nucleus [54]. As shown in Fig. 4B, iE-DAP–induced IκB-α phosphorylation was markedly reduced by concurrent overexpression of miR-139-5p in the cytoplasmic fraction, whereas iE-DAP reduced IκB-α, and it was dramatically restored by concurrent overexpression of miR-139-5p in the cytoplasmic fraction. We then explored the effect of miR-139-5p on NF-κB nuclear accumulation by immunocytochemistry. As shown in Fig. 4C, overexpression of miR-139-5p led to a reduction in nuclear p65 localization. These data suggest that the inhibitory role of Rg3–miR-139-5p in iE-DAP–induced EndMT may be due to inhibition of the NF-κB pathway by suppression of NF-κB nuclear translocation.
Fig. 4.

Rg3 suppresses iE-DAP–induced EndMT via Akt/NF-κB signaling. (A) Nuclear p65 protein expression in response to iE-DAP (20 μg/mL) with or without concurrent treatment with Rg3 (10 μg/mL) for 2 d. (B) Protein expression of nuclear p65, cytosolic IκB, and cytosolic p-IκB in response to iE-DAP (20 μg/mL) with or without miR-139-5p overexpression (12 nM) for 2 d. (C) Immunostaining images of nuclear p65 translocation in response to iE-DAP (20 μg/mL) with or without miR-139-5p overexpression (12 nM) for 2 d. Scale bar = 50 μm. (D) Importin-α3 and TAK1 protein expression in response to lentiviral miR-139-5p overexpression for 4 d. (E) Protein expression of p-Akt and Akt in response to iE-DAP (20 μg/mL) with or without miR-139-5p overexpression (12 nM) for 2 d. (F) Schematic outlining of the proposed mechanism in ECs. *P < 0.05, **P < 0.01, and ***P < 0.001 compared with the controls, as determined by the unpaired two-tailed Student t test. Error bars, s.e.m. N = 3 experiments per condition.
ECs, endothelial cells; EndMT, endothelial-to-mesenchymal transition; iE-DAP, γ-d-glutamyl-meso-diaminopimelic acid; s.e.m., standard error of the mean; Rg3, ginsenoside Rg3; TAK1, transforming growth factor beta-activating kinase 1.
To further investigate the direct target of miR-139-5p in this response, we used target prediction algorithm (TargetScan) to identify the potential targets of miR-139-5p associated with NF-κB signaling. Two candidates of importin-α3 and transforming growth factor beta-activating kinase 1 (TAK1) were identified as predicted targets associated with NF-κB signaling. First, we tested whether miR-139-5p can directly target importin-α3, a protein essential for NF-κB translocation to the nucleus. However, the overexpression of miR-139-5p did not affect importin-α3 expression in ECs, although NF-κB translocation to the nucleus was inhibited by Rg3 and miR-139-5p, suggesting that different mechanisms are involved in NF-κB translocation mediated by Rg3–miR-139-5p. Because activation of NF-κB was inhibited by TAK1 inhibitors those were predicted as a potential target of miR-139-5p [55], we suspected that TAK1 is the direct target of miR-139-5p. However, TAK1 expression was also not changed by miR-139-5p overexpression (Fig. 4D). It has been shown that miR-139-5p inhibits Akt signaling, which is potentially upstream of NF-κB [49], [50], [56], [57]. Thus, we examined the effect of miR-139-5p on the AKT/NF-κB pathway. Overexpression of miR-139-p led to a significant decrease in phosphorylated Akt (Fig. 4E) and inhibition of NF-κB translocation to the nucleus (Fig. 4B and C). These data indicate that the Rg3–miR-139-5p axis, but not importin-α3 and TAK1, blocked the NF-κB nuclear translocation through Akt signaling.
4. Discussion
Endothelial dysfunction under inflammatory conditions can lead to various diseases such as fibrosis, pathological angiogenesis, and PAH [1], [58], and EndMT in endothelial dysfunction is an emerging concept in the pathogenesis of vascular diseases [1], [12]. Here, we described an Rg3-mediated beneficial effect on EndMT via inhibition of Akt/NF-κB signaling mediated by miR-139-5p upregulation and provide valuable insights into a novel therapeutic strategy for many diseases caused by inflammation-induced EndMT.
Proinflammatory cytokines, tumor necrosis factor, interleukin-1 beta, and interleukin-6 induce EndMT in several ECs [12]. In addition, it has been identified that endotoxins, LPS, and septic serum induce EndMT [19], [20], [21], [22], [23], [59]. Intracellular pathogens are sensed by NLRs, followed by inflammatory gene expression triggered by activation of NOD1 and NOD2 signaling [60]. A recent study demonstrated that NOD2 promotes EndMT in glomerular ECs in cases of diabetic nephropathy [61]. However, the relation between NOD1 and EndMT has never been studied. We demonstrated that NOD1 activation by iE-DAP induces EndMT. Treatment with iE-DAP promotes a morphological change and expression of the stress fiber, F-actin. In addition, protein and RNA expressions of mesenchymal markers were upregulated, whereas endothelial markers were downregulated in response to iE-DAP. Moreover, iE-DAP–induced EndMT was inhibited by the NOD1 inhibitor, ML130.
Because EndMT is involved in various inflammation-associated diseases, it is considered a therapeutic target [1]. Many studies have shown that blocking of EndMT ameliorates fibrosis, PAH, and diabetic nephropathy, which could be driven by inflammation. For example, salvianolic acid A, a type of polyphenolic compound, inhibits EndMT and improves vascular function in animal models of PAH [62]. The dipeptidyl peptidase-4 (DPP4) inhibitor linagliptin alleviated kidney fibrosis by blocking EndMT in a diabetic nephropathy model [63]. It has been demonstrated that ginseng extracts and ginsenosides have antiinflammatory effects and act as vascular protective agents [64]. Among various ginsenosides, Rb1 and Rg3 exert vascular protective effects. Ginsenoside Rb1 ameliorates LPS-induced lung injury and vascular protein leakage in rats [65], [66]. Furthermore, it improves endothelial function by regulating the production of endothelial nitric oxide synthase (eNOS) [67]. Previous studies have demonstrated the antiinflammatory effect of Rg3 on epithelial cells, macrophages, and ECs [33], [34], [68], as well as on LPS-treated rats [67], [69]. As Rb1 and Rg3 exhibit a protective role against endothelial dysfunction and vascular injury, we examined the effects of both these ginsenosides on iE-DAP–induced EndMT. Rg3 treatment restored the expression of iE-DAP–induced EndMT markers to baseline levels; however, Rb1 had no such effect. Because it has been reported that ginsenoside Rb1 and Rg3 have similar chemical structures and cardioprotective effects, but only Rg3 has antitumor effect, our findings may be related to those of previous studies [70].
Although Rg3 has been approved for clinical applications, its relationship with EndMT has never been studied. Therefore, although we have shown the possibility of Rg3 being a therapeutic agent for inflammation-associated diseases, further investigations are needed. Moreover, studies on whether Rg3 treatment affects fibroblast proliferation are required. In addition, clinically efficacious doses of Rg3 for the treatment of EndMT-associated inflammatory diseases need to be determined.
In our study, we examined the molecular mechanism underlying iE-DAP–induced EndMT. Many studies have demonstrated that miR-139-5p inhibits EMT and enhances the drug sensitivity of carcinoma cells [48], [49], [71], [72]. Shear stress–responsive endothelial miR-139-5p is crucial for vascular maturation [47]. However, the role of miR-139-5p in the endothelium remains unclear with regard to EndMT. We demonstrated that miR-139-5p was downregulated by iE-DAP and recovered by Rg3 treatment. Overexpression of miR-139-5p suppressed the protein markers fibronectin and N-cadherin. Our findings suggest that iE-DAP–induced EndMT is partly mediated by miR-139-5p (Fig. 4F).
Inflammatory stimuli induce EndMT through NF-κB activation [12]. It has been reported that miR-139-5p is associated with inflammation. For example, deletion of miR-139-5p induces intestinal inflammation through NF-κB signaling in vivo [73]. Thus, we examined the correlation between NF-κB signaling and miR-139-5p under inflammatory conditions. Our findings revealed that miR-139-5p overexpression and Rg3 suppress NF-κB signaling. Because the mechanism of miR-139-5p–mediated regulation of NF-κB remains unclear, we investigated the predicted target of miR-139-5p. We discovered that both importin-α3 and TAK1 were not direct targets of miR-139-5p in ECs. Therefore, further studies are necessary to determine the direct targets involved in the regulation of EndMT by miR-139-5p, although we identified the regulation of the Akt/NF-κB signaling by the Rg3–miR-139-5p axis.
In conclusion, we demonstrated that NOD1 activation by iE-DAP induces EndMT through Akt/NF-κB signaling, which can be reversed by Rg3. Furthermore, we identified that miR-139-5p is involved in the regulation of iE-DAP–induced EndMT. Our findings suggest Rg3 can be a useful therapeutic target for many diseases caused by inflammation-induced EndMT (Fig. 4F).
Conflicts of interest
All authors have no conflicts of interest.
Acknowledgments
This work was supported by the 2016 grant from the Korean Society of Ginseng, the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Minister of Education, Science and Technology (NRF-2016R1A5A1011974 to J.K and NRF-2016R1C1B2006591 to J.K), Bio & Medical Technology Development Program of the National Research Foundation (NRF) funded by the Korean government (MSIT) (NRF-2015M3A9B6027818 to J.K), and the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (HI18C0321 to J.K.).
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jgr.2019.01.003.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
{kind=link}
References
- 1.Cho J.G., Lee A., Chang W., Lee M.S., Kim J. Endothelial to mesenchymal transition represents a key link in the interaction between inflammation and endothelial dysfunction. Front Immunol. 2018;9 doi: 10.3389/fimmu.2018.00294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lee A., Papangeli I., Park Y., Jeong H.N., Choi J., Kang H., Jo H.N., Kim J., Chun H.J. A PPARgamma-dependent miR-424/503-CD40 axis regulates inflammation mediated angiogenesis. Sci Rep. 2017;7(1):2528. doi: 10.1038/s41598-017-02852-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kim J. MicroRNAs as critical regulators of the endothelial to mesenchymal transition in vascular biology. Bmb Rep. 2018;51(2):65–72. doi: 10.5483/BMBRep.2018.51.2.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vanhoutte P.M., Shimokawa H., Feletou M., Tang E.H. Endothelial dysfunction and vascular disease - a 30th anniversary update. Acta Physiol (Oxf). 2017;219(1):22–96. doi: 10.1111/apha.12646. [DOI] [PubMed] [Google Scholar]
- 5.Sena C.M., Pereira A.M., Seica R. Endothelial dysfunction - a major mediator of diabetic vascular disease. Biochim Biophys Acta. 2013;1832(12):2216–2231. doi: 10.1016/j.bbadis.2013.08.006. [DOI] [PubMed] [Google Scholar]
- 6.Ghosh A.K., Quaggin S.E., Vaughan D.E. Molecular basis of organ fibrosis: potential therapeutic approaches. Exp Biol Med (Maywood). 2013;238(5):461–481. doi: 10.1177/1535370213489441. [DOI] [PubMed] [Google Scholar]
- 7.Zeisberg E.M., Potenta S., Xie L., Zeisberg M., Kalluri R. Discovery of endothelial to mesenchymal transition as a source for carcinoma-associated fibroblasts. Cancer Research. 2007;67(21):10123–10128. doi: 10.1158/0008-5472.CAN-07-3127. [DOI] [PubMed] [Google Scholar]
- 8.Good R.B., Gilbane A.J., Trinder S.L., Denton C.P., Coghlan G., Abraham D.J., Holmes A.M. Endothelial to mesenchymal transition contributes to endothelial dysfunction in pulmonary arterial hypertension. Am J Pathol. 2015;185(7):1850–1858. doi: 10.1016/j.ajpath.2015.03.019. [DOI] [PubMed] [Google Scholar]
- 9.Potenta S., Zeisberg E., Kalluri R. The role of endothelial-to-mesenchymal transition in cancer progression. Brit J Cancer. 2008;99(9):1375–1379. doi: 10.1038/sj.bjc.6604662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sanchez-Duffhues G., Orlova V., Ten Dijke P. In brief: endothelial-to-mesenchymal transition. J Pathol. 2016;238(3):378–380. doi: 10.1002/path.4653. [DOI] [PubMed] [Google Scholar]
- 11.Chen P.Y., Simons M. When endothelial cells go rogue. Embo Mol Med. 2016;8(1):1–2. doi: 10.15252/emmm.201505943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Perez L., Munoz-Durango N., Riedel C.A., Echeverria C., Kalergis A.M., Cabello-Verrugio C., Simon F. Endothelial-to-mesenchymal transition: cytokine-mediated pathways that determine endothelial fibrosis under inflammatory conditions. Cytokine Growth F R. 2017;33:41–54. doi: 10.1016/j.cytogfr.2016.09.002. [DOI] [PubMed] [Google Scholar]
- 13.Yamaguchi N., Suzuki Y., Mahbub M.H., Takahashi H., Hase R., Ishimaru Y., Sunagawa H., Watanabe R., Eishi Y., Tanabe T. The different roles of innate immune receptors in inflammation and carcinogenesis between races. Environ Health Prev Med. 2017;22(1):70. doi: 10.1186/s12199-017-0678-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yan R., Liu Z. LRRK2 enhances Nod1/2-mediated inflammatory cytokine production by promoting Rip2 phosphorylation. Protein Cell. 2017;8(1):55–66. doi: 10.1007/s13238-016-0326-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fernandez-Velasco M., Prieto P., Terron V., Benito G., Flores J.M., Delgado C., Zaragoza C., Lavin B., Gomez-Parrizas M., Lopez-Collazo E. NOD1 activation induces cardiac dysfunction and modulates cardiac fibrosis and cardiomyocyte apoptosis. PLoS One. 2012;7(9) doi: 10.1371/journal.pone.0045260. e45260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Caruso R., Warner N., Inohara N., Nunez G. NOD1 and NOD2: signaling, host defense, and inflammatory disease. Immunity. 2014;41(6):898–908. doi: 10.1016/j.immuni.2014.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tao Z., Zhu C., Song W., Xu W., Zhang S., Liu H., Li H. Inductive expression of the NOD1 signalling pathway in chickens infected with Salmonella pullorum. Brit Poultry Sci. 2017;58(3):242–250. doi: 10.1080/00071668.2017.1280771. [DOI] [PubMed] [Google Scholar]
- 18.Kang H., Park Y., Lee A., Seo H., Kim M.J., Choi J., Jo H.N., Jeong H.N., Cho J.G., Chang W. Negative regulation of NOD1 mediated angiogenesis by PPAR gamma-regulated miR-125a. Biochem Bioph Res Co. 2017;482(1):28–34. doi: 10.1016/j.bbrc.2016.11.032. [DOI] [PubMed] [Google Scholar]
- 19.Gasperini P., Espigol-Frigole G., McCormick P.J., Salvucci O., Maric D., Uldrick T.S., Polizzotto M.N., Yarchoan R., Tosato G. Kaposi sarcoma herpesvirus promotes endothelial-to-mesenchymal transition through Notch-dependent signaling. Cancer Res. 2012;72(5):1157–1169. doi: 10.1158/0008-5472.CAN-11-3067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cheng F., Pekkonen P., Laurinavicius S., Sugiyama N., Henderson S., Gunther T., Rantanen V., Kaivanto E., Aavikko M., Sarek G. KSHV-initiated notch activation leads to membrane-type-1 matrix metalloproteinase-dependent lymphatic endothelial-to-mesenchymal transition. Cell Host Microbe. 2011;10(6):577–590. doi: 10.1016/j.chom.2011.10.011. [DOI] [PubMed] [Google Scholar]
- 21.Echeverria C., Montorfano I., Tapia P., Riedel C., Cabello-Verrugio C., Simon F. Endotoxin-induced endothelial fibrosis is dependent on expression of transforming growth factors beta 1 and beta 2. Infect Immun. 2014;82(9):3678–3686. doi: 10.1128/IAI.02158-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Echeverria C., Montorfano I., Sarmiento D., Becerra A., Nunez-Villena F., Figueroa X.F., Cabello-Verrugio C., Elorza A.A., Riedel C., Simon F. Lipopolysaccharide induces a fibrotic-like phenotype in endothelial cells. J Cell Mol Med. 2013;17(6):800–814. doi: 10.1111/jcmm.12066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Echeverria C., Montorfano I., Hermosilla T., Armisen R., Velasquez L.A., Cabello-Verrugio C., Varela D., Simon F. Endotoxin induces fibrosis in vascular endothelial cells through a mechanism dependent on transient receptor protein melastatin 7 activity. PLoS One. 2014;9(4) doi: 10.1371/journal.pone.0094146. e94146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mohanan P., Subramaniyam S., Mathiyalagan R., Yang D.C. Molecular signaling of ginsenosides Rb1, Rg1, and Rg3 and their mode of actions. J Ginseng Res. 2018;42(2):123–132. doi: 10.1016/j.jgr.2017.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li Y., Lu J.Y., Bai F.R., Xiao Y.A., Guo Y.R., Dong Z.M. Ginsenoside Rg3 suppresses proliferation and induces apoptosis in human osteosarcoma. Biomed Res Int. 2018;2018 doi: 10.1155/2018/4306579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mochizuki M., Yoo Y.C., Matsuzawa K., Sato K., Saiki I., Tonooka S., Samukawa K., Azuma I. Inhibitory effect of tumor-metastasis in mice by saponins, ginsenoside-Rb2, 20(R)-Ginsenoside-Rg3 and 20(S)-Ginsenoside-Rg3, of red-ginseng. Biol Pharm Bull. 1995;18(9):1197–1202. doi: 10.1248/bpb.18.1197. [DOI] [PubMed] [Google Scholar]
- 27.Wang X., Chen L., Wang T., Jiang X., Zhang H., Li P., Lv B., Gao X. Ginsenoside Rg3 antagonizes adriamycin-induced cardiotoxicity by improving endothelial dysfunction from oxidative stress via upregulating the Nrf2-ARE pathway through the activation of akt. Phytomedicine. 2015;22(10):875–884. doi: 10.1016/j.phymed.2015.06.010. [DOI] [PubMed] [Google Scholar]
- 28.Pan X.Y., Guo H., Han J., Hao F., An Y., Xu Y., Xiaokaiti Y., Pan Y., Li X.J. Ginsenoside Rg3 attenuates cell migration via inhibition of aquaporin 1 expression in PC-3M prostate cancer cells. Eur J Pharmacol. 2012;683(1–3):27–34. doi: 10.1016/j.ejphar.2012.02.040. [DOI] [PubMed] [Google Scholar]
- 29.Kim J.W., Jung S.Y., Kwon Y.H., Lee J.H., Lee Y.M., Lee B.Y., Kwon S.M. Ginsenoside Rg3 attenuates tumor angiogenesis via inhibiting bioactivities of endothelial progenitor cells. Cancer Biol Ther. 2012;13(7):504–515. doi: 10.4161/cbt.19599. [DOI] [PubMed] [Google Scholar]
- 30.Keung M.H., Chan L.S., Kwok H.H., Wong R.N.S., Yue P.Y.K. Role of microRNA-520h in 20(R)-ginsenoside-Rg3-mediated angiosuppression. J Ginseng Res. 2016;40(2):151–159. doi: 10.1016/j.jgr.2015.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee B., Sur B., Park J., Kim S.H., Kwon S., Yeom M., Shim I., Lee H., Hahm D.H. Ginsenoside Rg3 alleviates lipopolysaccharide-induced learning and memory impairments by anti-inflammatory activity in rats. Biomol Ther. 2013;21(5):381–390. doi: 10.4062/biomolther.2013.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang L.P., Jiang Y.C., Yu X.F., Xu H.L., Li M., Zhao X.Z., Sui D.Y. Ginsenoside Rg3 improves cardiac function after myocardial ischemia/reperfusion via attenuating apoptosis and inflammation. Evid-Based Compl Alt. 2016;2016 doi: 10.1155/2016/6967853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shin Y.M., Jung H.J., Choi W.Y., Lim C.J. Antioxidative, anti-inflammatory, and matrix metalloproteinase inhibitory activities of 20(S)-ginsenoside Rg3 in cultured mammalian cell lines. Mol Biol Rep. 2013;40(1):269–279. doi: 10.1007/s11033-012-2058-1. [DOI] [PubMed] [Google Scholar]
- 34.Lee I.S., Uh I., Kim K.S., Kim K.H., Park J., Kim Y., Jung J.H., Jung H.J., Jang H.J. Anti-inflammatory effects of ginsenoside Rg3 via NF-kappa B pathway in A549 cells and human asthmatic lung tissue. J Immunol Res. 2016;2016 doi: 10.1155/2016/7521601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xing W., Yang L., Peng Y., Wang Q.L., Gao M., Yang M.S., Xiao X.Z. Ginsenoside Rg3 attenuates sepsis-induced injury and mitochondrial dysfunction in liver via AMPK-mediated autophagy flux. Bioscience Rep. 2017;37 doi: 10.1042/BSR20170934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhou Y.D., Hou J.G., Liu W., Ren S., Wang Y.P., Zhang R., Chen C., Wang Z., Li W. 20(R)-ginsenoside Rg3, a rare saponin from red ginseng, ameliorates acetaminophen-induced hepatotoxicity by suppressing PI3K/AKT pathway-mediated inflammation and apoptosis. Int Immunopharmacol. 2018;59:21–30. doi: 10.1016/j.intimp.2018.03.030. [DOI] [PubMed] [Google Scholar]
- 37.Kim N.D., Kang S.Y., Park J.H., Schini-Kerth V.B. Ginsenoside Rg3 mediates endothelium-dependent relaxation in response to ginsenosides in rat aorta: role of K+ channels. Eur J Pharmacol. 1999;367(1):41–49. doi: 10.1016/s0014-2999(98)00898-x. [DOI] [PubMed] [Google Scholar]
- 38.Kim N.D., Kim E.M., Kang K.W., Cho M.K., Choi S.Y., Kim S.G. Ginsenoside Rg3 inhibits phenylephrine-induced vascular contraction through induction of nitric oxide synthase. Br J Pharmacol. 2003;140(4):661–670. doi: 10.1038/sj.bjp.0705490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nagar H., Choi S., Jung S.B., Jeon B.H., Kim C.S. Rg3-enriched Korean Red Ginseng enhances blood pressure stability in spontaneously hypertensive rats. Integr Med Res. 2016;5(3):223–229. doi: 10.1016/j.imr.2016.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Choi S.H., Hong Z.Y., Nam J.K., Lee H.J., Jang J., Yoo R.J., Lee Y.J., Lee C.Y., Kim K.H., Park S. A hypoxia-induced vascular endothelial-to-mesenchymal transition in development of radiation-induced pulmonary fibrosis. Clin Cancer Res. 2015;21(16):3716–3726. doi: 10.1158/1078-0432.CCR-14-3193. [DOI] [PubMed] [Google Scholar]
- 41.Mina S.G., Wang W., Cao Q.F., Huang P., Murray B.T., Mahler G.J. Shear stress magnitude and transforming growth factor-beta eta 1 regulate endothelial to mesenchymal transformation in a three-dimensional culture microfluidic device. Rsc Adv. 2016;6(88):85457–85467. [Google Scholar]
- 42.Liu D.H., Chen Y.M., Liu Y., Hao B.S., Zhou B., Wu L., Wang M., Chen L., Wu W.K., Qian X.X. Rb1 protects endothelial cells from hydrogen peroxide-induced cell senescence by modulating redox status. Biol Pharm Bull. 2011;34(7):1072–1077. doi: 10.1248/bpb.34.1072. [DOI] [PubMed] [Google Scholar]
- 43.Kim M.K., Lee S.K., Park J.H., Lee J.H., Yun B.H., Park J.H., Seo S.K., Cho S., Choi Y.S. Ginsenoside Rg3 decreases fibrotic and invasive nature of endometriosis by modulating miRNA-27b: in vitro and in vivo studies. Sci Rep. 2017;7(1):17670. doi: 10.1038/s41598-017-17956-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li J., Lu J., Ye Z., Han X., Zheng X., Hou H., Chen W., Li X., Zhao L. 20(S)-Rg3 blocked epithelial-mesenchymal transition through DNMT3A/miR-145/FSCN1 in ovarian cancer. Oncotarget. 2017;8(32):53375–53386. doi: 10.18632/oncotarget.18482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wu B., Wang M., Ma Y., Yuan L., Lu S. High-throughput sequencing and characterization of the small RNA transcriptome reveal features of novel and conserved microRNAs in Panax ginseng. PLoS One. 2012;7(9) doi: 10.1371/journal.pone.0044385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mathiyalagan R., Subramaniyam S., Natarajan S., Kim Y.J., Sun M.S., Kim S.Y., Kim Y.J., Yang D.C. Insilico profiling of microRNAs in Korean ginseng (Panax ginseng Meyer) J Ginseng Res. 2013;37(2):227–247. doi: 10.5142/jgr.2013.37.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Papangeli I., Kim J., Maier I., Park S., Lee A., Kang Y.J., Tanaka K., Khan O.F., Ju H., Kojima Y. MicroRNA 139-5p coordinates APLNR-CXCR4 crosstalk during vascular maturation. Nat Commun. 2016;7 doi: 10.1038/ncomms11268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li Q.G., Liang X., Wang Y.W., Meng X.K., Xu Y., Cai S.J., Wang Z.M., Liu J.W., Cai G.X. miR-139-5p inhibits the epithelial-mesenchymal transition and enhances the chemotherapeutic sensitivity of colorectal cancer cells by downregulating BCL2. Sci Rep-Uk. 2016;6 doi: 10.1038/srep27157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jiang C., Tong Z., Fang W.L., Fu Q.B., Gu Y.J., Lv T.T., Liu D.M., Xue W., Lv J.W. Microrna-139-5p inhibits epithelial-mesenchymal transition and fibrosis in post-menopausal women with interstitial cystitis by targeting LPAR4 via the PI3K/Akt signaling pathway. J Cell Biochem. 2018;119(8):6429–6441. doi: 10.1002/jcb.26610. [DOI] [PubMed] [Google Scholar]
- 50.Kim B.M., Kim D.H., Park J.H., Surh Y.J., Na H.K. Ginsenoside Rg3 inhibits constitutive activation of NF-kappaB signaling in human breast cancer (MDA-MB-231) cells: ERK and akt as potential upstream targets. J Cancer Prev. 2014;19(1):23–30. doi: 10.15430/jcp.2014.19.1.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kim S.M., Lee S.Y., Yuk D.Y., Moon D.C., Choi S.S., Kim Y., Han S.B., Oh K.W., Hong J.T. Inhibition of NF-kappaB by ginsenoside Rg3 enhances the susceptibility of colon cancer cells to docetaxel. Arch Pharm Res. 2009;32(5):755–765. doi: 10.1007/s12272-009-1515-4. [DOI] [PubMed] [Google Scholar]
- 52.Maleszewska M., Moonen J.R., Huijkman N., van de Sluis B., Krenning G., Harmsen M.C. IL-1beta and TGFbeta2 synergistically induce endothelial to mesenchymal transition in an NFkappaB-dependent manner. Immunobiology. 2013;218(4):443–454. doi: 10.1016/j.imbio.2012.05.026. [DOI] [PubMed] [Google Scholar]
- 53.Arciniegas E., Carrillo L.M., De Sanctis J.B., Candelle D. Possible role of NFkappaB in the embryonic vascular remodeling and the endothelial mesenchymal transition process. Cell Adh Migr. 2008;2(1):17–29. doi: 10.4161/cam.2.1.5789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vallabhapurapu S., Karin M. Regulation and function of NF-kappaB transcription factors in the immune system. Annu Rev Immunol. 2009;27:693–733. doi: 10.1146/annurev.immunol.021908.132641. [DOI] [PubMed] [Google Scholar]
- 55.Gatheral T., Reed D.M., Moreno L., Gough P.J., Votta B.J., Sehon C.A., Rickard D.J., Bertin J., Lim E., Nicholson A.G. A key role for the endothelium in NOD1 mediated vascular inflammation: comparison to TLR4 responses. PLoS One. 2012;7(8) doi: 10.1371/journal.pone.0042386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang Z., Ding Q., Li Y., Liu Q., Wu W., Wu L., Yu H. Reanalysis of microRNA expression profiles identifies novel biomarkers for hepatocellular carcinoma prognosis. Tumour Biol. 2016;37(11):14779–14787. doi: 10.1007/s13277-016-5369-3. [DOI] [PubMed] [Google Scholar]
- 57.Maoa R., Zou F., Yang L., Lin S., Li Y., Ma M., Yin P., Liang X., Liu J. The loss of MiR-139-5p promotes colitis-associated tumorigenesis by mediating PI3K/AKT/Wnt signaling. Int J Biochem Cell Biol. 2015;69:153–161. doi: 10.1016/j.biocel.2015.10.008. [DOI] [PubMed] [Google Scholar]
- 58.Zhang C. The role of inflammatory cytokines in endothelial dysfunction. Basic Res Cardiol. 2008;103(5):398–406. doi: 10.1007/s00395-008-0733-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Huang X., Pan L., Pu H., Wang Y., Zhang X., Li C., Yang Z. Loss of caveolin-1 promotes endothelial-mesenchymal transition during sepsis: a membrane proteomic study. Int J Mol Med. 2013;32(3):585–592. doi: 10.3892/ijmm.2013.1432. [DOI] [PubMed] [Google Scholar]
- 60.Moreira L.O., Zamboni D.S. NOD1 and NOD2 signaling in infection and inflammation. Front Immunol. 2012;3:328. doi: 10.3389/fimmu.2012.00328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shang J., Zhang Y., Jiang Y., Li Z., Duan Y., Wang L., Xiao J., Zhao Z. NOD2 promotes endothelial-to-mesenchymal transition of glomerular endothelial cells via MEK/ERK signaling pathway in diabetic nephropathy. Biochem Biophys Res Commun. 2017;484(2):435–441. doi: 10.1016/j.bbrc.2017.01.155. [DOI] [PubMed] [Google Scholar]
- 62.Chen Y., Yuan T., Zhang H., Yan Y., Wang D., Fang L., Lu Y., Du G. Activation of Nrf2 attenuates pulmonary vascular remodeling via inhibiting endothelial-to-mesenchymal transition: an insight from a plant polyphenol. Int J Biol Sci. 2017;13(8):1067–1081. doi: 10.7150/ijbs.20316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kanasaki K., Shi S., Kanasaki M., He J., Nagai T., Nakamura Y., Ishigaki Y., Kitada M., Srivastava S.P., Koya D. Linagliptin-mediated DPP-4 inhibition ameliorates kidney fibrosis in streptozotocin-induced diabetic mice by inhibiting endothelial-to-mesenchymal transition in a therapeutic regimen. Diabetes. 2014;63(6):2120–2131. doi: 10.2337/db13-1029. [DOI] [PubMed] [Google Scholar]
- 64.Lee D.C., Lau A.S. Effects of Panax ginseng on tumor necrosis factor-alpha-mediated inflammation: a mini-review. Molecules. 2011;16(4):2802–2816. doi: 10.3390/molecules16042802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yuan Q., Jiang Y.W., Ma T.T., Fang Q.H., Pan L. Attenuating effect of Ginsenoside Rb1 on LPS-induced lung injury in rats. J Inflamm (Lond). 2014;11(1):40. doi: 10.1186/s12950-014-0040-5. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 66.Zhang Y., Sun K., Liu Y.Y., Zhang Y.P., Hu B.H., Chang X., Yan L., Pan C.S., Li Q., Fan J.Y. Ginsenoside Rb1 ameliorates lipopolysaccharide-induced albumin leakage from rat mesenteric venules by intervening in both trans- and paracellular pathway. Am J Physiol Gastrointest Liver Physiol. 2014;306(4):G289–G300. doi: 10.1152/ajpgi.00168.2013. [DOI] [PubMed] [Google Scholar]
- 67.Zhou W., Chai H., Lin P.H., Lumsden A.B., Yao Q., Chen C. Ginsenoside Rb1 blocks homocysteine-induced endothelial dysfunction in porcine coronary arteries. J Vasc Surg. 2005;41(5):861–868. doi: 10.1016/j.jvs.2005.01.054. [DOI] [PubMed] [Google Scholar]
- 68.Hien T.T., Kim N.D., Kim H.S., Kang K.W. Ginsenoside Rg3 inhibits tumor necrosis factor-alpha-induced expression of cell adhesion molecules in human endothelial cells. Pharmazie. 2010;65(9):699–701. [PubMed] [Google Scholar]
- 69.Kang K.S., Kim H.Y., Yamabe N., Park J.H., Yokozawa T. Preventive effect of 20(S)-ginsenoside Rg3 against lipopolysaccharide-induced hepatic and renal injury in rats. Free Radic Res. 2007;41(10):1181–1188. doi: 10.1080/10715760701581740. [DOI] [PubMed] [Google Scholar]
- 70.Jiang Y., Li M., Lu Z., Wang Y., Yu X., Sui D., Fu L. Ginsenoside Rg3 induces ginsenoside Rb1-comparable cardioprotective effects independent of reducing blood pressure in spontaneously hypertensive rats. Exp Ther Med. 2017;14(5):4977–4985. doi: 10.3892/etm.2017.5198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Qiu G., Lin Y., Zhang H., Wu D. miR-139-5p inhibits epithelial-mesenchymal transition, migration and invasion of hepatocellular carcinoma cells by targeting ZEB1 and ZEB2. Biochem Biophys Res Commun. 2015;463(3):315–321. doi: 10.1016/j.bbrc.2015.05.062. [DOI] [PubMed] [Google Scholar]
- 72.Shao Q., Zhang P., Ma Y., Lu Z., Meng J., Li H., Wang X., Chen D., Zhang M., Han Y. MicroRNA-139-5p affects cisplatin sensitivity in human nasopharyngeal carcinoma cells by regulating the epithelial-to-mesenchymal transition. Gene. 2018;652:48–58. doi: 10.1016/j.gene.2018.02.003. [DOI] [PubMed] [Google Scholar]
- 73.Zou F., Mao R., Yang L., Lin S., Lei K., Zheng Y., Ding Y., Zhang P., Cai G., Liang X. Targeted deletion of miR-139-5p activates MAPK, NF-kappaB and STAT3 signaling and promotes intestinal inflammation and colorectal cancer. FEBS J. 2016;283(8):1438–1452. doi: 10.1111/febs.13678. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.