

Abstract
Summary
Here we present Zombi, a tool to simulate the evolution of species, genomes and sequences in silico, that considers for the first time the evolution of genomes in extinct lineages. It also incorporates various features that have not to date been combined in a single simulator, such as the possibility of generating species trees with a pre-defined variation of speciation and extinction rates through time, simulating explicitly intergenic sequences of variable length and outputting gene tree—species tree reconciliations.
Availability and implementation
Source code and manual are freely available in https://github.com/AADavin/ZOMBI/.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Reconstructing the pattern of horizontal gene transfers between species can help us date the origin of different taxa (Davín et al., 2018; Wolfe and Fournier, 2018), understand the spread of genes of clinical importance (Lerminiaux and Cameron, 2019) and resolve difficult phylogenetic questions, such as inferring the rooting point of prokaryotic trees (Abby et al., 2012; Szöllősi et al., 2012; Williams et al., 2017) or the evolutionary position of certain lineages of unclear origin (Boussau et al., 2008). In the last decades, a large number of simulators have been developed to model a wide range of evolutionary scenarios (Beiko and Charlebois, 2007; Carvajal-Rodríguez, 2008; Dalquen et al., 2012; Kundu and Bansal, 2019; Mallo et al., 2016; Sjöstrand et al., 2013) but none so far have considered the existence of extinct lineages and the horizontal transmission of genes (by lateral gene transfers) involving species that are not represented in the phylogeny (Fournier et al., 2009; Szöllősi et al., 2013; Zhaxybayeva and Peter Gogarten, 2004). Zombi simulates explicitly the genome evolution taking place in these extinct lineages, which is expected to have an impact in extant lineages by means of Lateral Gene Transfers (Szöllősi et al., 2013). By not considering extinct lineages, other simulators make the implicit assumption that the transfer donor always leaves a surviving descendant among sampled species, while we know that this is most often not true (Szöllősi et al., 2013). Making this assumption may potentially hamper our ability to simulate realistic scenarios of evolution. In addition to considering evolution along extinct lineages, Zombi includes several features hitherto not found together in any other simulator (Supplementary Table S1).
2 Basic features of Zombi
Zombi is a multilevel simulator, where a species tree is first simulated, then genomes evolve along the branches of this species tree, and finally, sequences are generated for each genome. These three steps, depicted in Figure 1 and detailed hereafter, are controlled by three main ‘modes’, named T, G and S, for species Tree, Genome and Sequence, respectively.
Fig. 1.
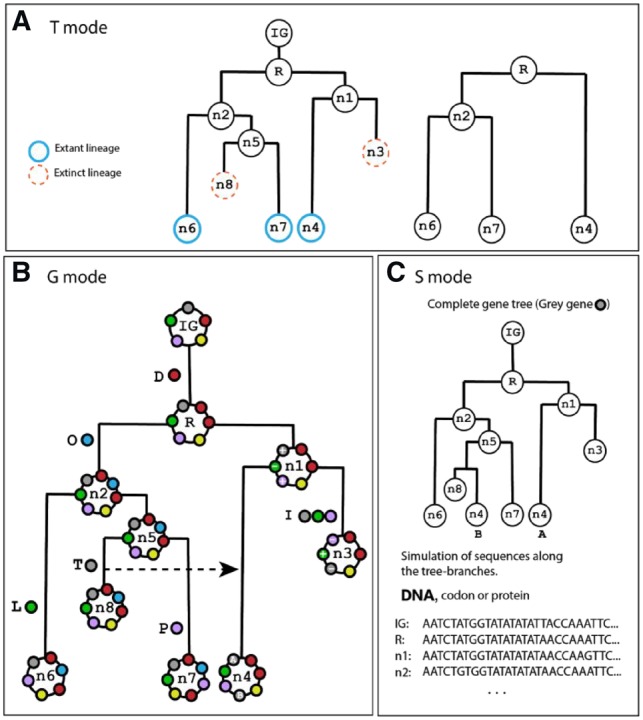
Overview of the three steps of the Zombi simulator. (A) In T mode, Zombi simulates a species tree using a birth-death process and outputs the pruned version of it by removing extinct lineages. In this example, lineages n3 and n8 go extinct before the simulation ends. (B) in G mode, a circular genome evolves within the branches of the complete species tree obtained with the T mode by Duplications (D), Originations (O), Inversions (I), Transpositions (P), Losses (L) and Transfers (T) of genes. The simulation starts with the initial genome (IG) containing a number of genes determined by the user (5 in this example, represented by the coloured circles). Each gene has an orientation (+ or -) that is determined randomly and represents the direction of the gene in the coding strand. Several events affecting different genes and their impact on the genome structure are indicated next to the branches where they occur. The inversion events not only modify the positions of the genes but also change their orientation. (C) In S mode, Zombi can be used to simulate codon, nucleotides and amino acids along the branches of the gene family trees. Here, the gene tree of the grey coloured gene family from B has been depicted
The T mode simulates a species tree under the birth-death model (Kendall, 1948), using the Gillespie algorithm (Gillespie, 1977), which is the standard method for simulating arbitrarily complex continuous time Markov processes (Supplementary Fig. S1). While more efficient and accurate methods exist to simulate the reconstructed tree (Hartmann et al., 2010), taking into consideration unrepresented (extinct and unsampled) species requires simulating the complete species tree, which includes all extinct and unsampled branches of the phylogeny (Szöllősi et al., 2013). This tree is subsequently pruned to obtain the reconstructed tree, by removing all the lineages that did not survive until the end of the simulation (Fig. 1A).
The G mode simulates the evolution of genomes within the branches of the complete species tree (Fig. 1B) using also the Gillespie algorithm (Supplementary Fig. S2) to account for six possible genome-level events: duplications, losses, inversions, transpositions, transfers and originations. Each of the first five events is characterized by two parameters: the first one is the effective rate, that controls the frequency and fixation probability; the second one controls the extension, i.e. the number of contiguous genes simultaneously affected by the event. Originations of new genes occurs one by one and therefore only a single effective rate parameter is needed. When a Transfer event occurs, the recipient lineage is randomly chosen from all the lineages alive at that time. The user can make the frequence of transfers to be higher between closely related lineages (Ochman et al., 2000) (Supplementary Fig. S3). Once the simulation reaches the end, Zombi outputs a list containing each event that has occurred in the simulation for every gene family (all genes that share a common origin). Besides, the gene trees of each family are reconstructed by combining both species-level events (Speciations and Extinctions) and genome-level events (Duplications, Transfers and Losses). Inversions and transpositions do not modify the topology of the tree but add an extra layer of complexity by changing the neighborhood of genes, which is especially relevant when genome-level events affect more than one gene at a time (Supplementary Fig. S4). The gene family trees are also pruned to present the user the trees that can be expected to be recovered from most real-data analyses, removing all extinct lineages and gene branches that do not arrive until the present time. The S mode, finally, simulates gene sequences (at either the codon, nucleotide or protein level) along the gene family trees (Fig. 1C). The user can modify the scaling of the tree to better control the number of substitutions that take place per unit of time, and thus simulate fast or slow-evolving genes.
3 Advanced features
In addition to the basic features presented above, ‘advanced’ modes of Zombi (listed in Supplementary Table S2) can be used to obtain richer and more realistic evolutionary scenarios. For example, it is possible to use a species tree input by the user, to generate species trees with variable extinction and speciation rates, or to control the number of living lineages at each unit of time (Supplementary Fig. S5). At the genome level, Zombi can simulate genomes using branch-specific rates (Gu mode, allowing the user to simulate very specific scenarios such as one in which a certain lineage experiences a massive loss of genes), gene-family specific rates (Gm mode, which makes easier the process of using rates estimated from real datasets) and genomes accounting for intergenic regions (Gf mode) of variable length [drawn from a flat Dirichlet distribution (Biller et al., 2016)]. At the sequence level the user can fine-tune the substitution rates to make them branch specific. Zombi provides the user with a clear and detailed output of the complete evolutionary process simulated, including the reconciled gene trees with the species tree in the RecPhyloXML reconciliation standard (Duchemin et al., 2018).
4 Performance and validation
Simulations with Zombi are fast: with a starting genome of 500 genes and a species tree of 2000 taxa (extinct + extant), it takes around 1 min on a 3.4Ghz laptop to simulate all the genomes (Supplementary Fig. S6).
We validated that the distribution of waiting times between successive events was following an exponential distribution (Supplementary Figs S7 and S8), that the distribution of intergene sizes at equilibrium was following a flat Dirichlet distribution, as expected from Biller et al. (2016) (Supplementary Fig. S9), that the number of events and their extension occur with a frequency according to their respective rates (Supplementary Fig. S10) and that the gene family size distribution followed a power-law when duplication rates are higher than loss rates and stretched-exponential in the opposite case (Reed and Hughes, 2003; Szöllosi and Daubin, 2011) (Supplementary Fig. S11). We also checked by hand the validity of many simple scenarios to detect possible inconsistencies in the algorithm.
5 Implementation
Zombi is implemented in Python 3.6. It relies on the ETE 3 toolkit (Huerta-Cepas et al., 2016) and the Pyvolve package (Spielman and Wilke, 2015). It is freely available at https://github.com/AADavin/ZOMBI along with detailed documentation and two tutorials in a wiki page.
Supplementary Material
Acknowledgements
We thank Vincent Daubin, Wandrille Duchemin, Nicolas Lartillot and Thibault Latrille for insightful discussions during the preparation of this manuscript.
Funding
A.A.D. and G.J.Sz. received funding from the European Research Council under the European Union’s Horizon 2020 research and innovation programme under grant agreement no. 714774., in addition, G.J.Sz. was supported by the grant GINOP-2.3.2.-15-2016-00057. T.T and D.M.d.V received funding from grant ANR-18-CE02-0007-01 (‘STHORIZ’).
Conflict of Interest: none declared.
References
- Abby S.S. et al. (2012) Lateral gene transfer as a support for the tree of life. Proc. Natl. Acad. Sci. USA, 109, 4962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beiko R.G., Charlebois R.L. (2007) A simulation test bed for hypotheses of genome evolution. Bioinformatics, 23, 825–831. [DOI] [PubMed] [Google Scholar]
- Biller P. et al. (2016) Breaking good: accounting for fragility of genomic regions in rearrangement distance estimation. Genome Biol. Evol., 8, 1427–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boussau B. et al. (2008) Accounting for horizontal gene transfers explains conflicting hypotheses regarding the position of aquificales in the phylogeny of bacteria. BMC Evol. Biol., 8, 272.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvajal-Rodríguez A. (2008) Simulation of genomes: a review. Curr. Genomics, 9, 155–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalquen D.A. et al. (2012) ALF—a simulation framework for genome evolution. Mol. Biol. Evol., 29, 1115–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davín A.A. et al. (2018) Gene transfers can date the tree of life. Nat. Ecol. Evol., 2, 904–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchemin W. et al. (2018) RecPhyloXML – a format for reconciled gene trees. Bioinformatics, 34, 3646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fournier G.P. et al. (2009) Horizontal gene transfer from extinct and extant lineages: biological innovation and the coral of life. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci., 364, 2229–2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillespie D.T. (1977) Exact stochastic simulation of coupled chemical reactions. J. Phys. Chem., 81, 2340–2361. [Google Scholar]
- Hartmann K. et al. (2010) Sampling trees from evolutionary models. Syst. Biol., 59, 465.. [DOI] [PubMed] [Google Scholar]
- Huerta-Cepas J. et al. (2016) ETE 3: reconstruction, analysis, and visualization of phylogenomic data. Mol. Biol. Evol., 33, 1635–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kendall D.G. (1948) On the generalized ‘Birth-and-Death’ process. Ann. Math. Stat., 19, 1–15. [Google Scholar]
- Kundu S., Bansal M.S. (2019) SaGePhy: an improved phylogenetic simulation framework for gene and subgene evolution. Bioinformatics, 35, 3496–3498. [DOI] [PubMed] [Google Scholar]
- Lerminiaux N.A., Cameron A.D.S. (2019) Horizontal transfer of antibiotic resistance genes in clinical environments. Can. J. Microbiol., 65, 34–44. [DOI] [PubMed] [Google Scholar]
- Mallo D. et al. (2016) SimPhy: phylogenomic simulation of gene, locus, and species trees. Syst. Biol., 65, 334–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochman H. et al. (2000) Lateral gene transfer and the nature of bacterial innovation. Nature, 405, 299–304. [DOI] [PubMed] [Google Scholar]
- Reed W.J., Hughes B.D. (2003) Power-law distribution from exponential processes: an explanation for the occurrence of long-tailed distributions in biology and elsewhere. Sci. Math. Jpn. http://www.math.uvic.ca/faculty/reed/JAMS_sub.pdf. [Google Scholar]
- Sjöstrand J. et al. (2013) GenPhyloData: realistic simulation of gene family evolution. BMC Bioinformatics, 14, 209.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spielman S.J., Wilke C.O. (2015) Pyvolve: a flexible python module for simulating sequences along phylogenies. PLoS One, 10, e0139047.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szöllősi G.J., Daubin V. (2011) The pattern and process of gene family evolution. arXiv preprint arXiv:1102.2331. [Google Scholar]
- Szöllősi G.J. et al. (2012) Phylogenetic modeling of lateral gene transfer reconstructs the pattern and relative timing of speciations. Proc. Natl. Acad. Sci. USA, 109, 17513–17518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szöllősi G.J. et al. (2013) Lateral gene transfer from the dead. Syst. Biol., 62, 386–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams T.A. et al. (2017) Integrative modeling of gene and genome evolution roots the archaeal tree of life. Proc. Natl. Acad. Sci. USA, 114, E4602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe J.M., Fournier G.P. (2018) Horizontal gene transfer constrains the timing of methanogen evolution. Nat. Ecol. Evol., 2, 897–903. [DOI] [PubMed] [Google Scholar]
- Zhaxybayeva O., Peter Gogarten J. (2004) Cladogenesis, coalescence and the evolution of the three domains of life. Trends Genet., 20, 182–187. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.