

Abstract
Stimuli-responsive nanogels are important drug and gene carriers that mediate the controlled release of therapeutic molecules. Herein, we report the synthesis of fully degradable disulfide cross-linked nanogel drug carriers formed by oxidative radical polymerization of 2,2’-(ethylenedioxy)diethanethiol (EDDET) as a monomer with different cross-linkers, including pentaerythritol tetramercaptoacetate (PETMA). Because the poly(EDDET) backbone repeat structure and cross-linking junctions are composed entirely of disulfide bonds, these nanogels specifically degrade to small molecule dithiols intracellularly in response to the reducing agent glutathione present inside of cells. Cross-linked nanogels were synthesized using controlled microfluidic mixing in the presence of a nonionic Pluronic surfactant PLU-127 to increase the nanogel stability. Adjusting the monomer to cross-linker ratio from 5:1 to 100:1 (mol/mol) tuned the cross-linking density, resulting in swelling ratios from 1.65 to >3. Increasing the amount of stabilizing Pluronic surfactant resulted in a decrease of nanogel diameter, as expected due to increased surface area of the resulting nanogels. The monomer to cross-linker ratio in the feed had no effect on the formed nanogel diameter, providing a way to control cross-linking density with constant nanogel size but tunable drug release kinetics. Nanogels exhibited an entrapment efficiency of up to 75% for loading of Rhodamine B dye. In vitro studies showed low cytotoxicity, quick uptake, and fast degradation kinetics. Due to the ease of synthesis, rapid gelation times, and tunable functionality, these non-toxic and fully degradable nanogels offer potential for use in a variety of drug delivery applications.
Graphical Abstract
Introduction
Controlled drug delivery carriers can improve the pharmacokinetic properties of a wide variety of drugs. In addition to controlled release of small molecules, such as in FDA-approved microparticle drug depots and chemotherapeutic drug-loaded liposomes,1 nanoparticle carriers are essential for the delivery of biomacromolecular drugs including nucleic acids that cannot cross cell membranes on their own.1–3 Embedding drugs into nanoparticles not only effectively suppresses interaction with blood components, but also enhances drug targeting specificity, lowers systemic drug toxicity, improves treatment absorption rates, and provides protection for pharmaceuticals against degradation.4–6
Polymer-based drug carriers are an important class of materials because of the ability to readily control their chemical and physical properties via chemical synthesis and their ease of processing. Furthermore, stimuli-responsive polymers enable targeted delivery and controlled release in response to biological stimuli changes, such as pH, temperature, or redox potential to trigger cargo release.7 Drug delivery systems (e.g. micelles, liposomes, dendrimers, nanogels, and hydrogels) composed of responsive polymers can release the cargo in response to specific triggers resulting in degradation or collapse and expansion of the network in an aqueous environment.8 Aliphatic polyesters, such as poly(lactic acid) (PLA), poly(glycolic acid) (PGA), poly(ε-caprolactone) (PCL), polycarbonates, and their copolymers degrade under physiological conditions, but are generally hydrophobic and lack the functional groups required for delivery of drugs that require electrostatic interactions (e.g. nucleic acids), bioconjugation reactions, and attachment of targeting ligands.9, 10 Also, ester bond degradation generates acidic products, which can cause an undesirable local decrease in pH.
Polydisulfides, on the other hand, can be degraded specifically in response to redox potential through thiol-disulfide exchange reactions.11 Intracellular compartments of cells are more reductive than the extracellular matrix, and the glutathione/glutathione disulfide (GSH/GSSG) couple is regarded as the representative cellular redox mechanism that plays a critical role in redox homeostasis.12 The concentration of GSH is found in millimolar concentrations inside of cells, and is 100–1000 times lower outside of cells.13 Therefore, polydisulfides can degrade in physiological settings (i.e., in cells), potentially with reduced cytotoxicity. It was also reported that the GSH level is related to many human diseases like neurodegenerative diseases, liver diseases, stroke, seizures, and diabetes.14–18 For example, an abnormally high concentration of GSH in cancerous cells protects the cells against the anti-cancer drugs and free radicals generated during radiation therapy, which results in multi-drug and radiation resistance.14, 16 This could provide a potential physiological trigger for polydisulfide degradation and drug delivery to diseased tissues.8
The significant difference in the redox environment has been explored for developing stimuli-responsive drug delivery systems. Disulfide bonds have been incorporated into polymeric materials in a variety of ways,5, 19 including the use of disulfide containing cross-linkers,20–28 redox-responsive self-assembly of amphiphilic polymers in the form of micelles or polymersomes,29, 30 biodegradable polymers, both linear and dendritic from disulfide-containing monomers,31–35 and redox-responsive drug/polymer conjugates or polymer prodrugs. Disulfide-containing polymers and nanogels have synthesized by controlled/living radical polymerization (CRP) methods as well.28
To date, the majority of these approaches have been limited to polymerization of vinyl monomers, cross-linked by disulfide containing cross-linkers (e.g., star polymers, micelles, branched polymers, and gels).28 These structures degrade to the original carbon-carbon bond-based polymer upon disulfide reduction, thus limiting the extent of degradation to long polymer chains. Direct incorporation of disulfides into the polymer backbone would allow for tunable levels of degradation and has been accomplished to some extent in a small number of examples.36–38 However, the preparation of linear polymers composed entirely by polydisulfide bonds (no vinyl comonomers) remains challenging.
We were attracted to recent reports on highly efficient oxidative systems for the polymerization of dithiols to high molecular weight polydisulfide polymers via a base-catalyzed thiol oxidation mechanism.39 Once sulfhydryl groups are deprotonated,40 the thiolate anion can undergo two separate processes that lead to disulfide formation. In one process, the nucleophilic thiolate anion may attack another disulfide bond resulting in the formation of a new disulfide and a new thiolate anion. This mechanism has been utilized in the context of cell-penetrating polydisulfides, which have been shown to efficiently internalize into cells and rapidly degrade in the cytosol by GSH-assisted depolymerization with minimal cytotoxicity.41–46 In an alternative process, a single electron transfer step leads to generation of thiyl radicals, which rapidly couple with each other to form disulfide bonds and polymers.47
Inspired by this thiyl radical process, we aimed to synthesize fully degradable disulfide cross-linked nanogel drug carriers formed by oxidative radical polymerization of 2,2’-(ethylenedioxy)diethanethiol (EDDET) as a monomer with different cross-linkers. Numerous studies show that nanogels are excellent drug carriers due to their high drug loading capacity, stability, and responsiveness to a wide variety of environmental stimuli.48, 49 Regarding disulfide redox-responsive nanogels, literature reports are limited to fabrication of the nanogels using polymer precursors and crosslinking the polymer strands.50–53 For example, nanogels were prepared using dextran-lipoic acid derivatives cross-linked by dithiothreitol (DTT).54 The nanogels showed a high drug loading efficiency and delivery of doxorubicin into cells. There remains a need to develop nanogels that have high stability, high loading efficiency, fast degradation kinetics, and the ability to completely degrade down to non-toxic small molecule components.
Herein, we report a facile method to prepare redox responsive nanogel drug carriers via dithiol oxidation. A dithiol monomer, 2,2’-(ethylenedioxy)diethanethiol (EDDET) was co-polymerized with the cross-linkers ethoxylated-trimethylolpropan tri(3-mercaptopropionate) (ETTMP), pentaerythritol tetra(3-mercaptopropionate) (PETMP), and pentaerythritol tetramercaptoacetate (PETMA) into nanogels via a base-catalyzed thiol oxidation mechanism. Mechanistic studies indicated that a single electron transfer step occurred, and disulfide formation was confirmed by FT-IR. Polymerization occurred in bulk or solution. Nanogels were engineered using microfluidic mixing and a non-ionic surfactant was incorporated to impart colloidal stability. Control over mesh size was achieved for both bulk and formulated nanogels. An assay using Ellman’s Reagent was employed to quantify the cross-linking density and number of free –SH groups. Nanogels were characterized by dynamic light scattering (DLS) and transmission electron microscopy (TEM). The weight percent loading and entrapment efficiency was also measured. In vitro cell viability assays and cellular uptake studies were performed in HeLa cells. Lastly, quenching studies and degradation kinetics were completed. These nanogel drug carriers provide a versatile platform that can be customized to fit a specific application (e.g. via incorporation of functional thiols). Furthermore, the free –SH groups could potentially be used to attach targeting moieties, drugs, or stabilizing chemical functionalities. Due to the rapid and facile synthesis with a variety of thiol building blocks, tunable physical properties, high biocompatibility, and fully degradable nature, the resulting nanogels are potentially useful in a wide variety of drug delivery applications.
Experimental
Materials.
Unless otherwise noted, all chemicals were purchased from Sigma-Aldrich and used as received. Organic solvents were purchased from Fisher Scientific and purified with a solvent purification system (Innovative Technology). The multifunctional THIOCURE cross-linkers were provided by Bruno Bock.
Methods.
Homopolymerization of EDDET.
2,2’-(ethylenedioxy) diethanethiol (EDDET) and trimethylamine (TEA) were dissolved in tetrahydrofuran (THF) at room temperature. While stirring, 3 weight (wt.) % hydrogen peroxide (aq.) was added dropwise. The same polymerization procedure could be performed in water instead of THF. After 10 minutes, the molecular weight and polydisperisity index (PDI) for the homopolymers were measured by GPC. An example synthesis is as follows: In a glass vial equipped with a stir bar, 600 μL EDDET (3.84 mmol, 700 mg) was dissolved in 1.2 mL TEA (8.45 mmol, 855 mg), then 2.3 mL 3 wt. % H2O2 (2.20 mmol, 68.2 mg) was added dropwise over 5 minutes.
Hydrogel polymerization.
EDDET, cross-linker (PETMA, PETMP, or ETTMP), and TEA were dissolved in water:acetone 1:1 (vol/vol). The solution was then added into excess 3 wt. % hydrogen peroxide solution (aq.) and gelation occurred within seconds to give a white or opaque hydrogel. A molar equivalence of the following was used CL:M:Base = 1:5:0.1, 1:10:0.1, 1:25:0.1, 1:50:0.1, 1:75:0.1, 1:100:0.1 and 3 wt. % H2O2 was used in excess. Depending on the type of cross-linker used, the hydrogel texture varied from soft (ETTMP) to spongy (PETMA) to rubbery (PETMP).
Gelation time mechanistic study.
Hydrogel synthesis was carried out in four different reaction vials for each PETMA, PETMP, and ETTMP gel. The first reaction flask observed normal conditions; the second, omitted the addition of TEA; the third, included the addition of 1:1 molar equivalence of hydroquinone as a free radical inhibitor; and the fourth, included the addition of 1:500 molar equivalence of hydroquinone. The gelation time was recorded.
Degree of swelling study.
Samples of PETMA, PETMP, and ETTMP hydrogels were completely dried for 48 hours under vacuum. The gel sample was allowed to fully swell for 4 hours in H2O and the wet mass was measured. The gel sample was again allowed to completely dry for 48 hours under vacuum and the dry mass was measured. The swelling ratio was calculated by the following: swelling ratio = [mass of swollen gel]/[mass of dry gel].
Quantification of free –SH groups.
5,5’-dithio-bis-(2-nitrobenzoic acid) (DTNB, Ellman’s Reagent) stock solutions in water were prepared and used to establish a standard curve of absorbance versus free moles of –SH, where absorbance was measured using Tecan HP8300 plate reader. Samples were prepared at a concentration located in the working range of standard curve. Samples were incubated with 1 μg/μL stock Ellman’s reagent for 15 minutes. Absorbance was measured at λ = 412 nm in a black 96-well clear bottom plate in triplicate. Moles of free –SH for each sample was determined using the standard curve.
Nanogel synthesis.
Nanogels were fabricated using a microfluidic mixing instrument with herringbone rapid mixing features (Precision Nanosystems NanoAssembler). EDDET, PETMA, and TEA were dissolved in water:acetone 1:1 (volume) and mixed with an aqueous phase containing different weight percentages of hydrogen peroxide (weight percentages tested included: 3%, 6%, 12%, 24%, and 30%) and surfactant (PLU-127) (weight percentage of PLU-127: 0%, 25%, 50%, and 75%). The optimized conditions included a 1:10 ratio of organic:aqueous phase (volume), 12 mL/min flow rate, and 12 wt. % of hydrogen peroxide. Different cross-linking densities (CL:M:Base = 1:5:0.1, 1:10:0.1, 1:25:0.1, 1:50:0.1, 1:75:0.1, 1:100:0.1) were obtained using the same 25% wt. percentage of surfactant and different cross-linker to monomer ratios.
Nanogel purification.
Nanogels were purified via dialysis using Spectrum Laboratories RC Dialysis Membrane with 1000 Dalton MWCO. The nanogel reaction mixture was placed into dialysis membrane tube and sealed at both ends. Dialysis was conducted against water:acetone 1:1 (volume) 3X for 4 hours, each run, and then against water 3X for 4 hours, each run. All water utilized was ultra-purified using a Milli-Q water purification system. After dialysis purification, an accurate small aliquot of nanogels solution was taken by pipette, placed in a pre-weighed glass vial, and allowed to dry for 24 hours under vacuum. The vial was re-weighed and the mass of the completely dried nanogels was calculated. The nanogels were re-dispersed in PBS (or another solvent for analysis) at determined concentrations.
Quantification of dye encapsulation.
Nanogels with different mesh sizes were synthesized by varying the cross-linker:monomer ratio, all with the same initial 5 wt. % loading in the feed of Rhodamine B dye (5 wt. % loading of the total mass of monomer + crosslinker). Using the same concentration of all nanogel samples within the working range of the standard curve, the nanogels were degraded using 11 mM dithiothreitol (DTT) for 24 hours to allow all Rhodamine B to be freely soluble and detectable. Fluorescence was measured using a Tecan HP8300 plate reader, λex = 540 nm and λem = 576 nm top-read in solid black 96-well plates in triplicate. The mass of Rhodamine B loaded for each nanogel sample was determined using the standard curve. The weight percent loading was calculated by [(actual dye encapsulation)/(monomer + crosslinker)] x 100. The percent entrapment efficiency was calculated by [(actual dye encapsulation)/(theoretical dye encapsulation)] x 100.
Degradation of PETMA Rhodamine B loaded nanogels.
5 wt. % Rhodamine B loaded nanogels were imaged on an EVOS optical fluorescence microscope. Thin films were drop cast onto a cleaned glass slide. Images were taken before and after the addition of 11 mM DTT.
Kinetic study.
Samples of 5 wt. % Rhodamine B dye loaded nanogels (CL:M = 1:25, 1:50, 1:70, and 1:100 (mol/mol)) were prepared using the experimentally determined percent efficiencies (45.32%, 51.24%, 56.38%, and 75.88%, respectively) to provide samples each containing the same amount of Rhodamine B loaded dye. A stock solution of 11 mM GSH was prepared using Milli-Q water. Nanogel samples and 11 mM GSH were added into an Amicon Ultra-0.5 mL centrifugal filter tubes and allowed to incubate for the following time points: 0 min, 5 min, 10 min, 15 min, 30 min, 45 min, and 60 min. After each time interval, the nanogels + GSH mixture was centrifuged at 4,000 rcf for 10 min at 4 °C. A fluorescence measurement of the filtrate (filtrate A) was taken via triplicate (Tecan HP8300 plate reader, λex = 540 nm and λem = 576). The percent release of Rhodamine B dye was quantified using a standard curve. The remaining nanogels + GSH mixture (filtrate B) was obtained by a reverse spinning step after collecting filtrate A. Reverse spinning step was centrifuged at 1,000 rfc for 2 min. DLS measurements were taken before addition GSH, filtrate A, and filtrate B.
Quenching study.
Nanogels were incubated with 11 mM GSH and a fluorescence time scan measurement was performed using a Hitachi F-7000 Fluorescence Spectrophotometer (λem = 590 nm λex = 550 nm). Fluorescence intensity was normalized to baseline. DLS measurements were made before and after the addition of GSH.
Cell culture.
HeLa cells (ATCC) were maintained in RPMI-1640 (Life Technologies) supplemented with 5% fetal calf serum at 37 °C in a humidified atmosphere containing 5% CO2 and 95% air.
Cell viability assay.
Cytotoxicity of nanogels (1:25, 1:50, 1:70, and 1:100 cross-linker:monomer ratios) and nanogels degraded by 11 mM GSH for 24 hours was assessed in HeLa cells (10,000 cells/well in 96-well plates seeded 12 h prior to experiments). HeLa cells were incubated in DMEM with 5% FBS for 24 hours with a series of total nanogels or degraded nanogels products of 0 ng/μL, 2.5 ng/μL, 5 ng/μL, 10 ng/μL, 25 ng/μL, 50 ng/μL, 75 ng/μL, and 100 ng/μL. Cell viability was measured using the CellTiter-Glo Luminescent Cell Viability Assay (Promega) following the recommended protocol.
Cellular uptake study:
HeLa cells were seeded at a density of 30,000 cells/well in 8-chambered cover glass slides (Nunc) and allowed to attach for 24 hours (37 °C, 5% CO2). 5 wt. % Rhodamine B dye loaded nanogels (1:25 cross-linker:monomer ratio) solutions were prepared via serial dilution from original stock using PBS (2.5 ng/μL, 5 ng/μL, 10 ng/μL, 25 ng/μL, 50 ng/μL, 75 ng/μL, and 100 ng/μL). After 24 h incubation, the cells were washed with PBS, fixed with 4% (volume) PFA, and stained with DAPI for confocal imaging. Confocal microscopy imaging was performed using a Zeiss Confocal Microscope at 40X magnification and images were analyzed using ImageJ (NIH).
Time course study.
HeLa cells were seeded at a density of 30,000 cells/well in 8-chambered cover glass slides (Nunc) and allowed to attach for 24 hours (37 °C, 5% CO2). 5 wt. % Rhodamine B dye loaded nanogels (1:25, 1:50, 1:70, and 1:100 cross-linker:monomer ratio) with a final dye concentration of 23 ng/μL were prepared from original stock using PBS. Incubation time points included the following: 0 min, 15 min, 1 hr, 3 hr, 6 hr, 15 hr, and 24 hr. After each respective incubation period, the cells were washed with PBS, fixed with 4% (volume) PFA, and stained with DAPI for confocal imaging. Confocal microscopy imaging was performed using a Zeiss Confocal Microscope at 40X magnification and images were analyzed using ImageJ (NIH).
Instrumental.
Molecular weight analysis.
Molecular weights were measured by a Gel Permeation Chromatography (GPC) (Viscotek) system equipped with a refractive index (RI) detector and ViscoGEL I-series columns (Viscoteck I-MBLMW-3078) using DMF as the eluent at 0.75 mL/min and 45 °C. The instrument was calibrated with a series of 10 narrow polydispersity polystyrene standards (500 to 200,000 g/mol).
Fourier Transform Infrared Spectroscopy (FTIR).
FTIR was performed on a Thermo Scientific Nicolet 380 FTIR instrument with an attenuated total reflation (ATR) accessory. After the universal diamond ATR top-plate was cleaned using a solvent soaked tissue and a background scan collected, a gel sample was placed onto the small crystal area. Enough sample to cover the crystal area and a height no more than a few millimeters was applied. Once the sold was placed on the crystal area, the pressure arm was positioned over the crystal/sample area. The pressure arm was locked into position above the diamond crystal and force was applied to the sample, pushing it onto the diamond surface. Spectra were collected using PerkinElmer’s Spectrum FT-IR software. The ATR top-plate was cleaned before and after each use.
Dynamic light scattering (DLS).
Hydrodynamic diameter analysis (particle sizes) were measured by Dynamic Light Scattering (DLS) using a Malvern Zetasizer Nano ZS (He-Ne laser, λ = 632 nm).
Transmission electron microscopy (TEM).
TEM was performed on a FEI Tecnai G2 Spirit Gatan Camera at 6,800X magnification and at an accelerated voltage of 120 kW. For sample preparation, a drop of nanogels stock was placed on a 200 mesh carbon film covered TEM grid using phosphotungstic (PTA) negative staining, excess liquid was then wicked by filter paper. The copper grid was then dried under vacuum for one hour.
Results and Discussion
Oxidative homopolymerization of EDDET produced high molecular weight polydisulfide chains.
Before preparing nanogels, we initially investigated whether EDDET could be homopolymerized by the proposed radical oxidative polymerization mechanism. EDDET was selected as the monomer due to the presence of ethylene oxide repeats, which would render the chemical structure of the resulting polydisulfides as poly(ethylene glycol) (PEG) mimics. Due to the extensive use of PEG in drug delivery and bioconjugation applications, we envisioned that poly(EDDET) would be highly biocompatible. To deprotonate the thiols in EDDET, trimethylamine (TEA) was employed. The resulting thiol anions then underwent a single electron transfer mechanism in the presence of 3 wt. % H2O2 to give thiyl radicals. It was found that the produced thiyl radicals quickly coupled together to produce high molecular weight poly(EDDET). Polymerization was successfully performed in either THF or H2O as a solvent. Although EDDET is not water soluble, it dissolves readily in water after deprotonation by TEA. The number average molecular weights (Mn) were 106,900 g/mol and 156,700 g/mol, the weight average molecular weights (Mw) were 197,900 g/mol and 280,900 g/mol, and the polydisperisity indexes (PDI) were 1.85 and 1.79 for representative homopolymers in THF and H2O solvent conditions, respectively (Figure 1). The large Mw and high PDI (typical for conventional radical polymerization) indicate that homopolymerization of EDDET occurred rapidly (within seconds), thus making the polymerization facile and convenient for bioengineering purposes. We therefore reasoned that oxidative polymerization of EDDET would be well suited for construction of cross-linked materials, particularly nanogels as potential drug carriers responsive to GSH inside of cells. We thus proceeded towards synthesis and characterization of polydisulfide hydrogels.
Figure 1.

GPC analysis of EDDET homopolymerization in THF (—) and H2O ().
Oxidative polymerization of EDDET in the presence of dithiol cross-linkers yielded hydrogels with tunable properties.
To investigate synthesis of hydrogels, we identified three thiol-based cross-linkers denoted as PETMA, PTMP, and ETTMP (Scheme 1). These tri- and tetra-functional cross-linkers repeat the chemical design themes of including ethers and esters in the hydrogel structure. Their low molecular weights ensure that the hydrogels will degrade to small molecules after disulfide bond cleavage in cells. Hydrogels with different degrees of cross-linking density were synthesized. EDDET and cross-linker thiols were deprotonated using TEA to give thiol anions, which generated thiyl radicals after addition of 3 wt. % H2O2 (aq.). The resulting thiyl radicals quickly coupled together to form disulfide cross-linked hydrogels. Materials with different cross-linking density was achieved by varying the crosslinker to monomer ratio (CL:M = 1:5, 1:10, 1:25, 1:50, 1:75, and 1:100). Depending on the chemical structure of the cross-linker used, the hydrogel texture varied from soft (ETTMP) to spongy (PETMA) to rubbery (PETMP) (Scheme 1). ETTMP was not used further because of prolonged degradation time exhibited by ETTMP due to the longer PEG chains that must completely swell before degradation begins, coupled to the inability to degrade to small molecules. Given that PETMA and PETMP are very similar in chemical structure, PETMA was used in all further experiments due to its preferable, gel-like physical property.
Scheme 1.

Polymerization scheme to form hydrogels. Images of dried hydrogels synthesized using EDDET, PETMA, PETMP, and ETTMP.
To prove that the hydrogels formed via a radical mechanism, we performed mechanistic studies by measuring gelation time under different conditions that would examine either an ionic or a radical process. Under normal reaction conditions using TEA and 3 wt. % H2O2 (aq.), all gels formed within approximately 5 seconds. If no base was added, no gelation occurred. This indicates that deprotonation involving formation of thiol anions is a required step for polymerization. Next, we examined if free radical inhibitors could slow or stop the polymerization. If a 1:1 molar equivalence of hydroquinone to monomer was added, then gelation did not occur for the ETTMP hydrogel and the gelation time was significantly delayed for the PETMA and PETMP gels. If a 1:500 molar equivalence of monomer to hydroquinone was added, then no hydrogels formed at all under any conditions with all three cross-linkers, even after an extended wait period (Figure 2A). The impeded gelation time by a radical inhibitor indicates that these gels form via a single electron transfer mechanism to provide a thiyl radical, which couple to form polydisulfides.
Figure 2.

Hydrogels form via a single-electron transfer radical mechanism. (A) Table of gelation time of ETTMP, PETMA, and PETMP hydrogels in the absence of base (TEA), and 1:1 and 1:500 molar equivalence of free –SH to radical inhibitor. (B) FTIR traces of monomer (—), crosslinker (), and hydrogel () for PETMA. (C) FTIR traces of PETMA synthesized in different weight percentages of H2O2 0.1% (—), 1% (), 3% (), and () 3% with the addition of PLU127 surfactant. Moles of free –SH decreased as mesh size increased. (D) Calculated moles of free -SH of PETMA nanogels with different cross-linking densities.
To further confirm disulfide bond formation, we employed Fourier Transform Infrared Spectroscopy (FTIR). Both 2,2’-(ethylenedioxy)diethanethiol monomer and PETMA crosslinker FTIR traces revealed a clear –SH peak, which disappeared after gel formation (Figure 2B). Furthermore, different weight percentages of H2O2 (0.1%, 1%, 3%) showed no change in FTIR traces, indicating minimal over oxidation to sulfones or sulfonates (Figure 2C). Because no change in FTIR traces occurred when using 3 wt. % H2O2, all further experiments utilized 3 wt. % of H2O2 to allow for greatest amount of disulfide bond formation to occur. The degree of swelling is used as a common measure of the degree of crosslinking, where the equilibrium degree of swelling will be smaller when the degree of cross-linking is higher. PETMA gels with different cross-linker to monomer ratios (CL:M = 1:5, 1:10, 1:25, 1:50, 1:75, and 1:100) were synthesized and showed a clear trend in crosslinking density. The measured swelling ratios were 1.65, 2.06, 2.24, 2.26, 2.32, and 3.03 for the 1:5, 1:10, 1:25, 1:50, 1:75, and 1:100 gels, respectively (Figure 3). Increasing the amount of monomer in the feed resulted in a larger equilibrium degree of swelling, hence a larger gel mesh size. We also verified degradation of cross-linked gels by DTT to the original monomers using 1H NMR. In addition to seeing the insoluble cross-linked gels physically degrade into completely soluble small molecules (transparent solution), we confirmed the presence of starting thiol monomers and cross-linkers by NMR (Figure S1).
Figure 3.

Decreasing the cross-linker to monomer ratio led to higher swelling of hydrogels. The equilibrium degree of swelling of different cross-linking densities of PETMA hydrogels.
After confirming that disulfide bond formation and no over oxidation of disulfides to sulfones and sulfonates occurs, as well as establishing an understanding of how cross-linker to monomer ratio can affect the cross-linking density via the swelling ratios, we wanted to see whether any free thiols remained in the gels. FTIR indicates that the free –SH peak disappears after gel formation, but a few moles of free –SH may remain and not be detected via FTIR due to sensitivity limitation. Quantification of these remaining free –SH is functionally important because free thiols could be used to click targeting moieties or therapeutic agents directly to the nanogels. 5,5’-dithio-bis-(2-nitrobenzoic acid) (DTNB or Ellman’s Reagent) reacts with free thiols to produce 2-nitro-5-thiobenzoic acid (TNB). TNB is a colored species, where the absorbance can be measured and then the amount of free –SH can be quantified when compared to a standard curve. We found that the percent of free –SH decreased as the amount of monomer in the feed increased, hence indicating a larger mesh size that corresponded with the previous degree of swelling results (Figure 2D).
Surfactant-stabilized, cross-linked nanogels were synthesized using controlled microfluidic mixing.
To formulate monodisperse nanogels, we employed microfluidic mixing, where optimized conditions resulted in >40% yield (mass of nanogels collected) and well dispersed nanogels with opaque blue sheen in solution, which is characteristic of nanoparticles in solution. Because we found that purifying bare nanogels via dialysis resulted in aggregation, we therefore incorporated PLU-127 during nanogel formation. This polymer surfactant coating prevented aggregation of nanogels from occurring during further isolation and purification. Before and after dialysis purification, nanogels possessed a hydrodynamic diameter between 60–70 nm. Increasing the weight percentage of PLU-127 (0%, 25%, 50%, and 100%) led to a smaller hydrodynamic diameter, as anticipated due to increased surface area of coated nanogels with more PLU-127 coating (Figure 4A). However, keeping 25 wt. % of PLU-127 constant and only adjusting the cross-linker to monomer ratio (CL:M = 1:0.5, 1:1, and 1:2) did not change nanogel hydrodynamic diameters (Figure 4B). Therefore, the weight percentage % of PLU-127 controlled the overall nanogel hydrodynamic diameter and the mesh size was tuned independently by altering the cross-linker to monomer ratio.
Figure 4.

Hydrodynamic diameter of nanogels after (A) varying the weight percent of surfactant PLU-127 and (B) varying the monomer:cross-linker ratio (with constant 25 wt. % PLU-127). TEM image of monodispersed disulfide cross-linked nanogels with (C) 1:25, (D) 1:50, (E) 1:75, and (F) 1:100 cross-linker:monomer (mol/mol) ratio. (G) Table of quantified dye encapsulation of different mesh size for 5 wt. % Rhodamine B loaded nanogels.
We then prepared uniformly sized nanogels with different mesh sizes (CL:M = 1:25, 1:50, 1:75, and 1:100) using a constant weight percentage of surfactant and different cross-linker to monomer ratios. Nanogels exhibited similar hydrodynamic diameters (Figure S3) and TEM images of monodisperse nanogels showed uniform particles (Figure 4C–F). We next examined dye encapsulation as a drug mimic and quantified the weight percent loading and percent entrapment efficiency. We used Rhodamine B to represent a model small molecule drug with 5 wt. % loading in the feed. Dye encapsulation was quantified using dithiothreitol (DTT). DTT reduces disulfide bonds by two sequential thiol-disulfide exchange reactions. The amount of dye released was quantified by using a standard curve. Both the weight percent loading and percent entrapment efficiency showed a similar trend: As mesh size increased (CL:M = 1:25, 1:50, 1:75, and 1:100), the weight percent loading and percent entrapment efficiency increased (Figure 4G). The ability for π stacking in the larger mesh sized nanogels could potentially account for the increased Rhodamine B dye loading. These results indicated that the synthesis and purification conditions led to stable and reproducible nanogels and that these nanogels impart the ability to control the mesh size and loading capacity.
To demonstrate that these nanoparticles enable triggerable release of the encapsulated Rhodamine B in a reducing environment through disulfide-thiol chemistry, thin films of nanogel solution were drop cast onto a cleaned glass slide and images were taken before (Figure 5A) and after (Figure 5B) the addition of 11 mM DTT. After the addition of the reducing agent, the entire imaging field was flooded with a diffuse fluorescent signal which indicates the release of the Rhodamine B molecules from the nanogels. Hence, the degradation of nanogels could trigger the controlled release of encapsulated drug therapeutics in a reducing environment.
Figure 5.

Optical fluorescence microscopy images of nanogels containing 5 wt. % Rhodamine B before (A) and after (B) the addition of 11 mM dithiothreitol (DTT). Scale bars = 100 μm.
Nanogels demonstrated low toxicity, quick uptake, and fast degradation kinetics in vitro.
To examine applicability as drug carriers, we measured potential cytotoxicity from both the nanogels themselves, as well as the ultimate nanogel degradation products. HeLa cervical cancer cells were incubated with PBS suspensions of intact nanogels (Figure 6A) or degraded nanogel products (Figure 6B). High cell viability was quantified under all conditions, indicating limited to no pronounced cytotoxicity. Degradation products were isolated following 24 hour incubation with 11 mM GSH. This concentration was chosen based on prior literature studying GSH-responsive materials and on physiological measurements of GSH in cells.24–28, 55, 56
Figure 6.

Cytotoxicity analysis of HeLa cells cultured with (A) PETMA nanogels and (B) degraded nanogel products. Results were normalized to untreated cells.
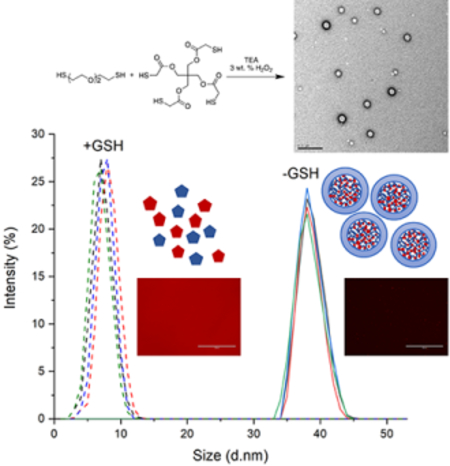
Next, we examined the cellular uptake behavior of nanogels formed with different cross-linking densities (Figure 7). Loaded Rhodamine B dye was used to track the nanogels. Disulfide-based materials are conceptually interesting because they may utilize conventional endocytosis pathways and/or specialized reaction with surface thiols to aid internalization.42, 46 In our experiments, we observed that all nanogels with different cross-linking densities (CL:M = 1:25, 1:50, 1:75, and 1:100) began internalizing within 15 min and completed uptake by 6 hours (Figure 7E). Red fluorescence was observed in both puncta and diffuse signal suggesting some nanogels may be in endosomes while some nanogels had degraded and released free dye (Figure 7A–D). To further confirm intracellular localization, we constructed overlay images of fluorescence with bright field, which clearly shows all dye within the cell membrane boundaries for all CL:M ratios (Figure S8). We also incubated HeLa cells with an equivalent concentration of free Rhodamine B to examine the differences with Rhodamine-B loaded nanogels at the same time points from 15 minutes to 24 hours (Figure S9). We found that the patterns were distinctly different, which is in agreement with other studies57 (Figure S10). Because high loading of Rhodamine B molecules within nanogels could potentially lead to quenched fluorescence emission by homo FRET, we decided to investigate this possibility by performing fluorescence time scan measurements before and after addition of GSH. Fluorescence intensity was normalized to CL:M (mol:mol) = 1:100 nanogel, which had the highest fluorescence intensity. Results indicated that no quenching occurred (Figure 8). Furthermore, DLS measurements taken before and after the addition of 11 mM GSH revealed that nanogels degrade down to their composition units (Figure 8B and 8D).
Figure 7.

HeLa cells were incubated with 5 wt. % Rhodamine B loaded nanogels with a final dye concentration of 23 ng/μL and incubated at different time points (0 min, 15 min, 1 hr, 3 hr, 6 hr, 15 hr, and 24 hr). HeLa cells were fixed using PFA and the nucleus stained with DAPI. Images taken at 40X magnification and scale bars represent 50 μm scale. Confocal images of (A) 1:25, (B) 1:50, (C) 1:75, and (D) 1:100, monomer:cross-linker (mol/mol) at the 24 hr time point. (E) The rate of internalization was quantified by tracking fluorescence intensity over time.
Figure 8.

(A) Change in fluorescence intensity over time as nanogels loaded with rhodamine dye degraded in 11 mM GSH and (B) respective hydrodynamic diameters. (C) Change in normalized fluorescence intensity over elapsed time and (D) respective hydrodynamic diameters.
To further understand these results and characterize the nanogels, we measured degradation kinetics by looking at the change in fluorescence intensity over time as nanogels, loaded with Rhodamine dye, were degraded in 11 mM GSH. The concentration of GSH is found in millimolar concentrations inside of cells, and is 100–1000 times lower outside of cells.13 All nanogels began to degrade down within the first ten minutes and exhibited a burst release profile due to disulfide bond breakage (Figure 8). Overall, in vitro studies show low toxicity, quick uptake, and fast degradation kinetics. Also, due to the ease of synthesis, rapid gelation times, and tunable functionality, these non-toxic and fully degradable nanogels offer excellent potential for use in a wide variety of drug delivery applications.
Conclusions
In this manuscript, we described the synthesis of fully degradable disulfide cross-linked nanogel drug carriers formed by oxidative radical polymerization of 2,2’-(ethylenedioxy)diethanethiol (EDDET) as a monomer with different cross-linkers, including pentaerythritol tetramercaptoacetate (PETMA). Because the poly(EDDET) backbone repeat structure and cross-linking junctions were composed entirely of disulfide bonds, these nanogels specifically degraded to small molecule dithiols intracellularly in response to the reducing agent glutathione present inside of cells. Cross-linked nanogels were synthesized using controlled microfluidic mixing in the presence of a nonionic Pluronic surfactant PLU-127 to increase the nanogel stability. Adjusting the monomer to cross-linker ratio from 1:5 to 1:100 (mol/mol) tuned the cross-linking density, resulting in swelling ratios from 1.65 to >3. Increasing the amount of stabilizing Pluronic-127 surfactant resulted in a decrease of nanogel diameter, as expected due to increased surface area of the resulting nanogels. The monomer to cross-linker ratio in the feed had no effect on the formed nanogel diameter, providing a way to control cross-linking density with constant nanogel size but tunable drug release kinetics. Nanogels exhibited an entrapment efficiency of up to 75% for loading of Rhodamine B dye. In vitro studies showed low cytotoxicity, quick uptake, and fast degradation kinetics. Due to the ease of synthesis, rapid gelation times, and tunable functionality, these non-toxic and fully degradable nanogels offer excellent potential for use in a variety of drug delivery applications. Future experiments will involve incorporating amino thiols into the polydisulfide nanogels to enable mRNA delivery. mRNA therapeutics have broad potential in applications such as protein replacement therapy, cancer immunotherapy, and genomic engineering, and effective intracellular delivery remains challenging.58–60 We envision applying the fundamental polydisulfide chemistry reported in this paper to additional applications, including mRNA delivery, in future reports.
Supplementary Material
Acknowledgements
D.J.S. acknowledges financial support from the Cancer Prevention and Research Institute of Texas (CPRIT) (R1212), the Welch Foundation (I-1855), the American Cancer Society (RSG-17-012-01), a National Institutes of Health (NIH) Cancer Center Support Grant (CCSG), the Mary Kay Foundation (049–15), and the NIH (R01 EB025192–01A1). S.A.E. acknowledges the National Science Foundation (NSF) (GRFP 1000198224) for fellowship support. The content is solely the responsibility of the authors and does not necessarily represent the official views of any of the above-mentioned funding agencies.
Footnotes
Conflicts of Interest
There are no conflicts to declare.
References
- 1.Anselmo AC and Mitragotri S, J. Controlled Release, 2014, 190, 15–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Davis ME, Chen ZG and Shin DM, Nat. Rev. Drug Discov, 2008, 7, 771–782. [DOI] [PubMed] [Google Scholar]
- 3.Jain RK and Stylianopoulos T, Nat. Rev. Clin. Oncol, 2010, 7, 653–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Peer D, Karp JM, Hong S, Farokhzad OC, Margalit R and Langer R, Nat. Nanotechnol, 2007, 2, 751–760. [DOI] [PubMed] [Google Scholar]
- 5.Cheng W, Gu L, Ren W and Liu Y, Mater. Sci. Eng. C Mater. Biol. Appl, 2014, 45, 600–608. [DOI] [PubMed] [Google Scholar]
- 6.Kanasty R, Dorkin JR, Vegas A and Anderson D, Nat. Mater, 2013, 12, 967–977. [DOI] [PubMed] [Google Scholar]
- 7.Lu Y, Sun W and Gu Z, J. Controlled Release, 2014, 194, 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sun W, Ji W, Hu Q and Gu Z, Physiol. Rev, 2017, 97, 189–225. [Google Scholar]
- 9.Albertsson AC and Varma IK, Adv. Polym. Sci, 2002, 157, 1–40. [Google Scholar]
- 10.Tian H, Tang Z, Zhuang X, Chen X and Jing X, Prog. Polym. Sci, 2012, 37, 237–280. [Google Scholar]
- 11.Bang E-K, Lista M, Sforazzini G, Sakai N and Matile S, Chem. Sci, 2012, 3, 1752–1763. [Google Scholar]
- 12.Cheng R, Feng F, Meng F, Deng C, Feijen J and Zhong Z, J. Controlled Release, 2011, 152, 2–12. [DOI] [PubMed] [Google Scholar]
- 13.Schafer FQ and Buettner GR, Free Radic. Biol. Med, 2001, 30, 1191–1212. [DOI] [PubMed] [Google Scholar]
- 14.Estrela JM, Ortega A and Obrador E, Crit. Rev. Clin. Lab. Sci, 2006, 43, 143–181. [DOI] [PubMed] [Google Scholar]
- 15.Djuric Z, Malviya VK, Deppe G, Malone JM Jr., McGunagle DL, Heilbrun LK, Reading BA and Lawrence WD, J. Cancer Res. Clin. Oncol, 1990, 116, 379–383. [DOI] [PubMed] [Google Scholar]
- 16.Balendiran GK, Dabur R and Fraser D, Cell Biochem. Funct, 2004, 22, 343–352. [DOI] [PubMed] [Google Scholar]
- 17.Franco R, Schoneveld OJ, Pappa A and Panayiotidis MI, Arch. Physiol. Biochem, 2007, 113, 234–258. [DOI] [PubMed] [Google Scholar]
- 18.Valko M, Leibfritz D, Moncol J, Cronin MT, Mazur M and Telser J, Int. J. Biochem. Cell Biol, 2007, 39, 44–84. [DOI] [PubMed] [Google Scholar]
- 19.Kharkar PM, Rehmann MS, Skeens KM, Maverakis E and Kloxin AM, ACS Biomater. Sci. Eng, 2016, 2, 165–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang YC, Li Y, Sun TM, Xiong MH, Wu J, Yang YY and Wang J, Macromol. Rapid Commun, 2010, 31, 1201–1206. [DOI] [PubMed] [Google Scholar]
- 21.Lv L-P, Xu J-P, Liu X-S, Liu G-Y, Yang X and Ji J, Macromol. Chem. Phys, 2010, 211, 2292–2300. [Google Scholar]
- 22.Koo AN, Lee HJ, Kim SE, Chang JH, Park C, Kim C, Park JH and Lee SC, Chem. Commun, 2008, DOI: 10.1039/b815918a, 6570–6572. [DOI] [PubMed] [Google Scholar]
- 23.Zhang A, Zhang Z, Shi F, Ding J, Xiao C, Zhuang X, He C, Chen L and Chen X, Soft Matter, 2013, 9, 2224–2233. [Google Scholar]
- 24.Oh JK, Siegwart DJ, Lee HI, Sherwood G, Peteanu L, Hollinger JO, Kataoka K and Matyjaszewski K, J. Am. Chem. Soc, 2007, 129, 5939–5945. [DOI] [PubMed] [Google Scholar]
- 25.Oh JK, Siegwart DJ and Matyjaszewski K, Biomacromolecules, 2007, 8, 3326–3331. [DOI] [PubMed] [Google Scholar]
- 26.Oh J, Drumright R, Siegwart D and Matyjaszewski K, Prog. Polym. Sci, 2008, 33, 448–477. [Google Scholar]
- 27.Siegwart DJ, Srinivasan A, Bencherif SA, Karunanidhi A, Oh JK, Vaidya S, Jin R, Hollinger JO and Matyjaszewski K, Biomacromolecules, 2009, 10, 2300–2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Siegwart DJ, Oh JK and Matyjaszewski K, Prog. Polym. Sci, 2012, 37, 18–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hu X, Zhang Y, Xie Z, Jing X, Bellotti A and Gu Z, Biomacromolecules, 2017, 18, 649–673. [DOI] [PubMed] [Google Scholar]
- 30.Kataoka K, Harada A and Nagasaki Y, Adv. Drug Deliv. Rev, 2001, 47, 113–131. [DOI] [PubMed] [Google Scholar]
- 31.Cheng W, Kumar JN, Zhang Y and Liu Y, Macromol. Biosci, 2014, 14, 347–358. [DOI] [PubMed] [Google Scholar]
- 32.Sun Y, Yan X, Yuan T, Liang J, Fan Y, Gu Z and Zhang X, Biomaterials, 2010, 31, 7124–7131. [DOI] [PubMed] [Google Scholar]
- 33.Luo P, Luo Y, Huang J, Lu W, Luo D, Yu J and Liu S, Colloids Surf. B Biointerfaces, 2013, 109, 167–175. [DOI] [PubMed] [Google Scholar]
- 34.Liu J, Pang Y, Huang W, Zhu Z, Zhu X, Zhou Y and Yan D, Biomacromolecules, 2011, 12, 2407–2415. [DOI] [PubMed] [Google Scholar]
- 35.Navath RS, Wang B, Kannan S, Romero R and Kannan RM, J. Controlled Release, 2010, 142, 447–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tsarevsky NV and Matyjaszewski K, Macromolecules, 2002, 35, 9009–9014. [Google Scholar]
- 37.Paulusse JM, Amir RJ, Evans RA and Hawker CJ, J. Am. Chem. Soc, 2009, 131, 9805–9812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chang C-C and Emrick T, Macromolecules, 2014, 47, 1344–1350. [Google Scholar]
- 39.Rosenthal-Kim EQ and Puskas JE, Pure Appl. Chem, 2012, 84, 2121. [Google Scholar]
- 40.Ballinger P and Long FA, J. Am. Chem. Soc, 1960, 82, 795–798. [Google Scholar]
- 41.Bang EK, Gasparini G, Molinard G, Roux A, Sakai N and Matile S, J. Am. Chem. Soc, 2013, 135, 2088–2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gasparini G, Bang EK, Molinard G, Tulumello DV, Ward S, Kelley SO, Roux A, Sakai N and Matile S, J. Am. Chem. Soc, 2014, 136, 6069–6074. [DOI] [PubMed] [Google Scholar]
- 43.Chuard N, Gasparini G, Roux A, Sakai N and Matile S, Org. Biomol. Chem, 2015, 13, 64–67. [DOI] [PubMed] [Google Scholar]
- 44.Gasparini G, Sargsyan G, Bang EK, Sakai N and Matile S, Angew. Chem. Int. Ed, 2015, 54, 7328–7331. [DOI] [PubMed] [Google Scholar]
- 45.Son S, Namgung R, Kim J, Singha K and Kim WJ, Acc. Chem. Res, 2012, 45, 1100–1112. [DOI] [PubMed] [Google Scholar]
- 46.Fu J, Yu C, Li L and Yao SQ, J. Am. Chem. Soc, 2015, 137, 12153–12160. [DOI] [PubMed] [Google Scholar]
- 47.Holmgren A and Bjornstedt M, Methods Enzymol, 1995, 252, 199–208. [DOI] [PubMed] [Google Scholar]
- 48.Sui X, Feng X, Hempenius MA and Vancso GJ, J. Mater. Chem. B, 2013, 1, 1658–1672. [DOI] [PubMed] [Google Scholar]
- 49.Zhang H, Zhai Y, Wang J and Zhai G, Mater. Sci. Eng. C Mater. Biol. Appl, 2016, 60, 560–568. [DOI] [PubMed] [Google Scholar]
- 50.Yang S, Tang Z, Zhang D, Deng M and Chen X, Biomater. Sci, 2017, 5, 2169–2178. [DOI] [PubMed] [Google Scholar]
- 51.Curcio M, Diaz-Gomez L, Cirillo G, Concheiro A, Iemma F and Alvarez-Lorenzo C, Eur. J. Pharm. Biopharm, 2017, 117, 324–332. [DOI] [PubMed] [Google Scholar]
- 52.Zhang X and Waymouth RM, J. Am. Chem. Soc, 2017, 139, 3822–3833. [DOI] [PubMed] [Google Scholar]
- 53.Cheng X, Jin Y, Qi R, Fan W, Li H, Sun X and Lai S, Polymer, 2016, 101, 370–378. [Google Scholar]
- 54.Li YL, Zhu L, Liu Z, Cheng R, Meng F, Cui JH, Ji SJ and Zhong Z, Angew. Chem. Int. Ed, 2009, 48, 9914–9918. [DOI] [PubMed] [Google Scholar]
- 55.Muller JG, Bucheler US, Kayser K, Schirmer RH, Werner D and Krauth-Siegel RL, Cell Mol. Biol, 1993, 39, 389–396. [PubMed] [Google Scholar]
- 56.Gamcsik MP, Kasibhatla MS, Teeter SD and Colvin OM, Biomarkers, 2012, 17, 671–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang JY, Wang Y and Meng X, Nanoscale Res. Lett, 2016, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Miller JB, Zhang S, Kos P, Xiong H, Zhou K, Perelman SS, Zhu H and Siegwart DJ, Angew. Chem. Int. Ed, 2017, 56, 1059–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yan Y, Xiong H, Zhang X, Cheng Q and Siegwart DJ, Biomacromolecules, 2017, 18, 4307–4315. [DOI] [PubMed] [Google Scholar]
- 60.Hajj KA and Whitehead KA, Nat. Rev. Mater, 2017, 2, 17056. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.