

Abstract
Background
The Mucopolysaccharidosis type VI (MPS VI), also known as Maroteaux-Lamy syndrome (OMIM 253200) is an autosomal recessive lysosomal disorder, caused by the deficiency of the enzyme N-acetylgalactosamine 4-sulfatase (also known as arylsulfatase B) due to mutations of the ARSB gene. Cardiologic features are well recognized, and are always present in MPS VI patients. Generally, the onset and the progression of the cardiologic symptoms are insidious, and just a few patients have developed a rapidly progressive disease. Cardiac involvement in MPS VI is a common and progressive feature. For MPS patients, cardiac evaluations are recommended every 1 to 2 years, including blood pressure measurement, electrocardiography and echocardiography. However, congestive heart failure and valvular surgical repair are not frequently seen, and if so, they are performed in adults. Here we report on an atypical MPS VI case with ascites fetalis and a rapidly progressive cardiac disease.
Case presentation
A 6-month-old Brazilian male, only child of a Brazilian healthy non-consanguineous couple. During pregnancy, second trimester ultrasonography observed fetal ascites and bilateral hydrocele. Physical exam at 6 months-old revealed a typical gibbus deformity and MPS was suspected. Biochemical investigation revealed a diagnosis of MPS type VI, confirmed by molecular test. Baseline echocardiogram revealed discrete tricuspid regurgitation and a thickened mitral valve with posterior leaflet prolapse, causing moderate to severe regurgitation. The patient evolved with mitral insufficiency and congestive heart failure, eventually requiring surgical repair by the first year of age.
Conclusions
We report the first case of MPS VI whose manifestations started in the prenatal period with fetal ascites, with severe cardiac valvular disease that eventually required early surgical repair. Moreover, in MPS with neonatal presentation, including fetal hydrops, besides MPS I, IVA and VII, clinicians should include MPS VI in the differential diagnosis.
Keywords: Mucopolysaccharidosis, Mucopolysaccharidosis type VI, Fetal ascites, Valvular disease, Inborn error of metabolism, Lysosomal disorder
Background
The Mucopolysaccharidosis type VI (MPS VI), also known as Maroteaux-Lamy syndrome (OMIM 253200) is an autosomal recessive lysosomal disorder, caused by the deficiency of the enzyme N-acetylgalactosamine 4-sulfatase (also known as arylsulfatase B) due to mutations of the ARSB gene [1, 2].
The incidence of MPS VI lays between 1 in 43,261 and 1 in 1,505,160 live births [3].
The French doctors Pierre Maroteaux and Maurice Lamy published the first description of MPS VI in 1963, focusing in the orthopedic features of this condition [4].
The age of onset of the symptoms varies and so does the phenotypical spectrum, from mild to severe. Sometimes the diagnosis in the mild form (also called slowly progressing) can be missed because the symptoms are attenuated. On the other hand, in the severe form (or rapidly progressing), in which the symptoms may be present at birth, usually diagnosis gets sooner than between the 2nd or 3rd birthday. Death occurs near the 2nd or 3rd decades, the majority being caused by cardiac failure [3, 5].
Azevedo et al. [6] collected data from 28 Latin American patients (majority of whom were Brazilians) and found that the mean age at diagnosis for MPS VI was 48.9 months. The typical phenotype of this syndrome is caused by the progressive deposition of glycosaminoglycan in various tissues: dysostosis multiplex with claw hands and short stature, facial dysmorphism/coarse facies, corneal clouding, enlarged visceral organs (liver, spleen), hearing loss, airway difficulties and hernias (inguinal, umbilical). Usually, there is no cognitive impairment [3].
Cardiologic features are well recognized since early 1940’s as described by Strauss [7], and are always present in MPS VI patients [6]. The left side of the heart is more severely affected than the right side, being the most frequent features mitral/aortic valve stenosis (60–90% of patients) [8], and cardiomyopathy, which are usually observed in adult age [9].
Infrequent presentations have been reported: a 5-month-old infant with MPS VI and cardiomyopathy and a 9-month-old infant with endocarditis fibroelastosis, both ending on cardiac failure [9–11].
Generally, the onset and the progression of the cardiologic symptoms are insidious, but some patients, as the two described above and the one reported here, have developed a rapidly progressive disease [10, 11].
Formerly, the cardiologic management was preferably clinical and palliative. With the development of the enzyme replacement therapy (ERT) and hematopoietic stem cell transplantation (HSCT), the range of possibilities became wider [5].
However, surgical possibilities have also been explored. Open-heart operations in patients with mucopolysaccharidoses are extremely rare because of multiple issues such as: poor life expectancy, multiple infiltrated organs (myocardial tissue included) and, especially, airway complications [12–14].
Considering all MPS types, there are less than 30 cases reported in the literature as having undergone successful cardiac surgery, approximately half was valvular surgery (12 out of 23). In this group of patients, 26% were MPS VI (6 out of 23) [9].
Here we report a case of MPS VI whose manifestations started in the prenatal period with fetal ascites, rapidly evolving with mitral insufficiency and congestive heart failure, eventually requiring surgical repair by the first year of age.
Case presentation
Male patient, only child of a Brazilian healthy non-consanguineous couple. Second trimester ultrasonography observed fetal ascites and bilateral hydrocele. Prenatal screening for infectious diseases was negative. There was no drug abuse during pregnancy.
The child was born at term, by cesarean section, with birth weight of 3400 g (p58), length of 48 cm (p20), and OFC 35.5 cm (p79). Clinical examination showed hydrocele, diastasis recti, and unilateral clubfoot. There were no signs of hepato or splenomegaly (and abdominal ultrasound was normal at birth). Echocardiogram at 4 days of life disclosed just patent foramen ovale. The patient was evaluated by the Genetics unit. No specific diagnosis was suspected, even though mild coarsening facies was already present. Skeletal survey performed at one-month-old, due to congenital clubfoot and dysmorphisms, revealed mild proximal misshapen metacarpals and thickening of the provisional cartilage.
When the child came back to the Genetics evaluation at 6 months-old, the mother reported she had noticed a progressive growing mass in the lower back since the patient was 2 months old. This had been investigated with X-rays and MRI in one of the patient’s visit in the Emergency Unit due to respiratory symptoms. The mother also reported that the patient had been suffering of frequent upper respiratory tract infections, needing hospitalization twice for wheezing crises.
At physical exam, the patient displayed a typical gibbus deformity (Fig. 1), which raised the suspicion of mucopolysaccharidosis. Since the patient had had fetal ascites, initially, MPS VII was suspected. Biochemical investigation revealed a diagnosis of MPS type VI (urinary glycosaminoglycans: 402 μg/mg Cr, reference value for age: 133–460 μg/mg Cr, with dermatan sulfate excretion, and enzymatic assay detected arylsulfatase B deficiency in white blood cells, with another sulfatase within the normal range).
Fig. 1.

a Patient in the newborn period. b At the age of 6 months, gibbous deformity in the lumbar region. c At 8 months with low nasal bridge and mild coarse facies
Sequencing of the ARSB gene showed two pathogenic variants in trans: c.944G > A (p.Arg315Gln) and c.1143-1G > C.
At the age of 7 months, the patient was brought to the emergency unit due to respiratory distress. A chest X-ray showed a possible lung congestion and echocardiogram revealed discrete tricuspid regurgitation and a thickened mitral valve with posterior leaflet prolapse, causing moderate to severe regurgitation. As a result, furosemide was prescribed. Due to the rapid cardiologic changes, captopril and spironolactone were added.
At 9 months of age, the patient was hospitalized again because of cardiac decompensation. Comparative chest X-ray showed an increased cardiac area, and echocardiogram indicated worsening of mitral regurgitation. Dobutamin and dopamine were initiated, and the patient was transferred to the intensive care unit (ICU). Dobutamin was progressively withdrawn and carvedilol was introduced. Progressive improvement of respiratory distress was seen. However, a few days later echocardiogram showed left atrium and left ventricle enlargement (Fig. 2), normal left ventricular systolic function, discrete tricuspid insufficiency, mitral valve with thickened leaflets, posterior prolapsed leaflet and severe regurgitation (Fig. 3), evident with Doppler (Fig. 4).
Fig. 2.

M mode of right and left ventricles (age: 9.5 months). Left ventricle is extremely enlarged (50.7 mm), with preserved ejection fraction (78.9%). RV: right ventricle; LV: left ventricle
Fig. 3.

Apical 4 chamber view, focused on mitral valve. Notice the large mitral regurgitation jet on color Doppler. LV: left ventricle; LA: left atrium; MV: mitral valve
Fig. 4.

Doppler profile obtained at the mitral valve, showing severe regurgitation
With the rapid progression of mitral regurgitation, at 10 months the patient developed congestive heart failure and dobutamine was resumed. After stabilization, the patient was discharged receiving furosemide, captopril, spironolactone, carvedilol, digoxin, aspirin, and domperidone.
A few days after discharge, the patient was readmitted to the emergency room due to hyporexia, irritability and vomiting. The physical exam showed tachycardia, hypoxemia, respiratory distress, and hepatomegaly. He was sent to ICU, with worsening of cardiac function. The patient used bilevel positive airway pressure (BiPAP). Because of significant mitral insufficiency, left ventricular dilatation and refractory cardiac failure, in a patient with a genetic multisystemic disorder, a multidisciplinary team met to discuss the management.
It was decided to perform cardiac surgery (valvuloplasty with mitral valve ring reduction) (Fig. 5). The patient was 11 months by the time of the surgical intervention. LV shortening fraction at 7, 9, 10 and 11 months were 70, 56, 79 and 58%, respectively. ECG showed left ventricular overload and normal sinus rhythm.
Fig. 5.
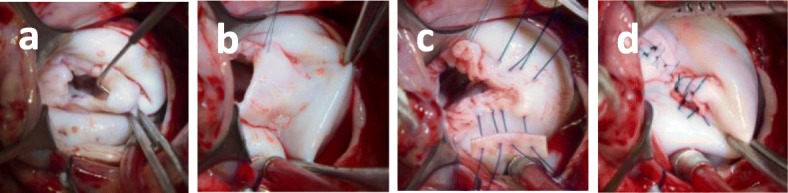
Mitral valvuloplasty, surgeon’s view: a Redundant mitral valve. P2 segment prolapse. b Quadrangular P2 resection of the mitral valve. c Annuloplasty with bovine pericardium patch. d Annuloplasty with valvuloplasty, final result
A month after the surgery, the patient began enzyme replacement therapy (ERT) with galsulfase weekly.
The last echocardiography shows discrete mitral insufficiency after valvuloplasty and last glycosaminoglycan measurement in urine was within normal range (246 μg/mg creatinine - Reference value for children under 2y: 79–256 μg/mg creatinine).
After the cardiac surgery, growth was improved (weight and height). The patient is currently with 2.5 years old and presents with mild motor delay (sat alone at 18 months and walked at 23 months old).
Discussion and conclusions
Cardiac involvement in MPS VI is a common and progressive feature. For MPS patients, cardiac evaluations are recommended every 1 to 2 years, including blood pressure measurement, electrocardiography and echocardiography [15].
However, congestive heart failure and valvular surgical repair are not frequently seen, and if so, they are performed in adults. The mean reported age for this group of patients is 30.9 years old, ranging from 3 to 62 years [7, 9].
Table 1 shows the few MPS VI patients reported in literature who had undergone cardiac surgery (valvuloplasty or valve replacement) in spite of the high surgical risk and mortality reported for this kind of patients (20% mortality for left heart valve disease) [14, 16–18].
Table 1.
Patients reported in the literature diagnosed with MPS VI who had undergone valvuloplasty
| Characteristics | Described cases | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Wilson et al., 1980 [16] | Tan et al., 1992 [17] | Marwick et al., 1992 [18] | Torre et al., 2016 [14] | Current case | |||||
| Male | Male | Female | Female | Female | Female | Female | Male | ||
| Age at diagnosis with MPS VI | 30y | +/−31y | N/A | +/−20y | Childhood | 34y | 7 m | ||
| Age of onset of cardiologic symptoms | 43y | 28y | +/−28y | 25y | +/−20y | +/− 20y | +/−37y | +/−7 m | |
| Age at surgery | N/A | 30y | 34y | 25y | 21y | 25y | 40y | 11 m | |
| Pre-operative Echocardiography findings | Aortic Valve | Severe stenosis | Stenosis, with mild regurgitation | Stenosis | – | – |
Mildly echo dense, with normal leaflet excursion. Minimal regurgitation |
Severe stenosis, calcified cusps, moderate regurgitation | Thick valve, discrete regurgitation |
| Mitral Valve | N/A | Thick and stenotic leaflets, with mild regurgitation | Stenosis | – | Severe regurgitation | Stenotic and rigid valve, commissural fusion, resembling a rheumatic valve; mild regurgitation | Thickened, with severe stenosis |
Thick, redundant, prolapsed posterior cusp, central valve coaptation failure. Chordae tendon mildly thickened, a broken chordae tendon was not excluded |
|
| Tricuspid Valve | N/A | Thick leaflets, no stenosis | – | – | Moderate regurgitation | – | – | Discrete-moderate regurgitation | |
| Other findings | N/A | – | – | – | Small left ventricular cavity | – | Mild left ventricular hypertrophy | Patent foramen ovale, large left auricle, Major dilatation of left ventricle | |
| Cardiac Surgery | AVR | AVR SJA 19 mm, MVR SJA 21 mm, aortic root enlargement | AVR SJA 19 mm, MVR SJA 21 mm, aortic root enlargement | None | AVR SJA 19 mm, MVR SJA 21 mm, aortic root enlargement | MVR size 2 M Starr-Edwards 6120 prosthesis. | AVR 19 mm mechanical prosthesis. | Mitral valvuloplasty, annuloplasty with bovine pericardium patch | |
N/A Not Available, AVR Aortic Valve Replacement, MVR Mitral Valve Replacement, AVR SJA 19 mm = Aortic Valve Replacement with St. Jude aortic prosthesis, size 19 mm; MVR SJA 21 mm = Mitral Valve Replacement with inverted St. Jude aortic prosthesis, size 21 mm
In contrast with most previous reports, our patient had minimal aortic and severe mitral valvular disease with onset before the first year of life. To our knowledge, this is the second patient with MPS who has undergone successful mitral valvuloplasty; the first case being a 6-year-old boy with MPS III [19].
After valvular surgery, the patients compiled in Table 1 experienced clinical improvement with minimal residual valvulopathy or, in the worst of the cases, palliation of symptoms for several years. These outcomes may suggest that performing open cardiac surgery for selected MPS patients could be beneficial. Also, in some MPS VI cases, especially those with the rapidly progressing type, it may be important to perform an early and more frequent cardiac follow-up in case there are symptoms of cardiac etiology. Noteworthy, cardiac disease may be one of the initial signs of MPS, as reported by Fong et al. [20], who diagnosed two siblings with MPS VI with dilated cardiomyopathy and autopsy showing endocardial fibroelastosis.
Some authors show stabilization or slower deterioration of valvular disease with ERT [9, 20–23]. In our patient, an early diagnosis was also important, because even though the cardiac disease was surgically assessed, other manifestations of MPS can be treated by ERT.
Regarding our patient’s genotype, c.944G > A (p.Arg315Gln) is a common described variant, with homozygous patients showing an intermediate or severe phenotype [24, 25]. The second variant (c.1143-1G > C) is common in Spanish and Argentinian patients with MPS VI [26]; our patient’s parents did not know their ancestral origin. Our patient’s variants are related to classical MPS VI and not with the non-classical cardiac phenotype [27, 28].
Newborn screening can lead in the future to early diagnosis of MPS [29] making it possible to start ERT within the first months of age, which may prevent cardiac valve involvement and other MPS manifestations [9, 21–23].
It is the first case of MPS VI with prenatal manifestation with ascites fetalis, with a few neonatal manifestations of MPS but precocious gibbous since 2 months, and severe progressive cardiac manifestation. Fetal hydrops has been detected mainly in patients with MPS I, IVA and VII [30–32]. There is one case reported by Choy et al. in 2015 with prenatal generalized edema, necessitating intrauterine drainage of pleural effusion, and eventual biochemical diagnosis of MPS VI at 13 months of age (genotype not reported in the publication). However, this patient presented with mild to moderate valve regurgitation and progressed to severe upper cervical cord compression in the first year of life [33]. The present case indicates that, in MPS with neonatal presentation, including fetal hydrops, besides MPS VII, clinicians should include MPS VI in the differential diagnosis. This can be an extremely valuable diagnostic clue to an early diagnosis so that specific therapy and management can be implemented [34–36].
Acknowledgements
Not applicable.
Consent to publish
The patient’s legal guardians (parents) have provided written consent to publish this case report, including medical data and images.
Abbreviations
- AVR SJA 19 mm
Aortic valve replacement with St. Jude aortic prosthesis, size 19 mm
- AVR
Aortic valve replacement
- BiPAP
Bilevel positive airway pressure
- ERT
Enzyme replacement therapy
- HSCT
Hematopoietic stem cell transplantation
- ICU
Intensive care unit
- LA
Left atrium
- LV
Left ventricle
- MPS III
Mucopolysaccharidosis type III
- MPS VI
Mucopolysaccharidosis type VI
- MPS VII
Mucopolysaccharidosis type VII
- MPS
Mucopolysaccharidosis
- MV
Mitral valve
- MVR SJA 21 mm
Mitral valve replacement with inverted St. Jude aortic prosthesis, size 21 mm
- MVR
Mitral valve replacement
- N/A
Not available
Authors’ contributions
RSH and ECNV wrote the main manuscript text, GNL performed the Echocardiograms and prepared Figs. 2, 3, and 4. DMA, NMI and MBJ were responsible for the patient’s clinical and surgical cardiac management. AMM provided infrastructural support for ERT and CAK reviewed the manuscript and contributed to the report organization. All authors read and approved the final manuscript.
Funding
The authors received financial support for the payment of the fee publication by BioMarin Pharmaceutical Inc. The funding body played no role in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Availability of data and materials
For further details regarding this case report, please contact Prof Chong Ae Kim, MD, PhD (chong.kim@hc.fm.usp.br).
Ethics approval and consent to participate
The patient was studied as part of a research protocol approved by the local Ethics Committee (139.728 – CAPPesq - Comissão de Ética para Análise de Projetos de Pesquisa). The patient’s legal guardians (parents) have signed informed consent to participate in this study.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Rachel Sayuri Honjo, Email: rachel.honjo@hc.fm.usp.br.
Evelyn Cristina Nuñez Vaca, Email: cristinanunezvaca@gmail.com.
Gabriela Nunes Leal, Email: gnleal@gmail.com.
Deipara Monteiro Abellan, Email: deipara.abellan@hc.fm.usp.br.
Nana Miura Ikari, Email: nana.miura@hc.fm.usp.br.
Marcelo Biscegli Jatene, Email: marcelo.jatene@hc.fm.usp.br.
Ana Maria Martins, Email: ni.martins10@gmail.com.
Chong Ae Kim, Email: chong.kim@hc.fm.usp.br.
References
- 1.Gitzelmann R, Steinmann B, Wiesmann U, Spycher M, Herschkowitz N, Marti H-R. Aldersche Granulationsanomalie: Albert Alders Patienten litten nicht an M. Pfaundler-Hurler. (Abstract) Helv Paediat Acta. 1987;42:90. [Google Scholar]
- 2.Wicker G, Prill V, Brooks D, Gibson G, Hopwood J, von Figura K, Peters C. Mucopolysaccharidosis VI (Maroteaux-Lamy syndrome): an intermediate clinical phenotype caused by substitution of valine for glycine at position 137 of arylsulfatase B. J Biol Chem. 1991;266:21386–21391. [PubMed] [Google Scholar]
- 3.Valayannopoulos V, Nicely H, Harmatz P, Turbeville S. Mucopolysaccharidosis VI. Orphanet J Rare Dis. 2010;5:5. doi: 10.1186/1750-1172-5-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Maroteaux P, Leveque B, Marie J, Lamy M. Une nouvelle dysostose avec elimination urinaire de chondroitine sulfate B [a new dysostosis with urinary elimination of chondroitin sulfate B] Presse Med. 1963;71:1849–1852. [PubMed] [Google Scholar]
- 5.Encarnacion CO, Hang D, Earing M, Mitchell ME. Mucopolysaccharidoses causing Valvular heart disease: report and review of surgical management. World J Pediatr Congenit Heart Surg. 2017;1:2150135117690105. doi: 10.1177/2150135117690105. [DOI] [PubMed] [Google Scholar]
- 6.Azevedo AC, Schwartz IV, Kalakun L, Brustolin S, Burin MG, Beheregaray AP, Leistner S, Giugliani C, Rosa M, Barrios P, Marinho D, Esteves P, Valadares E, Boy R, Horovitz D, Mabe P, da Silva LC, de Souza IC, Ribeiro M, Martins AM, Palhares D, Kim CA, Giugliani R. Clinical and biochemical study of 28 patients with mucopolysaccharidosis type VI. Clin Genet. 2004;66:208–213. doi: 10.1111/j.1399-0004.2004.00277.x. [DOI] [PubMed] [Google Scholar]
- 7.Strauss L. The pathology of gargoylism. Report of a case and review of the literature. Am J Pathol. 1948;24:855–887. [PMC free article] [PubMed] [Google Scholar]
- 8.Fesslová V, Corti P, Sersale G, Rovelli A, Russo P, Mannarino S, Butera G, Parini R. The natural course and the impact of therapies of cardiac involvement in the mucopolysaccharidoses. Cardiol Young. 2009;19:170–178. doi: 10.1017/S1047951109003576. [DOI] [PubMed] [Google Scholar]
- 9.Braunlin EA, Harmatz PR, Scarpa M, Furlanetto B, Kampmann C, Loehr JP, Ponder KP, Roberts WC, Rosenfeld HM, Giugliani R. Cardiac disease in patients with mucopolysaccharidosis: presentation, diagnosis and management. J Inherit Metab Dis. 2011;34:1183–1197. doi: 10.1007/s10545-011-9359-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hayflick S, Rowe S, Kavanaugh-McHugh A, Olson JL, Valle D. Acute infantile cardiomyopathy as a presenting feature of mucopolysaccharidosis VI. J Pediatr. 1992;120:269–272. doi: 10.1016/S0022-3476(05)80441-X. [DOI] [PubMed] [Google Scholar]
- 11.Miller G, Partridge A. Mucopolysaccharidosis type VI presenting in infancy with endocardial fibroelastosis and heart failure. Pediatr Cardiol. 1983;4:61–62. doi: 10.1007/BF02281012. [DOI] [PubMed] [Google Scholar]
- 12.Glober GA, Tanaka KR, Turner JA, Liu CK. Mucopolysaccharidosis, an unusual cause of cardiac valvular disease. Am J Cardiol. 1968;22:133–136. doi: 10.1016/0002-9149(68)90256-7. [DOI] [PubMed] [Google Scholar]
- 13.Chen MR, Lin SP, Hwang HK, Yu CH. Cardiovascular changes in mucopolysaccharidoses in Taiwan. Acta Cardiol. 2005;60:51–53. doi: 10.2143/AC.60.1.2005049. [DOI] [PubMed] [Google Scholar]
- 14.Torre S, Scarpelli M, Salviati A, Buffone E, Faggian G, Luciani GB. Aortic and mitral valve involvement in Maroteaux-Lamy syndrome VI: surgical implications in the enzyme replacement therapy era. Ann Thorac Surg. 2016;102:e23–e25. doi: 10.1016/j.athoracsur.2015.11.062. [DOI] [PubMed] [Google Scholar]
- 15.Giugliani R, Harmatz P, Wraith JE. Management guidelines for mucopolysaccharidosis VI. Pediatrics. 2007;120:405–418. doi: 10.1542/peds.2006-2184. [DOI] [PubMed] [Google Scholar]
- 16.Wilson CS, Mankin HT, Pluth JR. Aortic stenosis and mucopolysaccharidosis. Ann Intern Med. 1980;92:496–498. doi: 10.7326/0003-4819-92-4-496. [DOI] [PubMed] [Google Scholar]
- 17.Tan CT, Schaff HV, Miller FA, Jr, Edwards WD, Karnes PS. Valvular heart disease in four patients with Maroteaux-Lamy syndrome. Circulation. 1992;85:188–195. doi: 10.1161/01.CIR.85.1.188. [DOI] [PubMed] [Google Scholar]
- 18.Marwick TH, Bastian B, Hughes CF, Bailey BP. Mitral stenosis in the Maroteaux-Lamy syndrome: a treatable cause of dyspnoea. Postgrad Med J. 1992;68:287–288. doi: 10.1136/pgmj.68.798.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Muenzer J, Beekman RH, Profera LM, Bove EL. Severe mitral insufficiency in mucopolysaccharidosis type III-B (Sanfilippo syndrome) Pediatr Cardiol. 1993;14:130–132. doi: 10.1007/BF00796996. [DOI] [PubMed] [Google Scholar]
- 20.Fong LV, Menahem S, Wraith JE, Chow CW. Endocardial fibroelastosis in mucopolysaccharidosis type VI. Clin Cardiol. 1987;10:362–364. doi: 10.1002/clc.4960100612. [DOI] [PubMed] [Google Scholar]
- 21.Leal GN, de Paula AC, Morhy SS, Andrade JL, Kim CA. Advantages of early replacement therapy for mucopolysaccharidosis type VI: echocardiographic follow-up of siblings. Cardiol Young. 2014;24:229–235. doi: 10.1017/S1047951113000152. [DOI] [PubMed] [Google Scholar]
- 22.Scarpa M, Barone R, Fiumara A, Astarita L, Parenti G, Rampazzo A, Sala S, Sorge G, Parini R. Mucopolysaccharidosis VI: the Italian experience. Eur J Pediatr. 2009;168:1203–1206. doi: 10.1007/s00431-008-0910-z. [DOI] [PubMed] [Google Scholar]
- 23.Kampmann C, Lampe C, Whybra-Trümpler C, Wiethoff CM, Mengel E, Arash L, Beck M, Miebach E. Mucopolysaccharidosis VI: cardiac involvement and the impact of enzyme replacement therapy. J Inherit Metab Dis. 2014;37:269–276. doi: 10.1007/s10545-013-9649-4. [DOI] [PubMed] [Google Scholar]
- 24.Tomanin R, Karageorgos L, Zanetti A, Al-Sayed M, Bailey M, Miller N, Sakuraba H, Hopwood JJ. Mucopolysaccharidosis type VI (MPS VI) and molecular analysis: review and classification of published variants in the ARSB gene. Hum Mutat. 2018;39:1788–1802. doi: 10.1002/humu.23613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zanetti A, D'Avanzo F, Rigon L, Rampazzo A, Concolino D, Barone R, Volpi N, Santoro L, Lualdi S, Bertola F, Scarpa M, Tomanin R. Molecular diagnosis of patients affected by mucopolysaccharidosis: a multicenter study. Eur J Pediatr. 2019;178:739–753. doi: 10.1007/s00431-019-03341-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Garrido E, Chabás A, Coll MJ, Blanco M, Domínguez C, Grinberg D, Vilageliu L, Cormand B. Identification of the molecular defects in Spanish and Argentinian mucopolysaccharidosis VI (Maroteaux-Lamy syndrome) patients, including 9 novel mutations. Mol Genet Metab. 2007;92:122–130. doi: 10.1016/j.ymgme.2007.06.002. [DOI] [PubMed] [Google Scholar]
- 27.Jurecka A, Golda A, Opoka-Winiarska V, Piotrowska E, Tylki-Szymańska A. Mucopolysaccharidosis type VI (Maroteaux-Lamy syndrome) with a predominantly cardiac phenotype. Mol Genet Metab. 2011;104:695–699. doi: 10.1016/j.ymgme.2011.08.024. [DOI] [PubMed] [Google Scholar]
- 28.Jurecka A, Zakharova E, Cimbalistiene L, Gusina N, Kulpanovich A, Golda A, Opoka-Winiarska V, Piotrowska E, Voskoboeva E, Tylki-Szymańska A. Mucopolysaccharidosis type VI: a predominantly cardiac phenotype associated with homozygosity for p.R152W mutation in the ARSB gene. Am J Med Genet A. 2013;161A:1291–1299. doi: 10.1002/ajmg.a.35905. [DOI] [PubMed] [Google Scholar]
- 29.Tomatsu S, Kubaski F, Sawamoto K, Mason RW, Yasuda E, Shimada T, Montaño AM, Yamaguchi S, Suzuki Y, Orii T. Newborn screening and diagnosis of mucopolysaccharidoses: application of tandem mass spectrometry. Nihon Masu Sukuriningu Gakkai Shi. 2014;24:19–37. [PMC free article] [PubMed] [Google Scholar]
- 30.Moreno CA, Kanazawa T, Barini R, Nomura ML, Andrade KC, Gomes CP, Heinrich JK, Giugliani R, Burin M, Cavalcanti DP. Non-immune hydrops fetalis: a prospective study of 53 cases. Am J Med Genet A. 2013;161A:3078–3086. doi: 10.1002/ajmg.a.36171. [DOI] [PubMed] [Google Scholar]
- 31.Gimovsky AC, Luzi P, Berghella V. Lysosomal storage disease as an etiology of nonimmune hydrops. Am J Obstet Gynecol. 2015;212:281–290. doi: 10.1016/j.ajog.2014.10.007. [DOI] [PubMed] [Google Scholar]
- 32.Vianey-Saban C, Acquaviva C, Cheillan D, Collardeau-Frachon S, Guibaud L, Pagan C, Pettazzoni M, Piraud M, Lamazière A, Froissart R. Antenatal manifestations of inborn errors of metabolism: biological diagnosis. J Inherit Metab Dis. 2016;39:611–624. doi: 10.1007/s10545-016-9947-8. [DOI] [PubMed] [Google Scholar]
- 33.Choy YS, Bhattacharya K, Balasubramaniam S, Fietz M, Fu A, Inwood A, Jin DK, Kim OH, Kosuga M, Kwun YH, Lin HY, Lin SP, Mendelsohn NJ, Okuyama T, Samion H, Tan A, Tanaka A, Thamkunanon V, Thong MK, Toh TH, Yang AD, McGill J. Identifying the need for a multidisciplinary approach for early recognition of mucopolysaccharidosis VI (MPS VI) Mol Genet Metab. 2015;115:41–47. doi: 10.1016/j.ymgme.2015.03.005. [DOI] [PubMed] [Google Scholar]
- 34.McGill JJ, Inwood AC, Coman DJ, Lipke ML, de Lore D, Swiedler SJ, Hopwood JJ. Enzyme replacement therapy for mucopolysaccharidosis VI from 8 weeks of age--a sibling control study. Clin Genet. 2010;77(5):492–498. doi: 10.1111/j.1399-0004.2009.01324.x. [DOI] [PubMed] [Google Scholar]
- 35.Akyol MU, Alden TD, Amartino H, Ashworth J, Belani K, Berger K, Borgo A, Braunlin E, Eto Y, Gold JI, Jester A, Jones SA, Karsli C, Mackenzie W, Marinho DR, McFadyen A, McGill J, Mitchell JJ, Muenzer J, Okuyama T, Orchard PJ, Stevens B, Thomas S, Walker R, Wynn R, Giugliani R, Harmatz P, Hendriksz C, Scarpa M, MPS Consensus Programme Steering Committee; MPS Consensus Programme Co-Chairs Recommendations for the management of MPS VI: systematic evidence- and consensus-based guidance. Orphanet J Rare Dis. 2019;14(1):118. doi: 10.1186/s13023-019-1080-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Whybra C, Mengel E, Russo A, Bahlmann F, Kampmann C, Beck M, Eich E, Mildenberger E. Lysosomal storage disorder in non-immunological hydrops fetalis (NIHF): more common than assumed? Report of four cases with transient NIHF and a review of the literature. Orphanet J Rare Dis. 2012;7:86. doi: 10.1186/1750-1172-7-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
For further details regarding this case report, please contact Prof Chong Ae Kim, MD, PhD (chong.kim@hc.fm.usp.br).