

Abstract
Background:
We test if inhaled nitric oxide (NO) attenuates platelet functional and metabolic hyper-reactivity in subjects with submassive pulmonary embolism (PE).
Methods:
Participants with PE were randomized to either 50 ppm NO+O2 or O2 only for 24 h with blood sampling at enrollment and after treatment; results were compared with healthy controls. Platelet metabolic activity was assessed by oxygen consumption (basal and uncoupled) and reactivity was assessed with agonist-stimulated thromboelastography (TEG) and fluorometric measurement of agonist-stimulated cytosolic [Ca++] without and with pharmacological soluble guanylate (sGC) modulation.
Results:
Participants (N=38 per group) were well-matched at enrollment for PE severity, comorbidities as well as TEG parameters and platelet O2 consumption. NO treatment doubled the mean plasma [NO3-] (P<0.001) indicating successful delivery, but placebo treatment produced no change. After 24 hours, neither TEG nor O2 consumption parameters differed significantly between treatment groups. Platelet cytosolic [Ca++] was elevated with PE versus controls, and was decreased by treatment with cinaciguat (an sGC activator), but not riociguat (an sGC stimulator). Stimulated platelet lysate sGC activity was increased with PE compared with controls.
Conclusions:
In patients with acute submassive PE, despite evidence of adequate drug delivery, inhaled NO had no major effect on platelet O2 consumption or agonist-stimulated parameters on TEG. Pharmacological activation, but not stimulation, of sGC effectively decreased platelet cytosolic [Ca++], and platelet sGC activity was increased with PE, confirming the viability of sGC as a therapeutic target.
Keywords: Humans, Pulmonary Embolism, Blood Platelets, Nitric Oxide, Thrombelastography, Platelet Activation, riociguat, Pyrazoles, Pyrimidines, Oxygen Consumption, Comorbidity
Graphical Abstract

1.0. Introduction
This work examines the question of whether inhaled nitric oxide (NO) gas can reduce platelet hyper-reactivity in patients with pulmonary embolism. The conceptual model addressed is that PE causes platelet hyperactivation leading to activated platelets binding to endothelium of the microvasculature in the lung, ultimately increasing pulmonary vascular resistance in the setting of thrombotic pulmonary embolism (PE). The model asserts that the microvascular obstruction in the pulmonary vasculature contributes to the direct obstructive effect of large clots.
Experimental models of PE demonstrate that the majority of the observed increase in pulmonary vascular resistance during PE is not directly secondary to macrovascular clot burden. At least part of this abrupt increase in resistance is secondary to pulmonary vasoconstriction due to impaired endogenous NO bioavailability. (Watts et al., 2011b) This decreased bioavailability results both from hemolysis-induced NO scavenging by free hemoglobin and arginase-induced arginine consumption and decreased endogenous NO synthesis.(Kline et al., 2013; Watts et al., 2011a) Moreover, platelet hyper-reactivity has been previously demonstrated in acute PE, which may further increase pulmonary microvascular obstruction and contribute to this vicious cycle of hemolysis-induced impairment of NO bioavailability and increased pulmonary vascular resistance.(Chung et al., 2007; Helms et al., 2013; Rezania et al., 2017) Patients with PE have platelets that demonstrate increased oxygen consumption in both unstimulated conditions with glucose and octanoate as substrates, and after mitochondria are chemically uncoupled.(Rezania et al., 2017) Simultaneously, whole blood from these patients demonstrated increased thrombin-independent maximum amplitude (MA) on thromboelastography (TEG).(Rezania et al., 2017)
In addition to its effects on smooth muscle, NO decreases platelet aggregation in vitro via the sGC/cGMP/PKG pathway.(Gambaryan and Tsikas, 2015Pawloski, 1998 #3109Pawloski, 1998 #3109) Previous studies of inhaled NO in healthy human subjects has demonstrated a systemic inhibitory effect on fibrinogen binding, decreased aggregation, and prolongation of in vitro bleeding time.(Gries et al., 1998; Gries et al., 2000) These effects appear to have a threshold effect with maximal response notable even at ~10 ppm inhaled NO in healthy patients. Inhaled NO increases plasma nitrate/nitrite concentrations, both of which can serve as NO donors to produce sGC-mediated platelet inhibition independently of constitutive NO synthesis by the platelet.(Apostoli et al., 2014) Mechanistically, NO directly inhibits cytochrome c oxidase, the terminal enzyme in the electron transport chain (ETC) at physiologic concentrations in competition with oxygen, decreasing mitochondrial energy production and platelet secretory function.(Tomasiak et al., 2004) The loss of regulation of platelet ETC activity from reduced NO, therefore, would be expected to result in increased platelet oxygen consumption. Restoration of NO to platelets might restore this control and therefore show reduced platelet oxygen consumption.
To produce smooth muscle relaxation, NO must stimulate the soluble guanylate cyclase (sGC) enzyme.(Derbyshire and Marletta, 2012) However, oxidant stress to this enzyme can cause it to release its prosthetic heme group, rendering the enzyme unable to bind NO.(Derbyshire and Marletta, 2012; Evgenov et al., 2006) The oxidation status of sGC can determine the success of NO treatment, because oxidized sGC will not synthesize cyclic guanylate monophosphate, regardless of NO concentration, therefore precluding any relaxation effect of pulmonary arterial smooth muscle.(Stasch et al., 2011) To help differentiate the role of oxidized sGC in platelets, we used two drugs that modulate sGC, riociguat and cinaciguat, Riociguat has been cleared by the Food and Drug Administration for the treatment of chronic thromboembolic pulmonary hypertension and produces smooth muscle relaxation by NO-dependent stimulation of non-oxidized, heme-containing sGC.(Dasgupta et al., 2015) Cinaciguat, on the other hand, is an activator of sGC, a nomenclature that indicates its ability to increase the activity of the oxidized and heme-free sGC.(Ahrens et al., 2011; Mendes-Silverio et al., 2012) Riociguat and cinaciguat have been demonstrated to inhibit platelet responses to ADP.(Reiss et al., 2015; Roger et al., 2010; Stasch et al., 2002)
We sought to examine the question of whether exogenous supplementation of inhaled NO to patients with submassive pulmonary embolism reduces platelet activation.
2.0. Methods
2.1. Study design:
This study was a preplanned ancillary aim of a multicenter, randomized, double-blind, controlled trial of inhaled NO plus oxygen versus nitrogen plus oxygen in subjects with submassive PE and objective evidence of RV dysfunction.(Kline et al., 2019) The trial design, protocol, and aims have been published previously.(Kline et al., 2017; Kline et al., 2019) The study took place from February 2014-October 2016 at three hospitals in Indianapolis, IN (Methodist Hospital, the Sydney and Louis Eskanazi Hospital, and University Hospital) and one hospital in Jackson, MS (University of Mississippi Medical Center). The trial was conducted in accordance with the Code of Federal Regulations (21CFR312) was approved by the institution review boards of each of the participating institutions. All patients gave written, informed consent prior to study participation. The trial took place under an investigational new drug exemption from the Food and Drug Administration and was registered atClinicaltrials.gov () The data for this study were analyzed by MAP and JAK, and all authors had access to primary clinical trial data.
2.2. Inclusion / exclusion criteria:
Patients with imaging proven PE were screened by trained research coordinators and investigators for the presence of at least 1 predictor of RV dysfunction, defined as echocardiography with RV dilation or hypokinesis, estimated RV systolic pressure (RVSP) > 40 mmHg, RV > LV on computed tomography pulmonary angiography (CTPA), elevated troponin I (>0.1 ng/mL), elevated natriuretic peptide (>90 pg/mL), RV strain on electrocardiogram (Daniel score >8), or screening bedside ultrasonography with color flow with RV dysfunction (in the presence of another RV dysfunction predictor). Exclusion criteria of greatest relevance to this secondary aim included vasopressor support, completed or planned use of thrombolytics or any surgical or catheter based embolectomy (in order to minimize confounding), contraindication to anticoagulation, plan to utilize anti-platelet agents, or recent use of drugs known to increase cGMP or NO (such as nitroglycerin or sildenafil). Other exclusion criteria included pregnancy, altered mental status precluding informed consent, comfort care measures instituted, inability to use a nasal cannula, supplemental oxygen requirement greater than could be delivered via nasal cannula to maintain SpO2 >80%, pneumothorax with decompression, or at the discretion of the principal investigatior. All patients were treated with full dose unfractionated heparin to maintain the activated partial thromboplastin time between 80 and 120 seconds, or with full dose low molecular weight heparin.
Controls were healthy volunteer subjects enrolled as previously described, under a separate IRB-approved protocol, using explicit definitions of “healthy” which excluded major disease (e.g., cancer, organ failure, chronic inflammatory conditions) but did allow obesity, smoking or treated hypertension. Blood was drawn by qualified phlebotomists from fasting volunteers in the morning of two consecutive days, 24 hours apart and immediately centrifuged at 5,000 × g for 20 minutes to produce platelet poor plasma.
2.3. Intervention:
All patients with PE were randomized to either NO (50 ppm) or nitrogen plus oxygen delivered via a commercially available, FDA cleared device (INOvent; Mallinckrodt Pharmaceauticals; Clinton, NJ) equipped with a shield that blinded investigators to study group assignment. As described in detail in a methods publication, the dose of 50 ppm by nasal cannula was based upon results from case reports and the authors’ experience from a phase I study.(Kline et al., 2017; Kline et al., 2014) Patients received 24 hours of continuous treatment and were followed for clinical study outcomes. All patient assessments and study measurements were performed by study staff and investigators blinded to the treatment allocation.
Blood sampling:
Blood was drawn by qualified phlebotomists following a waste using 18 gauge needles into two (blue-top) tubes containing sodium citrate and 3 tubes containing K2EDTA at two time points; prior to initiation of treatment or placebo, and just prior to initiation of study drug weaning (24 ± 2 hours following initiation). Blood was transported at room temperature (~23 °C) and processed within 60 minutes.
2.4. NOx measurements:
Plasma concentrations of nitrite and nitrate were measured with high performance liquid chromatography (EiCOM ENO-30, Sand Diego, CA).(Jobgen et al., 2007) Our definition of “adequate” NO delivery was evidenced by a significant increase in either [NO2-] or [NO3=] (collectively abbreviated as NOx), indicating increased substrate for the multiple endogenous biological oxidation pathways that convert NO to NOx.(Apostoli et al., 2014; Gladwin et al., 2011)
2.5. Thromboelastography:
Whole-blood TEG was assessed using two 2-channel Haemoscope 5000 devices (Haemonetics Corperation, Braintree, MA). Blood protein and platelet contributions to coagulation were assessed by measuring time to coagulation (R time, protein only), K time and α angle (α, protein and platelet), and maximum amplitude (platelet) under the following conditions: 1. With heparin to inhibit native thrombin generation and coagulation initiation with reptilase; 2. With heparinase to assess platelet function in the presence of native thrombin generation; 3. With heparin and ADP (2 μM); 4. With heparin and arachidonic acid (AA, 1 μM). The reported data were derived from the manufacturer’s provided software.
2.6. Platelet oxygen consumption:
All chemicals were purchased from Sigma Aldrich (St. Louis, MO). Methods were consistent with prior investigations in our laboratories.(Rezania et al., 2017) Following centrifugation of 2 EDTA tubes at 300g for 15 minutes, the resultant platelet rich plasma was centrifuged at 4,600g for 5 minutes leaving a platelet pellet and platelet poor plasma. This platelet pellet was gently resuspended in ~100–200 μL platelet poor plasma and the platelet count of this concentrated suspension was measured. A sufficient volume of this suspension to achieve a desired final platelet count of ~400×106 platelets in the 2 mL O2k high resolution oximeter chamber (Oxygraph 2k Oroboros Instruments, Innsbruck, Austria) was determined, and transferred into calcium-free mitochondrial respiration medium MiR05 containing glucose (5.5 mM) and octanoate (0.05 μM), prewarmed to 37 °C. Following equilibration, the baseline oxygen consumption rate was determined. Oligomycin (2 μL of 4 μg/mL) was added to the chamber to inhibit ATP synthase and estimate a cellular state IV or “leak” respiration rate. Carbonylcyanide p-(trifluoromethoxy) phenylhydrazone (FCCP, 0.1 mM) aliquots were added to determine maximal uncoupled respiration (“ETS” state), followed by the addition of the complex I and III inhibitors rotenone (4 μL of 5 mM) and antimycin A (3 μL of 1 mM) to determine a residual oxygen consumption rate. These additions inhibit the electron train chain and provide an estimate of non-electron transport chain linked oxygen consumption. This residual rate was subtracted from all other measurements, and the resultant respiration rates were normalized to platelet count.
2.7. Platelet-specific efficacy outcomes:
Based upon preliminary work demonstrating an increase in intact platelet mitochondrial oxygen consumption and thrombin-independent MA on TEG in platelets from patients with PE compared with healthy controls, we hypothesized that NO treatment would reduce both of these parameters. The primary treatment efficacy outcome, detailed in a separate manuscript, required patients to have a normal troponin T and normal echocardiogram at 24 hours.(Kline et al., 2019)
2.8. Cytosolic calcium concentrations:
To further investigate the effect of PE on calcium flux, platelets from an additional group of 12 healthy volunteers (as previously defined(Rezania et al., 2017)) and a convenience subgroup of 12 PE patients from this trial underwent additional assessment. Following preparation of platelet rich plasma (PRP) from K2EDTA tubes, ~3×10^8 cells/mL were added to 1mL of Ca-Na buffer (138mM NaCl, 1mM MgCl2 hexahydrate, 5mM KCl, 10mM HEPES, and 10mM glucose, pH 7.4), followed by 1uL of 0.5 Units/mL Apyrase and 1 μL 0.1uM H-Gly-Pro-Arg-Pro-OH, 4.5uM Fluo-8, AM (calcium fluorophore). Following incubation, the suspension was centrifuged, supernatant removed, and platelets washed with CaNa buffer. Washed platelets were re-suspended for spectrofluorometric analysis (Spectramax M5; Molecular Devices, Sunnyvale, CA). Platelets were measured at rest and following stimulation with thrombin (5 μL 100 U/mL), ADP (2 μM). To test the responsiveness of platelet cytosolic calcium and membrane potential to sGC stimulation and activation, in separate wells, either riociguat or cinaciguat (0.1 μM or 1.0 μM) were added. Excitation and emission wavelengths were set at 490 and 520nm for Fluo-8, AM.
2.9. Guanylate cyclase activity.
Platelet lysates (approximately 108 cells/mL) were clarified by centrifugation and aliquots of homogenates equivalent to 2.5 million platelets were added to 200 uL (final volume) of sGC assay buffer (Tris pH 7.4, 5 mM GTP, 5 mM MgCl2, 1 mM EGTA, 3 mM IBMX [3-isobutyl-1-methyl xanthine]). Cinaciguat, riociguat or the NO donor, DEA/NO, 2(N,N-Diethylamino)-diazenolate-2-oxide. diethylammonium salt (NONO Enzo Life Sciences, cat # ALX-430–034-M010) were added to assay buffer to achieve concentrations of 0.4 uM for each. All ingredients of the assay were combined and equilibrated at 37°C before addition of platelets lysates, which were added last to initiate the enzyme reaction. Reactions proceeded at 37°C and 40ul aliquots were removed at appropriate times and added to 100ul ethanol containing 0.1% acetic acid to stop the reactions. Ethanol/acetic acid solutions were air-dried overnight at 37°C and the residue containing cGMP was resuspended in 200ul ELISA assay solution. Samples were assayed for cGMP using a commercially available ELISA (R&D Systems) and expressed as fold elevation with sGC modulators versus buffer.
2.10. Biomarkers of hemolysis:
We used the human lactate dehydrogenase-1 isoform as a primary indicator of intravascular hemolysis, and also measured haptoglobin and hemopexin. These were measured in controls and PE patients 24 hours apart in plasma using commercially available ELISA assays (Cat #s: LS-F11927; ABIN6574215).
2.10. Data analysis:
Summary data are presented using descriptive statistics with normally distributed data described as mean (standard deviation) and non-normal data as median (interquartile range); means or medians were compared with student’s t-test or Mann-Whitney-U tests, as appropriate (STATA 10.0, College Station, TX). To compare mean cytosolic [Ca++], in the presence of agonists or sGC modulators at individual times, we used a one-way analysis of variance (ANOVA), followed by Dunnett’s multiple comparison post-hoc test. To compare groups over time, we used a two-way, repeated measures ANOVA, and report the p-value from the time*group interaction. All tests were two-sided and p-values <0.05 were considered significant. Figures were produced using GraphPad Prism for Windows version 7.04.
3.0. Results
3.1. Patient characteristics.
We enrolled 76 patients into the trial with complete data, 38 in each treatment arm. Patients were well matched for baseline demographics, clinical characteristics, and PE severity as indicated by clinical laboratory and radiologic findings (Table 1). Baseline TEG measurements prior to protocol initiation did not differ significantly based on treatment allocation (Supplemental Table 1) nor did platelet oxygen consumption measurements. All patients completed treatment, and there were no cases or early withdrawal or mortality within the 24 hours treatment window. There was no difference in the primary composite clinical efficacy endpoint, though a post-hoc subgroup analysis of patients with matched pre- and post-treatment echocardiograms demonstrated an increased probability of resolving RV dilation and hypokinesis with NO treatment. The data shown for controls are from 20 non-smoking, healthy volunteers (6 females and 14 males), mean age 32 years who were taking no medications.
Table 1:
Baseline demographics, clinical characteristics, and pulmonary embolism severity in treatment (nitric oxide plus oxygen) versus placebo (nitrogen plus oxygen) arms
| Control (Nitrogen + O2) n = 38 | Interventional (NO + O2) n = 38 | p-value | |
|---|---|---|---|
| Age, years (median, IQR) | 58 (46, 72) | 59 (46, 68) | 0.38 |
| Gender, males (%) | 18 (47) | 17 (45) | 0.81 |
| Race, n (%) White African American |
20 (53) 18 (47) |
27 (71) 11 (29) |
0.09 0.09 |
| Ethnicity n (%) Hispanic or Latino Not Hispanic or Latino Not reported |
0 (0) 37 (97) 1 (3) |
1 (3) 35 (92) 2 (5) |
0.99 0.30 0.56 |
| Comorbidities n (%) Malignancy Prior venous thromboembolism Smoking Coronary artery disease Congestive heart failure |
9 (24) 11 (29) 4 (11) 5 (13) 1 (3) |
8 (21) 10 (26) 7 (18) 3 (8) 6 (16) |
0.79 0.80 0.33 0.45 0.05 |
| Parenteral anticoagulant n (%) Enoxaparin Unfractionated heparin |
17 (45) 21 (55) |
16 (42) 22 (58) |
0.81 0.81 |
| Pretreatment vital signs (mean, SD) Heart rate, beats / min Respiratory rate, breath / min Systolic blood pressure, mmHg Oxygen saturation, % |
94 (14) 21 (4) 126 (21) 96 (6) |
86 (13) 21 (5) 125 (19) 97 (3) |
0.02 0.87 0.20 0.73 |
| PE severity (medians and IQR) Borg dyspnea score (1–10) Brain natriuretic peptide, pg/mL Troponin T, pg/mL Right ventricular systolic pressure (mmHg)* |
1 (0.5, 3) 234 (84, 556) 51 (18, 101) 50 (47, 50) |
2 (0.5, 4) 206 (83, 336) 23 (12, 71) 54.5 (50, 62) |
0.21 0.40 0.09 0.17 |
Pre-treatment standard of care echocardiograms performed in 52/76 patients
3.2. Nitrates.
Baseline plasma nitrates did not differ significantly between treatment arms at baseline. There was no significant change in nitrates in patients enrolled in the placebo group at 24 hours. The median nitrate concentrations (μg/mL, 1st-3rd quartiles) in the Placebo group were 15 (9–19) and 15 (11–20) (P=0.113, sign-rank test) at enrollment and 24 hours, respectively and the median nitrate concentrations in the NO group (μg/mL, 1st-3rd quartiles) were 12 (8–16) and 27 (22–38) (P<0.001, sign rank-test, P=0.01 time*group). We found no change in plasma nitrites from baseline to 24 hours of treatment in either group. These data suggest adequate drug delivery and increased bioavailability in the treatment arm.
3.3. Biomarkers of hemolysis.
None of the data were normally distributed, and could not be transformed, therefore median values with interquartile ranges are shown in Table 2. Hemolysis is expected to decrease haptoglobin and hemopexin and increase LDH-1. Compared with health controls, patients with PE had higher haptoglobin and LDH-1 concentrations at both times. Using a two-way repeated measures Kruskal Wallis ANOVA, the LDH-1 concentrations were not significantly decreased by NO treatment (P=0.09). The somewhat paradoxical finding of increased haptoglobin is likely a result of increased production haptoglobin in response to the systemic inflammation associated with PE.(Nielsen and Moestrup, 2009)
Table 2.
Biomarkers of hemolysis
| Haptoglobin | Hemopexin | Lactate dehydrogenase-1 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Group | Value | T=0 hours | T=24 hours | T=0 hours | T=24 hours | T=0 hours | T=24 hours | ||||||
| Pooled | Pooled | Placebo | NO | Pooled | Pooled | Placebo | NO | Pooled | Pooled | Placebo | NO | ||
| PE | median | 29.0 | 46.5 | 31.2 | 49.5 | 21.2 | 22.7 | 23.3 | 22.0 | 92.6 | 90.8 | 93.0 | 67.7 |
| first | |||||||||||||
| quartile | 25.0 | 25.6 | 25.3 | 24.7 | 20.0 | 22.7 | 20.3 | 19.6 | 66.2 | 79.8 | 58.3 | 37.9 | |
| third | |||||||||||||
| quartile | 59.0 | 55.5 | 59.0 | 52.3 | 24.7 | 25.2 | 26.7 | 24.7 | 114.0 | 116.0 | 114.0 | 116.1 | |
| Controls | median | 16.6 | 15.7 | 20.6 | 20.0 | 1.3 | 0.5 | ||||||
| first | |||||||||||||
| quartile | 18.5 | 4.6 | 18.5 | 18.4 | 0.0 | 0.0 | |||||||
| third | |||||||||||||
| quartile | 23.5 | 24.8 | 23.5 | 24.0 | 4.4 | 6.0 | |||||||
| P value | 0.07* | 0.02 | 0.32** | 0.8 | 0.4 | 0.28 | 0.01 | 0.03 | 0.09 | ||||
P values comparing PE vs. Control from the Mann-Whitney U test;
P values comparing Placebo to NO from the Kruskal-Wallis repeated measures ANOVA
3.4. Thromboelastography results.
To demonstrate the overall effect of PE on TEG parameters, supplemental Table 1 compares TEG values for patients with PE with healthy controls. The primary differences were an increase in the angle and maximum amplitude in the presence of thrombin inhibition, and with coagulation initiated with reptilase.
There were no significant differences between treatment arms at 24 hours in any of the measured TEG parameters by any of the measured agonists (P>0.1 for all comparisons of mean K, MA, R and α values between groups). Because the MA is recognized as the TEG value most affected by platelet function, Figure 1 demonstrates the dot plots of the MA pre- and post-treatment by treatment allocation with each of the agonists with or without heparin, while Supplementary Table 2 compares means between placebo and NO treated groups and supplementary. Figures 1–3 provide dot plots of the K and R times and α angle.
Figure 1:

Thromboelastography data for patients with pulmonary embolism treated with placebo or inhaled nitric oxide. Maximum amplitude without (A and B) and with (C and D) heparinase, ADP (E and F) and AA (G and H) before and after treatment with placebo (A, C, E, and G) or nitric oxide (B, D, F, and H). No mean values were significantly different between treatments (P>0.1 by unpaired t-test).
Figure 3:
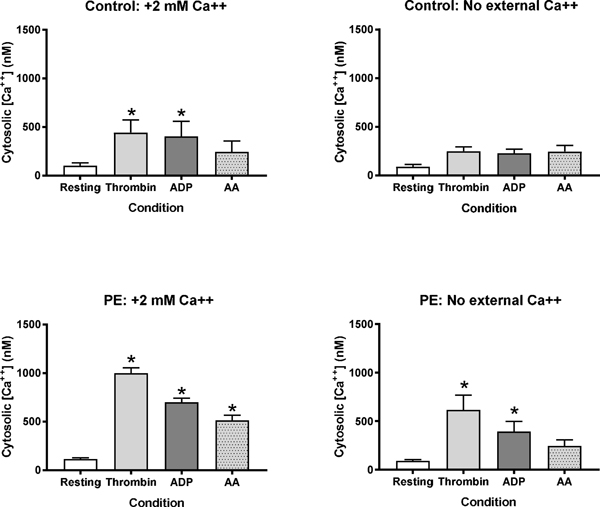
Platelet cytosolic calcium is similar between healthy volunteer (A) and pulmonary embolism (B) patients at rest, while pulmonary embolism platelets demonstrate hyper-responsiveness following agonist stimulation. Peak calcium concentrations are reduced, but not eliminated, following external calcium depletion (C-D). *indicates P<0.05 by one way analysis of variance with Dunnett’s post-hoc with resting as the reference value.
These findings were consistent whether the data were analyzed as a comparison of means between groups at 24 hours post-randomization (as shown in Figure 1, and Supplementary Figures 1–3), or by comparing the mean change in each parameter from baseline value (calculated as post-treatment value – pre-treatment value; data not shown). All time*group P values comparing changes between NO and placebo groups for all TEG from the repeated measures ANOVA were >0.05.
3.5. Platelet oxygen consumption.
Table 3 shows that platelets from patients with PE have approximately 25% higher oxygen consumption compared with healthy controls.
Table 3.
Comparison of platelet respirometry data between healthy controls and patients with PE (pmol O2/S/10^8 cells)
| PE | Control | PE | Control | PE | Control | |
|---|---|---|---|---|---|---|
| mean | 35.90 | 26.04 | 6.28 | 3.97 | 50.12 | 38.88 |
| SD | 9.49 | 4.51 | 1.71 | 0.99 | 9.36 | 10.37 |
| P value | 0.004 | 0.04 | 0.01 | |||
However, similar to the findings from TEG, in patients with PE, we found no significant differences in any of the measures of intact platelet oxygen consumption following completion of treatment between the two treatment groups. Specifically, we observed no difference in unstimulated, oligomycin-induced (leak), or maximal stimulated (ETS) oxygen consumption rates at 24 hours (Figure 2).
Figure 2:

Platelet oxygen consumption at enrollment and after 24 hours of treatment. From left to right, the bars show unstimulated (resting), oligomycin-induced (+oligo), and maximal uncoupled (+FCCP) platelet oxygen consumption before and after 24 hours of treatment with nitric oxide plus oxygen versus nitrogen plus oxygen (placebo). Data are means with standard errors of the mean. There were no significant differences in post-treatment oxygen consumption rates between treatment arms. The asterisks indicate P<0,05 from a paired t-test for values at 24 hours vs. enrollment for placebo, but not nitric oxide treatment.
Platelet oxygen consumption was not different between placebo and NO treatment under any conditions at either time point. The only consistent finding was that platelet oxygen consumption decreased significantly (P<0.05, paired t-test) in all states (resting, +oligomycin and +FCCP) between enrollment and 24 hours with placebo, but not with NO treatment.
3.6. Platelet cytosolic [Ca++].
To better understand the effect of PE stress on platelet hyperactivity, Figure 3 shows data from patients with PE enrolled in this study at enrollment compared with 12 healthy control patients. The resting cytosolic [Ca++] was similar in control and PE patients, while platelets from PE patients exhibited an exaggerated response with higher peak levels of cytosolic [Ca++]. The agonist-stimulated increase in cytosolic [Ca++] was partially reduced if the media lacked exogenous calcium, demonstrating partial dependence of external calcium for this process.
3.7. Effect of guanylate cyclase modulation on platelet [Ca++].
Treatment of PE platelets with the sGC activator cinciguat effectively lowered peak cytosolic [Ca++] following stimulation with either thrombin or ADP (Figure 4). This demonstrates as a positive control, the potential of guanylate cyclase modulation to influence cytosolic [Ca++].
Figure 4:

Effects of soluble guanylate cyclase modulation on platelet cytosolic [Ca++] (Y axis).Soluble guanylate cyclase activator cinaciguat, but not the stimulator riociguat attenuates cytosolic calcium flux of platelets of patients with PE following thrombin or adenosine diphosphate (ADP) stimulation. *indicates P<0.05 by one way analysis of variance with Dunnett’s post-hoc with resting as the reference value.
However, in separate platelet aliquots from the same patients, Figure 4 shows that stimulation of sGC with riociguat produced no effect on cytosolic [Ca++], suggesting that the sGC enzyme was predominately oxidized in these platelets. In platelets from control patients, treatment of platelets with cinaciguat and riociguat both effectively blocked increase in cytosolic [Ca++] with thrombin and ADP stimulation.
We then directly measured sGC activity in platelet lysates from healthy controls versus patients with PE. Table 4 shows the cGMP production in each lysate condition, and Figure 5 plots the ratio of activity with stimulation relative to control. Because the effect of stimulation is shown as ratios of the same lysate, no total protein correction was needed.
Table 4.
Guanylate cyclase activity* in platelet lysates
| Condition† | 2 min | 5 min | 10 min | |||||
|---|---|---|---|---|---|---|---|---|
| No PE | Mean | SEM | Mean | SEM | Mean | SEM | ||
| Unstimulated | 2.42 | 0.32 | 3.82 | 0.10 | 4.18 | 0.25 | ||
| NONO | 6.99 | 1.09 | 18.59 | 1.54 | 30.66 | 4.20 | ||
| Cinaciguat | 2.01 | 0.25 | 4.23 | 0.25 | 5.74 | 0.37 | ||
| Riociguat | 2.00 | 0.40 | 4.17 | 0.23 | 7.72 | 0.74 | ||
| Controls | Mean | SEM | Mean | SEM | Mean | SEM | ||
| Unstimulated | 4.62 | 0.38 | 7.90 | 1.70 | 12.45 | 2.46 | ||
| NONO | 30.34 | 1.36 | 76.19 | 5.58 | 132.43 | 8.59 | ||
| Cinaciguat | 5.20 | 0.41 | 11.86 | 1.19 | 24.52 | 3.39 | ||
| Riociguat | 4.92 | 0.47 | 10.34 | 1.33 | 21.65 | 2.31 | ||
pmol cGMP/10^6 cells
Concentrations of NON, Cinaciguate and Riociguat = 0.4 uM
Figure 5.

Activity of soluble guanylate cyclase (sGC) in platelet lysates from healthy volunteers (Control) compared to patients with submassive pulmonary embolism (PE). The Y axis shows the fold increase in cyclic guanosine monophosphate activity with the sGC modulator, relative to the same lysate with buffer added. Each time point was a unique measurement. A. Effect of 0.4 uM nitric oxide donor, DEA NONO; B. Effect of 0.4 uM cinaciguat; C. Effect of 0.4 uM riociguat. *P<0.001 by unpaired t-test at each time.
Unexpectedly, the stimulated sGC activity was higher in PE patients relative to controls when treated with the NO donor DEA NONO, and with the sGC activator, cinaciguat, but not riociguat. These data argue against any functional loss of sGC with PE, but show a paradoxical combination of sGC positive modulation with NO and cinaciguat but resistance to riociguat.
4.0. Discussion
In humans with submassive PE, compared with breathing nitrogen placebo, breathing 50 ppm of inhaled NO had no significant effects on platelet function as observed on TEG parameters as well as no significant effect on platelet mitochondrial oxygen consumption. Platelets from patients with PE were responsive to direct sGC modulation, and sGC activity was increased in platelets from patients with PE. Our data suggest that inhaled NO in unlikely to produce clinically important changes in platelet function or oxidative metabolism in the setting of acute PE, and provide mechanistic insight into the optimal pharmacological approach to leverage the NO/sGC pathway to control platelet hyperactivity with acute PE.
Rajendran and Chirkov previously described the observation of platelet hyporesponsiveness to NO in patients with chronic disease, and the authors hypothesized NO scavenging and oxidative stress to sGC as mechanisms.(Rajendran and Chirkov, 2008) We previously reported increased mitochondrial oxygen consumption and superoxide production in platelets from PE patients compared with controls, and here we extend that finding to show this increase in oxygen consumption occurs coincident with increased agonist-stimulated cytosolic [Ca++].(Rezania et al., 2017) Increase in platelet [cGMP] generally reduces voltage-sensitive calcium transients in platelets.(Smolenski, 2012) In our work, despite evidence of adequate NO delivery to the plasma, we observed no effect of this treatment on any of the measured parameters between treatment groups. However, cinaciguat, but not riociguat, effectively reduced agonist-stimulated cytosolic [Ca++] transients. This finding has mechanistic implications inasmuch as cinaciguat is thought to predominately activate heme-free (oxidized) guanylate cyclase, whereas riociguat exclusively stimulates the holoenzyme with its heme prosthetic intact.(Stasch et al., 2011) Accordingly, the present data, together with a prior report showing mitochondria derived reactive oxygen species production in platelets from the same patients as this study suggest oxidation of platelet sGC during PE, rendering it unresponsive to NO.(Rezania et al., 2017) However, the data in Figure 5 suggest a more complicated effect, given that sGC from PE platelets was responsive to DEA-NONO and cinaciguat, but not riociguat. Our data do not allow insight into the molecular mechanism of this mixed effect. One possibility is the presence of a mixture of the holo- and apo-sGC enzyme with PE, with both forms increased relative to the healthy state. Cinaciguat may also activate the sGC holoenzyme. Indeed, the view that cinaciguat exclusively activates heme-free sGC has been challenged by its ability to fully relax healthy arteries, and observations that cinaciguat can directly displace sGC’s heme group, therefore shifting the pool of sGC toward a larger fraction of heme-free, and therefore cinaciguat-sensitive sGC.(Kollau et al., 2018) The work is hypothesis-generating as to why riociguat would not increase PE platelet sGC activity in proportion to the increases seen with either DEA NONO or cinaciguat. The binding site of riociguat on sGC, as well as the charge and conformational requirements for its binding remain uncertain.(Montfort et al., 2017) It could be hypothesized that PE causes thiol group oxidation on sGC, leading to reduced riociguat binding, or the conformational change required to increase catalytic activity, therefore reducing riociguate potency.(Beuve, 2017; Derbyshire and Marletta, 2012) Accordingly, a therapeutic option may lie in strategies to prevent sGC inactivation and platelet resistance to NO. Treatment of patients with acute coronary syndrome with perhexiline (an inhibitor of fatty acid transport into the mitochondrion) quickly reversed their platelet resistance to inhibition by sodium nitroprusside in association with reduced neutrophil superoxide production.(Willoughby et al., 2002)
Current treatments for acute PE focus on prevention of propagation and reduction of clot burden, primarily through systemic anticoagulation to inhibit clotting proteins. However, experimental models suggest that a large proportion of the increased RV pressure of acute PE results from pulmonary vasoconstriction and microvascular obstruction.(Watts et al., 2011b) The reasons for this vasoconstriction originate with decreased bioavailability of NO locally in the pulmonary vasculature with more severe PE.(Watts et al., 2011a; Watts et al., 2011b) Ongoing hemolysis from the over pressured right heart provides the pathophysiological basis for NO, as suggested by the markedly elevated LDH-1 concentrations. Free hemoglobin is a potent scavenger of NO, and an activator of platelets, secondary to ADP release from red cell destruction from intra-cardiac shear stresses.(Helms et al., 2013; Kline et al., 2009; Kline et al., 2015; Zagorski et al., 2010) Moreover, mechanical shear is a well-known activator of platelets. However, prolonged shear forces may lead to a compensatory increase in sGC-cGMP activity to restrain platelet activation, which may explain the increase in sGC activity in Figure 5.(Wen et al., 2018) Provision of exogenous NO is a conceptually attractive therapeutic target to acutely reduce RV pressures through selective relaxation of the pulmonary vascular bed. Given the short half-life of NO, inhaled NO can lead to targeted drug delivery where it is most needed while minimizing systemic effects.
In addition to mediating vascular relaxation, NO also has direct effects on platelet activity. In a previous investigation, we observed that patients with acute PE exhibit increased thrombin-independent MA.(Rezania et al., 2017) These changes occur in tandem with increases in platelet cytosolic [Ca++] concentrations, mitochondrial oxygen consumption and apoptotic signaling pathways, as evidenced by cytochrome c mobilization, mitochondrial reactive oxygen species production, and platelet surface phosphatidylserine expression compared to control patients.(Rezania et al., 2017) In this study, platelet oxygen consumption normalized within 24 hours of acute PE independent of NO treatment, indicating the stimulating effects of PE on platelet mitochondria and apoptotic pathways is rapidly resolved with heparin anticoagulation alone. Therefore, interventions targeting these processes likely need to be administered early to maximize any chance of clinical efficacy.
With agonists ADP and arachidonic acid, no TEG parameters were significantly impacted by inhaled NO following 24 hours of treatment. With the possible exception of a smaller relative reduction in oxygen consumption with 24 hours of NO compared with placebo (Figure 2), NO had no comparative effect on platelet oxygen consumption. The data suggest that inhaled NO fails to stimulate platelet sGC sufficiently to substantially increase cGMP signaling in a substantial fraction of platelets. The responsiveness of PE platelets to the sGC activator cinaciguat (Figure 4), together the increased sGC activity in lysates (Figure 5) further suggest the enzyme and platelets from PE patients remain sufficiently responsive to an sGC-mediated therapy, even though inhaled NO failed to attenuate platelet hyper-reactivity. Reasons for failure could include concentration effects, excessive NO scavenging, or oxidative stress on sGC.(Ahrens et al., 2011; Evgenov et al., 2006)
There are several limitations to consider with this study. Only a single level of inhaled NO was tested, and it is possible larger doses would demonstrate more substantial effects. However, the dose was sufficient to demonstrate drug delivery and more than sufficient in previous literature in healthy patients to demonstrate platelet-specific effects.(Gladwin et al., 2011; Gries et al., 2000). We lack data to show the effect of NONO on platelet cytosolic [Ca++]. Also, we did not study activators of adenylate cyclase, such as prostacyclin or epoprostenol. Finally, it is possible other measures of platelet function, such as aggregometry or expression of activated surface receptors might demonstrate changes not detected by our study design. However, given the data demonstrated in this study that show no significant effect on whole-blood TEG and coagulation suggest such findings would be of questionable clinical relevance.
5.0. Conclusion
In humans with submassive pulmonary embolism, inhaled NO did not produce important changes in platelet function or bioenergetics, but direct pharmacological activation of platelet sGC effectively reduced agonist-stimulated calcium signaling, indicating a potentially novel method to control platelet hyperactivity in patients with PE.
Supplementary Material
Highlights.
Pulmonary embolism causes platelet hyperactivity that can worsen patient outcomes. Inhaled nitric oxide could reduce this effect. In a randomized trial, compared with placebo, inhaled NO did not did not affect platelet-specific thromboelastography, oxygen consumption. Agonist-stimulated platelet cytosolic calcium concentrations were elevated in PE patients compared with healthy controls, but were effectively suppressed with direct soluble guanylate cyclase (sGC) activation with cinaciguat but not stimulation with riociguat, suggesting that sGC oxidation may have impaired nitric oxide effect.
Significance statement:
Pulmonary embolism causes platelet hyperactivity that can worsen outcomes. Inhaled nitric oxide could reduce this effect. In a randomized trial, compared with placebo, inhaled NO did not did not affect platelet-specific thromboelastography, oxygen consumption. Agonist-stimulated platelet cytosolic calcium concentrations were elevated in PE patients compared with healthy controls, but were effectively suppressed with direct soluble guanylate cyclase (sGC) activation with cinaciguat but not stimulation with riociguat, indicating that sGC oxidation may have impaired nitric oxide effect.
Acknowledgments
Funding: This study was supported by NHLBI (1UM1HL113203-01A1) and. Dr. Puskarich received salary support from NIGMS for a study of platelet activation in septic shock (K23GM113041-01), as well as support from the NIH Loan Repayment Program. Bayer pharmaceuticals provided the author with cinaciguat in-kind.
Nonstandard abbreviations:
- NO
nitric oxide
- PE
pulmonary embolism
- sGC
soluble guanylate cyclase
- TEG
thromboelastography
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict-of-interest
No authors have a conflict of interest.
Reference List
- Ahrens I, Habersberger J, Baumlin N, Qian H, Smith BK, Stasch JP, Bode C, Schmidt HH and Peter K (2011) Measuring oxidative burden and predicting pharmacological response in coronary artery disease patients with a novel direct activator of haem-free/oxidised sGC. Atherosclerosis 218:431–434. [DOI] [PubMed] [Google Scholar]
- Apostoli GL, Solomon A, Smallwood MJ, Winyard PG and Emerson M (2014) Role of inorganic nitrate and nitrite in driving nitric oxide-cGMP-mediated inhibition of platelet aggregation in vitro and in vivo. J Thromb Haemost 12:1880–1889. [DOI] [PubMed] [Google Scholar]
- Beuve A (2017) Thiol-Based Redox Modulation of Soluble Guanylyl Cyclase, the Nitric Oxide Receptor. Antioxidants & redox signaling 26:137–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung T, Connor D, Joseph J, Emmett L, Mansberg R, Peters M, Ma D and Kritharides L (2007) Platelet activation in acute pulmonary embolism. JThrombHaemost 5:918–924. [DOI] [PubMed] [Google Scholar]
- Dasgupta A, Bowman L, D’Arsigny CL and Archer SL (2015) Soluble guanylate cyclase: a new therapeutic target for pulmonary arterial hypertension and chronic thromboembolic pulmonary hypertension. Clinical pharmacology and therapeutics 97:88–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derbyshire ER and Marletta MA (2012) Structure and regulation of soluble guanylate cyclase. Annual review of biochemistry 81:533–559. [DOI] [PubMed] [Google Scholar]
- Evgenov OV, Pacher P, Schmidt PM, Hasko G, Schmidt HH and Stasch JP (2006) NO-independent stimulators and activators of soluble guanylate cyclase: discovery and therapeutic potential. Nature reviews Drug discovery 5:755–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gambaryan S and Tsikas D (2015) A review and discussion of platelet nitric oxide and nitric oxide synthase: do blood platelets produce nitric oxide from L-arginine or nitrite? Amino acids 47:1779–1793. [DOI] [PubMed] [Google Scholar]
- Gladwin MT, Kato GJ, Weiner D, Onyekwere OC, Dampier C, Hsu L, Hagar RW, Howard T, Nuss R, Okam MM, Tremonti CK, Berman B, Villella A, Krishnamurti L, Lanzkron S, Castro O, Gordeuk VR, Coles WA, Peters-Lawrence M, Nichols J, Hall MK, Hildesheim M, Blackwelder WC, Baldassarre J and Casella JF (2011) Nitric oxide for inhalation in the acute treatment of sickle cell pain crisis: a randomized controlled trial. JAMA 305:893–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gries A, Bode C, Peter K, Herr A, Bohrer H, Motsch J and Martin E (1998) Inhaled nitric oxide inhibits human platelet aggregation, P-selectin expression, and fibrinogen binding in vitro and in vivo. Circulation 97:1481–1487. [DOI] [PubMed] [Google Scholar]
- Gries A, Herr A, Motsch J, Holzmann A, Weimann J, Taut F, Erbe N, Bode C and Martin E (2000) Randomized, placebo-controlled, blinded and cross-matched study on the antiplatelet effect of inhaled nitric oxide in healthy volunteers. ThrombHaemost 83:309–315. [PubMed] [Google Scholar]
- Helms CC, Marvel M, Zhao W, Stahle M, Vest R, Kato GJ, Lee JS, Christ G, Gladwin MT, Hantgan RR and Kim-Shapiro DB (2013) Mechanisms of hemolysis-associated platelet activation. JThrombHaemost 11:2148–2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jobgen WS, Jobgen SC, Li H, Meininger CJ and Wu G (2007) Analysis of nitrite and nitrate in biological samples using high-performance liquid chromatography. Journal of chromatography B, Analytical technologies in the biomedical and life sciences 851:71–82. [DOI] [PubMed] [Google Scholar]
- Kline JA, Hall CL, Jones AE, Puskarich MA, Mastouri RA and Lahm T (2017) Randomized trial of inhaled nitric oxide to treat acute pulmonary embolism: The iNOPE trial. Am Heart J 186:100–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kline JA, Hernandez-Nino J, Garrett JS and Jones AE (2014) Pilot study of a protocol to administer Inhaled nitric oxide to treat severe acute submassive pulmonary embolism. Emerg Med J 31:459–463. [DOI] [PubMed] [Google Scholar]
- Kline JA, Marchick MR and Hogg MM (2009) Reduction in plasma haptoglobin in humans with acute pulmonary embolism causing tricuspid regurgitation. JThrombHaemost 7:1597–1599. [DOI] [PubMed] [Google Scholar]
- Kline JA, Puskarich MA, Jones AE, Mastouri RA, Hall CL, Perkins A, Gundert EE and Lahm T (2019) Inhaled nitric oxide to treat intermediate risk pulmonary embolism: A multicenter randomized controlled trial. Nitric oxide : biology and chemistry 84:60–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kline JA, Steuerwald M, Watts JA, Courtney DM and Bonkovsky HL (2015) Leukocyte Expression of Heme Oxygenase-1 [hmox1] Varies Inversely with Severity of Tricuspid Regurgitation in Acute Pulmonary Embolism. Thromb Res 136:769–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kline JA, Watts J, Courtney D, Lee Y and Hwang S (2013) Severe pulmonary embolism decreases plasma L-arginine. EurRespirJ. [DOI] [PubMed] [Google Scholar]
- Kollau A, Opelt M, Wolkart G, Gorren ACF, Russwurm M, Koesling D, Mayer B and Schrammel A (2018) Irreversible Activation and Stabilization of Soluble Guanylate Cyclase by the Protoporphyrin IX Mimetic Cinaciguat. Molecular pharmacology 93:73–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendes-Silverio CB, Leiria LO, Morganti RP, Anhe GF, Marcondes S, Monica FZ, de NG and Antunes E (2012) Activation of haem-oxidized soluble guanylyl cyclase with BAY 60–2770 in human platelets lead to overstimulation of the cyclic GMP signaling pathway. PLoSOne 7:e47223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montfort WR, Wales JA and Weichsel A (2017) Structure and Activation of Soluble Guanylyl Cyclase, the Nitric Oxide Sensor. Antioxidants & redox signaling 26:107–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen MJ and Moestrup SK (2009) Receptor targeting of hemoglobin mediated by the haptoglobins: roles beyond heme scavenging. Blood 114:764–771. [DOI] [PubMed] [Google Scholar]
- Rajendran S and Chirkov YY (2008) Platelet hyperaggregability: impaired responsiveness to nitric oxide (“platelet NO resistance”) as a therapeutic target. Cardiovascular drugs and therapy 22:193–203. [DOI] [PubMed] [Google Scholar]
- Reiss C, Mindukshev I, Bischoff V, Subramanian H, Kehrer L, Friebe A, Stasch JP, Gambaryan S and Walter U (2015) The sGC stimulator Riociguat inhibits platelet function in washed platelets but not in whole blood. BrJPharmacol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rezania S, Puskarich MA, Petrusca DN, Neto-Neves EM, Rondina MT and Kline JA (2017) Platelet hyperactivation, apoptosis and hypercoagulability in patients with acute pulmonary embolism. Thromb Res 155:106–115. [DOI] [PubMed] [Google Scholar]
- Roger S, Paysant J, Badier-Commander C, Cordi A, Verbeuren TJ and Feletou M (2010) Anti-aggregating effect of BAY 58–2667, an activator of soluble guanylyl cyclase. VasculPharmacol 53:281–287. [DOI] [PubMed] [Google Scholar]
- Smolenski A (2012) Novel roles of cAMP/cGMP-dependent signaling in platelets. JThrombHaemost 10:167–176. [DOI] [PubMed] [Google Scholar]
- Stasch JP, Alonso-Alija C, Apeler H, Dembowsky K, Feurer A, Minuth T, Perzborn E, Schramm M and Straub A (2002) Pharmacological actions of a novel NO-independent guanylyl cyclase stimulator, BAY 41–8543: in vitro studies. BrJPharmacol 135:333–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stasch JP, Pacher P and Evgenov OV (2011) Soluble guanylate cyclase as an emerging therapeutic target in cardiopulmonary disease. Circulation 123:2263–2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomasiak M, Stelmach H, Rusak T and Wysocka J (2004) Nitric oxide and platelet energy metabolism. Acta biochimica Polonica 51:789–803. [PubMed] [Google Scholar]
- Watts JA, Gellar MA, Fulkerson MB, Das SK and Kline JA (2011a) Arginase depletes plasma l-arginine and decreases pulmonary vascular reserve during experimental pulmonary embolism. PulmPharmacolTher. [DOI] [PubMed] [Google Scholar]
- Watts JA, Gellar MA, F MB and Kline JA (2011b) Pulmonary vascular reserve during experimental pulmonary embolism: Effects of a soluble guanylate cyclase stimulator, BAY 41–8543. Crit Care Med 39:2700–2704. [DOI] [PubMed] [Google Scholar]
- Wen L, Feil S, Wolters M, Thunemann M, Regler F, Schmidt K, Friebe A, Olbrich M, Langer H, Gawaz M, de Wit C and Feil R (2018) A shear-dependent NO-cGMP-cGKI cascade in platelets acts as an auto-regulatory brake of thrombosis. Nature communications 9:4301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willoughby SR, Stewart S, Chirkov YY, Kennedy JA, Holmes AS and Horowitz JD (2002) Beneficial clinical effects of perhexiline in patients with stable angina pectoris and acute coronary syndromes are associated with potentiation of platelet responsiveness to nitric oxide. Eur Heart J 23:19461954. [DOI] [PubMed] [Google Scholar]
- Zagorski J, Marchick MR and Kline JA (2010) Rapid clearance of circulating haptoglobin from plasma during acute pulmonary embolism in rats results in HMOX1 up-regulation in peripheral blood leukocytes. Journal of Thrombosis & Haemostasis 8:289–296. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.