

Abstract
Technologies for imaging the pathophysiology of Alzheimer disease (AD) now permit studies of the relationships between the two major proteins deposited in this disease — amyloid-β (Aβ) and tau — and their effects on measures of neurodegeneration and cognition in humans. Deposition of Aβ in the medial parietal cortex appears to be the first stage in the development of AD, although tau aggregates in the medial temporal lobe (MTL) precede Aβ deposition in cognitively healthy older people. Whether aggregation of tau in the MTL is the first stage in AD or a fairly benign phenomenon that may be transformed and spread in the presence of Aβ is a major unresolved question. Despite a strong link between Aβ and tau, the relationship between Aβ and neurodegeneration is weak; rather, it is tau that is associated with brain atrophy and hypometabolism, which, in turn, are related to cognition. Although there is support for an interaction between Aβ and tau resulting in neurodegeneration that leads to dementia, the unknown nature of this interaction, the strikingly different patterns of brain Aβ and tau deposition and the appearance of neurodegeneration in the absence of Aβ and tau are challenges to this model that ultimately must be explained.
Multimodal imaging technologies have transformed research on human ageing and dementia by enabling the investigation of complex interrelated mechanisms that underlie the development of Alzheimer disease (AD). The two aggregated proteins implicated in the pathogenesis of AD — amyloid-β (Aβ) and tau — can be visualized with positron emission tomography (PET), and the proposed downstream consequence of neurodegeneration can be examined with structural MRI, functional MRI and glucose metabolism PET. Studies of cognitively healthy older people and those with cognitive impairment or dementia have partially elucidated these pathophysiological processes in an approach that could be a model for the investigation of many degenerative neurological diseases.
Aβ plaques.
Also termed neuritic or senile plaques, Aβ plaques are one of the pathological hallmarks of AD and are composed of aggregates of the Aβ protein that are found at postmortem examination of the brain.
AD is a slowly evolving disorder that usually manifests clinically with initial amnesia characterized by the inability to form new memories, reflecting dysfunction of the medial temporal lobe (MTL) episodic memory system1,2. In its late stages, AD is characterized by dementia and is associated with widespread Aβ plaques and tau aggregates as neurofibrillary tangles3,4. As plaque and neurofibrillary tangle pathology are often found in the brains of cognitively healthy older people, it is generally accepted that the biological processes underlying AD are present for decades before symptom expression5,6. Thus, imaging can potentially explain the evolution of AD from normal ageing, through the stage of mild cognitive impairment (MCI), to dementia. Such studies have driven a widely applied model of biomarker change, proposing a sequential series of empirically verifiable events7. This model places Aβ deposition at the start of this process, reflecting the amyloid cascade hypothesis of AD8. A major goal of this Review is to examine the imaging evidence collected to date in an effort to see how well these data conform to this model and how deviations from the model may, or may not, be accommodated. It is important to recognize that each imaging modality has been available for a different length of time; this is crucial because approaches that have been used for years fail to account for variables that have become measurable more recently. Similarly, studies with extensive longitudinal observations across multiple modalities are limited; therefore, many studies are cross-sectional or have used short periods of longitudinal observation across different age ranges in a ‘cohort-sequential’ design.
Neurofibrillary tangles.
The other major pathological hallmark of AD, they are composed of aggregated forms of hyperphosphorylated tau protein as intraneuronal paired helical filaments.
This Review examines how imaging has developed and tested models of the pathophysiology of AD in an effort to delineate disease mechanisms (the most widely applied imaging modalities are explained in Box 1). First, I discuss the aggregate data from AD and normal cognitive ageing to define the relationships between Aβ and tau, neurodegeneration and cognition, followed by an exploration of how different forms of AD and ageing can, or cannot, be used as interchangeable models. Then, I review the proposed underlying mechanisms that may drive pathological events. Last, I examine the data with regard to how well they conform to an amyloid-based model of sequential alterations underlying AD pathogenesis.
Box 1 |. Neuroimaging techniques.
Amyloid-PET
There are a number of positron emission tomography (Pet) radiopharmaceuticals that are available for imaging amyloid-β in the brain. On the basis of high sensitivity and specificity for the postmortem detection of amyloid plaques, three of these compounds have been approved by the US Food and Drug Administration203–205. The compounds all bind to aggregated fibrillar forms of amyloid-β and thus do not visualize soluble oligomeric forms of this peptide, which may be the most pathogenic. Amyloid-PET imaging has been available since approximately 2004; therefore, longitudinal human data from such imaging are increasingly available. Because amyloid-PET imaging predates tau-PET imaging by about 10 years, many amyloid imaging studies have failed to account for the possible effects of tau on neurodegeneration and cognition.
Tau-PET
The imaging agents for tau-PET have been available for only a few years. However, the development of tau-PET ligands is a very active research area, and at least five such ligands have been tested in humans. Tau occurs in six isoforms, labelled according to the number of microtubule-binding repeat sequences expressed; tau aggregates found in Alzheimer disease (AD) are a mixture of three-repeat (3R) and four-repeat (4R) isoforms. Current tau ligands label this 3R/4R mixture, and there are limited data indicating binding to other types of aggregates206. No large studies comparing Pet ligand binding with autopsy measures of tau are available, and although the former is actively being incorporated into many clinical studies, longitudinal data on changes in tau accumulation from this approach are still limited.
FDG-PET
Among the first PET techniques developed, this approach uses the glucose analogue 18F-fluorodeoxyglucose (FDG) to map brain glucose metabolism. FDG-PET has been used for decades to characterize metabolic deficits in AD, which are widely assumed to reflect loss of synaptic function in view of the energy budget of neurons. However, glucose metabolism is not specific to neurons; therefore, metabolic alterations can also reflect glial cell function, including their role in inflammation. Metabolic measurements show regional brain reductions in ageing and dementia but do not necessarily reveal anything specific about the underlying neurobiology; these metabolic alterations seem to reflect a fairly nonspecific neurodegeneration process.
Structural MRI
Structural MRI has a major advantage of contrast sensitivity that permits separation of brain tissue types, permitting accurate measurement of brain volumes. Structural measures that have proved most useful include measures of grey matter volume and cortical thickness, which can be evaluated at both a regional and global level. Similar to measures of glucose metabolism, regional tissue atrophy does not detect a single biological process but rather reflects the local impact of neurodegeneration that could result from many different possible mechanisms.
Functional MRI
This technique makes use of changes in the magnetic resonance signal that reflect tissue perfusion. The most common use of functional MRI in the ageing and dementia field is to examine brain networks. These networks are defined by examining the synchrony of the functional MRI signal across different brain regions; regions that show synchronous signal fluctuations are inferred to be part of the same network. Alterations in these measures of connectivity are interpreted as indicative of disruption or dysfunction of such networks and can be seen as reductions or increases in network connectivity. These networks also provide models to explain how pathology spreads through the brain.
Alzheimer disease
Individuals with AD can be differentiated into three widely accepted subgroups on the basis of their characteristic clinical phenotypes and genetic risks: those with autosomal dominant inheritance (autosomal dominant AD (ADAD)); those with early age at onset, generally before age 65 (early-onset AD (EOAD)); and those with typical late-onset AD (LOAD). Although EOAD and LOAD are associated with genetic risks, LOAD is probably a polygenic disorder (in contrast to the monogenic nature of ADAD) and is considered to be sporadic, whereas EOAD may be associated with recessive inheritance9,10. Each of these disorders provides a different window into the pathogenesis of AD. LOAD is by far the most common form of AD, representing a slowly progressive process beginning with amnesia in older people, and it is often pathologically complex, with plaque and tangle pathology accompanied by cerebrovascular disease and other protein aggregates such as α-synuclein and TAR DNA-binding protein 43 (TDP43) aggregates11,12. By contrast, EOAD is more likely to represent a relatively ‘pure’ plaque and tangle pathological process; furthermore, EOAD can present with strikingly focal neurobehavioural phenotypes reflecting dysfunction of specific neural systems. ADAD is also generally early in onset but is distinguished from EOAD by autosomal dominant inheritance, with mutations in the amyloid precursor protein (APP), presenilin 1 (PS1; also known as PSEN1) or PS2 (also known as PSEN2) genes13. Because the age at symptom onset is similar across generations14, studies of asymptomatic mutation carriers allow for the estimation of the timing of biochemical, structural and functional abnormalities before symptoms occur.
Mild cognitive impairment.
(MCI). An intermediate stage between normal cognition and dementia; individuals with MCI usually experience amnesia and are at increased risk of developing AD.
Despite their differences, ADAD, EOAD and LOAD share a number of common features. The distribution of Aβ deposition throughout the brain is similar, affecting large confluent areas of association cortex overlapping with a set of brain regions active at rest. This network, known as the default-mode network (DMN), comprises the medial frontal and parietal cortex and the lateral temporal and parietal cortex15 (Fig. 1). Other intrinsic connectivity networks also show substantial Aβ deposits16 and, in general, Aβ seems to accumulate in parts of the association cortex that show high structural and functional connectivity, characterized as ‘hubs’ or a ‘rich club’17,18. All forms of AD also show a pattern of tau accumulation that differs from that of Aβ accumulation (discussed below), as well as a typical pattern of glucose hypometabolism that predominates in the temporal and parietal cortices and brain atrophy in these same regions along with the MTL (Fig. 2). The similarities and differences between the patterns of Aβ deposition, tau deposition, brain atrophy and hypometabolism hold important and poorly understood clues about the aetiology of AD.
Fig. 1 |. Patterns of brain amyloid-β deposition.
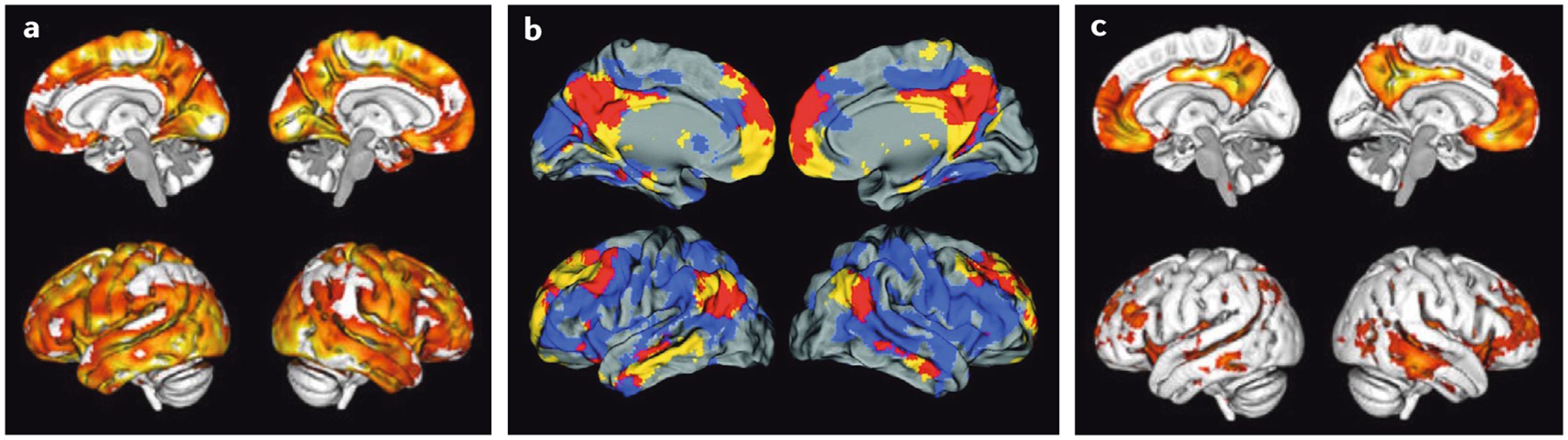
a | Brain map of amyloid-β (Aβ) accumulation, showing marked increases in Aβ (yellow denotes highest levels), as measured with the positron emission tomography (PET) tracer 18F-florbetapir in 191 amyloid-positive individuals without dementia compared with 218 amyloid-negative individuals. The brain regions susceptible to Aβ accumulation comprise large areas of the medial and lateral association cortex. b | Superimposition of the distribution of Aβ (in blue), as detected by 11C-Pittsburgh compound B- (PIB), onto the spatial location of the default mode network (DMN; in yellow) in the brains of cognitively healthy older people. The red brain regions reflect the spatial overlap between the DMN and Aβ accumulation. c | Brain map of Aβ accumulation in a group of 59 individuals without dementia who were defined as ‘early accumulators’ on the basis of evidence of abnormal cerebrospinal fluid levels of Aβ but normal 18F-florbetapir PET scans. The statistical maps show the areas of earliest accumulation of Aβ (yellow reflects areas of highest Aβ accumulation). Parts a and c are adapted from reF.77, Springer Nature Limited, CC By 4.0. Part b is adapted with permission from reF.96, Mormino, E. C. et al. Relationships between β-amyloid and functional connectivity in different components of the default mode network in aging. Cereb. Cortex (2011) 21(10), 2399–2407, by permission of Oxford University Press.
Fig. 2 |. Patterns of brain atrophy and glucose hypometabolism in Alzheimer disease.
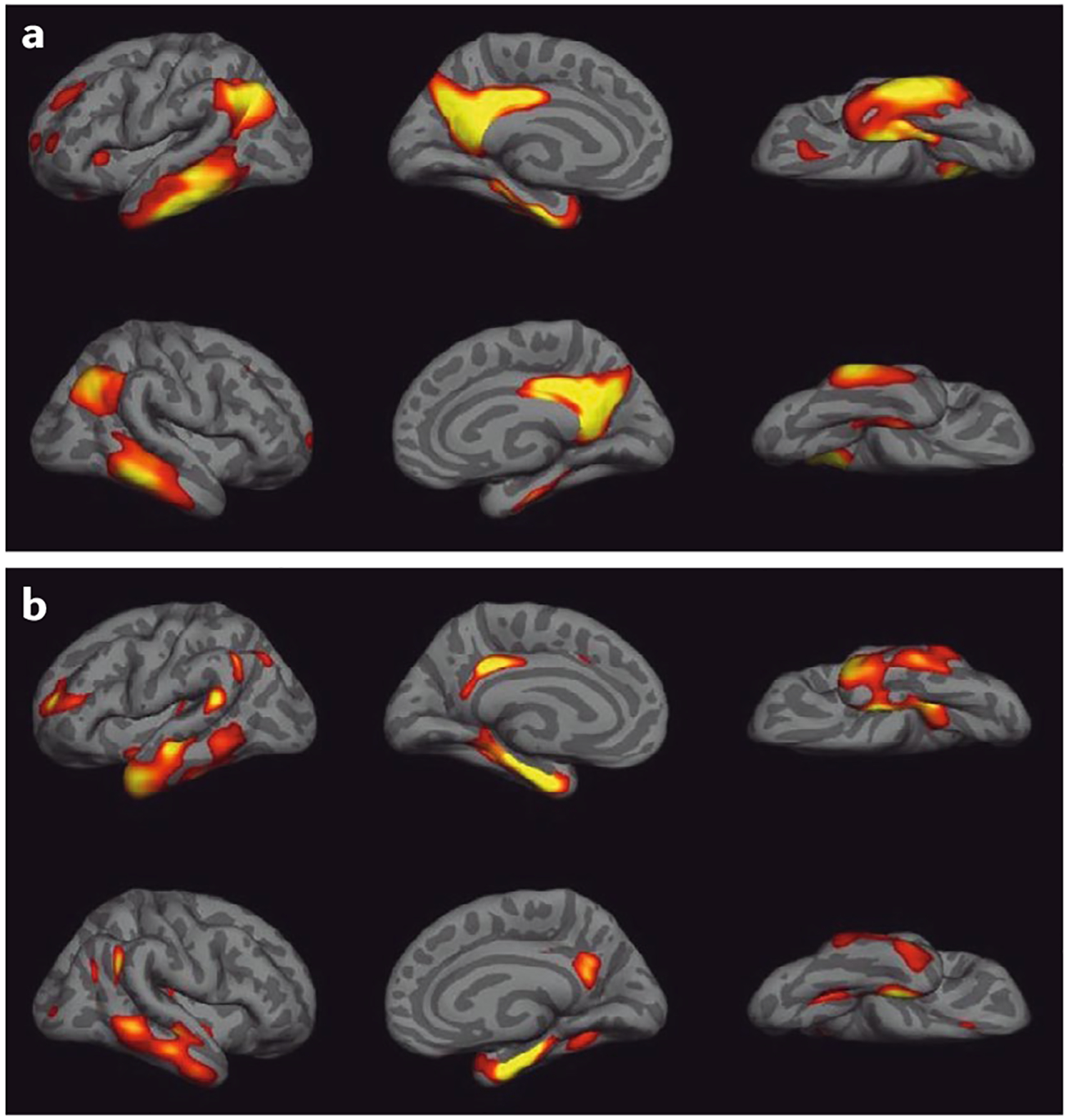
The maps were constructed by contrasting 18F-fluorodeoxyglucose-positron emission topography (FDG-PET) and MRI measures of cortical thickness for 50 individuals with Alzheimer disease (AD) and 39 cognitively healthy individuals from the Alzheimer’s Disease Neuroimaging Initiative. a | The map shows glucose hypometabolism in AD in the bilateral temporal and inferior parietal cortex, parts of the frontal cortex and the precuneus. b | Cortical thinning in AD occurs in comparable but somewhat smaller brain regions. Yellow regions indicate the areas of greatest hypometabolism or atrophy. Figure is adapted with permission from reF.207, republished with permission of Society for Neuroscience, from Alzheimer’s disease neurodegenerative biomarkers are associated with decreased cognitive function but not β-amyloid in cognitively normal older individuals, Wirth, M. et al., 33 (2013), permission conveyed through Copyright Clearance Center, Inc.
Autosomal dominant AD.
Because the longitudinal course of ADAD is so predictable, multiple investigators have used it as a model to investigate disease pathophysiology19–22. There is strong evidence from PET data in ADAD that brain Aβ deposition begins at least 15 years before expected disease onset21,22. Brain glucose metabolism declines later than the onset of Aβ deposition but still occurs at least 10 years before symptom onset, at about the same time that brain atrophy also appears23,24. Resting-state measures of functional connectivity show decreased connectivity of posterior nodes of the DMN about 12 years before symptoms25. The earliest stages of Aβ accumulation in ADAD sometimes show an unusual pattern affecting the striatum26,27. Longitudinal data show that initial Aβ deposition occurs in both the striatum and precuneus, and hypometabolism and atrophy also occur first in the precuneus28. By contrast, a recent report found that tau accumulated in the parahippocampal gyrus 6 years before expected symptom onset in cognitively unimpaired individuals carrying an AD-associated PS1 mutation and that more extensive tau accumulation in the MTL and the neocortex was more likely to occur later in the disease process in association with symptoms29. Hypometabolism and atrophy generally follow Aβ, but they are not in lockstep; for example, in the MTL, atrophy develops in the absence of Aβ deposition and hypometabolism, whereas the striatum, which often shows extensive Aβ deposition, does not show marked hypometabolism or atrophy until relatively late disease stages.
Amyloid cascade hypothesis.
A dominant hypothesis in the AD research field proposing that Aβ generation is the inciting event that leads to subsequent downstream processes of tau deposition and neurodegeneration, eventuating in dementia.
These data suggest that the first cortical abnormality in ADAD is Aβ deposition in the medial parietal lobe and that striatal Aβ accumulation is an equally early subcortical event. Aβ deposition appears to be followed by tau aggregation in the MTL. The spatiotemporal relationships between the timing and patterns of accumulation of these two proteins raise a profound question about the pathophysiology of AD, which is echoed in data from other aspects of the disorder.
Default-mode network.
(DMN). A canonical resting-state network of the brain that is active when individuals are not engaged in attending to or responding to external stimuli.
Early-onset AD.
Individuals with EOAD may present with striking neurobehavioural phenotypes reflecting damage to language systems30, visual systems31 or frontal-executive systems32. The focality and system-specific neurobehavioural features do not reflect regional accentuation of Aβ, but they do show strong correspondence to the pattern of glucose hypometabolism and atrophy33–36. Individuals with EOAD also show more severe temporoparietal hypometabolism than people with LOAD37,38, although the amount and pattern of Aβ deposition does not differ between these groups39. In contrast to imaging data for Aβ, the spatial pattern of tau distribution seen with PET imaging shows high correspondence with multiple disease features, reflecting symptom focality, greater severity with earlier onset and overlap with patterns of atrophy and glucose hypometabolism36,40–44. Thus, both the behavioural and neurodegenerative aspects of EOAD are explained better by the distribution and burden of tau than those of Aβ.
Hubs.
Brain regions (or nodes) that have many connections to other brain regions and serve as areas of convergence of information from multiple processing streams.
Late-onset AD.
As might be predicted from the EOAD data, individuals with LOAD show widespread deposition of Aβ that is weakly related to the severity of dementia symptoms45–47. Glucose hypometabolism reflects the temporoparietal regional predilection seen in patients with EOAD, although to a lesser extent, and the observed cortical atrophy largely recapitulates the hypometabolism in a pattern that has been referred to as an ‘AD signature’48,49 (Fig. 2). The evolution of brain atrophy from MCI to AD parallels the pattern of tau deposition from the medial temporal to lateral temporal and parietal cortices50. Deficits in cognition are better explained by changes in glucose metabolism and atrophy than by the deposition of Aβ51–55. Studies examining the relationships between Aβ and both glucose metabolism and atrophy have discovered associations ranging from absent to moderately strong47,56–60. Initial reports that AD is associated with DMN dysfunction61 have been widely confirmed and expanded, with evidence that Aβ alters connectivity in many large-scale brain networks62.
Rich club.
A group of brain regions (or nodes) that are highly connected to one another and that show a high degree of hub-like connectivity to many other brain regions.
Although the majority of available tau-PET imaging data are cross-sectional, they appear to be consistent with a pattern indicating a hierarchical evolution of tau deposition spreading from the MTL to the inferolateral temporal lobe and then to the medial and lateral parietal lobes in a distribution that largely recapitulates Braak neuropathological staging63 (Fig. 3); elevated tau is associated with both more Aβ deposition and worse cognitive function64–68. The topography of tau deposition in AD overlaps with brain regions that are particularly susceptible to atrophy and correlates with cortical thickness in these regions69; however, although the spatial distribution of tau appears to overlap with a number of intrinsic connectivity networks, it lacks the specificity for a single network and does not preferentially appear to involve the DMN70,71. Thus, the topography of tau accumulation overlaps with Aβ deposition but is not identical. Similar to the situation in EOAD, in LOAD, tau accumulation overlaps in topography and correlates with glucose hypometabolism — a relationship strengthened in the presence of Aβ72.
Fig. 3 |. Tau deposition in ageing and Alzheimer disease.
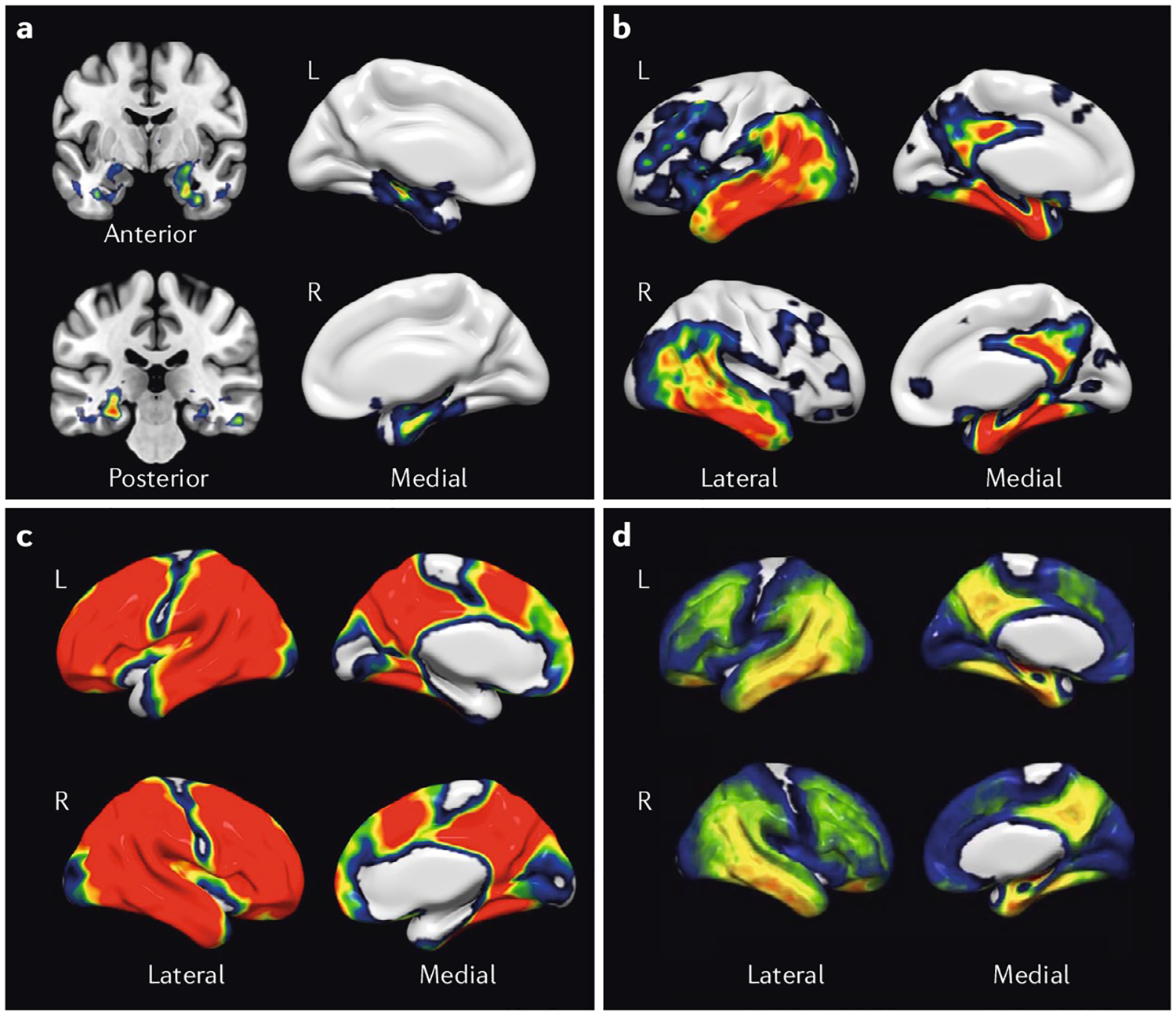
Using the positron emission topography (PET) radiotracer 18F-AV1451 (flortaucipir), a group of 216 individuals, including young and old cognitively healthy individuals and those with mild cognitive impairment and Alzheimer disease (AD), were staged with an adaptation of the Braak and Braak criteria63. Individuals were assigned to Braak stages I/II, III/IV or V/VI using PET imaging data as previously described101. a | The images show the contrast in tracer retention between those categorized as Braak stages I/II and those as stage 0, indicating that tau aggregation (yellow and red) begins in the medial temporal lobes. b | A contrast between stage III/IV and stage I/II indicates that subsequent progression of tau pathology is associated with tau aggregation in the inferolateral temporal and medial parietal lobes. c | A contrast between stage V/VI and III/IV indicates that the late stages of AD are characterized by widespread tau deposition (red). d | A map of all voxels in the brain over a threshold of 1.4 standard uptake value ratio (SUVR) units reveals the global distribution of tau in healthy individuals and those with advanced AD. L , left; R , right. Figure is adapted with permission from reF.208, Elsevier.
Braak neuropathological staging.
A widely adopted method of classification of tau pathology based on cross-sectional autopsy data that proposes a progression of tau neurofibrillary pathology from the MTL (Braak stages I/II) through a limbic stage (III/IV) to a diffuse neocortical stage (V/VI).
Normal ageing
Amyloid-β in the ageing brain.
Many cognitively healthy older people show substantial brain Aβ deposition that is similar in location and amount to that seen in AD. In such individuals, brain Aβ levels increase with age, with an overall rate of amyloid positivity of about 30% by age 80 (reF.73). Among these cognitively healthy older people, rates of amyloid positivity double in those carrying the apolipoprotein E (APOE) gene ε4 allele (APOE4)74, the major genetic risk factor for LOAD75. Similar to ADAD, in ageing, the earliest regions of Aβ deposition include the medial frontal and parietal cortices76,77 (Fig. 1). Although the extensive Aβ deposition seen in cognitively healthy older people appears to conflict with the amyloid cascade hypothesis, it is consistent with the weak or absent relationships seen between Aβ deposition and dementia symptoms in people with manifest AD.
Apolipoprotein E.
(APOE). A polymorphic gene with three alleles; the APOE ε4 allele is a risk factor for LOAD.
There are, however, weak relationships between Aβ deposition and cognition in ageing. The findings from many cross-sectional studies are inconsistent, but meta-analyses show a small reduction in cognitive function with more Aβ78,79. Longitudinal studies are more likely to show decline over time, but this decline is also quite small or even undetectable until about 4 years, when clinically relevant change may appear; this is probably for this reason that studies following participants for shorter periods have not found change80,81. Longitudinal cognitive decline is faster in those carrying APOE4 and in those with more Aβ deposition82,83.
Although cross-sectional data are conflicting, longitudinal data indicate small effects of Aβ deposition on brain atrophy in normal ageing84–87. The evidence that Aβ deposition reduces glucose metabolism in ageing is also mixed, probably because it is a small effect88, and it is complicated by the effects of the APOE4 genotype on metabolism, which are probably independent of those of Aβ89,90. Importantly, there is no relationship between the amount of Aβ deposition and the degree of hypometabolism in the same brain region91, raising the question as to whether an intermediary drives the association between Aβ deposition and metabolism. As described below, tau is again the likely candidate for this mediator. There are also reports that both healthy people and those with MCI show increased glucose metabolism with Aβ deposition that could reflect compensation as increased neural activity in response to Aβ-related brain injury or neural activity driving Aβ deposition92–94.
Aβ disrupts connectivity in the DMN, as well as other large-scale resting-state networks, and has been associated with loss of connectivity95 along with connec tivity increases in anterior components of the DMN96; one possibility is that connectivity patterns evolve over the course of the disease, first diminishing posteriorly and then increasing frontally97. Aβ deposition is also associated with changes in brain activation in the DMN and other components of the memory system during memory-encoding tasks, with evidence for increased activation, decreased activation and reduced deactivation98–100.
Tau in the ageing brain.
The deposition of tau in cognitively healthy older people revealed with imaging follows the staging pattern revealed through autopsy studies63, with the ubiquitous appearance of tau in the MTL (generally the entorhinal cortex typical of Braak stages I/II) and the frequent finding of tau in the inferolateral temporal cortex, consistent with Braak stages III/IV65,66,101. Postmortem data suggesting that tau pathology makes its earliest appearance in the locus coeruleus raise important questions about the onset of age-related tauopathy, but these questions cannot be answered by current PET imaging approaches102. Higher and more widespread tau accumulation is associated with Aβ deposition in cognitively healthy older individuals; even in people who are nominally amyloid negative, longitudinally increasing Aβ levels predict more tau deposition103,104. High levels of tau in widespread neocortical regions may not be compatible with normal cognition, but there is some evidence that some tau accumulation does occur in such regions with normal ageing, generally considered typical of Braak V/VI stages, usually in those with evidence of brain Aβ deposition but also to some extent in those without such deposition105. Longitudinal studies also indicate faster rates of accumulation in amyloid-positive than in amyloid-negative cognitively healthy people even in neocortical brain regions; the fastest rates are seen in cognitively impaired, amyloid-positive individuals106. The spatial relationships between Aβ and tau deposition are discordant; that is, more Aβ deposition, regardless of its location, is associated with greater accumulation of tau in the inferolateral temporal lobes107,108.
Suspected non-Alzheimer pathophysiology.
(SNAP). A descriptive term for evidence of neurodegeneration in the absence of biomarker evidence of Aβ.
In cognitively healthy older individuals, more tau deposition in the MTL is related to worse episodic memory performance and MTL atrophy over time109, and whole-brain measures of tau are related to temporoparietal atrophy both cross-sectionally and over time110. Similarly, tau accumulation in the entorhinal cortex is associated with a reduction in metabolism in the temporal lobe, and the tau accumulation in the inferior temporal lobe and MTL is associated with an AD-like pattern of hypometabolism when it is accompanied by Aβ deposition111,112. Tau accumulation also appears to result in loss of functional connectivity113 and increased neural activity during memory encoding114.
Primary age-related tauopathy.
(PART). An autopsy finding reflecting neurofibrillary tau pathology in the absence of Aβ pathology; fairly common in older people and with an unknown relationship to AD.
Neurodegeneration in the ageing brain.
Long before the advent of amyloid and tau imaging, brain atrophy and reductions in metabolism were identified as major aspects of brain ageing. There is some debate about the brain regions maximally affected by ageing-related atrophy, but both frontal and temporal lobes appear particularly susceptible to atrophy115,116. Glucose metabolism also declines with ageing in patterns similar to atrophy, but these PET measurements may be confounded by atrophy117. Most importantly, although LOAD most often occurs on a background of ageing, the patterns of atrophy and hypometabolism associated with AD are relatively distinct from those seen in ageing, and evidence of these AD-typical patterns confers an increased risk of cognitive decline over time in healthy older people118–121. More recent studies demonstrate that both hypometabolism and brain atrophy can occur independent of Aβ deposition122–124. This neurodegeneration in the absence of detectable Aβ has been referred to as suspected non-Alzheimer pathophysiology (SNAP)125. Although individuals with evidence of neurodegeneration but without Aβ deposition show risk of cognitive decline over time, the risk of such decline is higher in individuals with Aβ deposition and neurodegeneration125,126. The relationship between ‘amyloid-negative’ neurodegeneration and tau accumulation is currently unclear. Tau pathology in the absence of detectable Aβ is a well-known neuropathological entity that has been termed primary age-related tauopathy (PART)127. However, there is currently no evidence to suggest that neurodegeneration in the absence of Aβ is fully explained by PART128,129.
AD and ageing
As LOAD evolves in the setting of normal ageing, it is generally assumed that changes in the levels of Aβ, tau and neurodegeneration in cognitively healthy older people reflect the evolution of AD. However, because of the relative paucity of longitudinal data for changes in Aβ and tau deposition in the ageing brain, our knowledge of the risk of AD as informed by any biomarker or group of biomarkers is still imperfect. For example, it is certain that many cognitively unimpaired people with evidence of brain Aβ deposition will not live to express symptoms130. Nevertheless, measurement of aggregated proteins combined with longitudinal cognitive assessment provides both a useful model and a set of powerful tools to test ideas about the development of AD. In this regard, existing data suggest that the presence of Aβ increases the likelihood of subsequent cognitive decline in healthy older individuals80, although the lack of prospective data limits inferences about the role of tau in driving cognitive impairment. There is also a legitimate question as to how the less common forms of EOAD and ADAD are related to LOAD and normal ageing, from which they differ in many respects. It is useful in this situation to ask whether the imaging aspects of these three different forms of AD share commonalities and how they are related to pathological features seen in the ageing brain (TABLE 1).
Table 1 |.
Comparison of molecular and neurodegenerative pathologies in ageing and different Alzheimer disease syndromes
| Pathology | AD subtype or ageing | |||
|---|---|---|---|---|
| ADAD | EOAD | LOAD | Ageing | |
| Aβ pathology | Initial Aβ deposits form in the striatum and medial parietal lobe ~20 years before symptom onset22,28 |
|
|
|
| Tau pathology |
|
|
||
| Atrophy | The timing of atrophy onset is unknown, but the spatial location of atrophy is highly associated with tau deposition and symptoms36,40 | Typical ‘AD-signature’ pattern is frequently identified: atrophy predominates in the temporal and parietal cortex, including the MTL and medial parietal lobes48,49 |
|
|
| Hypometabolism | Hypometabolism begins in the medial parietal lobe (precuneus) ~18 years before symptom onset and spreads to the lateral parietal cortex23,28 | Similar to atrophy pattern, hypometabolism predominates in the temporoparietal cortex, particularly in the medial parietal lobe and MTL51 |
Aβ, amyloid-β; AD, Alzheimer disease; ADAD, autosomal dominant AD; EOAD, early-onset AD; LOAD, late-onset AD; MCI, mild cognitive impairment; MTL , medial temporal lobe.
There are important differences between ADAD and LOAD in terms of where pathologies, as determined by imaging, arise in the brain and how they are related to one another (TABLE 1). The early deposition of Aβ in the striatum is a striking feature of ADAD. However, both ADAD and LOAD are characterized by early onset of Aβ deposition in the medial parietal lobe, which is also the most susceptible site of hypometabolism and atrophy in both conditions28. The data on where tau deposition occurs in ADAD are limited, but they suggest that this process is similar to that in LOAD29. There are limited data on the earliest stages and presymptomatic evolution of EOAD for comparison with LOAD; however, focal neurodegeneration reflecting specific EOAD syndromes is detectable at the MCI stage131. In both EOAD and LOAD, there are strong relationships between tau deposition, neurodegeneration and cognition. Because LOAD arises in the setting of ageing, therefore almost invariably with MTL tau pathology, and usually presents with amnesia, it is reasonable to assume that MTL tau pathology is a substrate for the development of AD. The limited information about the evolution of EOAD makes it difficult to draw parallels with the evolution of LOAD; furthermore, the syndromic heterogeneity of EOAD phenotypes (language, visuospatial and executive dysfunction) is a barrier to defining a unitary theory of how the disorder develops. One possibility is that tau deposits early in brain regions that are uniquely susceptible in the EOAD syndromes (for example, in the left hemisphere in patients with early aphasia). Another possibility, based on evidence that all EOAD syndromes share functional disruption of the posterior DMN132, is that EOAD invariably arises from DMN pathology, which is also disrupted in LOAD; thus, EOAD and LOAD may in fact share a common network susceptibility. Why a common pathology affects the memory system in LOAD but other neural systems in EOAD is unknown.
In summary, ADAD, LOAD and EOAD all show similar distributions of Aβ and neurodegeneration (with the exception of Aβ deposition in the striatum in ADAD); however, the importance of tau in relation to neurodegeneration and symptoms has been established only in LOAD and EOAD. Major gaps in our knowledge concern how EOAD evolves over time, where and how tau deposits in ADAD and the relative importance of Aβ and tau pathologies in the transition from normal ageing to AD. Although it is unknown whether these three conditions reflect the same disease or a common response to different aetiologies, investigations with imaging approaches have revealed commonalities that support the likelihood of shared mechanisms, and each disorder opens a different window into AD pathogenesis.
Mechanisms and drivers of pathology
There is increasing information available on biological mechanisms that are associated with the protein aggregation and neurodegeneration of ageing and AD, particularly for both genetic and environmental factors in the aetiology of AD. However, the development of AD is complex, and many important aspects have not been explored or have been only touched upon using imaging approaches. For example, pathological studies have indicated that synapse loss is a major feature of AD133,134, but imaging of synapses has only recently been accomplished135. Similarly, changes in cholinergic function have received some attention with PET imaging but have largely been assumed to reflect secondary processes136. Although this Review focuses on Aβ and tau, we also know that other proteins such as α-synuclein and TDP43 aggregate in the brains of older people and those with dementia. The roles of these proteins have not yet been investigated during life because in vivo techniques for imaging them are not available; development of such new approaches is a high priority in the imaging field. Finally, because amyloid imaging was available years before the recent advent of tau imaging, disease mechanisms associated with tau are fairly unexplored, and most data reflect associations with Aβ.
Neural activity.
Preclinical data indicate that neural activity induces the extracellular release of Aβ through exocytosis137–139; indeed, manipulation of neural activity affects the regional localization of amyloid plaques in transgenic mouse models140. Neural activity can also lead to the extracellular release of tau141, enhancing its propagation and facilitating transcellular transfer142. These findings have different implications for the development of Aβ and tau pathologies.
Although the spatial overlap between the distribution of Aβ deposition and the DMN in particular is often cited, in reality, Aβ is found in regions where multiple networks converge and demonstrate hub-like qualities16,17,143 (Fig. 4). These regions demonstrate high degrees of connectivity with other brain regions, a characteristic that is energetically demanding144. Furthermore, the brain regions that are the most metabolically active in youth and that maintain this activity through adulthood show the greatest deposition of Aβ in ageing94. Reports showing that increased metabolism is associated with more Aβ are also consistent with a primary role for metabolism in driving Aβ deposition92,93. Brain regions showing Aβ deposition also demonstrate alterations in gene expression profiles for gene sets associated with mitochondrial respiration145. Higher metabolic activity could also be driven by uses of glucose in biosynthetic and non-oxidative pathways because maps of aerobic glycolysis correspond to the distribution of Aβ in the brain146.
Fig. 4 |. Relationships between canonical resting-state networks and amyloid-β deposition.
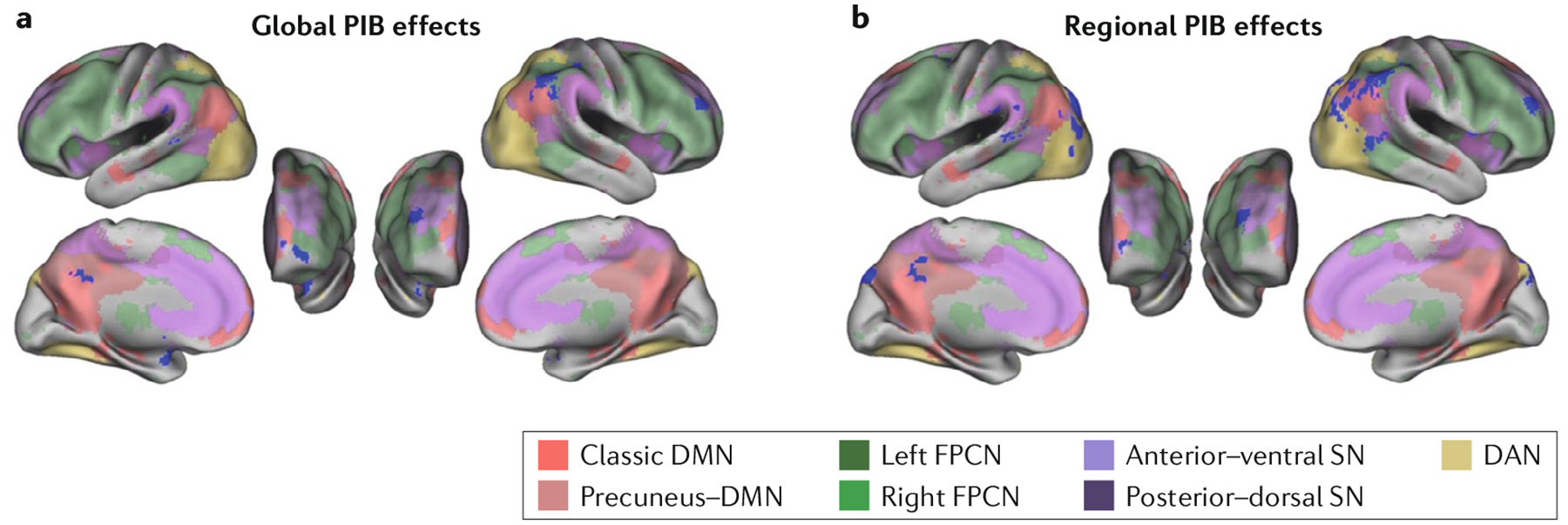
Brain maps show the topography of resting-state networks, defined using an independent component analysis with dual regression in a group of 92 cognitively healthy older individuals also imaged for brain amyloid-β (Aβ) with 11C-Pittsburgh compound B (PIB). Blue regions highlight where global brain Aβ deposition (part a) is associated with changes in resting network activity (either increases or decreases) or where local, or regional (part b), Aβ deposition is associated with network activity alterations. These regions are prominent in the default mode network (DMN) but are not isolated to this network: they occur in areas where networks converge that are also associated with high between-network and within-network connectivity. The affected regions are also similar to those affected by atrophy and hypometabolism (Fig. 2). The colours indicate the location of the networks. DAN, dorsal attention network; FPCN, frontoparietal control network; SN, salience network. Figure is reproduced with permission from reF.143, Elman, J. A. et al. Effects of beta-amyloid on resting state functional connectivity within and between networks reflect known patterns of regional vulnerability. Cereb. Cortex (2016) 26(2), 695–707 , by permission of Oxford University Press.
In addition to basal metabolism, Aβ deposition is associated with increased neural activity evoked by memory encoding in task-positive regions98 and reduced deactivation in task-negative regions99. Higher activation during memory encoding at baseline predicts greater subsequent deposition of Aβ over the next 4 years147. Most recently, cross-sectional data indicate associations between greater activation during memory encoding and more tau deposition in the MTL, where both tau and activation may help drive atrophy and cognitive decline114. Low levels of MTL tau pathology are associated with increased functional connectivity in cognitively healthy amyloid-positive individuals, whereas high levels of tau are associated with decreased connectivity, a finding that could explain the evolution of connectivity alterations during the progression of disease148. Administration of the anti-epileptic drug levetiracetam to individuals with MCI reduced this presumably excess activation and improved memory performance149. Taken together, these data support a role for increased neural activity in initiating and sustaining Aβ accumulation.
Networks as facilitators of pathological spread.
Patterns of brain atrophy in different neurodegenerative disorders correspond to distinct intrinsic connectivity networks, suggesting that specific neural systems are differentially vulnerable in these diseases150,151. The spatial relationships between canonical functional networks and neurodegenerative pathology converge with the preclinical data supporting the trans-synaptic spread and aggregation of proteins152. Network models using graph theoretical frameworks support the idea that neurodegeneration proceeds through epicentres and that strength of connectivity to these epicentres predicts the regional localization of atrophy153. Within EOAD subgroups, for example, patterns of regional atrophy parallel distinct networks in healthy individuals corresponding to language and visuospatial systems that converge in a common node in the posterior DMN–precuneus132. These networks also overlap with the syndrome-specific regional deposition of tau in EOAD. Evidence for the Aβ-facilitated spread of tau from the entorhinal cortex to the posterior cingulate via the cingulum bundle in humans is also an example of how a specific neural system, in this case one subserving memory, can be affected by a pathological protein154. Thus, large-scale networks appear to underlie patterns of tau and neurodegenerative pathology.
Network disruption may also play an aetiological role in disease pathology. In this model, the early failure of network components that are marked by reduced connectivity (the posterior DMN) is followed by increased connectivity in anterior components of the DMN; this increased connectivity, whether compensatory or not, drives further disease pathology97. Indeed, two studies have reported that cognitively healthy carriers of the APOE4 allele show signs of network dysfunction before amyloid deposition155,156, which is consistent with the idea that network failure itself may be a driving force behind Aβ aggregation. In this respect, this idea echoes theories related to regional metabolism-based vulnerability and neural activity.
Apolipoprotein E.
The APOE polymorphism affects the risk of AD in all its syndromic forms10. Carriage of the APOE4 allele results in increased brain Aβ deposition in normal ageing, as well as deposition at earlier ages74. Investigations of the effect of APOE4 on Aβ accumulation rates have been inconsistent157,158. Recent longitudinal data suggest that the APOE4 leads to faster accumulation of Aβ early in the pathophysiology of the disease in those who are nominally amyloid negative159, but there is also evidence that although APOE4 carriers deposit Aβ early (in their 50s in some brain regions), those with higher baseline levels of Aβ deposit Aβ more rapidly160. These data are consistent with preclinical data that APOE4 is crucial in seeding Aβ aggregation161. Other preclinical data indicate that the APOE4 allele promotes pathological tau accumulation and tau-related neurodegeneration162. These findings could explain how APOE is related to Aβ in producing cognitive decline82,163 and perhaps even how APOE4 could drive particular imaging phenotypes with accentuated MTL atrophy164. Similarly, these relationships could also be explained by presumably lifelong and Aβ-independent effects of APOE4 on brain structure, function and metabolism89,155,165.
Cerebrovascular disease.
There is extensive evidence that cerebrovascular disease can play an important role in the development of dementia symptoms, but its relationship to molecular processes is complex. Neuropathological studies have been instrumental in noting that dementia, especially in older people, frequently involves cerebral infarction in addition to plaque and tangle pathology; the additive nature of the association suggests that vascular insults lower the threshold for the expression of dementia symptoms but do not affect the protein aggregates themselves12,166. MRI is the most useful imaging tool for the investigation of cerebrovascular disease in vivo and has been used to quantify cerebral infarction, changes in white matter (denoted as white matter hyperintensities on T2 images) and microhaemorrhages. Not surprisingly, MRI evidence of cerebrovascular pathology is related to diminished cognitive function167,168. However, there is limited evidence for a link between Aβ and imaging measurements of vascular pathology, the exception being that individuals with cerebral amyloid angiopathy appear to have more white matter hyperintensities169. The prevailing imaging evidence supports a role whereby cerebrovascular brain injury and Aβ pathology are independent factors that combine to increase the likelihood of dementia170–172.
Relationships between cerebrovascular risk factors and Aβ are more complex. Several studies have shown associations between cardiovascular risk factors and brain Aβ deposition in cognitively healthy older people. These factors include elevated blood pressure, body mass index, smoking, hypercholesterolemia and diabetes in isolation and in various combinations173–176. Some of these factors have also been found to be associated with neurodegeneration but not Aβ deposition177, with some evidence for an association between vascular health and entorhinal cortical tau deposition seen on PET178. There is also extensive evidence that vascular risk factors, even in midlife, are associated with later-life brain atrophy, which in turn is associated with cognitive decline179,180.
Together, the findings support a role for cerebrovascular disease in leading to dementia. This role likely occurs because cerebrovascular disease results in tissue loss and changes in white matter that lower the threshold for cognitive dysfunction related to other pathological processes such as protein aggregation. However, there are also interesting associations between vascular risk and both Aβ deposition and brain atrophy that link vascular disease to molecular mechanisms associated with AD. These relationships require further study.
Environmental factors and lifestyle.
A wide range of lifestyle and environmental factors that are associated with many health outcomes are also associated with effects on the AD pathophysiological process. Importantly, there are extensive data linking resilience to AD pathology to lifestyle factors, invoking a process of cognitive reserve in compensating for AD pathology181. Although reserve and resilience are crucial concepts in understanding why individuals with similar amounts of disease pathology will express different levels of cognitive dysfunction, this section reviews evidence for effects on the pathological process itself.
Engagement in both cognitive and physical activity has been found to reduce brain Aβ levels. Cognitively healthy individuals who indicate greater levels of both past and current physical activity show lower levels of brain Aβ, an effect that is strongest in APOE4 carriers182,183. Evidence exists for a similar effect regarding cognitive activity, with individuals — particularly those carrying APOE4 — indicating higher lifelong cognitive engagement showing a reduction in brain Aβ levels184–186. Although these effects of both physical and cognitive activity converge with epidemiological associations with dementia risk, the mechanisms underlying a direct effect of these factors on Aβ are unclear.
There is a growing body of evidence that altered sleep contributes to pathological changes in AD. Cognitively healthy individuals reporting less or poorer sleep quality demonstrate increased brain Aβ deposition with PET imaging187,188; the early appearance of this association suggests it is not an epiphenomenon of late-stage dementia. Polysomnography shows that these associations are frequency specific in their relationship to slow-wave activity <1 Hz and topographically-specific, being associated with Aβ deposition in the medial prefrontal cortex, a site generating slow-wave activity189. These associations may reflect either a failure to adequately clear Aβ through the glymphatic system during sleep190 or higher levels of exposure to neural activity and oxidative stress. The observations that decreased sleep may reduce Aβ clearance and that increased medial prefrontal cortical Aβ may drive diminished slow-wave activity suggest the possibility of a vicious cycle in the relationship between sleep and Aβ.
Cognitive reserve.
A hypothetical construct proposing differences in individual susceptibility to account for why people with similar levels of disease pathology show different levels of cognitive ability.
Neuroinflammation.
Extensive data from basic cellular neuroscience and human genetics implicate inflammation as a key event in the pathogenesis of AD191–193. Investigation of neuroinflammation is amenable to in vivo study using various PET ligands that principally target the peripheral benzodiazepine receptor (PBR) by binding to the translocator protein (TSPO) on the outer mitochondrial membrane. PBR–TSPO imaging is useful because of increased expression of the TSPO protein in activated immune cells of the CNS, particularly micro-glia. Current TSPO ligands suffer from several shortcomings but nevertheless have been utilized in studies exploring the presence and time course of neuroinflammation in conjunction with other biomarkers of AD. The data obtained from such studies have been reasonably consistent in showing evidence of neuroinflammation in patients with AD but have been more inconsistent in showing relationships between neuroinflammation and other biomarkers and clinical syndromes, including MCI194–196. These studies have therefore been unable to explain the precise role of inflammation in the pathophysiology of AD and particularly the temporal relationship between inflammation, protein aggregates and cerebrovascular disease. Although it seems likely that PET imaging across the AD spectrum may ultimately help to reveal the role of neuroinflammation in the disorder, more definitive studies will probably require a new generation of PET ligands.
Does a model fit the data?
Is Aβ the start of a cascade of events leading to tau deposition, neurodegeneration and, eventually, dementia7? Do the relationships between Aβ, tau deposition, neurodegeneration and cognition support a multistage pathophysiological chain of events (Fig. 5)?
Fig. 5 |. Proposed relationships between pathological protein accumulation, neurodegeneration and drivers of the Alzheimer disease process.
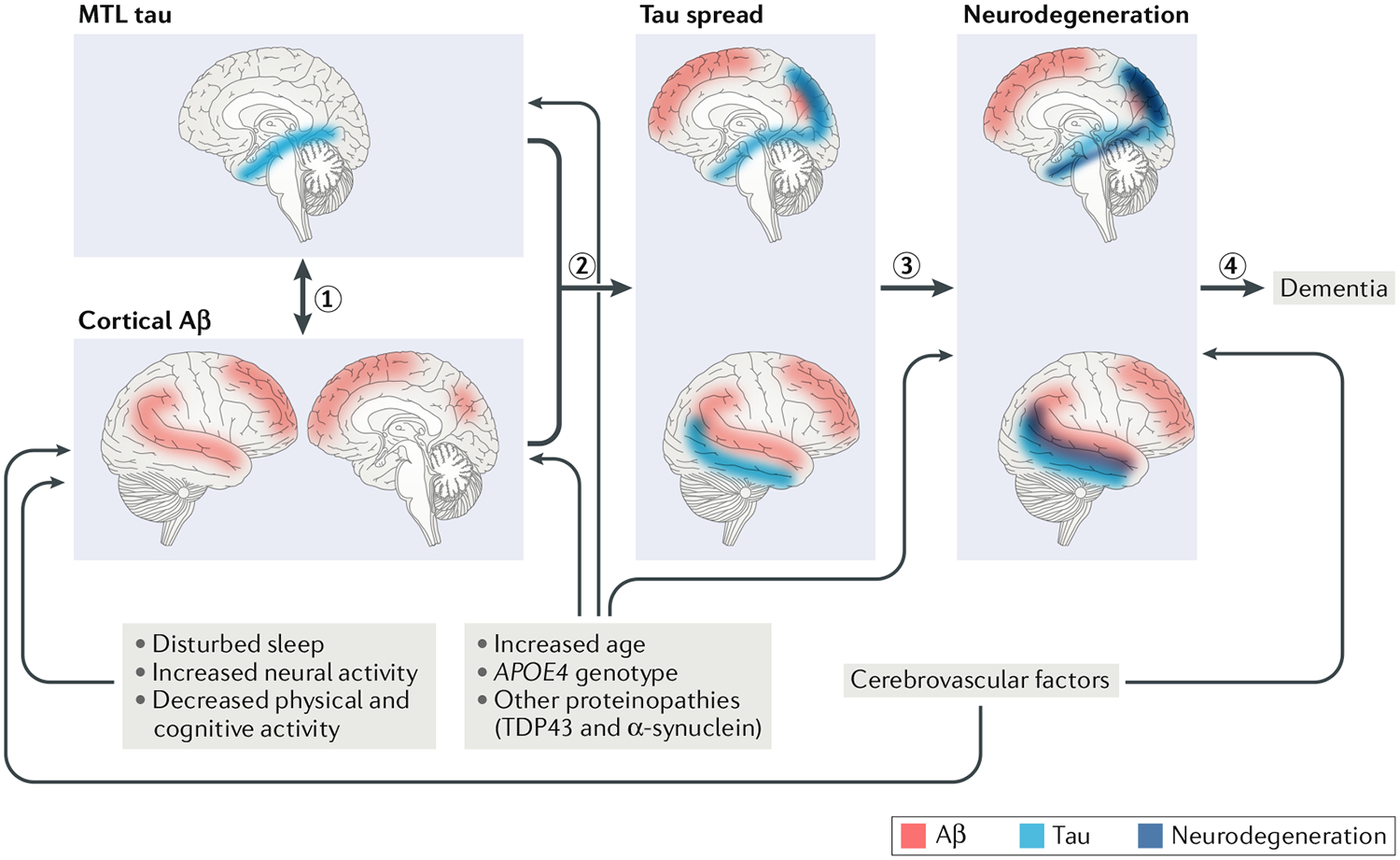
The initial stages of Alzheimer disease (AD) development reflect relationships between cortical amyloid-β (Aβ; red) and tau (blue) in the medial temporal lobe (MTL) (1). This process appears to begin with Aβ deposition in the cortex but could have a bidirectional nature as accumulation of MTL tau, which may or may not reflect AD, usually precedes cortical Aβ in cognitively healthy older people. The relationship between these two proteins is associated with spread of tau out of the MTL into the medial parietal, lateral parietal and temporal cortices (2). This tau spread is associated with neurodegeneration (dark blue) in a similar topography to tau deposition (3), which in turn is related to cognitive decline and, eventually , dementia (4). Drivers of Aβ include various processes and factors. Disturbed or diminished sleep, increased neural activity , reduced physical and cognitive activity , cerebrovascular disease, old age, the apolipoprotein E gene ε4 allele (APOE4) and other proteinopathies have been linked to Aβ deposition. Some of these associations have not been described for tau pathology , but this could reflect the relative novelty of in vivo tau imaging. Old age, APOE4 and other proteinopathies appear to be associated with tau deposition in the MTL. Neurodegeneration is also associated with these factors and with cerebrovascular disease. Increased age, APOE4, other proteinopathies and cerebrovascular disease may also affect neurodegeneration independent of their effects on tau or Aβ. TDP43, TAR DNA-binding protein 43.
Spatiotemporal associations and the beginning of AD.
Strong relationships between tau deposition, neurodegeneration and cognition are apparent in asymptomatic older people and in those with LOAD and EOAD. These data support an important role for tau in driving adverse events, but what is the role of Aβ in this process? The relationships between Aβ and both neurodegeneration and cognition are weak, but the relationships between Aβ and tau offer an explanation. There are strong cross-sectional and longitudinal associations between Aβ and tau, especially in the observation that very low, but rising Aβ levels are associated with tau deposition over years103,104. Aβ deposition facilitates the accumulation of tau in the posterior cingulate cortex via connections through the cingulum bundle154. Facilitation of tau spread into the medial parietal cortex by Aβ would explain why this region seems to be selectively vulnerable to hypometabolism and atrophy and defines relationships between Aβ and tau in this process. Associations between tau accumulation and both cortical atrophy and hypometabolism also seem more malignant in the presence of Aβ69,72,111,112. The observation that the rate of Aβ accumulation appears to slow as it reaches higher levels104,197,198 suggests that neurodegeneration and cognitive decline could eventually become uncoupled from Aβ levels but still be dependent on tau.
Thus, there is a compelling story that Aβ facilitates both the spread and pathogenicity of tau throughout the brain, which then drives neurodegeneration and dementia. This story seems to place Aβ at the start of a pathological cascade. However, in cognitively healthy older people, MTL tau accumulation precedes diffuse neocortical Aβ deposition. Does Aβ deposition result in the spread of this tau, or could this tau in the MTL drive neocortical Aβ buildup?
The findings in ADAD strongly implicate medial parietal cortical Aβ deposition as the primary measurable biochemical event in the development of AD28, which is supported by data from cognitively healthy older people76,77. Although MTL tau deposition may be an earlier event, Aβ deposition seems to increase MTL tau levels in cognitively healthy older people109 and in ADAD29. Thus, although it is appealing to consider age-related MTL tau accumulation as a substrate for Aβ-facilitated tau spread, the corollary of this event in EOAD is unknown, and it will be crucial to develop a better understanding of how tau deposition evolves in this form of AD. It is important to remember in this setting that PET can detect only aggregated fibrillar forms of Aβ; therefore, soluble or intracellular forms could play a role in this early tau deposition. At this time, a major unresolved question is how tau and Aβ interact — this is poorly understood from both the human imaging and molecular and cellular perspectives. Does isolated MTL tau accumulation represent a non-AD pathology (that is, PART) that may be exacerbated by Aβ build-up, or is it in fact the beginning of AD that in some way leads to Aβ deposition199? The data from ADAD indicate that Aβ pathology leads to tau deposition, but the situation in other forms of AD and ageing is unresolved. It is also possible that the relationships between these two molecules differ at different diseases stages, such that Aβ could initiate a transformation in tau, which then affects Aβ aggregation.
Spatiotemporal discrepancies.
The poorly understood relationships between Aβ and tau are reflected in the discordance between localization of these two proteins and their presumed downstream consequences. In normal ageing and all forms of AD, Aβ is diffusely present in the prefrontal cortex, but this region shows fairly little tau pathology, atrophy and hypometabolism; conversely, in ageing and LOAD, tau accumulation and atrophy are prominent in the MTL, a brain region with little Aβ200. Comparisons between EOAD and LOAD are particularly puzzling because, although both disorders show a generalized pattern of Aβ distribution, tau seems to accumulate preferentially in the memory system in ageing and LOAD but predominates in a number of other neural systems in EOAD. Even within EOAD, different syndromes have different distributions of tau despite similar distributions of Aβ. One possibility is that Aβ and tau accumulation develop independently and subsequently interact. It is also possible that Aβ effects occur at the synapse and, through an unknown process of retrograde signalling, alter tau residing in the soma, a hypothesis consistent with the spatially remote correlations between the two proteins108. The effects of Aβ may also be entirely related to soluble or intracellular forms undetectable by PET but lead to tau aggregation in specific patterns. Longitudinal studies of these pathological processes may help explain these confusing relationships.
Another important discrepancy is SNAP, which by definition is not related to Aβ and does not appear to be explained by tau. Cerebrovascular disease and other proteinopathies such as that associated with TDP43 could be drivers of brain atrophy in this situation11,179 so that multiple pathological processes may converge on neuro-degeneration. There is also evidence that different proteins may interact to induce aggregation; for example, α-synuclein may induce tau aggregation201. Furthermore, just as Aβ could remotely affect tau aggregation, pathological proteins in the MTL (including tau) can produce medial parietal hypometabolism in an AD-like pattern112,202. Neurodegeneration is not likely to reflect a single amyloid-dependent or tau-dependent process but rather involves multiple pathophysiological pathways that will eventually need to be incorporated into our understanding of AD pathogenesis.
Conclusions
In this Review, I have attempted to unify the sometimes conflicting and confusing data relating protein deposition to what have been termed ‘downstream’ processes in AD. Most of the conflicting data do not seem to contradict an underlying sequential model for this disease (Fig. 5), but they do raise gaps in our knowledge and indicate the likely importance of additional non-amyloid or tau-related disease pathways. Molecular mechanisms for many of these events are either poorly understood or entirely missing. The lack of large longitudinal data sets — with all of these processes measured over time — limits causal inferences, although these crucial data will probably begin to appear over the next few years. Associational studies also raise the possibility that unmeasured variables could play important or causal roles in driving the pathological processes we can measure. Most models of complex diseases are incorrect, and the current model of AD pathogenesis is probably no exception; the question is whether it provides reasonable therapeutic targets. Because the most persuasive test of causal association requires experimental intervention, the best experiment will be the removal of one or more of these putatively inciting proteins with the subsequent amelioration of neurodegeneration and cognitive decline. This experiment would be a positive clinical trial of an Aβ or tau-modifying therapy — something that the entire field awaits with anticipation and hope.
Acknowledgements
Research described in this article was supported in part by US National Institutes of Health grants AG034570, AG045611 and AG019724. The author is indebted to R. La Joie, S. Landau, A. Maass and G. Rabinovici for their thoughtful comments.
Footnotes
Competing interests
The author serves as a consultant to BioClinica, Novartis and Genentech.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Reviewer information
Nature Reviews Neuroscience thanks K. Josephs, P. Matthews and M. Rossor for their contribution to the peer review of this work.
References
- 1.Small SA, Schobel SA, Buxton RB, Witter MP & Barnes CA A pathophysiological framework of hippocampal dysfunction in ageing and disease. Nat. Rev. Neurosci 12, 585–601 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nelson PT et al. Correlation of Alzheimer disease neuropathologic changes with cognitive status: a review of the literature. J. Neuropathol. Exp. Neurol 71, 362–381 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ittner LM & Gotz J Amyloid-β and tau — a toxic pas de deux in Alzheimer’s disease. Nat. Rev. Neurosci 12, 65–72 (2011). [DOI] [PubMed] [Google Scholar]
- 4.Hyman BT et al. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimers Dement. 8, 1–13 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Price JL & Morris JC Tangles and plaques in nondemented aging and “preclinical” Alzheimer’s disease. Ann. Neurol 45, 358–368 (1999). [DOI] [PubMed] [Google Scholar]
- 6.Bennett DA et al. Neuropathology of older persons without cognitive impairment from two community-based studies. Neurology 66, 1837–1844 (2006). [DOI] [PubMed] [Google Scholar]
- 7.Jack CR Jr. et al. Tracking pathophysiological processes in Alzheimer’s disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol. 12, 207–216 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper proposes a model of the pathophysiology of AD that has been influential in guiding and interpreting human studies.
- 8.Hardy J & Selkoe DJ The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297, 353–356 (2002). [DOI] [PubMed] [Google Scholar]
- 9.Wingo TS, Lah JJ, Levey AI & Cutler DJ Autosomal recessive causes likely in early-onset Alzheimer disease. Arch. Neurol 69, 59–64 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cacace R, Sleegers K & Van Broeckhoven C Molecular genetics of early-onset Alzheimer’s disease revisited. Alzheimers Dement. 12, 733–748 (2016). [DOI] [PubMed] [Google Scholar]
- 11.Josephs KA et al. Rates of hippocampal atrophy and presence of post-mortem TDP-43 in patients with Alzheimer’s disease: a longitudinal retrospective study. Lancet Neurol. 16, 917–924 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schneider JA, Arvanitakis Z, Leurgans SE & Bennett DA The neuropathology of probable Alzheimer disease and mild cognitive impairment. Ann. Neurol 66, 200–208 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bertram L, Lill CM & Tanzi RE The genetics of Alzheimer disease: back to the future. Neuron 68, 270–281 (2010). [DOI] [PubMed] [Google Scholar]
- 14.Lopera F et al. Clinical features of early-onset Alzheimer disease in a large kindred with an E280A presenilin-1 mutation. JAMA 277, 793–799 (1997). [PubMed] [Google Scholar]
- 15.Buckner RL et al. Molecular, structural, and functional characterization of Alzheimer’s disease: evidence for a relationship between default activity, amyloid, and memory. J. Neurosci 25, 7709–7717 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grothe MJ, Teipel SJ & Alzheimer’s Disease Neuroimaging Initiative. Spatial patterns of atrophy, hypometabolism, and amyloid deposition in Alzheimer’s disease correspond to dissociable functional brain networks. Hum. Brain Mapp 37, 35–53 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Buckner RL et al. Cortical hubs revealed by intrinsic functional connectivity: mapping, assessment of stability, and relation to Alzheimer’s disease. J. Neurosci 29, 1860–1873 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Daianu M et al. Rich club analysis in the Alzheimer’s disease connectome reveals a relatively undisturbed structural core network. Hum. Brain Mapp 36, 3087–3103 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fox NC et al. Presymptomatic hippocampal atrophy in Alzheimer’s disease. A longitudinal MRI study. Brain 119, 2001–2007 (1996). [DOI] [PubMed] [Google Scholar]
- 20.Scholl M et al. Low PiB PET retention in presence of pathologic CSF biomarkers in Arctic APP mutation carriers. Neurology 79, 229–236 (2012). [DOI] [PubMed] [Google Scholar]
- 21.Bateman RJ et al. Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N. Engl. J. Med 367, 795–804 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]; This article is an examination of the sequence of pathophysiological events in the presymptomatic phase of ADAD.
- 22.Fleisher AS et al. Florbetapir PET analysis of amyloid-β deposition in the presenilin 1 E280A autosomal dominant Alzheimer’s disease kindred: a cross-sectional study. Lancet Neurol. 11, 1057–1065 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Benzinger TL et al. Regional variability of imaging biomarkers in autosomal dominant Alzheimer’s disease. Proc. Natl Acad. Sci. USA 110, E4502–E4509 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fleisher AS et al. Associations between biomarkers and age in the presenilin 1 E280A autosomal dominant Alzheimer disease kindred: a cross-sectional study. JAMA Neurol. 72, 316–324 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chhatwal JP et al. Impaired default network functional connectivity in autosomal dominant Alzheimer disease. Neurology 81, 736–744 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Klunk WE et al. Amyloid deposition begins in the striatum of presenilin-1 mutation carriers from two unrelated pedigrees. J. Neurosci 27, 6174–6184 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Villemagne VL et al. High striatal amyloid β-peptide deposition across different autosomal Alzheimer disease mutation types. Arch. Neurol 66, 1537–1544 (2009). [DOI] [PubMed] [Google Scholar]
- 28.Gordon BA et al. Spatial patterns of neuroimaging biomarker change in individuals from families with autosomal dominant Alzheimer’s disease: a longitudinal study. Lancet Neurol. 17, 241–250 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Quiroz YT et al. Association between amyloid and tau accumulation in young adults with autosomal dominant Alzheimer disease. JAMA Neurol. 75, 548–556 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gorno-Tempini ML et al. Classification of primary progressive aphasia and its variants. Neurology 76, 1006–1014 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mendez MF, Ghajarania M & Perryman KM Posterior cortical atrophy: clinical characteristics and differences compared to Alzheimer’s disease. Dement. Geriatr. Cogn. Disord 14, 33–40 (2002). [DOI] [PubMed] [Google Scholar]
- 32.Ossenkoppele R et al. The behavioural/dysexecutive variant of Alzheimer’s disease: clinical, neuroimaging and pathological features. Brain 138, 2732–2749 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rabinovici GD et al. Aβ amyloid and glucose metabolism in three variants of primary progressive aphasia. Ann. Neurol 64, 388–401 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.de Souza LC et al. Similar amyloid-β burden in posterior cortical atrophy and Alzheimer’s disease. Brain 134, 2036–2043 (2011). [DOI] [PubMed] [Google Scholar]
- 35.Lehmann M et al. Diverging patterns of amyloid deposition and hypometabolism in clinical variants of probable Alzheimer’s disease. Brain 136, 844–858 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ossenkoppele R et al. Tau PET patterns mirror clinical and neuroanatomical variability in Alzheimer’s disease. Brain 139, 1551–1567 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]; This report shows the strong relationships between tau, glucose metabolism and cognitive phenotype in EOAD.
- 37.Mielke R, Herholz K, Grond M, Kessler J & Heiss WD Differences of regional cerebral glucose metabolism between presenile and senile dementia of Alzheimer type. Neurobiol. Aging 13, 93–98 (1992). [DOI] [PubMed] [Google Scholar]
- 38.Kim EJ et al. Glucose metabolism in early onset versus late onset Alzheimer’s disease: an SPM analysis of 120 patients. Brain 128, 1790–1801 (2005). [DOI] [PubMed] [Google Scholar]
- 39.Rabinovici GD et al. Increased metabolic vulnerability in early-onset Alzheimer’s disease is not related to amyloid burden. Brain 133, 512–528 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xia C et al. Association of in vivo [18F]AV-1451 tau PET imaging results with cortical atrophy and symptoms in typical and atypical Alzheimer disease. JAMA Neurol. 74, 427–436 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schöll M et al. Distinct 18F-AV-1451 tau PET retention patterns in early- and late-onset Alzheimer’s disease. Brain 140, 2286–2294 (2017). [DOI] [PubMed] [Google Scholar]
- 42.Bejanin A et al. Tau pathology and neurodegeneration contribute to cognitive impairment in Alzheimer’s disease. Brain 140, 3286–3300 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Iaccarino L et al. Local and distant relationships between amyloid, tau and neurodegeneration in Alzheimer’s disease. Neuroimage Clin. 17, 452–464 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Whitwell JL et al. [18F]AV-1451 clustering of entorhinal and cortical uptake in Alzheimer’s disease. Ann. Neurol 83, 248–257 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Klunk WE et al. Imaging brain amyloid in Alzheimer’s disease with Pittsburgh compound-B. Ann. Neurol 55, 306–319 (2004). [DOI] [PubMed] [Google Scholar]; This article presents the initial report of the use of amyloid imaging in the study of ageing and dementia.
- 46.Rowe CC et al. Imaging β-amyloid burden in aging and dementia. Neurology 68, 1718–1725 (2007). [DOI] [PubMed] [Google Scholar]
- 47.Furst AJ et al. Cognition, glucose metabolism and amyloid burden in Alzheimer’s disease. Neurobiol. Aging 33, 215–225 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Singh V et al. Spatial patterns of cortical thinning in mild cognitive impairment and Alzheimer’s disease. Brain 129, 2885–2893 (2006). [DOI] [PubMed] [Google Scholar]
- 49.Dickerson BC et al. The cortical signature of Alzheimer’s disease: regionally specific cortical thinning relates to symptom severity in very mild to mild AD dementia and is detectable in asymptomatic amyloid-positive individuals. Cereb. Cortex 19, 497–510 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper reports on the use of MRI to identify a characteristic pattern of regional brain atrophy associated with AD and in asymptomatic older people.
- 50.Whitwell JL et al. 3D maps from multiple MRI illustrate changing atrophy patterns as subjects progress from mild cognitive impairment to Alzheimer’s disease. Brain 130, 1777–1786 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Silverman DH et al. Positron emission tomography in evaluation of dementia: regional brain metabolism and long-term outcome. JAMA 286, 2120–2127 (2001). [DOI] [PubMed] [Google Scholar]
- 52.Fouquet M et al. Longitudinal brain metabolic changes from amnestic mild cognitive impairment to Alzheimer’s disease. Brain 132, 2058–2067 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Landau SM et al. Associations between cognitive, functional, and FDG-PET measures of decline in AD and MCI. Neurobiol. Aging 32, 1207–1218 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.De Santi S et al. Hippocampal formation glucose metabolism and volume losses in MCI and AD. Neurobiol. Aging 22, 529–539 (2001). [DOI] [PubMed] [Google Scholar]
- 55.Jack CR Jr. et al. Comparison of different MRI brain atrophy rate measures with clinical disease progression in AD. Neurology 62, 591–600 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jack CR Jr. et al. Brain β-amyloid measures and magnetic resonance imaging atrophy both predict time-to-progression from mild cognitive impairment to Alzheimer’s disease. Brain 133, 3336–3348 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chetelat G et al. Relationship between atrophy and β-amyloid deposition in Alzheimer disease. Ann. Neurol 67, 317–324 (2010). [DOI] [PubMed] [Google Scholar]
- 58.Ewers M et al. CSF biomarker and PIB-PET-derived β-amyloid signature predicts metabolic, gray matter, and cognitive changes in nondemented subjects. Cereb. Cortex 22, 1993–2004 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tosun D et al. Spatial patterns of brain amyloid-β burden and atrophy rate associations in mild cognitive impairment. Brain 134, 1077–1088 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Archer HA et al. Amyloid load and cerebral atrophy in Alzheimer’s disease: an 11C-PIB positron emission tomography study. Ann. Neurol 60, 145–147 (2006). [DOI] [PubMed] [Google Scholar]
- 61.Greicius MD, Srivastava G, Reiss AL & Menon V Default-mode network activity distinguishes Alzheimer’s disease from healthy aging: evidence from functional MRI. Proc. Natl Acad. Sci. USA 101, 4637–4642 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper presents the initial report showing the effects of AD on resting-state connectivity in the DMN.
- 62.Badhwar A et al. Resting-state network dysfunction in Alzheimer’s disease: a systematic review and meta-analysis. Alzheimers Dement. 8, 73–85 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Braak H & Braak E Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 82, 239–259 (1991). [DOI] [PubMed] [Google Scholar]; This landmark report describes the results of autopsy studies of AD pathology that proposed a widely utilized staging scheme for tau pathology.
- 64.Schwarz AJ et al. Regional profiles of the candidate tau PET ligand 18F-AV-1451 recapitulate key features of Braak histopathological stages. Brain 139, 1539–1550 (2016). [DOI] [PubMed] [Google Scholar]
- 65.Johnson KA et al. Tau positron emission tomographic imaging in aging and early Alzheimer disease. Ann. Neurol 79, 110–119 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]; This report examines the use of tau-PET in individuals ranging from those who were cognitively healthy to those with dementia.
- 66.Cho H et al. In vivo cortical spreading pattern of tau and amyloid in the Alzheimer disease spectrum. Ann. Neurol 80, 247–258 (2016). [DOI] [PubMed] [Google Scholar]
- 67.Brier MR et al. Tau and Aβ imaging, CSF measures, and cognition in Alzheimer’s disease. Sci. Transl Med 8, 338ra66 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pontecorvo MJ et al. Relationships between flortaucipir PET tau binding and amyloid burden, clinical diagnosis, age and cognition. Brain 140, 748–763 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wang L et al. Evaluation of tau imaging in staging Alzheimer disease and revealing interactions between β-amyloid and tauopathy. JAMA Neurol. 73 1070–1077 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hansson O et al. Tau pathology distribution in Alzheimer’s disease corresponds differentially to cognition-relevant functional brain networks. Front. Neurosci 11, 167 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hoenig MC et al. Networks of tau distribution in Alzheimer’s disease. Brain 141, 568–581 (2018). [DOI] [PubMed] [Google Scholar]
- 72.Bischof GN et al. Impact of tau and amyloid burden on glucose metabolism in Alzheimer’s disease. Ann. Clin. Transl Neurol 3, 934–939 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jansen WJ et al. Prevalence of cerebral amyloid pathology in persons without dementia: a meta-analysis. JAMA 313, 1924–1938 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Morris JC et al. APOE predicts amyloid-β but not tau Alzheimer pathology in cognitively normal aging. Ann. Neurol 67, 122–131 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Farrer LA et al. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA 278, 1349–1356 (1997). [PubMed] [Google Scholar]
- 76.Villeneuve S et al. Existing Pittsburgh compound-B positron emission tomography thresholds are too high: statistical and pathological evaluation. Brain 138, 2020–2033 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Palmqvist S et al. Earliest accumulation of β-amyloid occurs within the default-mode network and concurrently affects brain connectivity. Nat. Commun 8, 1214 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hedden T, Oh H, Younger AP & Patel TA Meta-analysis of amyloid-cognition relations in cognitively normal older adults. Neurology 80, 1341–1348 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Jansen WJ et al. Association of cerebral amyloid-β aggregation with cognitive functioning in persons without dementia. JAMA Psychiatry 75, 84–95 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Donohue MC et al. Association between elevated brain amyloid and subsequent cognitive decline among cognitively normal persons. JAMA 317, 2305–2316 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; This large multisite study analyses the relationship between Aβ and cognitive decline in healthy older people.
- 81.Dubois B et al. Cognitive and neuroimaging features and brain β-amyloidosis in individuals at risk of Alzheimer’s disease (INSIGHT-preAD): a longitudinal observational study. Lancet Neurol. 17, 335–346 (2018). [DOI] [PubMed] [Google Scholar]
- 82.Mormino EC et al. Amyloid and APOE ε4 interact to influence short-term decline in preclinical Alzheimer disease. Neurology 82, 1760–1767 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Farrell ME et al. Association of longitudinal cognitive decline with amyloid burden in middle-aged and older adults: evidence for a dose-response relationship. JAMA Neurol. 74, 830–838 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Becker JA et al. Amyloid-β associated cortical thinning in clinically normal elderly. Ann. Neurol 69, 1032–1042 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Chetelat G et al. Accelerated cortical atrophy in cognitively normal elderly with high β-amyloid deposition. Neurology 78, 477–484 (2012). [DOI] [PubMed] [Google Scholar]
- 86.Dore V et al. Cross-sectional and longitudinal analysis of the relationship between Aβ deposition, cortical thickness, and memory in cognitively unimpaired individuals and in Alzheimer disease. JAMA Neurol. 70, 903–911 (2013). [DOI] [PubMed] [Google Scholar]
- 87.Villemagne VL et al. Amyloid β deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer’s disease: a prospective cohort study. Lancet Neurol. 12, 357–367 (2013). [DOI] [PubMed] [Google Scholar]
- 88.Lowe VJ et al. Association of hypometabolism and amyloid levels in aging, normal subjects. Neurology 82, 1959–1967 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Reiman EM et al. Functional brain abnormalities in young adults at genetic risk for late-onset Alzheimer’s dementia. Proc. Natl Acad. Sci. USA 101, 284–289 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Jagust WJ & Landau SM & Alzheimer’s Disease Neuroimaging Initiative. Apolipoprotein E, not fibrillar β-amyloid, reduces cerebral glucose metabolism in normal aging. J. Neurosci 32, 18227–18233 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Altmann A et al. Regional brain hypometabolism is unrelated to regional amyloid plaque burden. Brain 138, 3734–3746 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Cohen AD et al. Basal cerebral metabolism may modulate the cognitive effects of Aβ in mild cognitive impairment: an example of brain reserve. J. Neurosci 29, 14770–14778 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Johnson SC et al. Amyloid burden and neural function in people at risk for Alzheimer’s disease. Neurobiol. Aging 35, 576–584 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Oh H, Madison C, Baker S, Rabinovici G & Jagust W Dynamic relationships between age, amyloid-β deposition, and glucose metabolism link to the regional vulnerability to Alzheimer’s disease. Brain 139, 2275–2289 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hedden T et al. Disruption of functional connectivity in clinically normal older adults harboring amyloid burden. J. Neurosci 29, 12686–12694 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Mormino EC et al. Relationships between β-amyloid and functional connectivity in different components of the default mode network in aging. Cereb. Cortex 21, 2399–2407 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Jones DT et al. Cascading network failure across the Alzheimer’s disease spectrum. Brain 139, 547–562 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Mormino EC et al. Aβ deposition in aging is associated with increases in brain activation during successful memory encoding. Cereb. Cortex 22, 1813–1823 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Sperling RA et al. Amyloid deposition is associated with impaired default network function in older persons without dementia. Neuron 63, 178–188 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kennedy KM et al. Effects of β-amyloid accumulation on neural function during encoding across the adult lifespan. Neuroimage 62, 1–8 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Schöll M et al. PET imaging of tau deposition in the aging human brain. Neuron 89, 971–982 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Braak H & Del Tredici K Where, when, and in what form does sporadic Alzheimer’s disease begin? Curr. Opin. Neurol 25, 708–714 (2012). [DOI] [PubMed] [Google Scholar]
- 103.Tosun D et al. Association between tau deposition and antecedent amyloid-β accumulation rates in normal and early symptomatic individuals. Brain 140, 1499–1512 (2017). [DOI] [PubMed] [Google Scholar]
- 104.Leal SL, Lockhart SN, Maass A, Bell RK & Jagust WJ Subthreshold amyloid predicts tau deposition in aging. J. Neurosci 38, 4482–4489 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lowe VJ et al. Widespread brain tau and its association with ageing, Braak stage and Alzheimer’s dementia. Brain 141, 271–287 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Jack CR Jr. et al. Longitudinal tau PET in ageing and Alzheimer’s disease. Brain 141, 1517–1528 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sepulcre J et al. In vivo tau, amyloid, and gray matter profiles in the aging brain. J. Neurosci 36, 7364–7374 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Lockhart SN et al. Amyloid and tau PET demonstrate region-specific associations in normal older people. Neuroimage 150, 191–199 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Maass A et al. Entorhinal tau pathology, episodic memory decline, and neurodegeneration in aging. J. Neurosci 38, 530–543 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper reports on the structural and cognitive effects of MTL tau deposition in normal ageing.
- 110.LaPoint MR et al. The association between tau PET and retrospective cortical thinning in clinically normal elderly. Neuroimage 157, 612–622 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hanseeuw BJ et al. Fluorodeoxyglucose metabolism associated with tau-amyloid interaction predicts memory decline. Ann. Neurol 81, 583–596 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Adams JN, Lockhart SN, Li L & Jagust WJ Relationships between tau and glucose metabolism reflect Alzheimer’s disease pathology in cognitively normal older adults. Cereb. Cortex 10.1093/cercor/bhy078 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Sepulcre J et al. Tau and amyloid β proteins distinctively associate to functional network changes in the aging brain. Alzheimers Dement. 13, 1261–1269 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Marks SM, Lockhart SN, Baker SL & Jagust WJ Tau and β-amyloid are associated with medial temporal lobe structure, function, and memory encoding in normal aging. J. Neurosci 37, 3192–3201 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.DeCarli C et al. Measures of brain morphology and infarction in the framingham heart study: establishing what is normal. Neurobiol. Aging 26, 491–510 (2005). [DOI] [PubMed] [Google Scholar]
- 116.Gautam P, Cherbuin N, Sachdev PS, Wen W & Anstey KJ Relationships between cognitive function and frontal grey matter volumes and thickness in middle aged and early old-aged adults: the PATH Through Life study. Neuroimage 55, 845–855 (2011). [DOI] [PubMed] [Google Scholar]
- 117.Zuendorf G, Kerrouche N, Herholz K & Baron JC Efficient principal component analysis for multivariate 3D voxel-based mapping of brain functional imaging data sets as applied to FDG-PET and normal aging. Hum. Brain Mapp 18, 13–21 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Smith CD et al. Brain structural alterations before mild cognitive impairment. Neurology 68, 1268–1273 (2007). [DOI] [PubMed] [Google Scholar]
- 119.Dickerson BC et al. Alzheimer-signature MRI biomarker predicts AD dementia in cognitively normal adults. Neurology 76, 1395–1402 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.de Leon MJ et al. Prediction of cognitive decline in normal elderly subjects with 2-[(18)F]fluoro-2-deoxy-D-glucose/poitron-emission tomography (FDG/PET). Proc. Natl Acad. Sci. USA 98, 10966–10971 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Jagust W et al. Brain imaging evidence of preclinical Alzheimer’s disease in normal aging. Ann. Neurol 59, 673–681 (2006). [DOI] [PubMed] [Google Scholar]
- 122.Knopman DS et al. Brain injury biomarkers are not dependent on β-amyloid in normal elderly. Ann. Neurol 73, 472–480 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Bakkour A, Morris JC, Wolk DA & Dickerson BC The effects of aging and Alzheimer’s disease on cerebral cortical anatomy: specificity and differential relationships with cognition. Neuroimage 76, 332–344 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Fjell AM et al. Brain changes in older adults at very low risk for Alzheimer’s disease. J. Neurosci 33, 8237–8242 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Jack CR Jr. et al. Suspected non-Alzheimer disease pathophysiology — concept and controversy. Nat. Rev. Neurol 12, 117–124 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]; This article reviews the factors that may be associated with neurodegeneration in the absence of AD pathology.
- 126.Burnham SC et al. Clinical and cognitive trajectories in cognitively healthy elderly individuals with suspected non-Alzheimer’s disease pathophysiology (SNAP) or Alzheimer’s disease pathology: a longitudinal study. Lancet Neurol. 15, 1044–1053 (2016). [DOI] [PubMed] [Google Scholar]
- 127.Crary JF et al. Primary age-related tauopathy (PART): a common pathology associated with human aging. Acta Neuropathol. 128, 755–766 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Mormino EC et al. Heterogeneity in suspected non-Alzheimer disease pathophysiology among clinically normal older individuals. JAMA Neurol. 73, 1185–1191 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Jack CR Jr. et al. Age-specific and sex-specific prevalence of cerebral β-amyloidosis, tauopathy, and neurodegeneration in cognitively unimpaired individuals aged 50–95 years: a cross-sectional study. Lancet Neurol. 16, 435–444 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Brookmeyer R & Abdalla N Estimation of lifetime risks of Alzheimer’s disease dementia using biomarkers for preclinical disease. Alzheimers Dement. 14, 981–988 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Caminiti SP et al. FDG-PET and CSF biomarker accuracy in prediction of conversion to different dementias in a large multicentre MCI cohort. Neuroimage Clin. 18, 167–177 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Lehmann M et al. Intrinsic connectivity networks in healthy subjects explain clinical variability in Alzheimer’s disease. Proc. Natl Acad. Sci. USA 110, 11606–11611 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.DeKosky ST & Scheff SW Synapse loss in frontal cortex biopsies in Alzheimer’s disease: correlation with cognitive severity. Ann. Neurol 27, 457–464 (1990). [DOI] [PubMed] [Google Scholar]
- 134.Terry RD et al. Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann. Neurol 30, 572–580 (1991). [DOI] [PubMed] [Google Scholar]
- 135.Chen MK et al. Assessing synaptic density in Alzheimer disease with synaptic vesicle glycoprotein 2A positron emission tomographic imaging. JAMA Neurol. 10.1001/jamaneurol.2018.1836 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Kuhl DE et al. In vivo mapping of cerebral acetylcholinesterase activity in aging and Alzheimer’s disease. Neurology 52, 691–699 (1999). [DOI] [PubMed] [Google Scholar]
- 137.Nitsch RM, Farber SA, Growdon JH & Wurtman RJ Release of amyloid beta-protein precursor derivatives by electrical depolarization of rat hippocampal slices. Proc. Natl Acad. Sci. USA 90, 5191–5193 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Kamenetz F et al. APP processing and synaptic function. Neuron 37, 925–937 (2003). [DOI] [PubMed] [Google Scholar]
- 139.Cirrito JR et al. Synaptic activity regulates interstitial fluid amyloid-beta levels in vivo. Neuron 48, 913–922 (2005). [DOI] [PubMed] [Google Scholar]
- 140.Bero AW et al. Neuronal activity regulates the regional vulnerability to amyloid-β deposition. Nat. Neurosci 14, 750–756 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Pooler AM, Phillips EC, Lau DH, Noble W & Hanger DP Physiological release of endogenous tau is stimulated by neuronal activity. EMBO Rep. 14, 389–394 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Wu JW et al. Neuronal activity enhances tau propagation and tau pathology in vivo. Nat. Neurosci 19, 1085–1092 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Elman JA et al. Effects of beta-amyloid on resting state functional connectivity within and between networks reflect known patterns of regional vulnerability. Cereb. Cortex 26, 695–707 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Tomasi D, Wang GJ & Volkow ND Energetic cost of brain functional connectivity. Proc. Natl Acad. Sci. USA 110, 13642–13647 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Grothe MJ et al. Molecular properties underlying regional vulnerability to Alzheimer’s disease pathology. Brain 141, 2755–2771 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Vlassenko AG et al. Spatial correlation between brain aerobic glycolysis and amyloid-β (Aβ) deposition. Proc. Natl Acad. Sci. USA 107, 17763–17767 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Leal SL, Landau SM, Bell RK & Jagust WJ Hippocampal activation is associated with longitudinal amyloid accumulation and cognitive decline. Elife 8, e22978 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Schultz AP et al. Phases of hyperconnectivity and hypoconnectivity in the default mode and salience networks track with amyloid and tau in clinically normal individuals. J. Neurosci 37, 4323–4331 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Bakker A et al. Reduction of hippocampal hyperactivity improves cognition in amnestic mild cognitive impairment. Neuron 74, 467–474 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Seeley WW, Crawford RK, Zhou J, Miller BL & Greicius MD Neurodegenerative diseases target large-scale human brain networks. Neuron 62, 42–52 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper provides a description of how the patterns of neurodegeneration in different diseases reflect patterns of connectivity and large-scale neural systems.
- 151.Raj A, Kuceyeski A & Weiner M A network diffusion model of disease progression in dementia. Neuron 73, 1204–1215 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Kfoury N, Holmes BB, Jiang H, Holtzman DM & Diamond MI Trans-cellular propagation of tau aggregation by fibrillar species. J. Biol. Chem 287, 19440–19451 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Zhou J, Gennatas ED, Kramer JH, Miller BL & Seeley WW Predicting regional neurodegeneration from the healthy brain functional connectome. Neuron 73, 1216–1227 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Jacobs HLL et al. Structural tract alterations predict down-stream tau accumulation in amyloid positive older individuals. Nat. Neurosci 21, 424–431 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; This article provides compelling evidence for Aβ-facilitated spread of tau through the human episodic memory network.
- 155.Filippini N et al. Distinct patterns of brain activity in young carriers of the APOE-ε4 allele. Proc. Natl Acad. Sci. USA 106, 7209–7214 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Machulda MM et al. Effect of APOE ε4 status on intrinsic network connectivity in cognitively normal elderly subjects. Arch. Neurol 68, 1131–1136 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Grimmer T et al. Progression of cerebral amyloid load is associated with the apolipoprotein E ε4 genotype in Alzheimer’s disease. Biol. Psychiatry 68, 879–884 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Resnick SM et al. Changes in Aβ biomarkers and associations with APOE genotype in 2 longitudinal cohorts. Neurobiol. Aging 36, 2333–2339 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Lim YY, Mormino EC & Alzheimer’s Disease Neuroimaging Initiative. APOE genotype and early β-amyloid accumulation in older adults without dementia. Neurology 89, 1028–1034 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Mishra S et al. Longitudinal brain imaging in preclinical Alzheimer disease: impact of APOE ε4 genotype. Brain 141, 1828–1839 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Liu CC et al. ApoE4 accelerates early seeding of amyloid pathology. Neuron 96, 1024–1032.e3 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Shi Y et al. ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature 549, 523–527 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Kantarci K et al. APOE modifies the association between Aβ load and cognition in cognitively normal older adults. Neurology 78, 232–240 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Wolk DA, & Dickerson BC & Alzheimer’s Disease Neuroimaging Initiative. Apolipoprotein E (APOE) genotype has dissociable effects on memory and attentional-executive network function in Alzheimer’s disease. Proc. Natl Acad. Sci. USA 107, 10256–10261 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Chang L et al. Gray matter maturation and cognition in children with different APOE epsilon genotypes. Neurology 87, 585–594 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Schneider JA, Wilson RS, Bienias JL, Evans DA & Bennett DA Cerebral infarctions and the likelihood of dementia from Alzheimer disease pathology. Neurology 62, 1148–1155 (2004). [DOI] [PubMed] [Google Scholar]
- 167.de Groot JC et al. Cerebral white matter lesions and cognitive function: the Rotterdam Scan Study. Ann. Neurol 47, 145–151 (2000). [DOI] [PubMed] [Google Scholar]
- 168.Price TR et al. Silent brain infarction on magnetic resonance imaging and neurological abnormalities in community-dwelling older adults. The Cardiovascular Health Study. CHS Collaborative Research Group. Stroke 28, 1158–1164 (1997). [DOI] [PubMed] [Google Scholar]
- 169.Gurol ME et al. Cerebral amyloid angiopathy burden associated with leukoaraiosis: a positron emission tomography/magnetic resonance imaging study. Ann. Neurol 73, 529–536 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Hedden T et al. Cognitive profile of amyloid burden and white matter hyperintensities in cognitively normal older adults. J. Neurosci 32, 16233–16242 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Provenzano FA et al. White matter hyperintensities and cerebral amyloidosis: necessary and sufficient for clinical expression of Alzheimer disease? JAMA Neurol. 70, 455–461 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Vemuri P et al. Vascular and amyloid pathologies are independent predictors of cognitive decline in normal elderly. Brain 138, 761–771 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Langbaum JB et al. Blood pressure is associated with higher brain amyloid burden and lower glucose metabolism in healthy late middle-age persons. Neurobiol. Aging 33, 827.e11–827.e19 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Rodrigue KM et al. Risk factors for β-amyloid deposition in healthy aging: vascular and genetic effects. JAMA Neurol. 70, 600–606 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Reed B et al. Associations between serum cholesterol levels and cerebral amyloidosis. JAMA Neurol. 71, 195–200 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Gottesman RF et al. Association between midlife vascular risk factors and estimated brain amyloid deposition. JAMA 317, 1443–1450 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Vemuri P et al. Evaluation of amyloid protective factors and Alzheimer disease neurodegeneration protective factors in elderly individuals. JAMA Neurol. 74, 718–726 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Vemuri P et al. Age, vascular health, and Alzheimer disease biomarkers in an elderly sample. Ann. Neurol 82, 706–718 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.DeCarli C et al. Predictors of brain morphology for the men of the NHLBI twin study. Stroke 30, 529–536 (1999). [DOI] [PubMed] [Google Scholar]
- 180.Mungas D et al. MRI predictors of cognition in subcortical ischemic vascular disease and Alzheimer’s disease. Neurology 57, 2229–2235 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Stern Y What is cognitive reserve? Theory and research application of the reserve concept. JINS 8, 448–460 (2002). [PubMed] [Google Scholar]
- 182.Head D et al. Exercise engagement as a moderator of the effects of APOE genotype on amyloid deposition. Arch. Neurol 69, 636–643 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Brown BM et al. Physical activity and amyloid-β plasma and brain levels: results from the Australian Imaging, Biomarkers and Lifestyle Study of Ageing. Mol. Psychiatry 18, 875–881 (2013). [DOI] [PubMed] [Google Scholar]
- 184.Landau SM et al. Association of lifetime cognitive engagement and low β-amyloid deposition. Arch. Neurol 69, 623–629 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Wirth M, Villeneuve S, La Joie R, Marks SM & Jagust WJ Gene-environment interactions: lifetime cognitive activity, APOE genotype, and β-amyloid burden. J. Neurosci 34, 8612–8617 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Vemuri P et al. Effect of intellectual enrichment on AD biomarker trajectories: longitudinal imaging study. Neurology 86, 1128–1135 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Spira AP et al. Self-reported sleep and β-amyloid deposition in community-dwelling older adults. JAMA Neurol. 70, 1537–1543 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Sprecher KE et al. Amyloid burden is associated with self-reported sleep in nondemented late middle-aged adults. Neurobiol. Aging 36, 2568–2576 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Mander BA et al. β-amyloid disrupts human NREM slow waves and related hippocampus-dependent memory consolidation. Nat. Neurosci 18, 1051–1057 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]; This report describes relationships between slow-wave sleep, amyloid accumulation and memory loss that define a potential vicious cycle in an amyloid–sleep relationship.
- 190.Xie L et al. Sleep drives metabolite clearance from the adult brain. Science 342, 373–377 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Salter MW & Stevens B Microglia emerge as central players in brain disease. Nat. Med 23, 1018–1027 (2017). [DOI] [PubMed] [Google Scholar]
- 192.Jonsson T et al. Variant of TREM2 associated with the risk of Alzheimer’s disease. N. Engl. J. Med 368, 107–116 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Guerreiro R et al. TREM2 variants in Alzheimer’s disease. N. Engl. J. Med 368, 117–127 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Kreisl WC et al. In vivo radioligand binding to translocator protein correlates with severity of Alzheimer’s disease. Brain 136, 2228–2238 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Hamelin L et al. Early and protective microglial activation in Alzheimer’s disease: a prospective study using 18F-DPA-714 PET imaging. Brain 139, 1252–1264 (2016). [DOI] [PubMed] [Google Scholar]
- 196.Parbo P et al. Brain inflammation accompanies amyloid in the majority of mild cognitive impairment cases due to Alzheimer’s disease. Brain 140, 2002–2011 (2017). [DOI] [PubMed] [Google Scholar]
- 197.Villain N et al. Regional dynamics of amyloid-β deposition in healthy elderly, mild cognitive impairment and Alzheimer’s disease: a voxelwise PiB-PET longitudinal study. Brain 135, 2126–2139 (2012). [DOI] [PubMed] [Google Scholar]
- 198.Jack CR Jr. et al. Brain β-amyloid load approaches a plateau. Neurology 80, 890–896 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Duyckaerts C et al. PART is part of Alzheimer disease. Acta Neuropathol. 129, 749–756 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.La Joie R et al. Region-specific hierarchy between atrophy, hypometabolism, and β-amyloid (Aβ) load in Alzheimer’s disease dementia. J. Neurosci 32, 16265–16273 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Giasson BI et al. Initiation and synergistic fibrillization of tau and α-synuclein. Science 300, 636–640 (2003). [DOI] [PubMed] [Google Scholar]
- 202.Botha H et al. FDG-PET in tau-negative amnestic dementia resembles that of autopsy-proven hippocampal sclerosis. Brain 141, 1201–1217 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Clark CM et al. Cerebral PET with florbetapir compared with neuropathology at autopsy for detection of neuritic amyloid-β plaques: a prospective cohort study. Lancet Neurol. 11, 669–678 (2012). [DOI] [PubMed] [Google Scholar]
- 204.Sabri O et al. Florbetaben PET imaging to detect amyloid β plaques in Alzheimer’s disease: phase 3 study. Alzheimers Dement. 11, 964–974 (2015). [DOI] [PubMed] [Google Scholar]
- 205.Curtis C et al. Phase 3 trial of flutemetamol labeled with radioactive fluorine 18 imaging and neuritic plaque density. JAMA Neurol. 72, 287–294 (2015). [DOI] [PubMed] [Google Scholar]
- 206.Schonhaut DR et al. 18F-flortaucipir tau positron emission tomography distinguishes established progressive supranuclear palsy from controls and Parkinson disease: a multicenter study. Ann. Neurol 82, 622–634 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Wirth M et al. Alzheimer’s disease neurodegenerative biomarkers are associated with decreased cognitive function but not β-amyloid in cognitively normal older individuals. J. Neurosci 33, 5553–5563 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Maass A et al. Comparison of multiple tau-PET measures as biomarkers in aging and Alzheimer’s disease. Neuroimage 157, 448–463 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]