

Abstract
During cerebral cortex development, neural progenitors are required to elaborate a variety of cell differentiation signals to which they are continuously exposed. RA acid is a potent inducer of neuronal differentiation as it was found to influence cortical development. We report herein that TBR2, a transcription factor specific to Intermediate (Basal) Neural Progenitors (INPs), represses activation of the RA responsive element and expression of RA target genes in cell lines. This repressive action on RA signaling was functionally confirmed by the decrease of RA-mediated neuronal differentiation in neural stem cells stably overexpressing TBR2. In vivo mapping of RA activity in the developing cortex indicated that RA activity is detected in radial glial cells and subsequently downregulated in INPs, revealing a fine cell-type specific regulation of its signaling. Thus, TBR2 might be a molecular player in opposing RA signaling in INPs. Interestingly, this negative regulation is achieved at least in part by directly repressing the critical nuclear RA co-factor ZFP423. Indeed, we found ZFP423 to be expressed in the developing cortex and promote RA-dependent neuronal differentiation. These data indicate that TBR2 contributes to suppressing RA signaling in INPs, thereby enabling them to re-enter cell cycle and delay neuronal differentiation.
Keywords: Cortical development, TBR2, ZFP423
INTRODUCTION
Embryonic corticogenesis starts when highly proliferative neuroepithelial cells lose some of their epithelial features transforming into Radial Glial Cells (RGCs). While continuing to self-renew, RGCs also origin cortical neurons (Kriegstein and Götz, 2003; Malatesta et al., 2000; Noctor et al., 2001). Initially, RGCs lining the lateral ventricle in the ventricular zone (VZ) undergo asymmetric cell divisions, from which one daughter cells one of which will self-renew and the other will migrate out from the VZ differentiating in a neuronal cell that will constitute the future cortical plate (CP) (Götz et al., 2002; Kriegstein and Götz 2003; Malatesta et al., 2000. Soon after, RGCs undergo a different type of cell division giving rise to a RGC-like daughter cell and an intermediate (basal) neural progenitor (INP). INPs can proliferate and generate a second proliferative area abutting the VZ on the basal side, named the sub-ventricular zone (SVZ). In mice, the mitotic behavior of SVZ progenitors is different from RGCs. In fact, INPs undergo only one or few cell cycle(s) before differentiating and thereby increasing the final number of cortical neurons (Götz and Huttner, 2005; Haubensak et al., 2004; Miyata et al., 2004; Noctor et al., 2004). Recently, we and others demonstrated the critical role of TBR2, an INP-specific transcription factor, in specifying and maintaining the identity and behavior of these transits amplifying neural progenitors (Arnold et al., 2008; Sessa et al., 2008; Sessa et al., 2017). More generally, TBR2 directs both cell-autonomous and non cell-autonomous functions in the SVZ cell compartment to control the development of both glutamatergic and GABAergic neuronal populations, respectively (Sessa et al., 2010).
Recent works have also demonstrated that although RGCs and INPs are the two main neural progenitors in rodents, they are not the only progenitor cell types present in the VZ/SVZ, as mitotic Short Neural Progenitors (SNPs) and outer RGCs (oRGCs) have been described in just the last years (Gal et al., 2006; Lui et al., 2011; Shitamukai et al., 2011; Wang et al., 2011; Florio & Huttner, 2014). Interestingly, some of these recently described neural progenitors are poorly represented in the lissencephalic cortex (e.g. mouse) while very abundant and critical for the establishment of the gyrencephalic cortices (e.g. primate, human). Given this previously unexpected complexity in neuronal progenitor cell type architecture and their relative arrangements in the germinal layers, it is increasingly important to unravel the molecular repertoire of intrinsic and extrinsic cues that are responsible for the transition between different cell types and, in more general terms, for the emergence of neurons during corticogenesis. Several molecular signals have been discovered as modulators of the neuronal proliferation/differentiation balance, controlling relatively discrete steps of i) cell cycle and cell division (Konno et al., 2008; Lange et al., 2009; Postiglione et al., 2011); ii) cell polarity (Bultje et al., 2009; Costa et al., 2008; Götz and Huttner, 2005); iii) cell-cell interaction, e.g. through the Notch pathway (Muroyama and Saito, 2009; Ochiai et al., 2009; Shimojo et al., 2008); iv) neuronal migration (Heng et al., 2008; Kawauchi et al. 2015) and v) paracrine signaling, e.g. FGF signaling (Kang et al., 2009; Toyoda et al., 2010). One of the most potent neuronal differentiation signals during CNS development is Retinoic Acid (RA), a metabolite of vitamin A (retinol). However, the role of RA signaling in anterior territories and, in particular, in developing cortex has been underappreciated, perhaps because the enzymes required for RA synthesis were not detected there. This apparent conundrum has been resolved by the demonstration that the meninges overlaying the cortex are the cellular source of RA during cortical development (Siegenthaler et al., 2009). Mutant mice with disrupted meningeal differentiation undergo a severe RA deprivation, which results in severe craniofacial defects and impressive tangential enlargement and radial thinning of pallium, possibly promoted by an increase of RGC symmetric proliferative divisions due to a defective neuronal differentiation (Siegenthaler et al., 2009). This interpretation has been called into question by the analysis of Raldh2 and Rdh10 (encoding for enzymes critical for transforming retinol in RA) mutant mice that exhibit neither craniofacial defects nor abnormal cortical expansion (Chatzi et al., 2013; Chatzi et al., 2011). Nevertheless, RA seems have some roles in cortical development since meningeal Raldh2 KO displayed cortical defects (Haushalter et al., 2017a; Haushalter et al., 2017b).
Here, we show that RA signaling is kept low in INPs both in a cell culture model and during cortical development in situ, while it can have a role before (in RGCs) and after this step (in young neurons). Interestingly, we found that in INPs, TBR2 directly represses ZFP423, a co-factor of RA receptors that is expressed in RGCs. Of note, loss of ZFP423 results in severe neurodevelopmental defects in both mice (Alcaraz et al., 2006; Cheng et al., 2007) and humans (Chaki et al. 2012). Our data suggest that ZFP423 could act as differentiation factor, at least in part by modulating RA, during corticogenesis. These findings reveal a complex dynamic of RA signaling activity in neuronal progenitors whose tight control by a TBR2-related mechanism allows for the development of a balanced cerebral cortex.
MATERIALS AND METHODS
P19 Cell Culture and Transfection
P19 murine embryonic carcinoma cell line(McBurney MW, 1982) was purchased from the European Collection of Cell Culture (Sigma Life Science) and cultured at 37°C with 5% CO using MEM-α medium supplemented with 10% fetal bovine serum (FBS), Penicillin/Streptomycin, Glutamine and Sodium Pyruvate (Sigma). Once in semiconfluence, cells were washed with phosphate buffered saline (PBS) (EuroClone), subsequently dislodged with 0.05% Trypsin-EDTA (Gibco) and seeded in appropriate multiwells (Nunc-Thermo Scientific) for assays. Cells were transfected with the Lipofectamin reagent (Invitrogen) using (per 6-well) 1 µg of Luciferase reporter, 1 µg of expression construct and1 µg of Renilla-Luciferase as internal control of transfection efficiency. Cells were harvested 48 hrs after transfection (eventually treated with1μM Retinoic Acid (Sigma) for 1 day) and processed with Dual-Luciferase Reporter Assay System (Promega) following the manufacturer’s instruction. Cells induced to differentiate were treated with the medium described above supplemented with 1μM Retinoic Acid (Sigma) for 4 days.
Plasmids
Tbr2 (kind gift of J. Rubenstein and A. Bulfone), Zfp423 (kind gift of R. Reed), RARα and RARαDN (RARα aa1–374) coding sequences were cloned in the pCAGexpression vector (Niwa et al., 1991) upstream to an IRES-EGFP cassette. Tbr2∆Tbox was obtained by subcloning two PCR fragments from the full length, to exclude the aa codifying for the Tbox domain, using the following primers: Tbr2ATG_BglII: 5’- AGATCTatgcagttgggagagcagctcc-3’, Tbr2_N3’_Nhe: 5’-GCTAGCgggccggttgcacaggtagac-3’, Tbr2C5’_NheI: 5’-GCTAGCaactacgattccatgtacacggct-3’, Tbr2C3’_Xho: CTCGAGgggacttgtgtaaaaagcataataagc-3’. The RA-responding vector was created by cloning upstream to the luciferase three RARE elements (CGAGTGAACTTTCGGTGAACCCTACCC). The same RARE elements together with the TK minimal promoter were cloned into pCAG expression vector upstream to an IRES-dGFP cassette (destabilized GFP) (Matsuda and Cepko, 2007).
RNAi Consortium shRNA clones in pLKO-puro were obtained from Sigma. The backbone was modified to replace the puromycin marker with EGFP. Clones were validated in P19 cells and primary granule neurons for efficiency and specificity relative to multiple shRNA control clones by Dr. Chen-Jei Hong (Hong and Hamilton, 2016). Clone #84708 was used for electroporation.
Quantitative real time PCR
RNA from cells or cortical tissue was extracted with the RNeasy kit (QIAGEN) according to the manufacturer’s instructions. 1 μg of the total extracted RNA was reverse transcribed using a Thermoscript RetroTranscriptase (Invitrogen). The oligonucleotides used for the amplification of selected genes are the following: Atcb F: GTGACGTTGACATCCGTAAAGA – R: GCCGGACTCATCGTACTCC; Gapdh F: TGACCTCAACTACATGGTCTACA – R: CCGTGAGTGGAGTCATACTGG; Cyp26a1 F: TGGGACCTGTACTGTGTGAG – R: CCAAACAGATGCGTCTTGTAGA; Crabp2: F: ATGCCTAACTTTTCTGGCAACT – R: CCTGTTTGATCTCGACTGCTG; Zfp423 F: AGCAAGGATGTTGCGTCAC – R: GTCTCGGCTCCTCTTGTGT; Rai14 F: CGATACAAACGAGTGGAACAAGA – R: TCGCTGTCATGCTTCGTGG; Ntrk2 F: CTGGGGCTTATGCCTGCTG – R: AGGCTCAGTACACCAAATCCTA; Gadd45a: F: CCGAAAGGATGGACACGGTG – R: TTATCGGGGTCTACGTTGAGC; Hes6: F: ACCACCTGCTAGAATCCATGC – R: GCACCCGGTTTAGTTCAGC. qPCRs were carried out in a final volume of 25 μl, containing a concentration of 100 nM of each primer, 1x EvaGreen mix (Bio-Rad Laboratories) and 2 μl of the RT products. Thermal cycling was performed using a CFX96 Real Time System (Bio-Rad Laboratories). Melting curve analysis was performed for each primer reaction to ensure a single peak and amplicons were visualized after electrophoresis on 2% agarose gel to ensure the presence of a single PCR product.
The Livak method was applied for quantification (Schmittgen and Livak, 2008), using β-actin as normalizer. Briefly, the expression of each gene either in control or in experimental samples was normalized to that of the housekeeping gene β-actin: to this purpose a ∆CT, Ctrl = (CT, gene – CT, Act)Ctrl and a ∆CT,Exp =(CT, gene – CT, Act)Exp were calculated for each amplified gene and the results were reported as fold change (2−∆∆CT) in gene expression of experimental condition samples to the control ones, where –DDCT = (∆CT,Exp - ∆CT, ctrl).
Generation, maintenance, and differentiation of stable NSC line expressing Tbr2
Cerebral cortices were dissected from E13.5 CD1 mice and digested with papain (10 U/ml, Worthington) in Hanks’ balanced saline solution, containing 1 mM cysteine and 0.5 mM EDTA and subsequently triturated with glass pipettes (Colombo et al., 2006; Ferrari et al., 2010). Cell suspension was washed twice and finally cultured in sterile flasks. NSCs medium contains DMEM-F12 (Invitrogen) supplemented with: 0.66% Glucose 30%, 5.5mM Hepes Buffer (Sigma), 2mM glutamine, 1% penicillin/streptomycin (Sigma), 4 μg/mL Heparin (Sigma), 0.2% BSA (Bovine Serum Albumin) (Sigma-Aldrich), 20 ng/ml bFGF (basic Fibroblast Growth Factor, 20 ng/ml EGF (Epidermal Growth Factor) and 0,1% of Hormone Mix (6% glucose 30%, 5mM Hepes Buffer (Sigma), 250μg/ml insulin (Sigma), 97μg/ml putrescine (Sigma), 1mg/ml Apotransferrin (Sigma), 0.2μM Sodium Selenite and 0.3μM progesterone (Sigma). NSCs were transfected with pCAG-loxpGFPloxp-Tbr2-IRES-bgal-prom-Neo (Fig. 2A) using AMAXA-system according to manufacturer’s instructions (Lonza). Transfected cells were selected by Neomycin resistance until pure transgenic NSC lines were obtained.
Fig. 2. Generation and characterization of Tbr2 Cre inducible transgenic neural stem cell lines.

(A) Drawing of the construct for the conditional expression of Tbr2 where the Tbr2-IRES-GFP sequence is downstream to a constitutive promoter followed by a floxed GFP-Stop cassette. A Neomycin resistance cassette is also included. (B) Schematic representation of the generation of transgenic neural stem cells (NSCs). Wild-type NSCs from E14.5 cerebral cortices are propagated as neurospheres and nucleofected with the Tbr2 conditionally-expressing construct and selected for Neomycin resistance. When the population is composed by only GFP+ cells, then, a single infection with Cre-expressing or mock lentivirus is performed to induce the excision of GFP and release expression of Tbr2. (C) Neurospheres grown after a pulsed Cre expression showing the loss of the GFP fluorescent signal; immunocytochemistry on either mock or Cre-infected NSCs for GFP and TBR2 showing the strong activation of Tbr2 concomitant with the GFP downregulation; bars = 100 µm (left), 20 µm (right). (D) Neurospheres from both conditions were dissociated and plated in adhesion at single cell density to evaluate proliferation at single clone level. After three days in culture the Tbr2-overexpressing clones showed a decrease in clonal dimension as show in graph (Ctrl: 4.87±1.9% vs Tbr2-overexpressing: 2.53±0.9%). Quantification shown as mean + S.D. with dots representing 15 clones per condition, **** p < 0.0001. All measurements statistically compared using unpaired t test. bar = 10 µm. (E) The growth curve on bulk NSC cultures indicates that Tbr2-overexpression reduces cell growth (black line) compared to the control NSCs (green line). Quantification (shown as growth curve that links mean with dots representing individual samples): n = 3, day 20 **** p < 0.0001, differences between groups: F1,20 = 449.2, **** p < 0.0001; all measurements statistically compared using two-way ANOVA, Sidak’s’s multiple comparisons test. (F) Immunocytochemistry on cryostat section of neurospheres for the NSC marker NESTIN, TBR2 and the neuronal marker βIII-tubulin (TUBB3), shows that the control GFP+ NSC are exclusively NESTIN+, with no signal for TBR2 or TUBB3. Conversely, in Cre-infected NSCs TBR2 expression is activated and NESTIN staining is decreased while TUBB3 expression is turned on indicating premature neuronal differentiation; bar = 100 µm.
To passage neurosphere: cells were precipitated, mechanically dissociated in single cells and subsequently seeded in T-75 culture flasks (Nunc Thermo-Scientific). To induce differentiation, NSCs were cultured in adherent conditions on Matrigel (BD)-coated glass dishes and differentiated using NSC medium depleted of EGF for 2 days, followed by 4 days of culture in N2 medium in presence or in absence of 1 μM retinoic acid. N2 medium (DMEM-F12 supplemented with 1% N2 (Invitrogen), 1% penicillin/streptomycin, 1% glutamine and 1% Ascorbic Acid (Sigma)). Growth curve was performed counting the cell number after sphere dissociation over subsequent passages.
Lentivirus production and usage
The lentiviral constructs were produced as previously describe (Indrigo et al., 2010). Briefly 7.5x10^6 HEK 293 cells per 15mm petri dish were seeded depending on the quantity of lentiviral vector needed. The next day cells were transfected with the packaging plasmid and the plasmid containing the coding sequence using CaCl2 transfection. The next day media was changed. After at least 16 hours after media change, the supernatant in each petri dish was filtered and centrifuge for 2 hours at 20000 rpm in an ultracentrifuge. The lentiviral particles were then suspended in PBS and stored at −80° until ready to use.
Transgenic NSCs were seeded in suspension at density of 3x10^5 cells in a 6 wells or 5x10^4 cells in a 24 wells plate. The next day half the medium was removed and cells were infected with either Cre-expressing or mock lentivirus (expressing tTA transactivator protein) using 2–6µl of viral preparation for a 6 wells plate, or 0.25–1.5 µl of viral preparation for a 24 wells plate depending on viral titer. The next day media was changed. NSCs were maintained in culture using the NSCs medium described above.
Immunofluorescence
Cells were fixed with 4% paraformaldehyde for 20 min at RT while tissues were fixed O/N with 4% paraformaldehyde at 4° and processed with sucrose 30% O/N at 4° before the inclusion in OCT for cryosectioning. Immunochemistry analyses were performed as described previously (Colombo et al., 2004). Briefly, fixed cells or frozen section were blocked in 10% normal goat serum (Sigma) and 0.1% Triton X-100 (Sigma) for 1 hr at RT. Cells or frozen sections were then left overnight at 4°C in blocking solution containing the primary antibody. Secondary antibodies were applied to cells or sections for 2 hrs at RT. The primary antibodies utilized were as follows: rabbit anti-Tbr2 (1:300, Abcam), rabbit anti-GFP (1:500, Molecular Probes), chicken anti-GFP (1:500, Molecular Probes), rabbit anti-nestin (1:250, Chemicon), rabbit anti-TuJ1 (1:500, Covance), mouse anti-TuJ1 (1:500, Covance), rabbit anti-dsRed (1:200, Clonetech), rabbit anti-Pax6 (1:200, Covance) rabbit ant-phospho(Ser10) histone H3 (Millipore). Secondary antibodies (Molecular Probes) were conjugates of Alexa Fluor 488, Alexa Fluor 594, and Alexa Fluor 647 (1:500). DAPI (40,60-diamidino-2- phenylindole) was used as nuclear counterstaining. Finally, cells-coated glass dishes, or brain sections were mounted in fluorescent mounting medium (Dako Cytomation).
Quantification: details on number of biological and technical replicates are inserted in figure legends. Biological replicates are usually different cell preparation/mutant embryos/electroporated embryos while technical ones are represented by counts of different slides (at least 200 cells each). Counts are made on blind images taken either on Nikon Eclipse NI straight fluorescent microscope or Leica SP5 or SP8 confocal microscopes. Used objectives are 10X, 20X, 40X (air) and 60X (oil) depending on the single experiment, however scale bars are provided in each panel.
Animals
Tbr2 mutants (Sessa et al., 2008) were obtained crossing Tbr2flox/flox (Mao et al., 2008) and FoxG1-Cre animals (Hebert and McConnell, 2000) and maintained by backcrossing with C57BL/6 animals. Genotyping to distinguish wild-type, floxed and deleted Tbr2 alleles were performed as previously described (Mao et al., 2008). Mice for electroporation were outbreed CD1 (Charles River). The embryonic stage was calculated starting from the day of vaginal plug as E0.5. Congenic BALB/c–Zfp423nur12 null mutant mice (Alcaraz et al., 2011) were maintained at the UC San Diego School of Medicine. All other mice were maintained at the San Raffaele Scientific Institute institutional mouse facility. Experiments were performed in accordance with protocols approved by local Institutional Animal Care and Use Committee (IACUC) of each institution.
In Utero Electroporation
Electroporation was used to deliver expression vectors or reporter constructs to the ventricular RGCs in utero as previously described (Saito, 2001; Saito, 2006). Briefly, uterine horns of E13.5 pregnant dams were exposed by midline laparotomy after anesthetization with Avertin (312 mg/kg). 1 µl of DNA plasmid corresponding to 3 µg mixed with 0.03% fast-green dye in PBS was injected in the telencephalic vesicle using a pulled micropipette through the uterine wall and amniotic sac. 7 mm platinum tweezer-style electrodes were placed outside the uterus over the telencephalon and four pulses of 40 mV for 50 ms were applied at 950 ms intervals by using a BTX square wave electroporator. The uterus was then replaced within the abdomen, the cavity was filled with warm sterile PBS, and the abdominal muscle and skin incisions were closed with silk sutures.
In Situ RNA Hybridization
ISHs on frozen sections were performed as previously described (Schaeren-Wiemers and Gerfin-Moser, 1993) with the following modifications. Sections were fixed for 30 min at room temperature in 4% paraformaldehyde in PBS and treated for 5min with 1 µg/ml proteinase K in 1 mM EDTA, 20 mM Tris- HCl (pH 7.0). Prior to hybridization, they were washed twice in 2X SSC for 15 min and incubated in 0.1 M Tris/0.1 M glycine for at least 30 min. The hybridization solution (60 µl/slide) contained 50% formamide, 5X SSC (pH adjusted with citric acid to pH 6.0), 5% dextran sulfate, 2 mg/ml heparin, 100 mg/ml tRNA, and from 1:100 to 1:50 dilution of the riboprobes, and was performed overnight at 65°C under cover slips. Next, the sections were washed for 1–2 hrs in 0.5X SSC, 20% formamide at 65°C. Subsequently, they were treated with 10 µg/ml RNaseA for 30 min at 37°C in NTE, then washed for 4 hrs in 0.5X SSC, 20% formamide at 65°C and for 30 min in 2X SSC, and blocked for 1 hr at room temperature in 1% blocking reagent (Roche, Switzerland) in MABT. A 1:5000 dilution of anti-digoxigenin-AP conjugated antibody (Roche) was pre- incubated for at least 1 hr in 1% blocking reagent in MABT at 4°C. Slices were incubated with the antibody overnight at 4°C, washed for 6 hr in TBST, for 30 min in NTMT, and stained using centrifuged BM purple AP substrate (Roche) in 0.3% Tween-20 for 12–36 hr at 4°C or room temperature. They were washed in NTMT, then in distilled water, and mounted in Aquamount (Polysciences). The probe for Zfp423 was obtained by PCR amplification (NM_033327.2 nt 3460–4500).
Chromatin Immunoprecipitation
Developing cortices from E14.5 CD1 mouse embryos were fixed using a two steps protocol. Initially the tissue was washed with PBS and fixed in PBS containing 2 mM di-succinimidyl-glutarate (DSG, Sigma) at RT. After 45 min, DSG-fixed tissue was washed thoroughly with PBS and protein-DNA interactions were fixed by 10 min incubation in 1% formaldehyde at RT. Crosslinking was then quenched by addition of Glycine (final concentration 125 mM). Subsequently, tissue was dissociated and lysed in 1% SDS, 10 mM EDTA and 50mM Tris. Sonication was performed using a Bioruptor (Diagenode) at high power setting for 80 cycles (30 sec on/30 sec off). Immunoprecipitations with rabbit anti-TBR2 (Abcam) or unrelated control rabbit antibodies were carried out using 80 µg of chromatin per assay. DNA sequences were quantified by real time PCR (primers are listed in Table S1) by using CFX96 Real Time System (Bio-Rad) and Eva Green-based kit for quantitative PCR (Bio-Rad). Quantities of immunoprecipitated DNA were calculated by comparison with a standard curve generated by serial dilutions of input DNA.
RESULTS
TBR2 Moderates RA signaling In Vitro
Considering the possible role of Retinoic Acid (RA) signaling for the differentiation of cortical RGCs (Siegenthaler et al., 2009) we asked how this signaling could be interpreted by INPs. To tackle this question, we initially tested whether TBR2, a critical INP molecular determinant, could modulate RA signaling. Taking advantage of the Retinoic Acid Responsive Element (RARE) of the retinoic acid receptor (RAR) α, cloned upstream to the luciferase reporter gene, we performed an assay of RA-mediated promoter activation. Expression constructs harboring GFP, TBR2 or TBR2∆Tbox were co-transfected with RARE-Luc in P19 murine carcinoma cell line (McBurney MW, 1982). TBR2 inhibited reporter gene activation by RA 5-fold compared to the control GFP (Fig. 1A), indicating that TBR2 downregulates RA receptor-mediated transcriptional activation. Of note, the absence of the DNA binding ability of TBR2 through the removal of Tbox domain, abolished this effect (Fig. 1A). To further investigate the effect of TBR2 overexpression on the RA response, we evaluated the gene expression levels of Cyp26a and Crabp2, two RA target genes whose protein products are involved in the RA metabolism (Maden, 2007). Both genes were strongly induced (<100-fold) by RA treatment in GFP-transfected control cells (Fig. 1B, green bars), but this response was dramatically reduced by Tbr2 overexpression (Fig. 1B, red bars).
Fig. 1. TBR2 moderates retinoic acid response in vitro.

(A) Luciferase assays for the activation of the retinoic acid responsive element (RARE) in P19 cells reveal a strong downregulation induced by TBR2 when retinoic acid (RA) is added to the media (black bars) while no effect is present without RA (grey bars); this effect is abolished removing the Tbox domain from TBR2. Quantification shown as mean + S.D. with dots representing the five biological replicates (independent cell lysates) (each is the mean of five measurements, technical replicates): -RA, GFP vs TBR2 p = 0.9738, GFP vs TBR2∆Tbox p = 0.9979, TBR2 vs TBR2∆Tbox p = 0.999; +RA, GFP vs TBR2 **** p < 0.0001, GFP vs TBR2∆Tbox p = 0.9023, TBR2 vs TBR2∆Tbox p < 0.0001. All measurements statistically compared using two-way ANOVA, Sidak’s multiple comparisons test. (B) Histograms reveals the expression fold change, obtained by RT-qPCR, of the Cyp26a and Crabp2 genes when RA is added to the medium compared whit the RA-free medium for GFP transfected (green bars) and TBR2 transfected (red bars) cells. Quantification shown as mean + S.D. with dots representing the six biological replicates (independent RNA) (each is the mean of four PCR, technical replicates): Cyp26a, **** p < 0.0001; Crabp2, **** p < 0.0001. All measurements statistically compared using unpaired t test. (C) Immunocytochemistry for GFP and βIII-tubulin (TUBB3) on P19 cells, transfected with GFP mock or Tbr2, after 4 days of RA-induced neural differentiation (10 nM); bar = 100 µm. (D) Graph shows the counts for the βIII-tubulin+ cells among all nuclei for the wild-type condition (black bar) and for all GFP+ (green bar) or Tbr2-transfected (red bar) cells over the GFP− un-transfected cells using 10 nM and 100 nM of RA. Quantification shown as mean + S.D. with dots representing the five biological replicates (independent differentiations) (each is the mean of four quantifications, technical replicates): 10 nM, WT vs GFP p = 0.5612, WT vs TBR2 * p = 0.0132, GFP vs TBR2 ** p = 0.0021. 100 nM, WT vs GFP p = 0.5630, WT vs TBR2 **** p < 0.0001, GFP vs TBR2 **** p < 0.0001. All measurements statistically compared using one-way ANOVA, Tukey’s multiple comparisons test.
P19 cells undergo neural differentiation upon RA treatment (Chen and Reese, 2011). We therefore asked whether TBR2 can modulate this RA-dependent neuronal induction. After four days of RA exposure (10 nM or 100uM) the number of GFP-transfected P19 cells expressing βIII tubulin (TUBB3), an early neuronal marker, was highly comparable to the non-transfected cells (Fig. 1D). In contrast, Tbr2-transfected cells generated proportionately fewer neurons (∼4–5%) (Fig. 1C and D). These findings indicate that, in addition to individual genes, Tbr2 expression suppresses RA induction of neural differentiation in P19 cells. Both results indicate that Tbr2 inhibits RA signaling in vitro.
Tbr2 Overexpressing NSCs Are Resistant to RA-Induced Neuronal Differentiation
To employ a more physiological system for studying neuronal differentiation, we moved from P19 teratocarcinoma cells to a neural stem cell (NSC) culture system (Ferrari et al., 2010). We derived NSC lines from wild-type murine E14.5 cerebral cortices, using neurosphere culture protocol. To conditionally overexpress the Tbr2 gene, we generated a vector in which the Tbr2 cDNA was placed downstream to a LoxP-GFP-Stop-LoxP cassette, together with a neomycin resistant gene cassette (Fig. 2A). NSCs were nucleofected and selected with G418 up to obtaining homogeneous and stable GFP expressing cell lines (Fig. 2B). To overexpress Tbr2, NSCs were infected with a CRE-expressing lentivirus in order to delete the GFP-stop cassette and thereby activating Tbr2 expression (Fig. 2B). Indeed, after infection, the absence of the GFP expression was tightly associated with the presence of TBR2 (Fig. 2C). TBR2+/GFP− NSCs were maintained as neurosphere growing cultures similarly to the control (GFP+/TBR2−) cells (transgenic NSC infected with the same lentivirus lacking the CRE) (Fig. 2C). However, the Tbr2+ overexpressing cells showed reduced proliferation kinetics compared to control cells as assessed by counting both single cell clone progenies (Fig. 2D) and total bulk cell population over different passages in vitro (Fig. 2E). Considering the role of TBR2 during cortical development, we reasoned that this loss of proliferative potential might reflect an increased predisposition to cell differentiation. In fact, Tbr2 overexpressing neurospheres show a reduced expression of the neural stem cell marker NESTIN and a simultaneous increase of the neuronal marker TUBB3 (Fig. 2F), while the control NSCs presented an intensive and homogeneous NESTIN staining without evident expression of TBR2 and TUBB3 (Fig. 2F).
To keep investigating TBR2’s ability to repress the RA cell differentiation signaling, we employed the transgenic NSCs and carried out neural differentiation assays in vitro with and without RA treatment. When we differentiated NSCs without RA, a significantly higher proportion of TUBB3+ neuronal cells was observed in GFP−/TBR2+ cells compared to the GFP+/Tbr2− population (30.6% vs 22.9%, Fig. 3A–D and I, black bar). This finding indicates that Tbr2+ NSCs are more prone to differentiate compare to the control. Interestingly, when RA was added to the same differentiation protocol, GFP−/TBR2+ cells displayed a lower fraction of differentiated neurons compared with the control NSCs (13.6% vs 26.8%, Fig. 3E–H and J, black bar).
Fig. 3. TBR2 reduces RA-induced neural differentiation.

(A-D) Immunocytochemistry for GFP (A and C) and βIII-tubulin (TUBB3, B and D) on wild-type (A and B) and Cre-infected (C and D) NSCs after 10 days of culture in the differentiation medium without RA. (E-H) Immunocytochemistry for GFP (E and G) and βIII-tubulin (TuJ1, F and H) on wild-type (E and F) and Cre-infected (G and H) NSCs in the same culture medium usupplemented with RA for 10 days; bar = 50 µm. (I) Graph shows counts for TUBB3+ neurons in RA-free medium. Control, GFP+/TBR2− cells (green bar): 22.87±5.9% TUBB+/GFP+ vs GFP+/TBR2− (black bar): 30.64±5.5% TUBB3+/GFP−/DAPI+. Quantification shown as mean + S.D. with dots representing the seven biological replicates (independent differentiations) (each is the mean of three quantifications, technical replicates): **** p < 0.0001. All measurements statistically compared using unpaired t test. (L) Counts for TUBB3+ neurons in RA supplied medium. Control, GFP+/TBR2− cells (green bar): 26.82±2.6% TUBB3+/GFP+ vs GFP+/TUBB3− (black bar): 13.62±3.2% TUBB3+/GFP−/DAPI+. Quantification shown as mean + S.D. with dots representing the seven biological replicates (independent differentiations) (each is the mean of three quantifications, technical replicates): * p = 0.0267. All measurements statistically compared using unpaired t test.
These results suggest that Tbr2 activation reduces cell proliferation and expression of the stem cell-associated marker NESTIN enhancing neuronal differentiation in normal culture conditions, but counteracting RA signaling dependent cell differentiation process at least in vitro.
The RA dependent neuronal differentiation signaling is quiescent in Tbr2+ INPs
The results obtained so far indicate that TBR2 prevents the RA-dependent neuronal differentiation activity in two different cell lines. However, neither the P19 neurogenic tumor cell line nor the NSCs fully model the complexity of the differentiation processes occurring during corticogenesis, where different neuronal cell types form over time within an organized and well-defined tissue architecture (Götz and Huttner, 2005; Kriegstein and Alvarez-Buylla, 2009; Lehtinen and Walsh, 2011).
Therefore, we set out to decipher the involvement of TBR2 in controlling the RA signalling during corticogenesis. We first checked whether the expression of Rara was detectable in developing cortex by using Genepaint.org (Visel et al., 2004) (Fig S1A) and that its responsive elements were functional by targeting cortex, and basal ganglia as positive control, through in utero electroporation system (Fig S1B) (Saito, 2006). Then, we developed a molecular sensor of the RA activity in vivo by assembling the RARE promoter upstream to a highly-destabilized form of eGFP (2 hr. half-life, dGFP) (Matsuda and Cepko, 2007), which provides a reliable and steady-state estimation of the promoter activity. To assess this new RA reporter, RARE-dGFP or pCAG-dGFP (constitutive active promoter) were co-transfected with the pCAG-dsRed vector (as marker for transfected cells) in P19 cells (Fig. S1C–H). GFP expression was then monitored in dsRed+ cells before, during or after adding RA to the medium (Fig. S1C). In the case of pCAG-dGFP, the protein was constitutively present in transfected cells and its expression was not altered by the presence of RA (Fig. S1 D and E). Conversely, when the dGFP was controlled by the RARE promoter its expression was detectable only in the presence of RA (Fig S1G), while remaining at very low level or undetectable before RA administration (Fig S1F) or after RA washing out (Fig S1H).
Next, we employed this RA reporter to detect endogenous RA activity in the developing cerebral cortex at single cell level. Using in utero electroporation (Saito, 2006) we introduced the RARE-dGFP or pCAG-dGFP together with the pCAG-dsRed in E13.5 RGCs and evaluated reporter activity after 24 hrs (Fig. 4A), a time sufficient for transfected RGCs to divide and generate daughter INPs (Haubensak et al., 2004; Noctor et al., 2008; Noctor et al., 2004). In this timeframe, we observed that almost all red+ cells were co-stained with GFP and vice-versa when the pCAG-dGFP was used, confirming that the great majority of transfected cells took up and expressed both plasmids (Fig. 4B and E). By contrast, in the case of the RA reporter, while all the GFP+ cells were also red+, confirming the co-electroporation, they represent only 33.6% of the entire red+ cell population (Fig. 4C arrowheads and E). These findings indicate that only a fraction of electroporated cells in the developing cortex respond to RA signaling (Fig. 4C arrows). We previously demonstrated that TBR2 is necessary and sufficient to promote generation of INPs at the expense of RGCs (Sessa et al., 2008). To evaluate the activity of RA in INPs, we co-electroporated RARE-dGFP and pCAG-Tbr2-IRES-dsRed in E13.5 developing cortices. In this experiment, the fraction of the cells showing GFP expression was significantly reduced compared with the co-electroporation of RARE-dGFP + pCAG-Red, with only few double positive cells (14.4 ± 6.9%, Fig. 4D arrowheads and E).
Fig. 4. RA endogenous activity in RGCs and INPs.
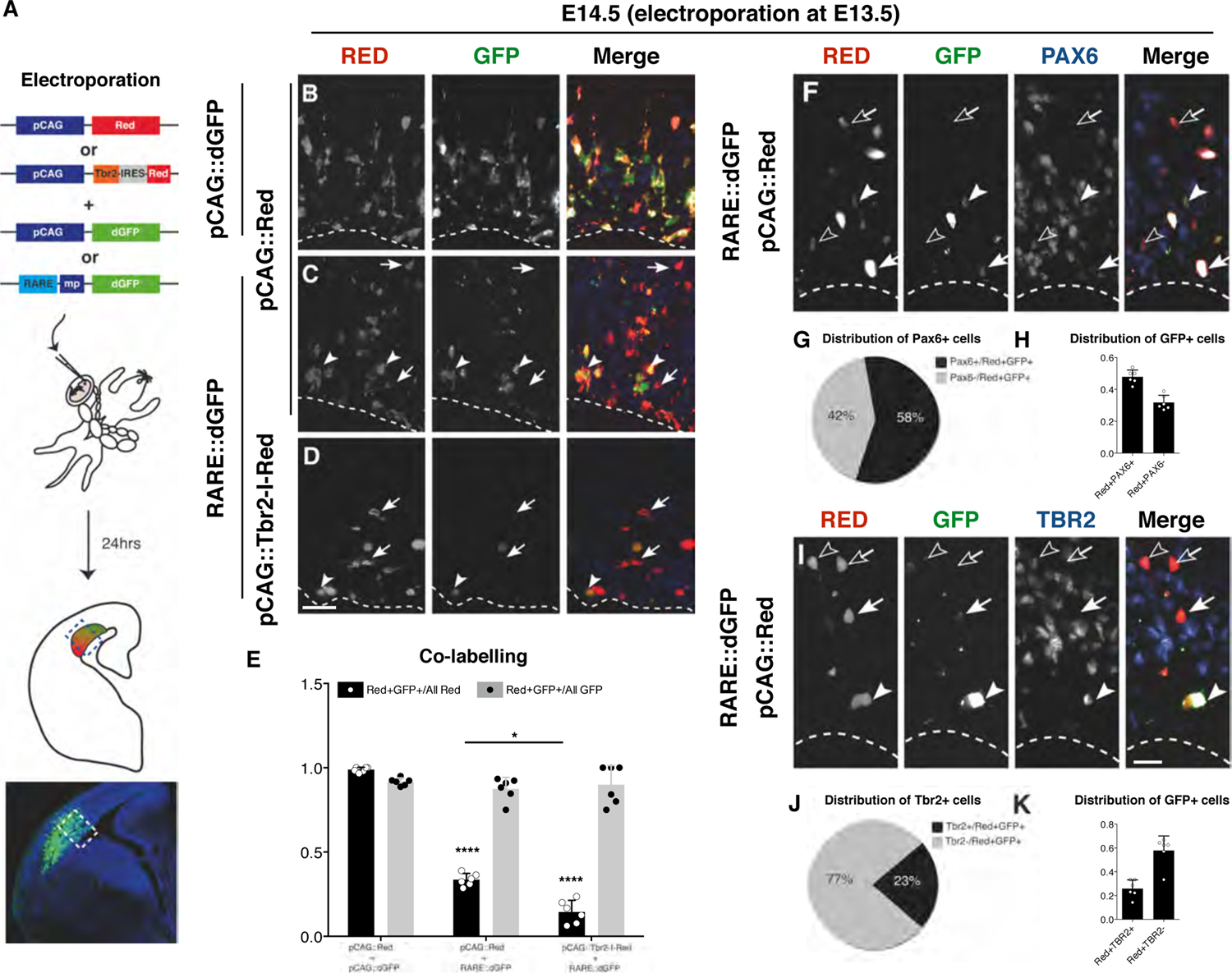
(A) Schematic representation of the experimental strategy. By in utero electroporation the combination of construct indicated were inserted in E13.5 developing cortices and their expression evaluated 24hrs later through immunohistochemistry. (B-D) dsRed and GFP immunohistochemistry to evaluate the co-expression of both dsRed and destabilizedGFP (dGFP) (B). (C) Constitutive dsRed and RARE::dGFP, arrows indicate cells electroporated (Red+) without RA activity (GFP−), while arrowheads indicate double positive cells. (D) Constitutively expressed Tbr2-IRES-Red and RARE::dGFP, arrows indicate cells electroporated (Red+) without RA activity (GFP−) while arrowhead indicates double positive cells; bar = 20 µm. (E) Graph illustrating the co-labelling rate. The double positive cells are the vast majority if normalized on GFP+ cells in all the condition (gray bars). However, when the GFP+ are compared with the Red+ cells (black bars), they are equal only for the control case (98.82±1.4%) indicating a high rate of co-electroporation, but only a subset when the RARE::dGFP (33.55±3.7%) and the pCAG::Tbr2-I-Red (14.43±6.9%) are used. Quantification shown as mean + S.D. with dots representing the six biological replicates (independent electroporated embryos) (each is the mean of three quantifications, technical replicates): Red+GFP+/All Red: pCAG::Red + pCAG::dGFP vs pCAG::Red + RARE::dGFP **** p < 0.0001, pCAG::Red + pCAG::dGFP vs pCAG::Tbr2-I-Red + RARE::dGFP **** p < 0.0001, pCAG::Red + RARE::dGFP vs pCAG::Tbr2-I-Red + RARE::dGFP * p = 0.0153. Red+GFP+/All GFP: pCAG::Red + pCAG::dGFP vs pCAG::Red + RARE::dGFP p = 0.8388, pCAG::Red + pCAG::dGFP vs pCAG::Tbr2-I-Red + RARE::dGFP p = 0.9977, pCAG::Red + RARE::dGFP vs pCAG::Tbr2-I-Red + RARE::dGFP p = 0.9863. All measurements statistically compared using two-way ANOVA, Sidak’s multiple comparisons test. (F) Immunohistochemistry for dsRed, GFP and PAX6 in pCAG::Red + RARE::dGFP co-electroporated progenitors: filled arrowhead, example of a triple positive cell; empty arrowhead, example of a RED+/GFP−/PAX6+ cell; filled arrowhead, example of RED+/GFP+/PAX6− cell; empty arrow = example of RED+/GFP−/PAX6− cell. (G) Pie chart showing the percentage of the PAX6+ (grey) over all the RED+/GFP+ cells. (H) Histogram representing the distribution of GFP+ in electroporated (Red+) cells positive (first bar) or negative for Pax6 (second bar). Quantification shown as mean + S.D. with dots representing the six biological replicates (independent electroporated embryos) (each is the mean of three quantifications, technical replicates). (I) Immunohistochemistry for dsRed, GFP and TBR2 in pCAG::Red + RARE::dGFP co-electroporation: filled arrowhead, example of a triple positive cell; empty arrowhead, example of a RED+GFP−TBR2+ cell; filled arrow, example of a Red+GFP+TBR2−cell; empty arrow, example of a Red+GFP−TBR2− cell; bar = 20 µm.. (J) Pie chart showing the percentage of the TBR2+ (grey) over all the RED+GFP+ cells. (K) Histogram representing the distribution of GFP+ in electroporated (RED+) cells positive (first bar) or negative for TBR2 (second bar). Quantification shown as mean + S.D. with dots representing the six biological replicates (independent electroporated embryos) (each is the mean of three quantifications, technical replicates).
To assess exactly in which cell type the RA signaling is endogenously active, we analyzed the RARE-dGFP, pCAG-Red double electroporated cells by immunohistochemistry for the expression of PAX6 and TBR2, two highly specific molecular markers for RGCs and INPs, respectively. Staining for PAX6 showed that 58% of the positive cells for the RA activity reporter were RGCs (Fig. 4F filled arrowheads, G). By contrast only 23% of the GFP+ cells were stained with the INP marker TBR2 (Fig. 4 filled arrowheads, J). To exclude that these results could be influenced by the relative composition of the electroporated cell types (e.g. more RGCs than INPs,) we evaluated the ratio of the electroporated RA responding cells (GFP+) on either electroporated RGCs or INPs (red+ and marker+). GFP+ cells accounted for the 47.9 ± 4.3% among all the electroporated presumptive RGCs (PAX6+) and the 31.8±4.6% among the electroporated non-RGCs (PAX6−) (Fig. 4H). Interestingly, RA responding (GFP+) cells accounted only for the 25.8 ± 7.4% among all the electroporated presumptive INPs (TBR2+) and the 57.7 ± 12.7% among the electroporated non-INPs (Tbr2−) (Fig. 4K).
Taken together, these results indicate that the RA signaling is active in cortical progenitors, with a prevalence for RGCs rather than INPs. Moreover, our data confirm that TBR2 is sufficient to downgrade RA signaling in vivo as well as in cell culture models.
TBR2 Binds and Regulates Zfp423 locus
Since neither TBR2 nor other T-box transcription factors are known as direct modulators of retinoic acid receptors (Conlon et al., 2001), we wondered whether TBR2 might directly regulate genes required for RA signaling. We therefore sought to identify TBR2 target genes in an unbiased way by profiling the transcriptome of E14.5 Tbr2 mutant and wild-type cortices (Sessa et al., 2017). Intriguingly, one of the most upregulated genes in the mutant mouse was Zfp423, encoding a zinc finger protein identified as transcriptional cofactor of RA in several cell types, including P19 and neuroblastomas (Huang et al., 2009).
In agreement with microarray data, we observed an upregulation of Zfp423 gene expression in Tbr2 cKO cortices, obtained both with Foxg1::Cre and Emx1::Cre drivers, as assessed by both in situ hybridization and qPCR (3.5 fold compare to the control) (Fig. 5A and B). Furthermore, Tbr2 overexpression in P19 cells performed in normal culture condition (without RA) was able to downregulate the expression of endogenous Zfp423 (38.46 ± 9.12% compared to the GFP transfected cells) (Fig. S2A).
Fig. 5. TBR2 binds and regulates Zfp423 expression.

(A) Zfp423 in situ hybridization on Tbr2 mutant or control E14.5 forebrain coronal sections; Zfp423 expression results upregulated in proliferative areas of mutants (arrowheads) compared to control cortices (insets are magnification of cortical areas of main images); bars = 100 µm (main panel), 50 µm (insets). Abbreviation: Ctx = Cortex; BG = Basal Ganglia. (B) Graph showing the relative fold increase by qPCR of the Zfp423 expression in Tbr2 mutant respect to control E14.5 cortices tissues. Quantification shown as mean + S.D. with dots representing the four biological replicates (independent RNA) (each is the mean of four PCR, technical replicates): Foxg1::cre ** p = 0.0078, Emx1::cre ** p = 0.0078. All measurements statistically compared using unpaired t test. (C) Left, genome browser view of TBR2 ChIP-seq track (Sessa et al., 2017) shows a clear peak in Zfp423 intron III. Right, validation (represented as % of ChIP input) by qPCR on independent TBR2 ChIP samples of both identified peak (Z023 amplicon) and three nearby regions located in intron I (Z004), II (Z019) and exon IV (Zorf) as negative controls. Quantification shown as mean + S.D. with dots representing the six independent ChIP experiments (each is the mean of three PCR, technical replicates): Zorf p = 0.7119, Z004 p = 0.1203, Z019 p = 0.0534, Z023 **** p < 0.0001. All measurements statistically compared using unpaired t test. (D) The intronic region containing the amplicon Z023 has been cloned into the luciferase promoter vector. (E) Luciferase assay shows that TBR2 is able to strongly downregulate the activity of the Zfp423 Z023 enhancer activity (6.35±4.6). Quantification shown as mean + S.D. with dots representing the five biological replicates (independent cell lysates) (each is the mean of five measurements, technical replicates): **** p < 0.0001. All measurements statistically compared using unpaired t test. (F) Venn diagram shows overlap between genes up-regulated (red) in Tbr2 mutant, genes associated with regulatory regions presenting RARα/RXRα binding motifs (orange) and genes associated with regulatory regions presenting ZFP423 binding motifs (blue). (G) Validation by RT-qPCR (represented as relative expression) of examples of genes contained in all three datasets in independent RNA samples from control and Tbr2 mutant cortices. Quantification shown as mean + S.D. with dots representing the six biological replicates (independent RNA) (each is the mean of four PCR, technical replicates): Crabp2 *** p = 0.0002, Nrtk2 ** p = 0.0033, Rai14 ** p = 0.0016, Gadd45a * = 0.0174, Hes6, **** p < 0.0001. All measurements statistically compared using unpaired t test.
To test whether Zfp423 could be a direct transcriptional target of TBR2, we analyzed previously generated ChIP-seq data for TBR2 occupancy in cortical cells previously generated (Sessa et al, 2017), looking at Zfp423 locus. We found, and independently validate, a clear TBR2 peak located at the intron 3 of the Zfp423 gene locus (Fig. 5C) an evolutionary conserved region between mouse and human (Ovcharenko et al. 2004) with three T-box core motif sites (Fig. S2B). This genomic region, cloned in a luciferase vector (Fig. 5D), was able to dramatically inhibit (>15-fold) luciferase expression when this vector was transfected in P19 cells together with a Tbr2 expression plasmid, compared to the vector alone (Fig. 5E). Interestingly,∼45% of the upregulated genes in Tbr2 mutant cortex (Sessa et al., 2017) including Crabp2, Ntrk2, Rai14, Gadd45a and Hes6, are both ZFP423-and RARA/RXRA-putative targets based on the presence of their binding motifs, (Fig. 5F and G) again indicating the possible TBR2 role in attenuating ZFP423-mediated RA signaling.
In order to evaluate the ability of TBR2 to downregulate Zfp423 in vivo, we evaluated RNA expression of the latter by in situ hybridization (ISH) and qPCR following Tbr2 overexpression by in utero electroporation (Fig. 6A). In these conditions, we noted an appreciable downregulation of both the Zfp423 ISH probe signal and its expression levels by qPCR in the Tbr2- compared to the GFP-electroporated area (Fig. 6B and C). Interestingly, three RA responsive genes, Crabp2, Cyp26a1 and Rai14, were downregulated as well (Fig. 6C). Strikingly, the concomitant overexpression of Zfp423 itself was able to revert the downregulation of these genes (Fig. 6C). These data strongly indicate that TBR2 negatively regulates Zfp423 expression through the direct binding to an evolutionary conserved intronic region causing a decrease in genes that are responsive to RA signaling.
Fig. 6. TBR2 downregulates Zfp423 in vivo.
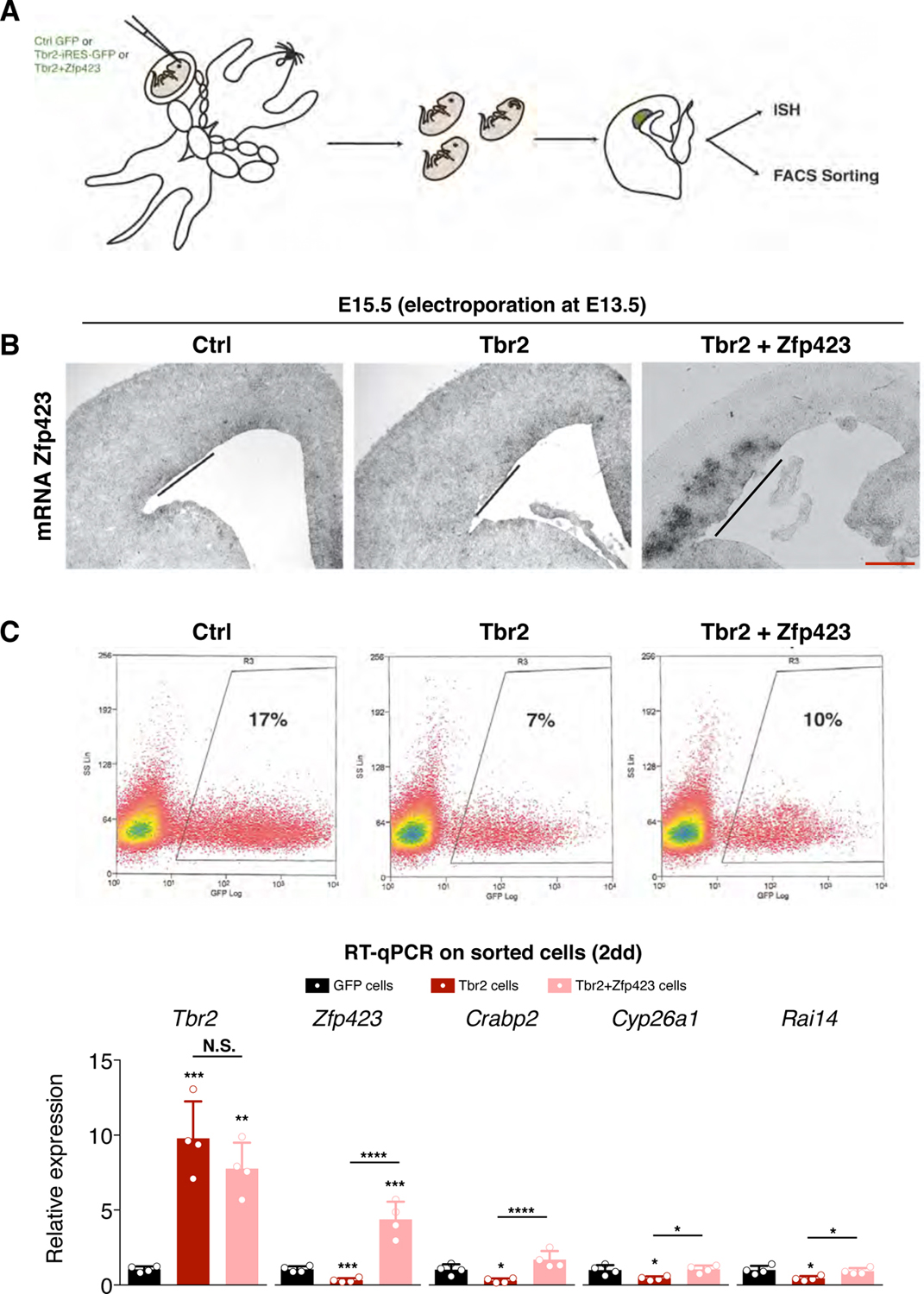
(A) Schematic representation of in utero electroporation; Ctrl GFP, Tbr2-IRES-GFP or Tbr2-IRES-GFP + pCAG-Zfp423 were targeted in E13.5 developing cortices and their effects evaluated few days later. (B) In situ hybridization for Zfp423 mRNA on coronal section of electroporated embryos. Zfp423 expression results downregulated in the misexpression area (line) upon Tbr2 overexpression compared to the control electroporation area (line) while its expression is strongly upregulated in Tbr2+Zfp423 experiments; bar = 100 µm. (C)Top, FACS analyses of GFP, Tbr2 and Tbr2+Zfp423 electroporated tissue, dissected after 2 days from surgery. Bottom, RT-qPCR (represented as relative expression) analyses of Tbr2, Zfp423 and RA target genes in RNA from Tbr2- (red bar) and Zfp423-electroporated samples (pink bar). Quantification shown as mean + S.D. with dots representing the four biological replicates (independent electroporated embryos) (each is the mean of four PCR, technical replicates): Tbr2: GFP cells vs Tbr2 cells *** p = 0.0001, GFP cells vs Tbr2+Zfp423 cells ** p = 0.0010, Tbr2 cells vs Tbr2+Zfp423 cells p = 0.2781; Zfp423: GFP cells vs Tbr2 cells *** p = 0.0003, GFP cells vs Tbr2+Zfp423 cells *** p = 0.0002, Tbr2 cells vs Tbr2+Zfp423 cells **** p < 0.0001; Crabp2: GFP cells vs Tbr2 cells * p = 0.0398, GFP cells vs Tbr2+Zfp423 cells p = 0.0981, Tbr2 cells vs Tbr2+Zfp423 cells **** p < 0.0001; Cyp26a1: GFP cells vs Tbr2 cells * p = 0.0292, GFP cells vs Tbr2+Zfp423 cells p = 0.9530, Tbr2 cells vs Tbr2+Zfp423 cells * p = 0.0185; Rai14: GFP cells vs Tbr2 cells * p = 0.0161, GFP cells vs Tbr2+Zfp423 cells p = 0.8842, Tbr2 cells vs Tbr2+Zfp423 cells * p = 0.0336. All measurements statistically compared using one-way ANOVA, Tukey’s multiple comparisons test.
ZFP423 regulates cortical development
To test whether ZFP423 acts in embryonic cortical development in a manner compatible with it being a major effector of TBR2 in INPs, we examined neural precursors in coronal sections of congenic Zfp423nur12 null mutant (Alcaraz et al., 2011; Alcaraz et al., 2006) and littermate controls at E14.5, E16.5 and E18.5 (Fig. 7). The nur12 mutation places a stop codon in the sequence encoding the second of 30 zinc finger domains of Zfp423 protein. In late embryogenesis Zfp423 mutant cortex tends to be thinner than littermates (Fig. 7A–C). We reasoned that this difference could be due to dynamics of proliferation and differentiation; to address this issue we estimated the number of cells in S-phase by CldU incorporation and cells entering mitosis by phospho-Ser10 histone H3 (PH3) immunoreactivity in multiple, non-overlapping sections from three littermate pairs at each age (Fig. 7D–E and data not shown). The most pronounced differences occurred at E16.5, where loss of Zfp423 results in reduced numbers of cells positive for each proliferation marker (77–88% of control values) (Fig. 7F) as well as the double-positive fraction (cells that progressed from S-phase to mitosis during labeling, 75% of control values). Proliferation measures were significant over the full experiment and independently at E16.5. In addition, we asked whether ZFP423 might reciprocally regulate TBR2 or block differentiation by examining expression in Zfp423-mutant cortex (Fig. S3 and Fig. 7G–I). While ZFP423 does not appear to have a reciprocal relationship with TBR2 in terms of cell number or intensity of staining, TBR2+ INPs appear less frequently in the intermediate zone (IZ) at E14.5, suggesting a delay in differentiation or migration (Fig. S3). Differentiation also appears delayed at E14.5 with respect to appearance of TBR1+ cells (Fig. 7G–I), with a statistically robust difference under the assumption of independence for cell-level responses (Hong and Hamilton 2016). All these data suggest that loss of Zfp423 function could result in diminished responsiveness of cortical progenitors by E14.5, resulting in diminished subsequent proliferation, differentiation and, ultimately, cortical volume.
Figure 7. Endogenous Zfp423 is required for normal cortical development.

(A-B) Zfp423 null (nur12, n in figure) cortex appears thinner than control by E18.5. Radial thickness of developing cortex was measured radially from the dorsal exterior tip of the ventricle (yellow dashed line) for littermate samples sectioned at matched rostral caudal levels. Scale bar, 500 µm. (C) Graphs summarize replicate measures from three littermate pairs at each of three ages. Littermates are encoded by color intensity. Paired measurements averaged 15% thinner cortex in mutant samples at E18.5, with a trend toward thinner cortex (6%) at E16.5. (D-E) Zfp423 null (nur12) cortex shows less proliferation of neural precursors at E16.5. Frequencies of PH3 immunoreactivity and 1hr. CldU incorporation were monitored in alternate sections from the same animals in (C). Representative fields are shown. Scale bar, 100 µm. (F) Distribution of counts per field at E16.5 are shown. Pooled across all ages, number of positive cells per field were not normally distributed, but significant by non-parametric Wilcoxon rank sum tests for PH3 (p=0.003), CldU (p=0.05), either label (p=0.03), and double-positive cells (p=0.007). Significance was uniquely attributable to differences at E16.5 (p=0.002, 0.0006, 0.0004, and 0.007, respectively), accounting for 17% fewer proliferating cells in mutants at this age. (G-H) Matched sections from the same animals were stained for TBR1 expression (red). Indicated layers are marginal zone (MZ), cortical plate (CP) and intermediate zone (IZ). (I) Numbers of TBR1+ nuclei per field were significant across the full data set (p=0.027, ANOVA). Although no single age was independently significant by genotype after correcting for multiple comparisons, a 19% difference at E14.5 approached conventional significance (p=0.06, Tukey HSD).
ZFP423 transduces the RA dependent neuronal differentiation effect in the developing cortex
Next, we asked whether ZFP423 is specifically involved in processing the RA molecular signaling cascade in the developing cortex. First, we assessed the ability of ZFP423 to enhance the RA response in P19 cells. As expected, we observed that Zfp423 overexpression was able to increase the activity of the RARE-Luc at levels comparable to those triggered by the overexpression of the RA receptor alpha (RARα) (Fig. S4A). ZFP423 enhanced RA signaling, most likely by acting as a RARα co-factor (Huang et al., 2009). In fact, ZFP423 was able to counteract the loss of the RA reporter activity when expressed together with the dominant negative form of RARα (RARαDN) (Fig. S4A), suggesting that it competes with RARαDN for limiting components of activating complexes.
To assess whether ZFP423 has a similar effect on RA signaling in vivo, we overexpressed it in E13.5 developing cortex. To start with, we observed that Zfp423 was able to enhance the activity RA sensor in vivo compared to control (Fig. S4B, C). The phenotypic consequences of the Zfp423 overexpression were comparable with the effects triggered by either the activation or inhibition of the RA signaling pathway as induced by overexpressing RARα or its dominant negative form (RARαDN), respectively (Fig. 8). Two days after overexpressing GFP only, few GFP+ cells were positive for TUBB3, a marker of post-mitotic neurons (7.3±4.6%), while the majority of the electroporated cells remained in the proliferative areas and stained for the RGC marker PAX6 (70.7±6.5%), and the INP marker TBR2 (38.2±9.8%) (Fig. 8B and C). Conversely, when Zfp423 was co-expressed with GFP the number of electroporated cells positive for TUBB3 strongly increased (35.8±9.7%) at the expense of PAX6+ RGCs (34±6.3%), while the total number of INPs were not significantly affected (29.9±7.9%) (Fig. S5, Fig. 8B and C). Importantly, the enhancement in cortical neurogenesis triggered by Zfp423 overexpression is in line with the possible neuronal differentiation activity of RA signaling during cortical development (Siegenthaler et al., 2009; Siegenthaler and Pleasure, 2011). In fact, overexpression of RARα induced a similar increase of the neuronal cell compartment with a concomitant loss of RGCs. Conversely, blunting RA signaling activity by overexpressing RARαDΝ caused RGCs to be unable to initiate radial migration toward the cortical plate although the relative composition of cortical cell type was not different to that obtained by electroporating the GFP only (Fig. 8C). We then asked whether the ZFP423-dependent neurogenic effect could be mitigated by concomitantly decreasing RA signaling activity. Interestingly, the simultaneous expression of RARαDN with Zfp423 was able to partially rescue the number of PAX6+ RGCs (59.9±2.7%) (Fig. S5, Fig. 8C) while limiting the increase in TUBB3+ neurons (17.6±2.7%) (Fig. 8B and C). Moreover, the block in neuronal migration associated with RARαDN expression was overcome by Zfp423 co-expression (Fig. 8B). These results indicate that the ZFP423 dependent premature neuronal differentiation is significantly ameliorated by downregulating RA signaling and further suggests a competitive relationship between ZFP423 and RARαDN activities.
Fig. 8. In vivo Zfp423 overexpression causes premature differentiation.

(A) Schematic representation of the experimental strategy. By in utero electroporation the different combinations of construct indicated were inserted in E13.5 developing cortices and their phenotype evaluated two days later via immunohistochemistry. (B) Immunohistochemistry for GFP and βIII-tubulin (TUBB3) shows peculiar ectopic TUBB3+ neurons in ventricular zones of cortices electroporated with Zfp423 and Rarα compared with the GFP or RarαDN alone or the co-electroporation of Zfp423 and RarαDN. The cells electroporated with RarαDN fail to reach the cortical plate (arrowheads); bar = 100 µm. (C) The histograms display the percentage of the cell double positive for GFP and TUBB3 (black bars) or PAX6 (dark grey bars) or TBR2 (light grey bars) counterstained with the GFP+ cells present in the proliferative area of the cortex (morphologically separated from IZ). Quantification shown as mean + S.D. with dots representing the five biological replicates (independent electroporated embryos) (each is the mean of 3 quantification, technical replicates): TUBB3+: GFP vs Zfp423 **** p < 0.0001, GFP vs Rarα **** p < 0.0001, GFP vs RarαDN p > 0.9999, GFP vs Zfp423+RarαDN p = 0.3615; PAX6+: GFP vs Zfp423 **** p < 0.0001, GFP vs Rarα **** p < 0.0001, GFP vs RarαDN p = 0.9194, GFP vs Zfp423+RarαDN p = 0.4775; TBR2+: GFP vs Zfp423 p = 0.8036, GFP vs Rarα p = 0.9956, GFP vs RarαDN p = 0.9996, GFP vs Zfp423+Rarα p > 0.9999. All measurements statistically compared using two-way ANOVA, Sidak’s multiple comparisons test. (D) Immunohistochemistry for GFP and Bromodeoxyuridine (BrdU) showing the fraction of proliferating cells undergoing S-phase during the 1hr BrdU pulse. (proliferative area, morphologically separated from IZ). Forced expression of Zfp423, Rarα and Zfp423+RarαDN reduced proliferative cells in the electroporated VZ compared to GFP or RarαDN electroporated cells; bar = 20 µm. (E) The graph indicates the percentage of BrdU+ cells over the total GFP+ electroporated cells. Quantification shown as mean + S.D. with dots representing the five biological replicates (independent electroporated embryos) (each is the mean of 3 quantification, technical replicates): GFP vs Zfp423 **** p < 0.0001, GFP vs Rarα **** p < 0.0001, GFP vs RarαDN p = 0.1018, GFP vs Zfp423+RarαDN p = 0.2323, Zfp423 vs Zfp423+RarαDN * p = 0.0429. All measurements statistically compared using one-way ANOVA, Tukey’s multiple comparisons test.
Next, 1-hr pulse of BrdU incorporation in the cells electroporated with the different gene combinations described above was evaluated. As expected, both Zfp423 and RARα induced a decrease in BrdU+ cell numbers (Fig. 8D and E). Conversely, an increase in proliferation was triggered by RARαDN activation (Fig. 8D, E). Notably, the co-expression of Zfp423 together with RARαDN triggered a significant increase in BrdU+ nuclei compared to either gene alone (Fig. 8D, E). Collectively these data suggested that ZFP423 is able to co-regulate the effects of RA signaling both in vitro and in vivo, and support the hypothesis that changes in this signaling may cause alterations in the percentage of proliferative progenitors in developing cortex.
We then asked whether ZFP423 and TBR2 might functionally interact in regulating the RA activity. Tbr2 expression suppresses the RARE-Luc activity in P19 cells (Fig. 1A). However, this repression was relieved by co-transfecting growing amount of Zfp423 in a dose-dependent manner (Fig S4D). Considering that TBR2 directly represses endogenous Zfp423 expression (Fig. S2 and Fig. 5F), these observations confirm ZFP423 as one RA modulator directly regulated by TBR2.
Acute loss of Zfp423 impairs cortical development
Given the impact into neuronal differentiation of both Zfp423 overexpression and null mutation, we sought to evaluate whether it is acutely required in cortical progenitors by knocking-down (KD) its expression during corticogenesis. To this end, we introduced a Zfp423 shRNA or a control scramble shRNA construct into E13.5 cortical progenitors by in utero electroporation. Two days after, the Zfp423 KD cells displayed small but significant differences compared to the control cells in proliferation index, as assessed by the increase of Ki67 labeling (shZfp423: 51.8 ± 2% vs shScramble 47.9 ± 2.1%, p < 0.001 unpaired t test), and in the relative content of PAX6+ cells (shZfp423: 41.9 ± 2% vs shScramble 38.5 ± 1.2%, p < 0.05 unpaired t test) but not of TBR2+ INPs (shZfp423: 31.1 ± 2% vs shScramble 30 ± 2.3%) (Fig. S6A–C). These findings point to a change in the ratio between cell proliferation and differentiation upon Zfp423 KD. However, the overall small effect could indicate the presence of significant redundant molecular pathways.
We then assessed the general distribution of the GFP+ cells in the cortical tissue. Cells were counted in five bins along the cortical thickness (Fig. 9A–C). We found that Zfp423 KD cells were preferentially localized in the most apical bins (bin 1 (VZ): 29.7 ± 1.2% shZfp423 vs 21.4 ± 0.7% shScramble; bin 3 (apical IZ) 37.4 ± 0.9% shZfp423 vs 28 ± 1.5% shScramble) (Fig. 9C) while many control cells have already reached the most basal areas of the cortex (bin 4 (basal IZ): 5.3 ± 0.6% shZfp423 vs 14.5 ± 0.8% shScramble; bin 5 (CP): 2.1 ± 0.7% shZfp423 vs 8.7 ± 1.0% shScramble; unpaired t test p<0.001) (Fig. 9C). To further investigate this issue, we carried out immunostaining for the neuronal marker TUBB3 (Fig. S7) in the electroporated cortical tissues detecting significantly fewer GFP+ neuronal cells located in the TUBB3+ cortical plate in shZfp423 compared to control shRNA experiments (Fig. S7A–F). Collectively, these data indicate that Zfp423 loss-of-function inhibits or delays correct neuronal migration in the cortical plate. This impairment in neuronal positioning might indicate either a physical impairment of KD Zfp423 neuronal cells to migrate or a block in neuronal maturation to a migratory state. To address this issue, we performed immunostaining for SATB2 (Britanova et al., 2008) an early marker of post-mitotic neuronal cells committed to a cortical upper layer identity (Fig.S6 F). GFP/SATB2 double positive neuronal cells located in the SVZ-IZ compartment accounted for a smaller proportion of cells after Zfp423 silencing (64.6 ± 4.8% vs shScramble 75.7 ± 8.2%)(Fig. S6). This result strongly suggests that Zfp423 deficiency could delay the commitment to subtype identity as well as the neuronal cell migration.
Fig. 9. In vivo Zfp423 downregulation impairs neuronal maturation and migration.

(A-B) GFP immunolabelled coronal sections of E15.5 developing cortices targeted at E13.5 with control shRNA (shScramble) (A), Zfp423 shRNA (B). Sections were subdivided (red dashed lines) in five bins from apical to basal side to evaluate the radial migration of the GFP+ cells; bar = 50 µm. (C) The graph represents the percentage of GFP+ in each bin on the total for control (black bars) or knock down experiments (gray bars) shown as mean + S.D. with dots representing the eight biological replicates (independent electroporated embryos) (each is the mean of 3 quantification, technical replicates): bin 1 (VZ) **** p < 0.0001, bin 2 (SVZ) p = 0.2016, bin 3 (apical IZ) **** p < 0.0001; bin 4 (basal IZ) **** p < 0.0001, bin 5 (CP) **** p < 0.0001. All measurements statistically compared using unpaired t test. (D-F) GFP stained coronal sections of E17.5 developing cortices targeted at E13.5 with control shRNA (shScramble) (D), Zfp423 shRNA (E) or Zfp423 shRNA + Zfp423 full length (F). Sections were subdivided (red dashed lines) in six parts from apical to basal side to evaluate the migration of GFP+ cells; bar = 100 µm. (G) The graph represents the percentage of GFP+ in each bin on the total for control (black bars), knock down (gray bars) or rescue experiments (empty bars) shown as mean + S.D. with dots representing the eight biological replicates (independent electroporated embryos) (each is the mean of 3 quantification, technical replicates): bin 1: shScramble vs shZfp423 **** p < 0.0001, shScramble vs shZfp423+Zfp423 **** p < 0.0001, shZfp423 vs shZfp423+Zfp423 *** p = 0.0002; bin 2: shScramble vs shZfp423 **** p < 0.0001, shScramble vs shZfp423+Zfp423 ** p = 0.0012, shZfp423 vs shZfp423+Zfp423 **** p < 0.0001; bin 3: shScramble vs shZfp423 **** p < 0.0001, shScramble vs shZfp423+Zfp423 **** p < 0.0001, shZfp423 vs shZfp423+Zfp423 **** p < 0.0001; bin 4: shScramble vs shZfp423 *** p = 0.0007, shScramble vs shZfp423+Zfp423 **** p < 0.0001, shZfp423 vs shZfp423+Zfp423 * p = 0.0373; bin 5: shScramble vs shZfp423 **** p < 0.0001, shScramble vs shZfp423+Zfp423 **** p < 0.0001, shZfp423 vs shZfp423+Zfp423 p = 0.5628; bin 6: shScramble vs shZfp423 **** p < 0.0001, shScramble vs shZfp423+Zfp423 **** p < 0.0001, shZfp423 vs shZfp423+Zfp423 **** p < 0.0001; all measurements statistically compared using two-way ANOVA, Tukey’s multiple comparisons test.
We further assessed the distribution of Zfp423 KD cells four days after in utero electroporation (E18.5) (Fig. S8 and 9D–G). As seen after two days, also after four days from the surgery Zfp423 KD cells showed marked impairment in their outward migration towards cortical domains characteristic of differentiated cells (bins 4–6) and conversely an accumulation in the most internal parts (bins 1–3) (Fig. 9D, E, G). This effect is specifically due to loss of Zfp423 since the co-electroporation of Zfp423 full length (rescue) mitigate the phenotype (Fig. 9F, G) Intriguingly, the few Zfp423 KD cells that reached the cortical plate showed a strong nuclear labeling for the cortical upper layer neuronal marker SATB2 (Britanova et al., 2008) (Fig. S8B) similar to control cells (Fig. S8A). Conversely, the vast majority of the Zfp423 KD cells entrapped in the IZ showed a weak or absent SATB2 staining (Fig. S8B).
All together, these findings fit well with a general model in which ZFP423 promotes cortical formation and its loss leads to impairment in proliferation/differentiation and radial migration.
DISCUSSION
In this study, we identified the role played by TBR2 in repressing the RA signaling both in vitro and during corticogenesis. Interestingly, this agrees with the low RA activity displayed by TBR2+ INPs as scored by mapping the activity of a RA molecular sensor introduced in the cortical tissues by in utero electroporation. These findings reveal how different levels of RA activity can be associated to distinct cell types indicating a fine tuning of this signaling during corticogenesis.
Since Retinol (Vitamin A), the precursor of RA, is an essential nutrient in vertebrates and its deprivation during pregnancy causes dramatic problems to the fetus (Duester, 2008; Niederreither and Dollé, 2008), much interest has been focused on understanding the role of RA during development (Ross et al., 2000; Clagett-Dame et al., 2002). The relevance of RA for the activation of those genes responsible for the neuronal differentiation program has been demonstrated both cell lines and in progenitor cells resident in distinct organs (Novitch et al., 2003). Furthermore, RA-induced differentiation has been confirmed in many regions of developing central nervous system (CNS) and recently its role has suggested also in the cortex (Siegenthaler et al., 2009; Haushalter et al., 2017).
Since for technical reasons RA cannot be directly visualized, its concentration at anatomical level can only be inferred from the map of its source (e.g. retinaldehyde dehydorgenases, RALDHs, positive cells) (Crandall et al., 2011; Duester, 2008; Smith et al., 2001) or activation of the ligand binding domain of its receptor (Urquiza et al., 1999). Moreover, being a small amphipathic lipid, RA can rapidly diffuse over large distances, thus the utilization of target gene reporter activity can be the solution to evaluate the presence of RA signaling in a given cell or area. The two RARE-LacZ transgenic mouse lines available (RARE-hsp68lacZ (Rossant et al., 1991) and RARE/TK/β-gal (Balkan et al., 1992) displayed a low but reliable reporter activity in developing cortex (Haskell and LaMantia, 2005). However, because of the poor correlation between LacZ RNA initiation and steady-state β-galactosidase abundance, these reporters are not ideal to evaluate fine and highly dynamic changes in RA activity as it appears to happen during corticogenesis. By contrast, the approach of using a highly destabilized-GFP conveyed acutely by in utero electroporation provides a temporally resolved and sensitive tool to obtain a cell type specific map of dynamic RA signaling. It has been demonstrated that meninges secrete RA which could play a role in regulating developmental processes of the underlying cortex (Siegenthaler et al., 2009; Smith et al., 2001). Mouse with defective meningeal development (Foxc1) or RA synthesis (Rdh10) feature reduced neuronal rate production during corticogenesis coupled with an increase on RGC pool (Siegenthaler et al., 2009). Subsequent studies suggested that the heavy craniofacial defects in Rdh10 and Foxc1 are the direct effect of RA depletion, on cranial neural crest cells, that lead to cortical phenotype as consequence (Chatzi et al., 2013; Chatzi et al., 2011). Finally, recent works indicated that meningeal RA can be effective on RGC progenitors and neuronal cells stimulating their proliferation and migration towards their final site in the cortex, respectively (Haushalter et al., 2017a). Further investigation is needed to determine the roles of RA, from meninges but possibly from other sources like cerebrospinal fluid (Lehtinen and Walsh, 2011), in the regulation of corticogenesis.
Our RA mapping experiments provided evidence that, while a fraction of RGCs activate the RA sensor, only a minor subset of INPs does so. INPs are fated to further proliferate after RGC differentiation and therefore need to block strong neuronal commitment and radial migration. One possibility is that RA must be restrained at SVZ level because it is contributing to these late events. Based on our data, it is conceivable that INPs might be less sensitive to RA signaling trough a TBR2-dependent intrinsic mechanism and indeed, we found that TBR2, a highly specific INP transcription factors acts as repressor of RA activity and RA related target genes both in vitro and in vivo. Interestingly, our data are perfectly in accordance with a recently published work in which it has been demonstrated that absence of RA in Rdh10 null embryonic cortex resulted in alteration of RGCs in particular, with downstream effect on INP and neuron production (Haushalter et al., 2017b). Moreover, RA signaling has been shown in the adult lateral ventricle SVZ niche, the adult counterpart of the proliferative areas of the developing cortex (Haskell and LaMantia, 2005; Kriegstein and Alvarez-Buylla, 2009). However, transit amplifying cell progenitors, which share features of INPs including Tbr2 expression, were negative for the RA activity reporter (Haskell and LaMantia, 2005). This might suggest a consistent pattern of RA signaling conserved in the dorsal forebrain both in embryogenesis and adulthood.
Looking for the TBR2-dependent molecular mechanism(s) controlling RA signaling responsiveness, we identified Zfp423 as one of the most strongly de-repressed gene in the Tbr2 mutant mouse cerebral cortex (Sessa et al., 2017). ZFP423 is a 30-Zn-finger transcription factor that has been suggested as a critical component of the molecular machinery required for the RA-induced differentiation (Huang et al., 2009). In fact, ZFP423 associates with the RARα/RXRα nuclear receptor complex and is essential for target gene activation in response to retinoids. Silencing of ZNF423 (the human orthologue of murine Zfp423) in neuroblastoma cells results in resistance to RA-induced differentiation, whereas its overexpression leads to growth inhibition and enhanced differentiation (Huang et al., 2009). For these reasons, ZFP423 appeared as a strong candidate as a molecular linker between TBR2 and RA signaling.
Interestingly, we found Zfp423 to be expressed in cerebral cortex RGCs, specifically, those progenitor cells that could respond to RA signaling. Therefore, we investigated its role in this cellular context by examining cortical development in Zfp423 mutant embryos and by manipulating its expression acutely using in utero electroporation. Brain development in Zfp423 mutants shows variation across samples (Alcaraz et al., 2011), but showed consistent loss of proliferation markers by E16.5 and reduced cortical thickness by E18.5, with evidence of diminished or delayed differentiation. Zfp423 forced expression in the developing VZ leads to neuronal differentiation at the expense of mitotically active RGCs. Importantly, this effect was rescued by a concomitant block of RA signaling indicating that the function of ZFP423 is closely associated with this molecular pathway even in this cellular context. On the other hand, acute downregulation of Zfp423 by shRNA caused only a minimal effect in RGC proliferation activity suggesting either incomplete loss of ZFP423 protein in some cells during this time course or the presence of redundant molecular pathways to maintain RA signaling once established or a minimal role of it in this compartment. We also noted that a significant number of Zfp423-silenced neuronal progenitors failed to activate SATB2 a marker for cortical upper layer neuronal commitment. Thus, ZFP423 is necessary for correct RGC proliferation and differentiation possibly through a RA-dependent mechanism. However, ZFP423 has been shown in different biological systems to modulate other signaling pathways like the SMAD/BMP, NOTCH and SHH pathways (Hata et al., 2000; Ku et al., 2003; Masserdotti et al., 2010, Hong and Hamilton, 2016) and appears to sit at a nexus of several developmental processes based on its number of identified genetic interactions (Alcaraz et al., 2011). Moreover, it has been recently suggested that ZFP423 mutant forms, lacking either Zn finger 9–20 or Zn finger 28–30 and downstream C-term region, can induce impairment of cell cycle progression and increase in DNA damage in cerebellar progenitors (Casoni et al., 2017). We cannot exclude at this moment that Zfp423 might also act on these pathways during corticogenesis. Its importance is witnessed by the fact that ZNF423 mutations results ciliopathies showing either cerebellar vermis hypoplasia, Joubert Syndrome or nephronophthisis (Chaki et al., 2012). The defects in cortical development we describe here have further implications for human patients.
We confirmed Zfp423 dysregulation by in situ hybridization and qPCR in both Tbr2 loss- and gain-of-function approaches, providing strong evidence for the repressing and non-reciprocal activity of TBR2 on ZFP423. Moreover, we showed that TBR2 is directly repressing Zfp423 expression by binding to an evolutionary conserved intronic regulatory region (Z023), which contains 3 canonical T-box binding sites. In previous reports, TBR2 has been shown to act as a transcriptional activator as for instance switching on the expression of the Il2rb gene in the immune system (Intlekofer et al., 2005) or Mesp1 during cardiac development (Costello et al., 2011). However, it has also been shown to behave as a repressor of both pluripotency gene expression (e.g. Oct4 and Sox2) during definitive endoderm differentiation (Teo et al., 2011), Sox2 during hippocampal neurogenesis (Hodge, 2012) and neurodevelopmental related gens by the cooperation with NEUROG2 and KDM6B in embryonic cortex (Sessa et al., 2017). Overall, these observations suggest that TBR2 belongs to a subgroup of T-box containing transcription factors that have the potential to act as activator or repressor of gene transcription depending by the cellular context and the associated molecular partners (Naiche et al., 2005).
In conclusion, we suggested that RA signaling is differentially regulated in distinct cellular contexts during corticogenesis, namely VZ, SVZ, and neuronal cells. In this scenario, SVZ reduced responsiveness to RA is promoted by INP specific TBR2 expression, which modulates the pathway by repressing a co-activator of RA nuclear receptors, Zfp423. This function allows for RGC progeny to transit into an intermediate state in which cell division is permitted by transiently repressing RA-mediated events such as shape changing and migration behavior of the cells as recently reported (Haushalter et al., 2017). The mechanism herein proposed is likely only one of several pathways controlling the RA-induced neuronal differentiation and maturation, which await to be investigated. Future studies will also help to clarify the real and direct contributions of retinoic acid to cortical development, since both TBR2 and ZFP423 intersect other signaling pathways that may impact cellular responses to RA.
Supplementary Material
Table S1. List of primers for ChIP-qPCR.
Fig. S1. Mapping the activity of the RA sensor in P19 cells.
(A) In situ hybridization of Rara mRNA in developing mouse forebrain at E14.5 (from www.genepaint.org). (B) Luciferase assay in vitro in P19 cells and in extract of cortical (Ctx) and basal ganglia (BG) regions after transfection (P19 cells) or in vitro electroporation (brain regions) of either empty- and RARE-Luciferase construct. Quantification shown as mean + S.D. with dots representing the five biological replicates (independent cell or brain region lysates) (each is the mean of five measurements, technical replicates). (C) Outline of the in vitro experiment to test the RA sensor (RARE::dGFP) compared with a constitutive vector (pCAG::dGFP) in P19 cells before, during and after RA induction. (D-E) The activity of the constitutive pCAG::dGFP vector was not influenced by the presence of RA in the medium. (F-H) RARE controlled dGFP is undetectable before (F), and one day after the wash out of the RA addiction (H) while is evidently notable with the red labelling when RA was present in the medium (G); bar = 5 µm.
Fig. S2. TBR2 downregulates Zfp423 in vitro through binding an intronic ECR.
(A) Graph showing the percentage of the Zfp423 expression in P19 cells upon Tbr2 overexpression relative to control GFP. Quantification shown as mean + S.D. with dots representing the four biological replicates (independent RNA) (each is the mean of four PCR, technical replicates): Zfp423 *** p = 0.0004. All measurements statistically compared using unpaired t test. (B) Snapshot of the Evolutionary Conserved Regions (ECR) representation (ECR browser: http://ecrbrowser.dcode.org/)(Ovcharenko et al., 2004) of the Zfp423 genomic region. The alignment human-mouse of the amplicon Z023 is shown and the three putative T-box binding sites are highlighted by boxed.
Fig. S3. ZFP423 regulates location but not intensity of TBR2+ positive cells.
(A-B) TBR2 nuclear staining intensities are similar in cells from littermate control (+/+) and Zfp423 null (nur12/nur12) cortex at E14.5. Scale bars, 200 µm. (C-F) TBR2+ cells appear less frequent in intermediate zone (IZ) and cortical plate (CP) of Zfp423 null brains, especially at E14.5 (C-D). Reduced frequency of IZ Tbr2+ cells in Zfp423 mutant remains, but is less striking at E16.5 (E-F). Scale bars, 100 µm.
Fig. S4. TBR2-dependent downregulation of RA signaling is rescued by ZFP423 in P19 cells.
(A) Graph shows the luciferase activity of the RARE promoter without (grey bar) or with (black bar) RA normalized on the basal activity in GFP transfected cells with RA. Notably, ZFP423 is able to upregulate the RARE promoter as well as RARα. Quantification shown as mean + S.D. with dots representing the five biological replicates (independent cell lysates) (each is the mean of five measurements, technical replicates): -RA, GFP vs ZFP423 p > 0.9999, GFP vs RARα p > 0.9999, GFP vs RARαDN p > 0.9999; GFP vs RARαDN+ZFP423 p > 0.9999; +RA, GFP vs ZFP423 ****p < 0.0001, GFP vs RARα ****p < 0.0001, GFP vs RARαDN ****p < 0.0001, GFP vs RARαDN+ZFP423 p = 0.9545. All measurements statistically compared using two-way ANOVA, Sidak’s multiple comparisons test. (B) dsRed and GFP immunohistochemistry to evaluate the co-expression of both dsRed and destabilizedGFP (dGFP) in in utero electroporation of Zfp423-I-dsRed and RARE::dGFP, arrowheads indicate some double positive cells. (C) Graph illustrating the co-labelling rate. The double positive cells are the vast majority if normalized on total GFP+ cells in all the condition (gray bars). Conversely, double positive cells are only a subset of the total Red+ cells (black bars). However, the overexpression of Zfp423 grants an high percentage of RA responding cells compared to the control. Quantification shown as mean + S.D. with dots representing the six biological replicates (independent electroporated embryos) (each is the mean of three quantifications, technical replicates): Red+GFP+/All Red: pCAG::Red + RARE::dGFP vs pCAG::Zfp423-I-Red + RARE::dGFP **** p < 0.0001. pCAG::Red + RARE::dGFP vs pCAG::Zfp423-I-Red + RARE::dGFP p = 0.2304. All measurements statistically compared using two-way ANOVA, Sidak’s multiple comparisons test. (D) ZFP423 neutralizes the TBR2-dependent downregulation of the RARE promoter in presence of RA. Quantification (value relative to GFP only cells) shown as mean + S.D. with dots representing the five biological replicates (independent cell lysates) (each is the mean of five measurements, technical replicates): GFP(2µg) vs GFP(1µg)+Tbr2(1µg) ****p < 0.0001, GFP(2µg) vs GFP(0.5µg)+Tbr2(1µg)+Zfp423(0.5µg) p = 0.5461, GFP(2µg) vs Tbr2(1µg)+Zfp423(1µg) p = 0.9834, GFP(2µg) vs GFP(0.5µg)+Tbr2(0.5µg)+Zfp423(1µg) ** p < 0.0088, GFP(2µg) vs GFP(1µg)+Zfp423(1µg) *** p < 0.0001. All measurements statistically compared using one-way ANOVA, Dunnett’s multiple comparisons test.
Fig. S5. In vivo Zfp423 overexpression promotes RGC differentiation.
(A-E) Immunohistochemistry for GFP and PAX6 shows loss of PAX6+ cells in the ventricular zone electroporated with Zfp423 (B) and Rarα (C) differently from GFP (A) or RarαDN (D) alone and the Zfp423/RarαDN co-electroporation (E). (F-J) Zfp423, Rarα and RarαDN electroporations induce a disorganization of the INP layer as assessed by double GFP and TBR2 immunohistochemistry However the total number of Tbr2+ INPs is not statistically affected (Fig 7E); bar = 50 µm..
Fig. S6. Zfp423 downregulation slightly enhances proliferation of cortical progenitors.
(A-B) Cortical areas targeted with control shRNA (shScramble) (A) or Zfp423 shRNA (B) immunostained for GFP two days after electroporations. Magnifications of the dashed boxes of panel A and B represent areas of ventricular zone (VZ), subventricular zone (SVZ) and inner zone (IZ); GFP, PAX6 and KI67 immunostainings are shown in VZ; GFP, TBR2 and KI67 immunostainings are shown in the SVZ and finally GFP and Satb2 antibody labellings are shown for the intermediate zone IZ. (C) The graph indicates the percentage of the GFP+ double positive for KI67, PAX6 and TBR2 in total cortex of shScramble (black bars) or shZfp423 (gray bars) electroporated samples. Quantification shown as mean + S.D. with dots representing the eight biological replicates (independent electroporated embryos) (each is the mean of three quantifications, technical replicates): Ki67 *** p = 0.0002, PAX6 ** p = 0.0090, TBR2 p = 0.3166. All measurements statistically compared using unpaired t test. (D) The graph indicates the fraction of double positive cells for GFP and PAX6 or KI67 on the total GFP+ cells in ventricular zones of shScramble (black bars) or shZfp423 (gray bars) electroporated. Quantification shown as mean + S.D. with dots representing the ten biological replicates (independent electroporated embryos) (each is the mean of three quantifications, technical replicates): PAX6 p = 0.1256, Ki67 p = 0.9235. All measurements statistically compared using unpaired t test. (E) The graph indicates the fraction of double positive cells for GFP and TBR2 or KI67 over the total GFP+ cells in subventricular zones of shScramble (black bars) or shZfp423 (gray bars) electroporated. Quantification shown as mean + S.D. with dots representing the ten biological replicates (independent electroporated embryos) (each is the mean of three quantifications, technical replicates): TBR2 p = 0.5469, Ki67 p = 0.0580. All measurements statistically compared using unpaired t test. (F) The graph indicates the fraction of double positive cells for GFP and SATB2 in subventricular and intermediate zones of shScramble (black bars) or shZfp423 (gray bars) electroporated cortices. Quantification shown as mean + S.D. with dots representing the ten biological replicates (independent electroporated embryos) (each is the mean of three quantifications, technical replicates): SATB2 ** p = 0.0016. All measurements statistically compared using unpaired t test.
Fig. S7. Zfp423 downregulation does not induce ectopic neuronal differentiation
(A-F) Immunohistochemistry for GFP (A and D) and βIII-tubulin (TUBB3) (B and E) and their merge (C and F) in shScramble (A-C) or shZfp423 (D-E) electroporated cortices don’t show ectopic neural differentiation in VZ. To note, only in the control-electroporation experiment many GFP+ cells have reached the TUBB3+ cortical plate (C); bar = 50 µm.
Fig. S8. Zfp423 knock-down cells fail to complete neuronal maturation
(A-B) Cortical targeted areas with control shRNA (shScramble) (A) or Zfp423 shRNA (B) labelled with a GFP antibody four days after electroporations; bar = 100 µm.. Magnifications of the dashed boxes in panels A and B, GFP and SATB2 immunostainings in both cortical plate (CP) and intermediate zone (IZ) where SATB2+ staining results strong or weak, respectively; bar = 50 µm..
Highlights.
Retinoic acid signaling is low in SVZ of developing cortex
TBR2 suppresses Zfp423 expression, a RA co-factor, in SVZ progenitors
TBR2 transcription factor regulates retinoic acid signaling in cortical development
Acknowledgements
We thank R. Reed and V. Zappavigna for the Zfp423 cDNA and the RARE-luciferase reporter vector, respectively. A. Lombardo and L. Naldini are acknowledged for the CRE-expressing lentivirus. We thank Vania Broccoli and the members of his laboratory for helpful discussions. We thank Kevin Cao, Weica Xie and Isabella Sanchez for assistance with fluorescence imaging and quantification of cortical cells.
Funding
This work was supported by Telethon grant (GGP15096) and the Italian Ministry of Health (Young investigator grant # GR-2013-02355540) to A.S. grants from the National Institute of Neurological Disorders and Stroke (R01 NS060109 and R01 NS097534) to B.A.H.
Footnotes
Competing interests statement
The authors declare no competing financial interests.
References
- Alcaraz WA, Chen E, Valdes P, Kim E, Lo YH, Vo J and Hamilton BA Modifier genes and non-genetic factors reshape anatomical deficits in Zfp423-deficient mice. Human Molecular Genetics 20 (2011) 3822–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alcaraz WA, Gold DA, Raponi E, Gent PM, Concepcion D and Hamilton BA Zfp423 controls proliferation and differentiation of neural precursors in cerebellar vermis formation. Proc Natl Acad Sci USA 103 (2006), 19424–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold S, Huang G, Cheung A, Era T, Nishikawa S, Bikoff E, Molnar Z, Robertson E and Groszer M The T-box transcription factor Eomes/Tbr2 regulates neurogenesis in the cortical subventricular zone. Genes & Development 22 (2008)., 2479–2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balkan W, Klintworth G, Bock C and Linney E Transgenic mice expressing a constitutively active retinoic acid receptor in the lens exhibit ocular defects. Dev Biol 151(1992), 622–5. [DOI] [PubMed] [Google Scholar]
- Britanova O, de Juan Romero C, Cheung A, Kwan KY, Schwark M, Gyorgy A, Vogel T, Akopov S, Mitkovski M, Agoston D et al. Satb2 is a postmitotic determinant for upper-layer neuron specification in the neocortex. Neuron 57 (2008), 378–92. [DOI] [PubMed] [Google Scholar]
- Bultje R, Castaneda-Castellanos D, Jan L, Jan Y, Kriegstein A and Shi S Mammalian Par3 Regulates Progenitor Cell Asymmetric Division via Notch Signaling in the Developing Neocortex. Neuron 63 (2009), 189–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casoni F, Croci L, Bosone C, D’Ambrosio R, Badaloni A, Gaudesi D, et al. Zfp423/ZNF423 regulates cell cycle progression, the mode of cell division and the DNA-damage response in Purkinje neuron progenitors. Development 144 (2017), 3686–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaki M, Airik R, Ghosh AK, Giles RH, Chen R, Slaats GG, Wang H, Hurd TW, Zhou W, Cluckey A et al. Exome capture reveals ZNF423 and CEP164 mutations, linking renal ciliopathies to DNA damage response signaling. Cell 150 (2012), 533–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatzi C, Brade T, Duester G Retinoic acid functions as a key GABAergic differentiation signal in the basal ganglia. PLoS Biol (2011), April;9(4) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatzi C, Cunningham TJ, Duester G Investigation of retinoic acid function during embryonic brain development using retinaldehyde-rescued Rdh10 knockout mice. Dev Dyn (2013), September;242(9):1056–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y and Reese DH The retinol signaling pathway in mouse pluripotent P19 cells. J. Cell. Biochem 112 (2011), 2865–2872. [DOI] [PubMed] [Google Scholar]
- Cheng LE, Zhang J and Reed RR The transcription factor Zfp423/OAZ is required for cerebellar development and CNS midline patterning. DevBiol 307 (2007), 43–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clagett-Dame M and Deluca HF The role of vitamin A in mammalian reproduction and embryonic development. Annu Rev Nutr 22 (2002), 347–81. [DOI] [PubMed] [Google Scholar]
- Colombo E, Galli R, Cossu G, Gecz J and Broccoli V Mouse orthologue of ARX, a gene mutated in several X-linked forms of mental retardation and epilepsy, is a marker of adult neural stem cells and forebrain GABAergic neurons. Dev. Dyn 231(2004), 631–639. [DOI] [PubMed] [Google Scholar]
- Colombo E, Giannelli SG, Galli R, Tagliafico E, Foroni C, Tenedini E, Ferrari S, Ferrari S, Corte G, Vescovi A et al. Embryonic Stem-Derived Versus Somatic Neural Stem Cells: A Comparative Analysis of Their Developmental Potential and Molecular Phenotype. Stem Cells 24 (2006), 825–834. [DOI] [PubMed] [Google Scholar]
- Conlon FL, Fairclough L, Price BM, Casey ES and Smith JC Determinants of T box protein specificity. Development 12 (2001), 8, 3749–58. [DOI] [PubMed] [Google Scholar]
- Costa MR, Wen G, Lepier A, Schroeder T and Götz M Par-complex proteins promote proliferative progenitor divisions in the developing mouse cerebral cortex. Development 135 (2008), 11–22. [DOI] [PubMed] [Google Scholar]
- Costello I, Pimeisl I-M, Dräger S, Bikoff EK, Robertson EJ and Arnold SJ The T-box transcription factor Eomesodermin acts upstream of Mesp1 to specify cardiac mesoderm during mouse gastrulation. Nat Cell Biol 13(9) (2011),1084–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crandall JE, Goodman T, Mccarthy DM, Duester G, Bhide PG, Dräger UC and Mccaffery P Retinoic acid influences neuronal migration from the ganglionic eminence to the cerebral cortex. Journal of Neurochemistry, 119(4) (2011), 723–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duester G Retinoic acid synthesis and signaling during early organogenesis. Cell 134 (2008), 921–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari D, Binda E, De Filippis L and Vescovi AL Isolation of neural stem cells from neural tissues using the neurosphere technique. Curr Protoc Stem Cell Biol (2010) Chapter 2, Unit2D.6. [DOI] [PubMed]
- Florio M, Huttner WB Neural progenitors, neurogenesis and the evolution of the neocortex. Development 141(11) (2014), 2182–94 [DOI] [PubMed] [Google Scholar]
- Fietz S, Kelava I, Vogt J, Wilsch-Bräuninger M, Stenzel D, Fish J, Corbeil D, Riehn A, Distler W, Nitsch R et al. OSVZ progenitors of human and ferret neocortex are epithelial-like and expand by integrin signaling. Nat Neurosci, 13(6) (2010), 690–9 [DOI] [PubMed] [Google Scholar]
- Fietz SA and Huttner WB Cortical progenitor expansion, self-renewal and neurogenesis—a polarized perspective. Curr Opin Neurobiol 21 (2011), 23–35. [DOI] [PubMed] [Google Scholar]
- Gal JS, Morozov YM, Ayoub AE, Chatterjee M, Rakic P and Haydar TF Molecular and morphological heterogeneity of neural precursors in the mouse neocortical proliferative zones. J Neurosci 26 (2006), 1045–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Götz M, Hartfuss E and Malatesta P Radial glial cells as neuronal precursors: A new perspective on the correlation of morphology and lineage restriction in the developing cerebral cortex of mice. Brain Research Bullettin 57 (2002), 777–88. [DOI] [PubMed] [Google Scholar]
- Götz M and Huttner WB The cell biology of neurogenesis. Nat Rev Mol Cell Biol 6 (2005), 777–88. [DOI] [PubMed] [Google Scholar]
- Hansen DV, Lui JH, Parker PR and Kriegstein A Neurogenic radial glia in the outer subventricular zone of human neocortex. Nature 464 (2010), 554–561. [DOI] [PubMed] [Google Scholar]
- Haskell GT and LaMantia A-S Retinoic acid signaling identifies a distinct precursor population in the developing and adult forebrain. J Neurosci 25 (2005), 7636–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hata A, Seoane J, Lagna G, Montalvo E, Hemmati-Brivanlou A and Massagué J OAZ uses distinct DNA- and protein-binding zinc fingers in separate BMP-Smad and Olf signaling pathways. Cell 100 (2000), 229–40. [DOI] [PubMed] [Google Scholar]
- Haubensak W, Attardo A, Denk W and Huttner WB Neurons arise in the basal neuroepithelium of the early mammalian telencefalon: a major site of neurogenesis. PNAS, 101(9) (2004), 3196–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haushalter C, Schuhbaur B, Dollé P, Rhinn M Meningeal retinoic acid contributes to neocortical lamination and radial migration during mouse brain development. Biol Open 6(2) (2017), 148–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haushalter C, Asselin L, Fraulob V, Dollé P, Rhinn M Retinoic acid controls early neurogenesis in the developing mouse cerebral cortex. Dev Biol 430 (2017), 129–141. [DOI] [PubMed] [Google Scholar]
- Hebert J and Mcconnell SK Targeting of cre to the Foxg1 (BF-1) Locus Mediates loxP Recombination in the Telencephalon and Other Developing Head Structures. Dev Biol 222 (2000), 296–306. [DOI] [PubMed] [Google Scholar]
- Heng JI, Nguyen L, Castro DS, Zimmer C, Wildner H, Armant O, et al. Neurogenin 2 controls cortical neuron migration through regulation of Rnd2. Nature 455(7209) (2008), 114–8. [DOI] [PubMed] [Google Scholar]
- Hodge RD, Nelson BR, Kahoud RJ, Yang R, Mussar KE, Reiner SL, Hevner RF Tbr2 Is Essential for Hippocampal Lineage Progression from Neural Stem Cells to Intermediate Progenitors and Neurons. J Neurosci 32(18) (2012), 6275–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong CJ, Hamilton BA Zfp423 Regulates Sonic Hedgehog Signaling via Primary Cilium Function. PLoS Genet 12(10) (2016), e1006357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S, Laoukili J, Epping MT, Koster J, Hölzel M, Westerman BA, Nijkamp W, Hata A, Asgharzadeh S, Seeger RC et al. ZNF423 is critically required for retinoic acid-induced differentiation and is a marker of neuroblastoma outcome. Cancer Cell 15 (2009), 328–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Intlekofer AM, Takemoto N, Wherry EJ, Longworth SA, Northrup JT, Palanivel VR et al. Effector and memory CD8+ T cell fate coupled by T-bet and eomesodermin. Nat Imm 6 (2005), 1236. [DOI] [PubMed] [Google Scholar]
- Kang W, Wong L, Shi S and Hebert J The Transition from Radial Glial to Intermediate Progenitor Cell Is Inhibited by FGF Signaling during Corticogenesis. J Neurosci 29 (2009), 14571–14580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawauchi T Cellullar insights into cerebral cortical development: focusing on the locomotion mode of neuronal migration. Front Cell Neurosci 9 (2015), 394 Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konno D, Shioi G, Shitamukai A, Mori A, Kiyonari H, Miyata T and Matsuzaki F Neuroepithelial progenitors undergo LGN-dependent planar divisions to maintain self-renewability during mammalian neurogenesis. Nat Cell Biol 10 (2008), 93–101. [DOI] [PubMed] [Google Scholar]
- Kriegstein A and Götz M Radial glia diversity: A matter of cell fate. Glia 43 (2003), 37–43. [DOI] [PubMed] [Google Scholar]
- Kriegstein AR and Alvarez-Buylla A The Glial Nature of Embryonic and Adult Neural Stem Cells. Annual Rev Neurosci 32 (2009), 149–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ku M. c., Stewart S and Hata A Poly(ADP-ribose) polymerase 1 interacts with OAZ and regulates BMP-target genes. Biochem Biophys Res Commun 311 (2003), 702–7. [DOI] [PubMed] [Google Scholar]
- Lange C, Huttner W and Calegari F Cdk4/CyclinD1 Overexpression in Neural Stem Cells Shortens G1, Delays Neurogenesis, and Promotes the Generation and Expansion of Basal Progenitors. Cell Stem Cell 5 (2009), 320–331. [DOI] [PubMed] [Google Scholar]
- Lehtinen MK and Walsh CA Neurogenesis at the Brain-Cerebrospinal Fluid Interface. Annu Rev Cell Dev Biol 27 (2011), 25.1–25.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visel A, Thaller C and Eichele G Genepaint.org: an atlas of gene expression patterns in the mouse embryo. Nucleic Acids Res 32 (2004), D552–D556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lui JH, Hansen DV and Kriegstein AR Development and evolution of the human neocortex. Cell 146 (2011), 18–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maden M Retinoic acid in the development, regeneration and maintenance of the nervous system. Nat Rev Neurosci 8 (2007), 755–65. [DOI] [PubMed] [Google Scholar]
- Malatesta P, Hartfuss E and Götz M Isolation of radial glial cells by fluorescent-activated cell sorting reveals a neuronal lineage. Development 127 (2000), 5253–63. [DOI] [PubMed] [Google Scholar]
- Mao C-A, Kiyama T, Pan P, Furuta Y, Hadjantonakis A-K and Klein WH Eomesodermin, a target gene of Pou4f2, is required for retinal ganglion cell and optic nerve development in the mouse. Development 135 (2008), 271–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masserdotti G, Badaloni A, Song Green Y, Croci L, Barili V, Bergamini G, Vetter M and Consalez G (2010). ZFP423 coordinates Notch and BMP signaling, selectively upregulating Hes5 gene expression. Journal of Biological Chemistry, 285(40):30814–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda T and Cepko CL Controlled expression of transgenes introduced by in vivo electroporation. Proc Natl Acad Sci USA 104 (2007), 1027–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBurney MW RB Isolation of male embryonal carcinoma cells and their chromosome replication patterns. Dev Biol 89 (1982), 503–8. [DOI] [PubMed] [Google Scholar]
- Miyata T, Kawaguchi A, Saito K, Kawano M, Muto T and Ogawa M Asymmetric production of surface-dividing and non-surface-dividing cortical progenitor cells. Development 131 (2004), 3133–45. [DOI] [PubMed] [Google Scholar]
- Muroyama Y and Saito T Identification of Nepro, a gene required for the maintenance of neocortex neural progenitor cells downstream of Notch. Development 136 (2009), 3889–3893. [DOI] [PubMed] [Google Scholar]
- Naiche LA, Harrelson Z, Kelly RG and Papaioannou VE T-box genes in vertebrate development. Annu Rev Genet 39 (2005), 219–39. [DOI] [PubMed] [Google Scholar]
- Niederreither K and Dollé P Retinoic acid in development: towards an integrated view. Nat Rev Genet 9 (2008), 541–53. [DOI] [PubMed] [Google Scholar]
- Niwa H, Yamamura K and Miyazaki J Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene 108 (1991), 193–9. [DOI] [PubMed] [Google Scholar]
- Noctor S, Martínez-Cerdeño V and Kriegstein A Distinct behaviors of neural stem and progenitor cells underlie cortical neurogenesis. J Comp Neurol 508 (2008), 28–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noctor SC, Flint AC, Weissman TA, Dammerman RS and Kriegstein AR Neurons derived from radial glial cells establish radial units in neocortex. Nature 409 (2001), 714–20. [DOI] [PubMed] [Google Scholar]
- Noctor SC, Martínez-Cerdeño V, Ivic L and Kriegstein A Cortical neurons arise in symmetric and asymmetric division zones and migrate through specific phases. Nat Neurosci 7 (2004), 136–44. [DOI] [PubMed] [Google Scholar]
- Novitch BG, Wichterle H, Jessell TM and Sockanathan S A requirement for retinoic acid-mediated transcriptional activation in ventral neural patterning and motor neuron specification. Neuron 40 (2003), 81–95. [DOI] [PubMed] [Google Scholar]
- Ochiai W, Nakatani S, Takahara T, Kainuma M, Masaoka M, Minobe S, Namihira M, Nakashima K, Sakakibara A, Ogawa M et al. Periventricular notch activation and asymmetric Ngn2 and Tbr2 expression in pair-generated neocortical daughter cells. Mol Cell Neurosci 40 (2009), 225–33. [DOI] [PubMed] [Google Scholar]
- Ovcharenko I, Nobrega MA, Loots GG and Stubbs L ECR Browser: a tool for visualizing and accessing data from comparisons of multiple vertebrate genomes. Nucleic Acids Res 32 (2004), W280–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postiglione MP, Jüschke C, Xie Y, Haas GA, Charalambous C and Knoblich JA Mouse inscuteable induces apical-basal spindle orientation to facilitate intermediate progenitor generation in the developing neocortex. Neuron 72 (2011), 269–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reillo I, De Juan Romero C, Garcia-Cabezas MA and Borrell V A Role for Intermediate Radial Glia in the Tangential Expansion of the Mammalian Cerebral Cortex. Cereb Cortex 21(7) (2010), 1674–94 [DOI] [PubMed] [Google Scholar]
- Ross SA, McCaffery PJ, Drager UC and De Luca LM Retinoids in embryonal development. Physiol Rev 80 (2000), 1021–54. [DOI] [PubMed] [Google Scholar]
- Rossant J, Zirngibl R, Cado D, Shago M and Giguère V Expression of a retinoic acid response element-hsplacZ transgene defines specific domains of transcriptional activity during mouse embryogenesis. Genes & Development 5 (1991), 1333–44. [DOI] [PubMed] [Google Scholar]
- Saito T Efficient Gene Transfer into the Embryonic Mouse Brain Using in Vivo Electroporation. Developmental Biology 240 (2001), 237–246. [DOI] [PubMed] [Google Scholar]
- Saito T In vivo electroporation in the embryonic mouse central nervous system. Nat Protoc 1 (2006), 1552–8. [DOI] [PubMed] [Google Scholar]
- Schaeren-Wiemers N and Gerfin-Moser A A single protocol to detect transcripts of various types and expression levels in neural tissue and cultured cells: in situ hybridization using digoxigenin-labelled cRNA probes. Histochemistry 100 (1993), 431–40. [DOI] [PubMed] [Google Scholar]
- Schmittgen T and Livak K Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc 3 (2008), 1101–8. [DOI] [PubMed] [Google Scholar]
- Sessa A, Mao C, Colasante G, Nini A, Klein W and Broccoli V Tbr2-positive intermediate (basal) neuronal progenitors safeguard cerebral cortex expansion by controlling amplification of pallial glutamatergic neurons and attraction of subpallial GABAergic interneurons. Genes & Dev 24 (2010), 1816–1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sessa A, Mao C, Hadjantonakis A, Klein W and Broccoli V Tbr2 Directs Conversion of Radial Glia into Basal Precursors and Guides Neuronal Amplification by Indirect Neurogenesis in the Developing Neocortex. Neuron 60 (2008), 56–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sessa A, Ciabatti E, Drechsel D, Massimino L, Colasante G, Giannelli S, Satoh T, et al. The Tbr2 Molecular Network Controls Cortical Neuronal Differentiation Through Complementary Genetic and Epigenetic Pathways. Cereb Cortex 27(6) (2017), 3378–3396. [DOI] [PubMed] [Google Scholar]
- Shimojo H, Ohtsuka T and Kageyama R Oscillations in Notch Signaling Regulate Maintenance of Neural Progenitors. Neuron 58 (2008), 52–64. [DOI] [PubMed] [Google Scholar]
- Shitamukai A, Konno D and Matsuzaki F Oblique radial glial divisions in the developing mouse neocortex induce self-renewing progenitors outside the germinal zone that resemble primate outer subventricular zone progenitors. J Neurosci 31 (2011), 3683–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegenthaler J, Ashique A, Zarbalis K, Patterson K, Hecht J, Kane M, Folias A, Choe Y, May S and Kume T Retinoic Acid from the Meninges Regulates Cortical Neuron Generation. Cell 139 (2009), 597–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegenthaler JA and Pleasure SJ We have got you ‘covered’: how the meninges control brain development. Curr Opin Gen & Dev 21, 249–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith D, Wagner E, Koul O, McCaffery P and Dräger UC Retinoic acid synthesis for the developing telencephalon. Cereb Cortex 11 (2011), 894–905. [DOI] [PubMed] [Google Scholar]
- Teo AKK, Arnold SJ, Trotter MWB, Brown S, Ang LT, Chng Z, Robertson EJ, Dunn NR and Vallier L Pluripotency factors regulate definitive endoderm specification through eomesodermin. Genes & Development 25(3) (2011), 238–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyoda R, Assimacopoulos S, Wilcoxon J, Taylor A, Feldman P, Suzuki-Hirano A, Shimogori T and Grove EA FGF8 acts as a classic diffusible morphogen to pattern the neocortex. Development 137 (2010), 3439–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urquiza A. M. d., Solomin L and Perlmann T Feedback-inducible nuclear-receptor-driven reporter gene expression in transgenic mice. Proc Natl Acad Sci USA 96 (1999), 13270–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Tsai J-W, Lamonica B and Kriegstein AR A new subtype of progenitor cell in the mouse embryonic neocortex. Nat Neurosci 14(5) (2011), 555–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. List of primers for ChIP-qPCR.
Fig. S1. Mapping the activity of the RA sensor in P19 cells.
(A) In situ hybridization of Rara mRNA in developing mouse forebrain at E14.5 (from www.genepaint.org). (B) Luciferase assay in vitro in P19 cells and in extract of cortical (Ctx) and basal ganglia (BG) regions after transfection (P19 cells) or in vitro electroporation (brain regions) of either empty- and RARE-Luciferase construct. Quantification shown as mean + S.D. with dots representing the five biological replicates (independent cell or brain region lysates) (each is the mean of five measurements, technical replicates). (C) Outline of the in vitro experiment to test the RA sensor (RARE::dGFP) compared with a constitutive vector (pCAG::dGFP) in P19 cells before, during and after RA induction. (D-E) The activity of the constitutive pCAG::dGFP vector was not influenced by the presence of RA in the medium. (F-H) RARE controlled dGFP is undetectable before (F), and one day after the wash out of the RA addiction (H) while is evidently notable with the red labelling when RA was present in the medium (G); bar = 5 µm.
Fig. S2. TBR2 downregulates Zfp423 in vitro through binding an intronic ECR.
(A) Graph showing the percentage of the Zfp423 expression in P19 cells upon Tbr2 overexpression relative to control GFP. Quantification shown as mean + S.D. with dots representing the four biological replicates (independent RNA) (each is the mean of four PCR, technical replicates): Zfp423 *** p = 0.0004. All measurements statistically compared using unpaired t test. (B) Snapshot of the Evolutionary Conserved Regions (ECR) representation (ECR browser: http://ecrbrowser.dcode.org/)(Ovcharenko et al., 2004) of the Zfp423 genomic region. The alignment human-mouse of the amplicon Z023 is shown and the three putative T-box binding sites are highlighted by boxed.
Fig. S3. ZFP423 regulates location but not intensity of TBR2+ positive cells.
(A-B) TBR2 nuclear staining intensities are similar in cells from littermate control (+/+) and Zfp423 null (nur12/nur12) cortex at E14.5. Scale bars, 200 µm. (C-F) TBR2+ cells appear less frequent in intermediate zone (IZ) and cortical plate (CP) of Zfp423 null brains, especially at E14.5 (C-D). Reduced frequency of IZ Tbr2+ cells in Zfp423 mutant remains, but is less striking at E16.5 (E-F). Scale bars, 100 µm.
Fig. S4. TBR2-dependent downregulation of RA signaling is rescued by ZFP423 in P19 cells.
(A) Graph shows the luciferase activity of the RARE promoter without (grey bar) or with (black bar) RA normalized on the basal activity in GFP transfected cells with RA. Notably, ZFP423 is able to upregulate the RARE promoter as well as RARα. Quantification shown as mean + S.D. with dots representing the five biological replicates (independent cell lysates) (each is the mean of five measurements, technical replicates): -RA, GFP vs ZFP423 p > 0.9999, GFP vs RARα p > 0.9999, GFP vs RARαDN p > 0.9999; GFP vs RARαDN+ZFP423 p > 0.9999; +RA, GFP vs ZFP423 ****p < 0.0001, GFP vs RARα ****p < 0.0001, GFP vs RARαDN ****p < 0.0001, GFP vs RARαDN+ZFP423 p = 0.9545. All measurements statistically compared using two-way ANOVA, Sidak’s multiple comparisons test. (B) dsRed and GFP immunohistochemistry to evaluate the co-expression of both dsRed and destabilizedGFP (dGFP) in in utero electroporation of Zfp423-I-dsRed and RARE::dGFP, arrowheads indicate some double positive cells. (C) Graph illustrating the co-labelling rate. The double positive cells are the vast majority if normalized on total GFP+ cells in all the condition (gray bars). Conversely, double positive cells are only a subset of the total Red+ cells (black bars). However, the overexpression of Zfp423 grants an high percentage of RA responding cells compared to the control. Quantification shown as mean + S.D. with dots representing the six biological replicates (independent electroporated embryos) (each is the mean of three quantifications, technical replicates): Red+GFP+/All Red: pCAG::Red + RARE::dGFP vs pCAG::Zfp423-I-Red + RARE::dGFP **** p < 0.0001. pCAG::Red + RARE::dGFP vs pCAG::Zfp423-I-Red + RARE::dGFP p = 0.2304. All measurements statistically compared using two-way ANOVA, Sidak’s multiple comparisons test. (D) ZFP423 neutralizes the TBR2-dependent downregulation of the RARE promoter in presence of RA. Quantification (value relative to GFP only cells) shown as mean + S.D. with dots representing the five biological replicates (independent cell lysates) (each is the mean of five measurements, technical replicates): GFP(2µg) vs GFP(1µg)+Tbr2(1µg) ****p < 0.0001, GFP(2µg) vs GFP(0.5µg)+Tbr2(1µg)+Zfp423(0.5µg) p = 0.5461, GFP(2µg) vs Tbr2(1µg)+Zfp423(1µg) p = 0.9834, GFP(2µg) vs GFP(0.5µg)+Tbr2(0.5µg)+Zfp423(1µg) ** p < 0.0088, GFP(2µg) vs GFP(1µg)+Zfp423(1µg) *** p < 0.0001. All measurements statistically compared using one-way ANOVA, Dunnett’s multiple comparisons test.
Fig. S5. In vivo Zfp423 overexpression promotes RGC differentiation.
(A-E) Immunohistochemistry for GFP and PAX6 shows loss of PAX6+ cells in the ventricular zone electroporated with Zfp423 (B) and Rarα (C) differently from GFP (A) or RarαDN (D) alone and the Zfp423/RarαDN co-electroporation (E). (F-J) Zfp423, Rarα and RarαDN electroporations induce a disorganization of the INP layer as assessed by double GFP and TBR2 immunohistochemistry However the total number of Tbr2+ INPs is not statistically affected (Fig 7E); bar = 50 µm..
Fig. S6. Zfp423 downregulation slightly enhances proliferation of cortical progenitors.
(A-B) Cortical areas targeted with control shRNA (shScramble) (A) or Zfp423 shRNA (B) immunostained for GFP two days after electroporations. Magnifications of the dashed boxes of panel A and B represent areas of ventricular zone (VZ), subventricular zone (SVZ) and inner zone (IZ); GFP, PAX6 and KI67 immunostainings are shown in VZ; GFP, TBR2 and KI67 immunostainings are shown in the SVZ and finally GFP and Satb2 antibody labellings are shown for the intermediate zone IZ. (C) The graph indicates the percentage of the GFP+ double positive for KI67, PAX6 and TBR2 in total cortex of shScramble (black bars) or shZfp423 (gray bars) electroporated samples. Quantification shown as mean + S.D. with dots representing the eight biological replicates (independent electroporated embryos) (each is the mean of three quantifications, technical replicates): Ki67 *** p = 0.0002, PAX6 ** p = 0.0090, TBR2 p = 0.3166. All measurements statistically compared using unpaired t test. (D) The graph indicates the fraction of double positive cells for GFP and PAX6 or KI67 on the total GFP+ cells in ventricular zones of shScramble (black bars) or shZfp423 (gray bars) electroporated. Quantification shown as mean + S.D. with dots representing the ten biological replicates (independent electroporated embryos) (each is the mean of three quantifications, technical replicates): PAX6 p = 0.1256, Ki67 p = 0.9235. All measurements statistically compared using unpaired t test. (E) The graph indicates the fraction of double positive cells for GFP and TBR2 or KI67 over the total GFP+ cells in subventricular zones of shScramble (black bars) or shZfp423 (gray bars) electroporated. Quantification shown as mean + S.D. with dots representing the ten biological replicates (independent electroporated embryos) (each is the mean of three quantifications, technical replicates): TBR2 p = 0.5469, Ki67 p = 0.0580. All measurements statistically compared using unpaired t test. (F) The graph indicates the fraction of double positive cells for GFP and SATB2 in subventricular and intermediate zones of shScramble (black bars) or shZfp423 (gray bars) electroporated cortices. Quantification shown as mean + S.D. with dots representing the ten biological replicates (independent electroporated embryos) (each is the mean of three quantifications, technical replicates): SATB2 ** p = 0.0016. All measurements statistically compared using unpaired t test.
Fig. S7. Zfp423 downregulation does not induce ectopic neuronal differentiation
(A-F) Immunohistochemistry for GFP (A and D) and βIII-tubulin (TUBB3) (B and E) and their merge (C and F) in shScramble (A-C) or shZfp423 (D-E) electroporated cortices don’t show ectopic neural differentiation in VZ. To note, only in the control-electroporation experiment many GFP+ cells have reached the TUBB3+ cortical plate (C); bar = 50 µm.
Fig. S8. Zfp423 knock-down cells fail to complete neuronal maturation
(A-B) Cortical targeted areas with control shRNA (shScramble) (A) or Zfp423 shRNA (B) labelled with a GFP antibody four days after electroporations; bar = 100 µm.. Magnifications of the dashed boxes in panels A and B, GFP and SATB2 immunostainings in both cortical plate (CP) and intermediate zone (IZ) where SATB2+ staining results strong or weak, respectively; bar = 50 µm..