

Abstract
Messenger RNA (mRNA) has recently emerged as a promising class of nucleic acid therapy with the potential to induce protein production to treat and prevent a range of diseases. While significant progress has been made in the design of in vitro-transcribed mRNA with high potency, low-cost manufacturing, and low innate immunogenicity, the widespread use of mRNA as a therapeutic requires safe and effective in vivo delivery technologies. Libraries of ionizable lipid nanoparticles (LNPs) have been designed to encapsulate mRNA, prevent its degradation, and mediate intracellular delivery. However, these LNPs are typically characterized and screened in an in vitro setting, which may not fully replicate the biological barriers that they encounter in vivo. Therefore, new technologies that facilitate the screening of LNPs in an in vivo setting can enhance our fundamental understanding of how LNP structure affects in vivo mRNA delivery to target cells and tissues. Here, an in vivo platform to accelerate mRNA delivery screening was designed, consisting of a library of engineered LNPs that encapsulate functional, custom-designed barcoded mRNA (b-mRNA). These b-mRNA are similar in structure and function to regular mRNA and contain barcodes that enable their delivery to be quantified via deep sequencing. Using a mini-library of b-mRNA LNPs formulated via microfluidic mixing, we show that these different formulations can be pooled together, administered intravenously into mice as a single pool, and their delivery to multiple organs (liver, spleen, brain, lung, heart, kidney, pancreas, and muscle) can be quantified simultaneously using deep sequencing. In the context of liver and spleen delivery, LNPs that exhibited high b-mRNA delivery also yielded high luciferase expression, indicating that this platform can identify lead LNP candidates as well as optimal formulation parameters for in vivo mRNA delivery. Interestingly, LNPs with identical formulation parameters that encapsulated different types of nucleic acid barcodes (b-mRNA versus a DNA barcode) altered in vivo delivery, suggesting that the structure of the barcoded nucleic acid affects LNP in vivo delivery. This platform, which enables direct barcoding and subsequent quantification of a functional mRNA itself, can accelerate the in vivo screening and design of LNPs for mRNA therapeutic applications such as CRISPR/Cas9 gene editing, mRNA vaccination, and other mRNA-based regenerative medicine and protein replacement therapies.
Keywords: mRNA, nanoparticle, gene delivery, gene therapy, nucleic acid delivery
Graphical abstract
INTRODUCTION
Messenger RNA (mRNA), which offers amplified production of therapeutic proteins through rapid and repeated translation in cells, has recently garnered significant attention as a therapeutic for the treatment or prevention of a range of diseases1–7. This is due, in part, to significant improvements in in vitro transcription that has enabled the development of mRNA with high potency, low-cost manufacturing, and low innate immunogenicity for in vivo delivery8–9. mRNA offers several advantages over the delivery of DNA to produce therapeutic proteins. One major benefit is that mRNA does not need to cross the nuclear barrier in cells to induce protein expression9. Therefore, mRNA can be transfected more efficiently than plasmid DNA, especially for slowly dividing cells10. Additionally, by not needing to reach to the cell nucleus, mRNA does not bear the risk of insertional mutagenesis and carcinogenesis associated with genomic integration6. mRNA delivery also offers several advantages over the direct delivery of proteins, as the large size, instability, and high production costs of proteins hinder their use in vivo11–12. The promise of mRNA as a new class of genetic medicine has led to significant investments in its commercial development – including companies such as Moderna, CureVac AG, and BioNTech11, 13–14 – with ongoing clinical trials focused on vaccination, cancer immunotherapy, and protein replacement15–17. While significant progress has been made in the design of in vitro-transcribed mRNA, the widespread use of mRNA as a therapeutic requires safe and effective delivery technologies6. mRNA is 105–106 Dalton in size and approximately three to four orders of magnitude larger than small molecules that diffuse into cells6. Furthermore, mRNA is highly negatively charged and thus repulses the anionic cell membrane8. Naked mRNA is also inherently unstable and quickly degraded by RNases8, 18–19.
Ionizable lipid nanoparticles (LNPs) have been engineered to encapsulate and protect nucleic acids – including mRNA – from degradation and mediate their intracellular delivery20. Of note, an LNP-based small interfering RNA (siRNA) drug developed by Alnylam was approved by the U.S. Food and Drug Administration in 201821. Several potent ionizable lipids have been synthesized using various approaches, including rational design approaches where the lipid head and tail structures are systematically varied22–26, as well as through the creation of large combinatorial libraries of lipid-like materials27–29. In addition to an ionizable lipid, LNPs are commonly formulated with three excipients: (i) cholesterol, which enhances the stability of the LNP bilayer and promotes membrane fusion30; (ii) a phospholipid, which fortifies the LNP bilayer structure and also aids in endosomal escape31; and (iii) a lipid-polyethylene glycol (PEG) conjugate, which inserts into the LNP bilayer and provides a PEG coating that reduces LNP aggregation and nonspecific endocytosis by immune cells32.
While LNPs have demonstrated significant promise for nucleic acid delivery applications, their therapeutic potential is limited by inefficient delivery to target cells and tissues in vivo. This is due, in part, to an incomplete understanding of how LNP physicochemical properties affect in vivo delivery33. The effects of LNP physicochemical properties are typically characterized and screened in an in vitro or ex vivo setting, and LNP structural and pKa criteria have been shown to predict delivery to particular organs in vivo20, 24. However, it is challenging to fully replicate the biological barriers that affect the biological fate of LNPs in vivo – including anatomical structures, circulating cells, formation of the nanoparticle protein corona, and physiological forces – in in vitro and ex vivo experiments33–34. Therefore, testing LNP formulations in vivo, rather than in vitro or ex vivo, is generally considered the standard for identifying lead LNP formulations for different applications. To quantify LNP delivery in vivo, one approach is to use LNPs that encapsulate fluorescently labeled nucleic acids22, 35–37. After LNP administration, tissue biodistribution of those fluorescently labeled nucleic acids from different tissue samples can be identified via fluorescence readouts. However, due to the limitations in spectral resolution for microscopy, the number of fluorophores - and thus the number of LNP types - that can be quantified in single animals is limited to only a few. Therefore, new approaches that facilitate the high-throughput screening of LNPs in an in vivo setting can enhance our fundamental understanding of how LNP structure affects in vivo mRNA delivery to target cells and tissues.
Recently, novel approaches have emerged to facilitate the high-throughput screening of nanoparticles (NPs) in an in vivo setting, leveraging various technologies including mass cytometry, DNA barcoding (b-DNA), and high-throughput sequencing38–40. In the context of mass cytometry approaches - where current instruments permit up to 50 metal isotope labels to be detected simultaneously in single cells – have been developed to enable high-throughput quantification of gold nanoparticles in single cells as a means to identify novel NP-based vaccines to target dendritic cells in vivo38. In addition to mass cytometry, b-DNA in tandem with PCR and deep sequencing has been utilized to accelerate drug discovery41. Rather than testing compounds individually, many DNA-tagged compounds can be administered in a single pool, and compounds that interact with the target can be identified by their b-DNA using deep sequencing41. This b-DNA concept has recently been applied to LNP delivery in the context of identifying barcoded NPs that target tumors39 as well as those that deliver nucleic acid therapeutics in vivo40, 42.
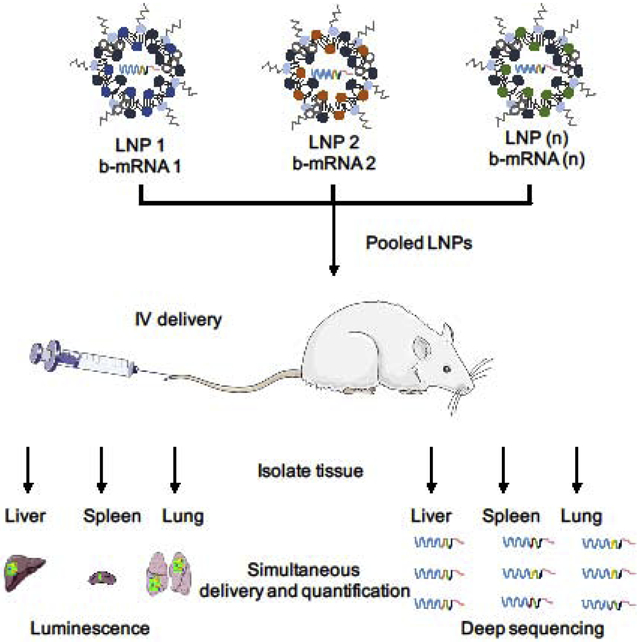
In the context of mRNA LNP delivery screening in vivo, an ideal approach would be to leverage a functional mRNA with a barcoded region in its 3’ untranslated region (UTR) that can be quantified directly using deep sequencing, rather than the encapsulation of additional b-DNA that may potentially alter LNP structure and subsequent in vivo delivery42–43. Towards this end, we have designed an in vivo delivery platform consisting of a library of engineered LNPs that encapsulate functional, custom-designed barcoded mRNA (b-mRNA, Fig. 1). These b-mRNA are similar in structure and function to regular mRNA, and contain barcodes and unique molecular identifier (UMI) that enables LNP in vivo delivery to be quantified via deep sequencing (Fig. 1B). We formulated a mini-library of LNPs via microfluidic mixing, where each LNP formulation encapsulated a unique b-mRNA (Fig. 1C). We show that different b-mRNA LNP formulations can be pooled together, simultaneously administered intravenously into mice, and LNP delivery to multiple organs can be quantified using deep sequencing (Fig. 1D). Our deep sequencing results were validated via LNP delivery of commercially available luciferase mRNA, indicating that this platform can be utilized to effectively identify lead LNP formulations for mRNA delivery in vivo to organs such as the liver and spleen. Additionally, we show that different types of nucleic acid cargo (b-mRNA versus a DNA barcode) altered LNP delivery in vivo, suggesting that the incorporation of different nucleic acid barcode structures within LNPs can affect their in vivo fate. This delivery platform, where functional mRNAs are barcoded and encapsulated in LNPs, can accelerate in vivo screening and the design of LNPs for mRNA gene therapy.
Fig. 1. Schematic of lipid nanoparticles (LNPs) encapsulating barcoded mRNA (b-mRNA) for accelerated in vivo delivery screening.
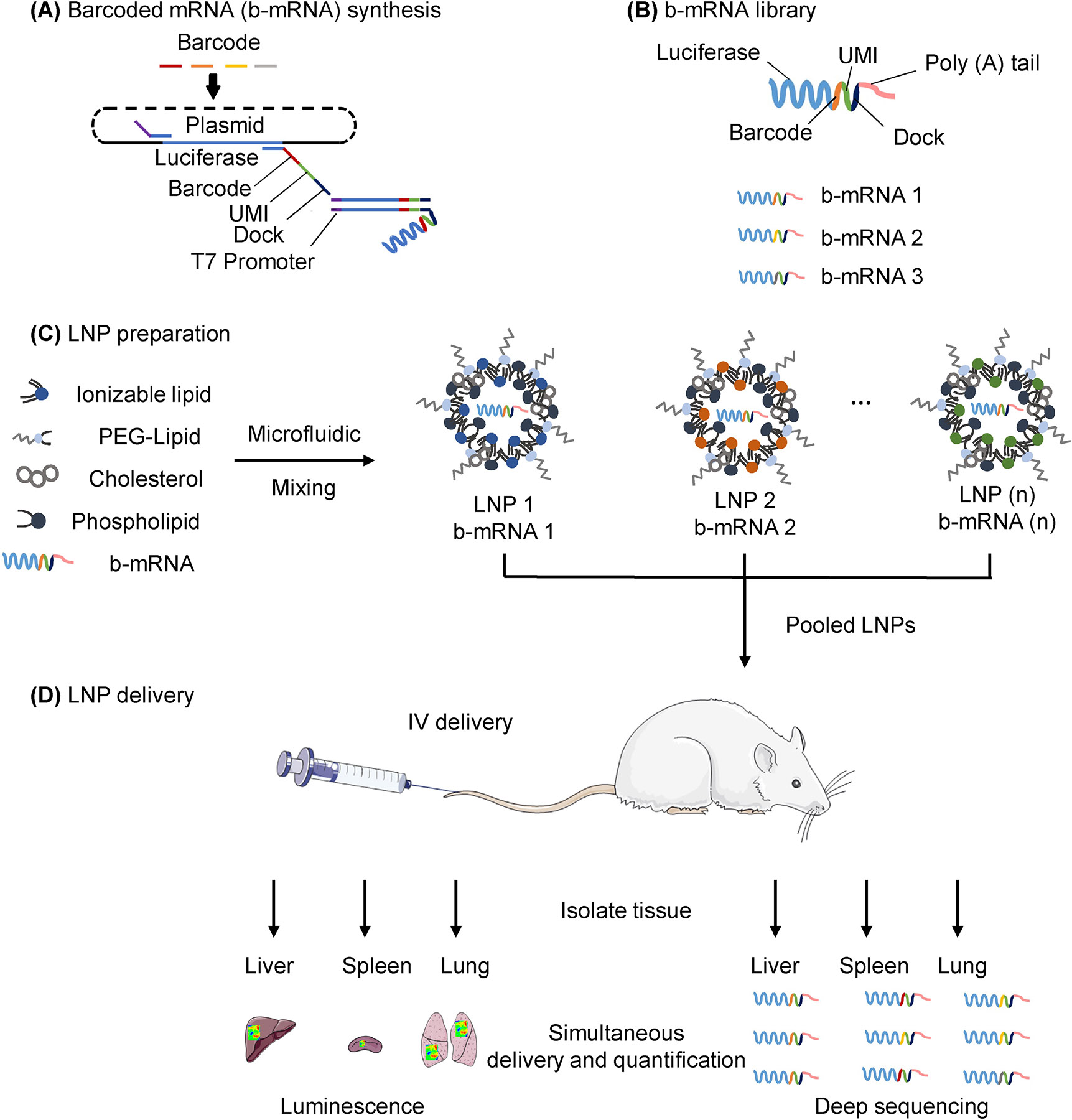
(A) b-mRNA templates were generated by polymerase chain reaction (PCR) using a plasmid vector template coding for the luciferase reporter gene luc2. For downstream processing, a T7 promoter sequence was added to 5’ end of the luc2 template on the forward primer, and a LNP-specific barcode, a unique molecular identifier (UMI), and a PCR handle/dock were added to the 3’ UTR of luc2 on the reverse primer. Subsequently, a library of b-mRNA was generated by in vitro transcription (IVT) using those b-mRNA templates. (B) b-mRNA includes a region coding for luciferase, a barcode sequence, a 10-nucleotide unique molecular identifier (UMI), and a poly(A) tail. (C) LNPs were formulated via microfluidic mixing, and each LNP formulation encapsulated unique b-mRNA. (D) Different LNP formulations were then pooled together and administered intravenously to C57BL/6 mice. Organs were harvested 4 hours post injection, and b-mRNA delivery was quantified using both whole-organ luminescence imaging and deep sequencing.
RESULTS AND DISCUSSION
Barcoded mRNA (b-mRNA) Synthesis
To synthesize in vitro transcribed b-mRNA (Fig. 1A), DNA templates were designed that included the following four necessary components: (i) a T7 promoter in the 5’ untranslated region (UTR) to initiate in vitro transcription, (ii) a PCR handle at the 3’ UTR for downstream polymerase chain reaction (PCR) amplification, (iii) a barcode sequence for quantification of in vitro transcribed b-mRNA during analysis by deep sequencing, and (iv) a unique molecular identifier (UMI) to avoid duplication during deep sequencing (Fig. 1A). These DNA templates were used for in vitro transcription to produce b-mRNA with dual functions: (i) the luciferase sequence enables b-mRNA to be translated and produce luciferase protein, (ii) the barcode and UMI sequences enable identification and quantification of b-mRNA through deep sequencing (Fig. 1B). Due to the ease of output measurements, luciferase mRNA has become one of the most commonly utilized sequences for gene delivery24, 44–48. Therefore, luciferase mRNA was chosen as a target sequence for the b-mRNA design.
Previous research demonstrates that mRNA modifications can enhance stability while suppressing innate immune responses and subsequently improving transfection49–52. Therefore, to assess the potency of b-mRNA with various modifications, we performed in vitro transcription to produce b-mRNA containing two different modifications, pseudouridine (ψ) or 5-methylcytosine (m5C). To compare the transfection efficiency between ψ modified b-mRNA and m5C modified b-mRNA, both b-mRNA were encapsulated in a previously optimized LNP for mRNA delivery53. As a positive control, Trilink-mRNA, the commercially available gold standard mRNA24, 45–47, was encapsulated in the identical formulation. In vitro transfection results showed that m5C modified b-mRNA induced higher luciferase expression than ψ modified b-mRNA (Fig. S1A). Therefore, we performed all subsequent experiments using m5C modified b-mRNA, unless otherwise noted. The b-mRNA synthesis protocol was shown to be reproducible, and in vitro transcribed m5C b-mRNA consistently produced full-length polyadenylated transcripts with minimum batch-to-batch variability (Fig. S1B and Fig. S1C).
b-mRNA LNP Formulation and Characterization
To determine whether b-mRNA can be encapsulated within LNPs, we utilized a well-characterized LNP formulation previously reported for mRNA delivery to the liver53. In this study, this formulation is referred to as F13 (Table 1). In brief, the formulation consisted of a well-characterized ionizable lipid (C12–200), helper lipid (DOPE), cholesterol, and a PEG-lipid conjugate (C14-PEG2000), which was mixed with the acidic aqueous phase containing b-mRNA in a microfluidic channel54 (Fig. 2A). Cryogenic-transmission electron microscopy (Cryo-TEM) micrographs confirmed that LNPs had a spherical shape and consist of a multilamellar structure (Fig. 2B). Furthermore, dynamic light scattering (DLS) indicated that LNPs with a mean hydrodynamic diameter of 83.36 nm were formed after microfluidic mixing (Fig. 2C), which was similar in size to previously reported LNPs encapsulating luciferase mRNA53. Collectively, these results indicate that b-mRNA can be encapsulated within LNPs that are similar in structure and size to previously reported mRNA delivery systems.
Table 1:
Formulation details for all LNPs
| Formulation | lonizable lipid:mRNA ratio | ||||
|---|---|---|---|---|---|
| Ionizable lipid | Phospholipid | Cholesterol | PEG-Lipid | ||
| F01 | 2.5:1 | 40 | 4 | 53.5 | 2.5 |
| F02 | 2.5:1 | 60 | 10 | 29.5 | 0.5 |
| F03 | 7.5:1 | 40 | 28 | 28.5 | 3.5 |
| F04 | 10:1 | 40 | 28 | 29.5 | 2.5 |
| F05 | 12.5:1 | 40 | 22 | 35.0 | 3.0 |
| F06 | 7.5:1 | 35 | 22 | 40.0 | 3.0 |
| F07 | 10:1 | 35 | 22 | 39.5 | 3.5 |
| F08 | 10:1 | 30 | 16 | 51.0 | 3.0 |
| F09 | 5:1 | 30 | 16 | 51.5 | 2.5 |
| F10 | 7.5:1 | 40 | 16 | 42.5 | 1.5 |
| F11 | 5:1 | 35 | 16 | 46.5 | 2.5 |
| F12 | 7.5:1 | 35 | 16 | 46.5 | 2.5 |
| F13 | 10:1 | 35 | 16 | 46.5 | 2.5 |
| F14 | 12.5:1 | 35 | 16 | 46.5 | 2.5 |
| F15 | 15:1 | 35 | 16 | 46.5 | 2.5 |
| F16 | 25:1 | 35 | 16 | 46.5 | 2.5 |
Fig. 2. Formulation and characterization of b-mRNA LNPs.
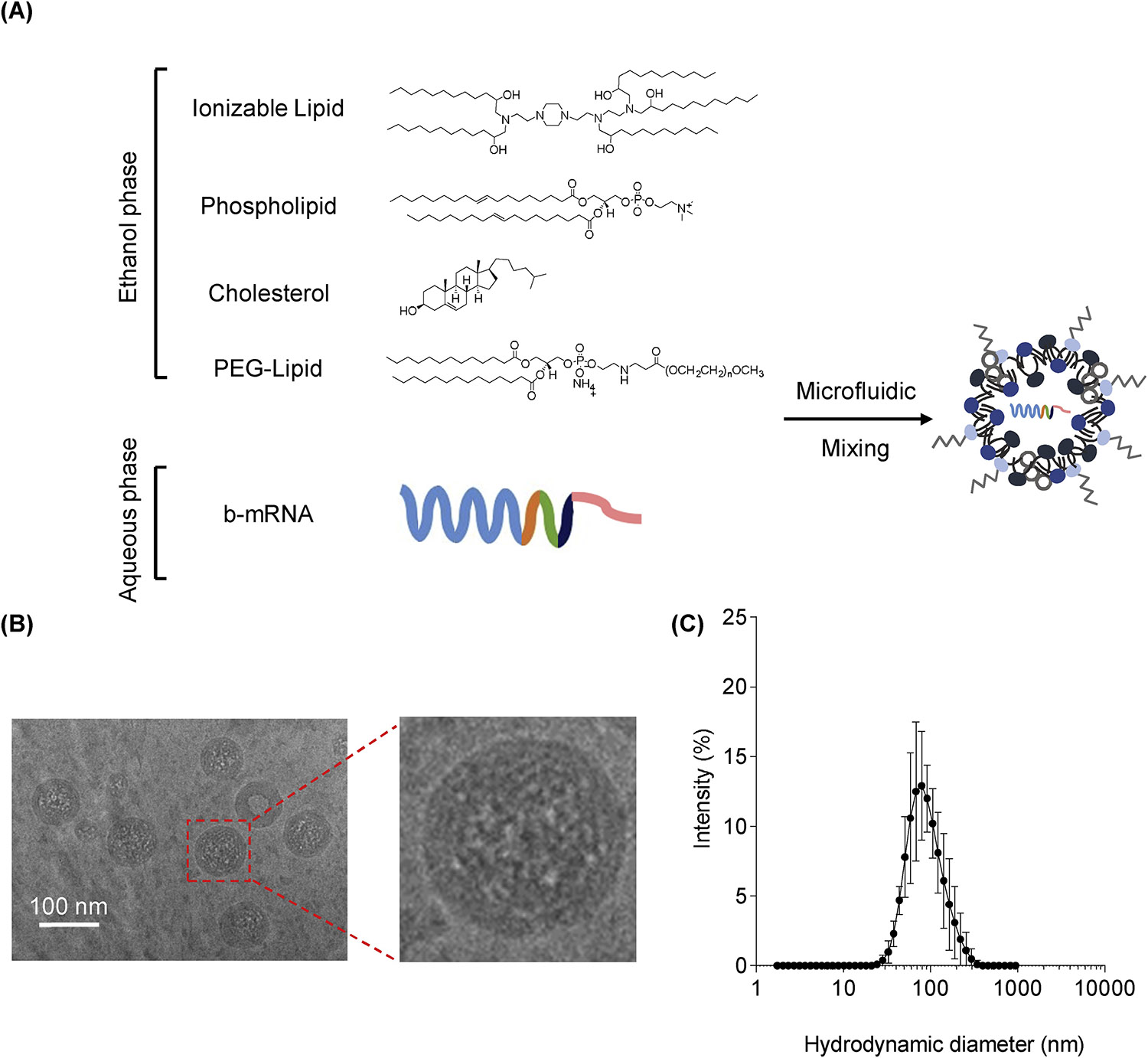
(A) LNPs were formulated via microfluidic mixing of an aqueous phase of b-mRNA and an ethanol phase of ionizable lipids, phospholipids, cholesterol, and a lipid-polyethylene glycol (PEG) conjugate. (B) Representative cryogenic-transmission electron microscopy image of LNPs encapsulating b-mRNA (scale bar = 100 nm). (C) Hydrodynamic diameter measurements of LNPs encapsulating b-mRNA quantified by dynamic light scattering.
In Vivo LNP Delivery and Dose-Dependent b-mRNA Detection
To assess whether b-mRNA can be delivered in vivo to the liver of mice and quantified using deep sequencing, five identical LNP formulations that each encapsulated different b-mRNA were pooled together - at different mRNA doses for each LNP formulation (17–1000 ng mRNA per LNP formulation) - and administered intravenously via tail vein injection into mice. 4 hours post-injection, the liver was harvested from mice and LNP b-mRNA delivery was quantified by deep sequencing. Doses as low as 17 ng of total b-mRNA were detected using deep sequencing (Fig. 3A), indicating that b-mRNA delivered using LNPs can be quantified at low doses. In a recent study, LNPs were systemically injected into mice at total mRNA doses as high as ~80 μg (4 mg/kg)22. Given that we were able to detect LNP doses as low as 17 ng total b-mRNA via deep sequencing, this platform can potentially allow for several thousand unique b-mRNA LNP formulations to be administered into mice and screened for delivery. b-mRNA LNP delivery was also shown to be dose-dependent, as LNPs delivered at a lower total b-mRNA dose resulted in lower sequencing reads and overall delivery to the liver, while LNPs delivered at higher doses resulted in higher sequencing reads and overall delivery (Fig. 3A). Furthermore, a linear correlation (R2 = 0.9646) between barcode delivery to the liver and total b-mRNA dose administered was found (Fig. 3B), further indicating that the b-mRNA quantification using deep sequencing is accurate. These results indicate that (i) unique b-mRNA LNPs can be pooled together, delivered in vivo, and quantified using deep sequencing, and (ii) deep sequencing can be used to accurately quantify b-mRNA LNP delivery across a broad range of injected b-mRNA doses.
Fig. 3. b-mRNA LNPs accelerate in vivo liver and spleen delivery screening and the identification of lead formulations.
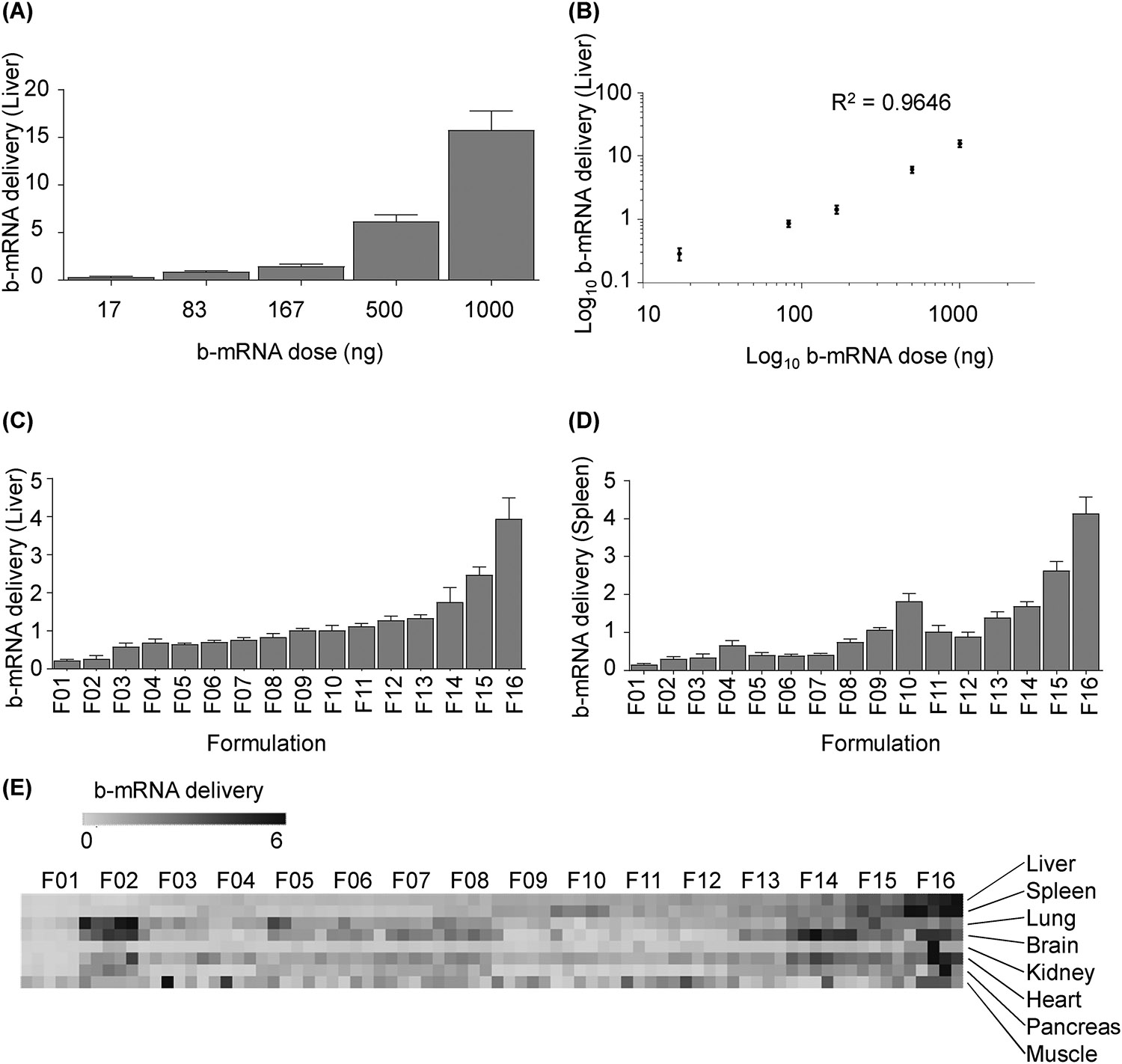
(A) LNP formulations with identical lipid and excipient composition but different b-mRNA were pooled at varying dosages and administered intravenously to C57BL/6 mice. 4 hours post injection, delivery of each b-mRNA LNP to the liver was quantified. N = 4 mice per group. (B) In vivo standard curve of b-mRNA delivery to the liver at a range of dosages showed a linear regression (R2 = 0.9646). (C-E) 16 LNP formulations (F01-F16) were engineered by varying the content of ionizable lipid, phospholipid (DOPE), cholesterol, and lipid-anchored PEG (C14-PEG2000). A 0.25 μg dose of each b-mRNA LNP was pooled and administered intravenously as a single injection. 4 hours post injection, b-mRNA delivery to the liver (C), spleen (D), and other organs (E) were quantified. N = 4 mice per group. Heat map (E) representing delivery to different tissue samples were created using Morpheus software. Darker clusters were designated as higher delivery whereas lighter clusters were designated as lower delivery. Within the heat map, the delivery of different LNP formulations within the same organ (left to right) can be compared, but the delivery of the same LNP formulation across different organs (top to bottom) cannot be compared. Data plotted as mean ± SD. Method to calculate b-mRNA delivery is explained in detail in the experimental section. R2 value was calculated based on linear regression model.
Simultaneous Delivery Screening of Multiple b-mRNA LNP Formulations In Vivo
After demonstrating the feasibility of the delivery system, we investigated whether several different b-mRNA LNP formulations can be screened simultaneously for in vivo delivery to various organs in mice. We formulated a mini-library of 16 different LNPs that were previously evaluated for in vivo mRNA delivery53 - now encapsulating b-mRNA instead of mRNA - as a means to validate the b-mRNA LNP screening platform (Table 1). 16 different b-mRNA LNP formulations were characterized by hydrodynamic diameter, polydispersity, and encapsulation efficiency (Table 2). The hydrodynamic diameter of all LNPs were between 74.42 nm and 90.77 nm, while their polydispersity ranged from 0.174 to 0.233 (Table 2). 13 of the 16 formulations possessed surface charge values between 0 mV and −10 mV, while the remaining 3 formulations had greater negative charge values (between −10 mV and −20 mV) (Table 2). Additionally, efficient b-mRNA encapsulation rates (over 85%) were observed in 11 of the 16 LNP formulations (Table 2). The 16 LNP formulations, each containing a unique b-mRNA, were then pooled and injected intravenously into mice at a dose of 0.25 μg total b-mRNA for each LNP formulation. 4 hours post-injection, organs (the liver, spleen, lung, brain, kidney, heart, pancreas, and muscle) were harvested from mice and LNP b-mRNA delivery was quantified using deep sequencing. We found a broad range in delivery of different b-mRNA LNPs to the liver (Fig. 3C), spleen (Fig. 3D), and other tissues (Fig. 3E and Fig. S2). The method of b-mRNA quantification was adapted from a previous report42 and explained in more detail in the MATERIALS AND METHODS. In brief, counts for each LNP formulation, per tissue, were normalized to the non-injected LNP pool. By using this quantification method, it is important to note that the delivery of different LNP formulations within the same organ can be compared, but the delivery of the same LNP formulation across different organs cannot be compared. Therefore, in the heat map (Fig. 3E), it is possible to compare delivery of different LNPs to a single tissue and determine the top-performing LNPs for each organ. However, it is not possible to assess if a given LNP formulation exhibits superior delivery to one organ compared to another.
Table 2:
Characterization of b-mRNA encapsulated LNPs
| Formulation | Hydrodynamic Diameter (nm) | PDI | Zeta Potential (mV) | Encapsulation efficiency (%) |
|---|---|---|---|---|
| F01 | 82.40 | 0.198 | −14.70 | 67.5 |
| F02 | 90.77 | 0.226 | −16.70 | 33.7 |
| F03 | 78.53 | 0.224 | −2.65 | 48.9 |
| F04 | 81.49 | 0.202 | −5.76 | 92.8 |
| F05 | 74.42 | 0.226 | −2.10 | 95.7 |
| F06 | 82.03 | 0.221 | −3.39 | 90.7 |
| F07 | 79.23 | 0.212 | −4.34 | 49.9 |
| F08 | 83.25 | 0.205 | −5.08 | 91.3 |
| F09 | 81.40 | 0.174 | −9.04 | 92.7 |
| F10 | 75.58 | 0.208 | −8.16 | 89.1 |
| F11 | 83.55 | 0.203 | −11.70 | 89.8 |
| F12 | 88.09 | 0.224 | −7.53 | 88.4 |
| F13 | 83.36 | 0.204 | −3.77 | 87.4 |
| F14 | 77.32 | 0.214 | −4.23 | 76.6 |
| F15 | 84.71 | 0.233 | −4.22 | 88.7 |
| F16 | 80.85 | 0.223 | −4.01 | 86.1 |
Some LNPs demonstrated similar behavior regarding b-mRNA delivery to different tissues. For example, F14-F16 showed higher b-mRNA delivery to most tissues (the liver, spleen, brain, heart, kidney, and pancreas) compared to other LNPs. Additionally, F11 to F16 were formulated with C12–200:b-mRNA weight ratios varying between 5:1 to 25:1, and we observed enhanced b-mRNA delivery to the liver and spleen with increased C12–200:mRNA ratios. Together, these data confirm that the b-mRNA platform can be used to screen several different LNP formulations in vivo simultaneously, and potentially identify lead LNPs for optimal mRNA delivery. While this mini-library of 16 LNP formulations serves as a proof-of-concept to validate the in vivo screening approach, the ease of synthesizing unique b-mRNA can be exploited to formulate and screen larger libraries of b-mRNA LNPs in vivo.
b-mRNA LNP Delivery Measurements Are Comparable to LNP-Mediated mRNA Transfection In Vivo
Given that b-mRNA was also designed to encode for the reporter protein firefly luciferase, we next assessed whether our relative b-mRNA LNP delivery measurements from deep sequencing were similar to functional in vivo luciferase expression readouts in mice. Two LNP formulations (F01 and F13) were selected for in vivo luciferase expression studies (Fig. 4), as F13 had higher relative b-mRNA delivery to the liver and spleen than F01 (Fig. 3C). Therefore, if the b-mRNA in vivo screen is accurate in terms of relative LNP delivery measurements, mice injected with F13 should have higher luciferase expression in the liver and spleen than those injected with F01 at the same total b-mRNA dose. F01 and F13 were separately administrated to two groups of mice, and 4 hours post-injection luciferase expression from different organs was quantified by an in vivo imaging system (IVIS, Fig 4A). Similar to a previous study53, high luciferase expression was observed in the liver and spleen (Fig. 4A). In some organ samples, luminescence signal was below the instrument threshold and was not captured by the IVIS28, 53, 55. Luciferase expression in the liver (Fig. 4B) and spleen (Fig. 4C) was higher in mice treated with F13 – which had higher relative b-mRNA delivery to the liver and spleen – than mice treated with F01.
Fig. 4. Lead LNPs identified from the delivery screen induce greater in vivo luciferase expression in the liver and spleen, and greater EPO production in mice.
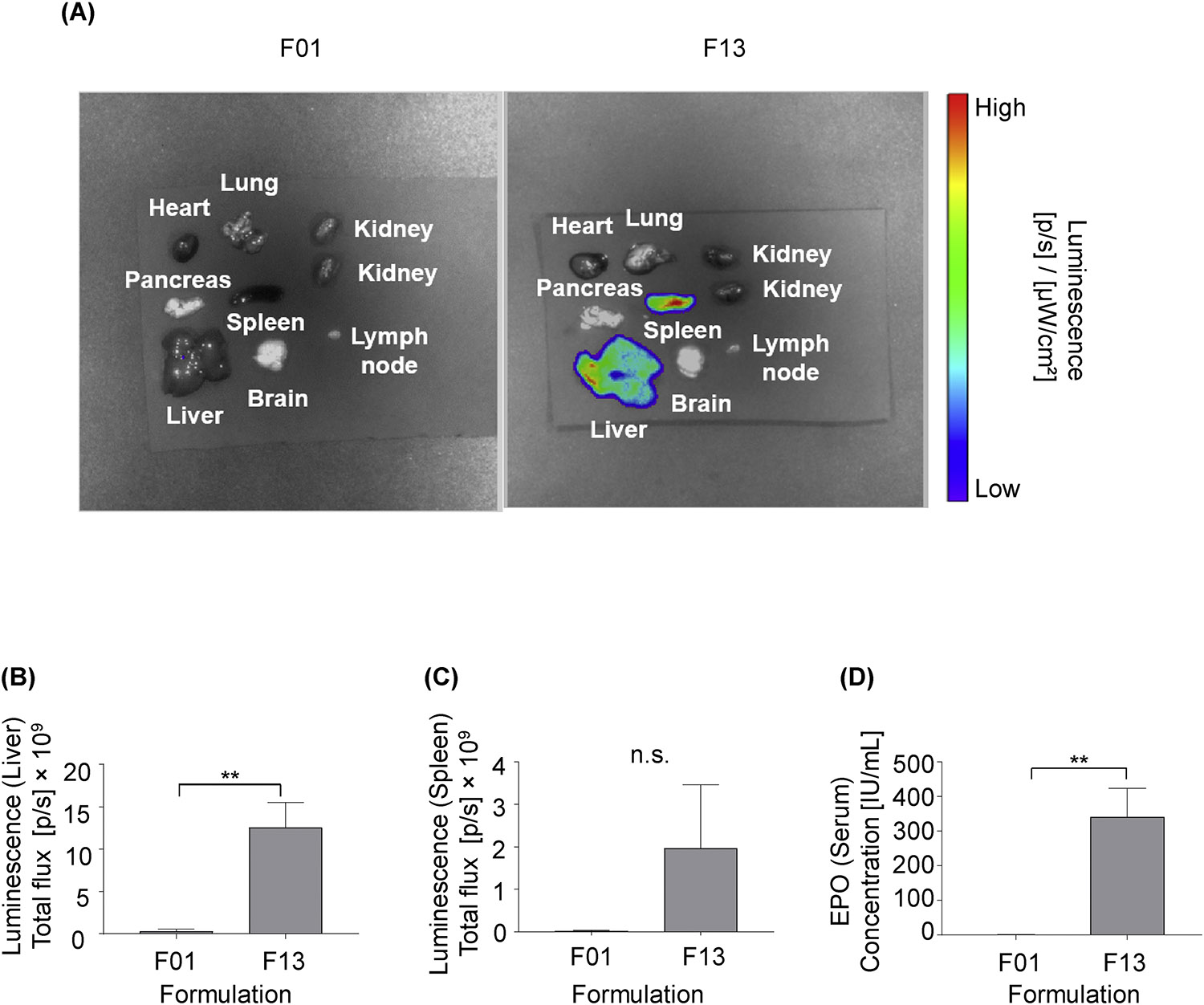
(A-C) C57BL/6 mice were intravenously injected with either LNP formulations F01 or F13 (5 μg b-mRNA per injection). 4 hours post administration, organs were harvested from mice, and their luminescence was measured by IVIS imaging. N = 3 mice per group. (A) Representative images of luminescence detection in organs from mice treated with either LNP formulations F01 or F13. (B, C) Total luminescent flux from two organs of interest, the liver and spleen, were quantified in (B) and (C) respectively. (D). C57BL/6 mice were intravenously injected with either the F01 or F13 LNP formulation that encapsulated human erythropoietin (EPO) mRNA (5 μg EPO mRNA per injection). 4 hours post injection, serum samples were collected from mice and their EPO concentrations were determined by an enzyme-linked immunosorbent assay (ELISA). N = 3 mice per group. Data were plotted as mean ± SD. N.S. denotes not significant, **P < 0.01 by t-test
To assess if the b-mRNA deep sequencing screening results were similar to the delivery of mRNAs beyond those encoding for luciferase, we next tested our system using another mRNA, human erythropoietin (EPO) mRNA. We chose EPO mRNA because systemic administration of LNPs encapsulating EPO mRNA results in a sustained expression of EPO protein in the serum53, 56; therefore, it potentially has therapeutic potential to be used as an alternative approach to EPO protein infusion for the treatment of anemia, myelodysplasia, and schizophrenia52, 57–58. To assess for EPO mRNA delivery, the same two LNP formulations (F1 and F13) used in the luciferase mRNA delivery assay were formulated with EPO mRNA and were separately injected into two groups of mice. 4 hours after administration, serum samples were collected and EPO concentrations were determined using an enzyme-linked immunosorbent assay (ELISA). Results indicated that F13 was significantly more potent than F1 (Fig. 4D), similar to the previous luciferase mRNA delivery results (Fig. 4A & Fig. 4B). Collectively, these results indicate that (i) b-mRNA LNPs can be quantified for relative delivery using deep sequencing and functional luciferase expression using IVIS, (ii) LNPs that yield higher relative delivery - measured using deep sequencing - also induced higher luciferase expression, and (iii) this screening platform can potentially be utilized to predict the potency of LNPs that encapsulate different therapeutic mRNAs.
To further validate our screening platform for functional mRNA delivery, LNPs from the initial screen were formulated with a commercially available mRNA encoding for luciferase (Trilink-mRNA)24, 45–47. Two LNPs with low relative b-mRNA delivery (F01, F06) and three LNPs with high relative delivery (F09, F13, F16) were then formulated with Trilink-mRNA encoding for firefly luciferase and were injected separately into 5 groups of mice. After 4 hours, 8 tissue samples (liver, spleen, kidney, lung, brain, pancreas, heart, and muscle) were harvested and luciferase expression was measured by IVIS (Fig. 5A). Similar to previous in vivo luciferase studies with b-mRNA LNPs (Fig. 4), F13 encapsulating Trilink-mRNA induced higher luciferase expression than F01 encapsulating Trilink-mRNA in both the liver (Fig 5B) and spleen (not statistically significant, P = 0.149; Fig 5C). These results suggest that LNPs encapsulating commercially available mRNA are comparable to b-mRNA LNPs in terms of function, and can potentially be utilized to validate results from in vivo b-mRNA LNP screening.
Fig. 5. LNPs encapsulating widely used, commercially available luciferase mRNA are comparable in vivo to b-mRNA LNPs.
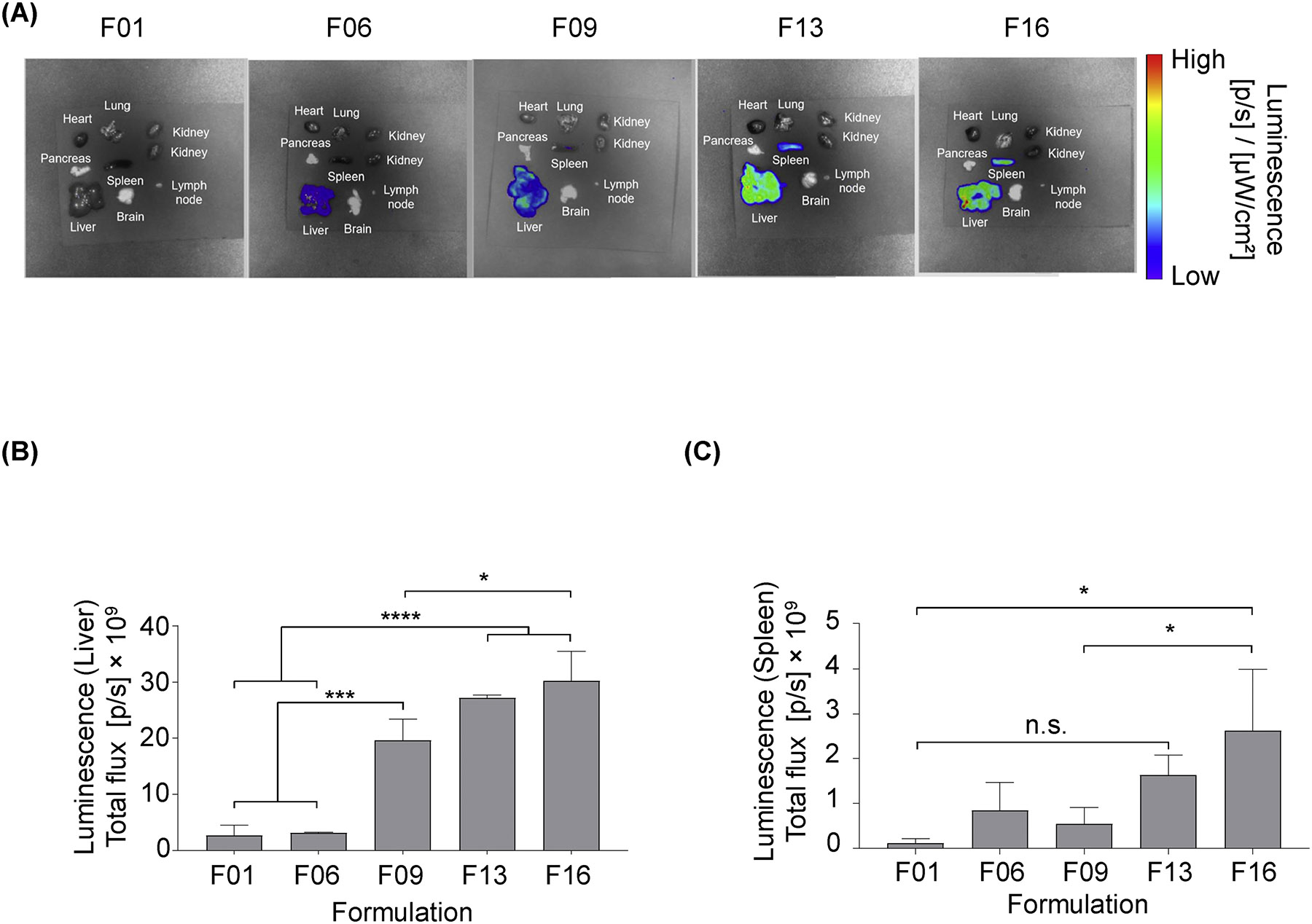
5 different LNP formulations (F01, F06, F09, F13, F16) were formulated with commercially available luciferase mRNA (TrilinkmRNA). C57BL/6 mice were intravenously injected with individual LNP formulations (5 μg Trilink-mRNA per injection). 4 hours post administration, organs were harvested from mice, and their luminescence was measured by IVIS imaging. N = 3 mice per group. (A) Representative images of luminescence detection in organs from mice treated with 5 different LNP formulations (F01, F06, F09, F13, F16). (B,C) Total luminescent flux from two organs of interest, the liver and spleen, were quantified in (B) and (C) respectively. Data were plotted as mean ± SD. N.S. denotes not significant, *P < 0.05, ***P < 0.001, ****P < 0.0001 by ANOVA with post-hoc Tukey-Kramer.
Barcoded Nucleic Acid Structure Impacts LNP Biodistribution In Vivo
The structure of different nucleic acid cargo (e.g. DNA, siRNA, mRNA) encapsulated within LNPs has previously been shown to play an important role in the LNP formulation process, requiring different types and ratios of ionizable lipids and excipients that consequently affect the physical properties of LNPs24, 59–61. A recent study showed that when different nucleic acid cargo (i.e. siRNA or mRNA) were encapsulated in the same LNP formulation, dramatic changes were found in terms of LNP size as well as the spatial location of various components (e.g. cholesterol, helper lipid, and PEG)43, indicating that the structure of the nucleic acid cargo encapsulated within LNPs ultimately affects LNP structure. However, how these structural changes can affect LNP delivery in vivo was not studied in the same report. Therefore, we compared our b-mRNA system to a previously studied b-DNA system to assess how different therapeutic cargos (i.e. b-DNA versus b-mRNA) affect LNP delivery62.
To assess this, LNPs used for b-mRNA delivery were formulated with b-DNA used in a previous report40. In brief, b-DNA included universal primer sites, a 10-nucleotide barcode region, and a 10-nucleotide UMI region to minimize PCR bias (Fig. 6A). We formulated the 16 LNPs that were used previously to encapsulate b-mRNA (Table 1), now encapsulating 16 different b-DNAs (Fig. 6A). As anticipated, switching nucleic acid cargo from b-mRNA to b-DNA in LNPs altered their hydrodynamic diameter and PDI for all 16 formulations (Fig. S3). To evaluate the delivery of b-DNA LNPs, all 16 b-DNA LNPs were pooled and administrated to mice intravenously. 4 hours post-injection, both the liver and spleen were isolated, and delivery of each b-DNA LNP formulation was quantified using deep sequencing in a similar manner to b-mRNA LNP delivery discussed previously (Fig. 6B and Fig. 6C).
Fig. 6. Encapsulation of barcoded DNA (b-DNA) versus b-mRNA in LNPs alters in vivo delivery.
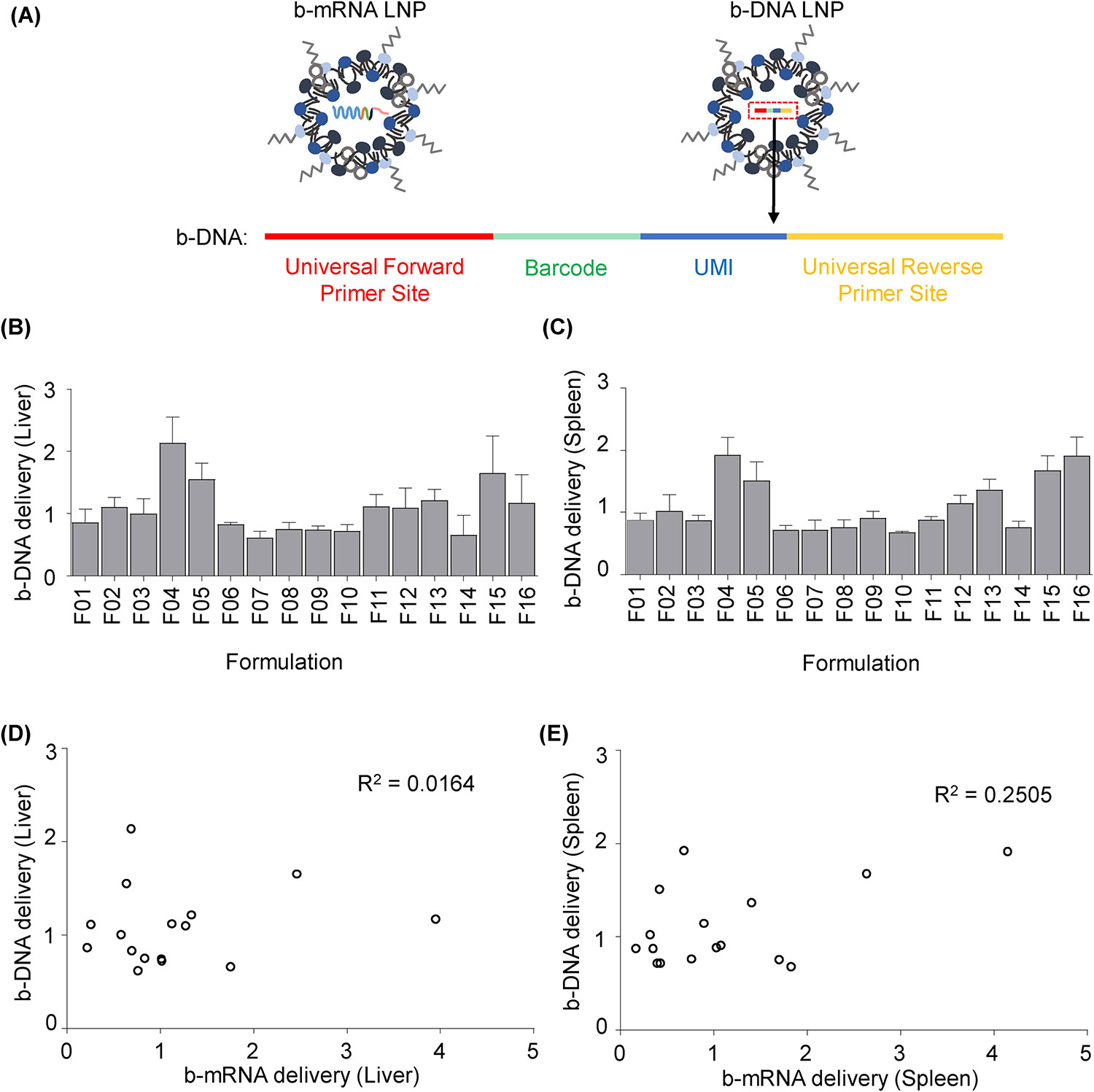
(A) 16 LNP formulations used in this study were now used to each encapsulate unique b-DNA instead of b-mRNA. b-DNA contained universal primer sites, a 10-nucleotide barcode sequence, and a 10-nucleotide UMI region to minimize polymerase chain reaction (PCR) bias. (B-C) 16 b-DNA LNP formulations were pooled (1 μg b-DNA per injection for each formulation) and administered to C57BL/6 mice intravenously. 4 hours post injection, b-DNA delivery to the liver (B) and spleen (C) was quantified. N = 4 mice per group. (D-E) In vivo delivery of 16 b-mRNA LNP formulations was plotted against the delivery of 16 b-DNA LNP formulations. Method to calculate b-DNA delivery is explained in detail in the experimental section. R2 values were calculated based on a linear regression model. Data were plotted as mean ± SD.
When b-DNA was encapsulated in LNPs, F04 was identified as one of the lead formulations for both liver and spleen delivery (Fig. 6B and Fig. 6C). However, when b-mRNA was encapsulated in the LNP, F04 exhibited lower delivery compared to several other formulations (Fig. 3C and Fig. 3D). In order to better understand these differences, delivery of 16 b-mRNA LNPs was plotted against the delivery of 16 b-DNA LNPs to the liver (Fig. 6D) and spleen (Fig. 6E). Weak correlations between b-DNA delivery and b-mRNA delivery to both the liver (R2 = 0.0164) and spleen (R2 = 0.2505) were found based off a linear regression model, demonstrating that differences in nucleic acid cargo can alter LNP delivery in vivo. This result was supported in recent studies, as altering LNP nucleic acid cargo has been shown to dramatically impact LNP physical characteristics43. For instance, the location of all components (ionizable lipid, excipient, and nucleic acid) within LNPs may affect the amount of PEG chains exposed on the LNP surface, thus affecting LNP interactions with serum proteins and ultimately altering LNP delivery32, 43, 63.
The use of b-DNA has previously enabled rapid, high-throughput in vivo screening of LNPs for small nucleic acid delivery, such as siRNA and sgRNA42, 64. However, predicting the functionality of a therapeutic mRNA using LNPs containing small nucleic acid has potential challenges. One potential challenge is that b-DNA is relatively similar in length to siRNA and sgRNA but is orders of magnitude smaller than mRNA. Therefore, an alteration in cargo from a shorter b-DNA to a longer mRNA can alter the fundamental structure and physical properties of the LNP formulation43. By contrast, b-mRNA by design has a similar size to a therapeutic mRNA, and therefore may minimize changes in LNP physical properties and ultimately delivery. To assess this, we directly compared the predictability of Trilink-mRNA LNP delivery to either b-mRNA LNP delivery or b-DNA LNP delivery, for both the liver (Fig. 7A and Fig. 7C) and spleen (Fig. 7B and Fig. 7D). By plotting b-mRNA delivery versus mRNA transfection in vivo, we found a strong correlation for the b-mRNA system, where higher b-mRNA delivery generally corresponded to higher mRNA transfection (Fig 7A and Fig. 7B). By contrast, the correlation was less clear for the b-DNA system by plotting b-DNA delivery versus mRNA transfection (Fig. 7C and 7D).
Fig. 7. Comparison of b-mRNA system versus b-DNA system to predict functional mRNA delivery in vivo.
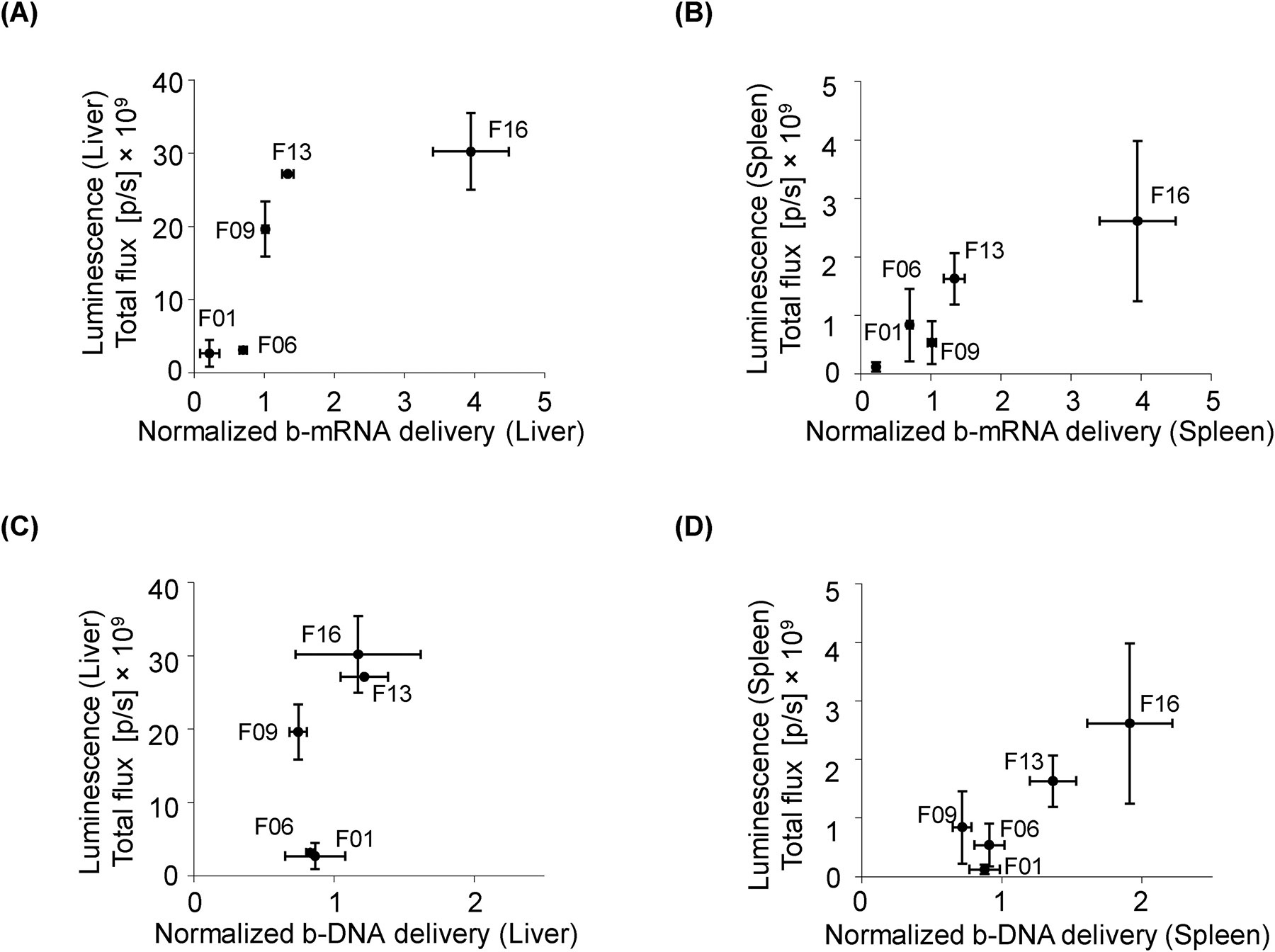
(A, B) b-mRNA LNP delivery was plotted against luciferase expression in the liver (A) and spleen (B) of luciferase mRNA LNP-treated mice. (C, D) Similarly, b-DNA LNP delivery was plotted against luciferase expression in the liver (C) and spleen (D) of luciferase mRNA LNP-treated mice. Data were plotted as mean ± SD.
After comparison of the b-mRNA (Fig. 3C,D) and b-DNA screening results (Fig. 6B,C), we identified two formulations (F4 and F13) that showed contradictory results in terms of delivery (Fig. 8 A–D). Based on the b-mRNA screening results, F13 delivery to the liver (Fig. 8A) and spleen (Fig. 8B) was significantly greater than F04. By contrast, the b-DNA screening results demonstrated that F13 delivery to the liver (Fig. 8C) and spleen (Fig. 8D) were significantly less than F04. To determine which screening platform provided a more reliable prediction for functional mRNA delivery, F4 and F13 LNPs were formulated with EPO mRNA and were separately injected to two groups of mice. Quantification of serum EPO concentration 4 hours after administration showed that F13 was significantly more potent than F4 in terms of increasing EPO production (Fig. 8E), indicating that b-mRNAs – which more closely mimic the structure of therapeutic mRNAs – can potentially better predict functional mRNA delivery in vivo. Collectively, our b-mRNA system, either alone or in combination with other novel systems that co-deliver b-DNA with mRNA65, may provide an accurate means for accelerated in vivo mRNA delivery screening.
Fig. 8. b-mRNA LNPs predict functional mRNA delivery.
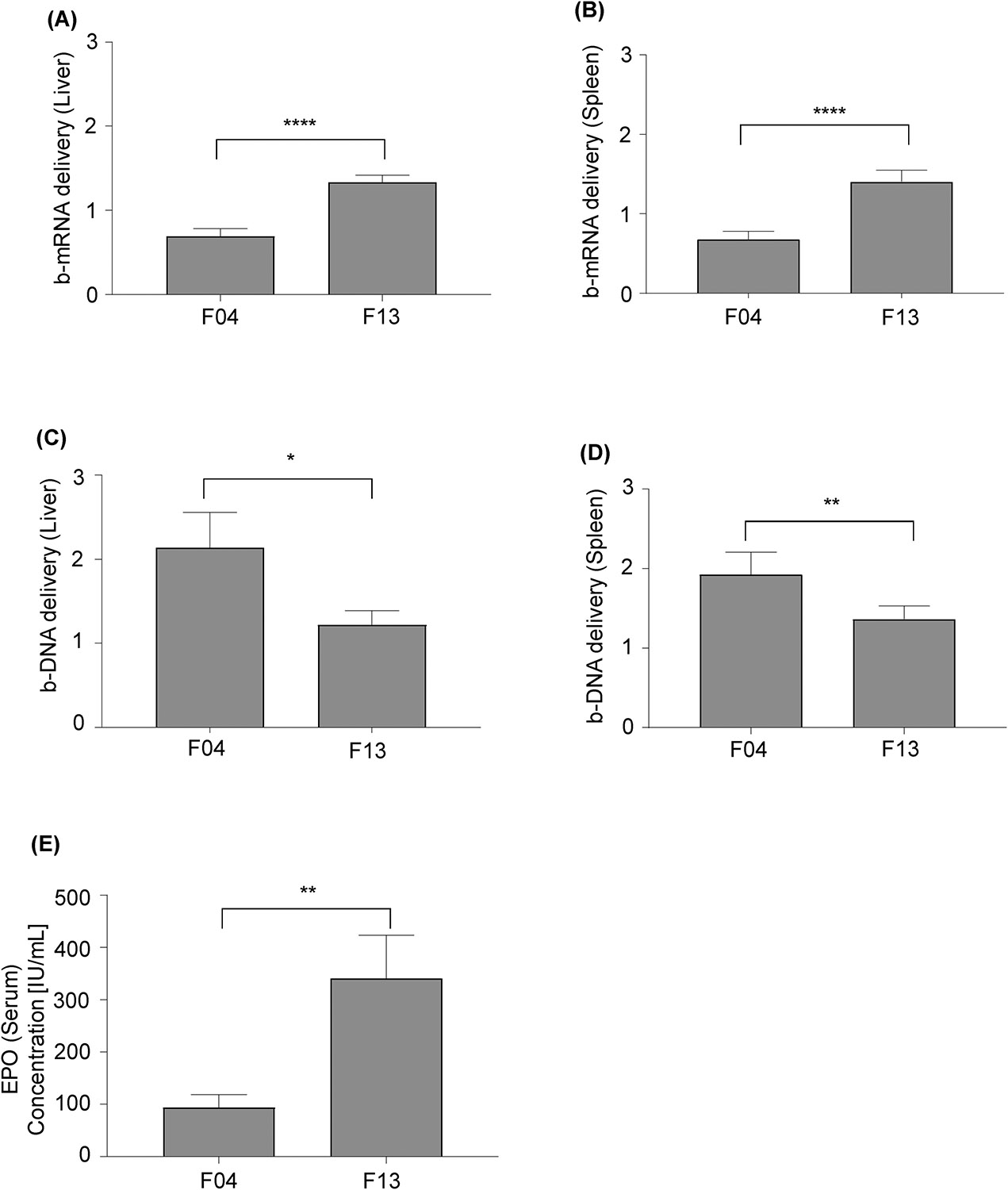
(A, B) Comparison of LNP formulations F4 and F13 for b-mRNA delivery to the liver (A) and spleen (B). (C, D) Comparison of LNP formulations F4 and F13 for b-DNA delivery to the liver (C) and spleen (D). (E) C57BL/6 mice were intravenously injected with either the F04 or F13 LNP formulation that encapsulated EPO mRNA (5 μg EPO mRNA per injection). Serum EPO concentrations at 4 hour post-intravenous injection were determined using ELISA. Data were plotted as mean ± SD. *P < 0.05, **P < 0.01, ****P < 0.0001 by t-test.
CONCLUSION
In this study, we demonstrated that b-mRNA LNPs are a potential high-throughput tool for tracking tissue-specific delivery of a functional mRNA. Furthermore, our studies comparing b-mRNA LNPs and b-DNA LNPs indicated that the structure of different nucleic acid cargo (i.e. b-DNA versus b-mRNA) can affect LNP physical properties and subsequently alter their in vivo delivery. Therefore, the inclusion of a nucleic acid barcode that is similar in size and structure to the therapeutic cargo is a potentially important factor for predicting therapeutic mRNA delivery. Since b-mRNA has a similar structure to functional mRNA, b-mRNA may provide an optimal “first-pass” delivery screen to identify lead formulations for mRNA delivery. The flexible design of b-mRNA allows them to be utilized as proxies for many other mRNA sequences with different sizes, such as Cas9 mRNA (4,521 nucleotides), or the smaller human erythropoietin (EPO) mRNA (858 nucleotides). In summary, this proof-of-concept study described a high-throughput screening method to rapidly identify lead LNP formulations for mRNA delivery. Future efforts will focus on incorporating this platform into mRNA related therapeutic applications, such as CRISPR/Cas9 gene editing, mRNA vaccines, and other mRNA-based immunotherapies.
MATERIALS AND METHODS
Barcoded in vitro transcription (IVT) template
Barcoded templates for IVT were constructed via PCR from a plasmid, pGL4.10[luc2] (Promega, E1751) using the following primers:
Luc_T7_F2:
5’- TAATACGACTCACTATAgggCATTCCGGTACTGTTGG
Luc_BC_R(N):
5’-GCCCAGTCATAGCCGAATAGNNNNNNNNNN
[Barcode]CCGCCCCGACTCTAGAATTA
A full list of IVT templates can be found in Table S2. All oligonucleotides were purchased from Integrated DNA Technologies with standard desalting. PCR was conducted using 1X Phusion HF buffer containing a final concentration 0.5 μM Miseq primer (Table S1), 200 μM dNTPs, and 0.4 U Phusion High-Fidelity DNA Polymerase (New England BioLabs, M0530S). The samples were denatured at 98 °C for 30 seconds then run for 35 cycles through the following conditions: 98 °C for 10 seconds, 65 °C for 30 seconds, and 72 °C for 2 minutes. This was followed by a final 10-minute extension at 72 °C. Templates were separated using 1% agarose gel (Universal Medical, IB70060), and 1.7kb products were excised and purified via Zymoclean Gel DNA Recovery Kit (Zymo Research, D4007) per the manufacturer’s protocol.
IVT
Uncapped RNA was synthesized via IVT using a modified HiScribe T7 High Yield RNA Synthesis Kit (New England Biolabs, E2040S) containing 100 ng of purified template in 20 μL reactions. The manufacturer’s protocol was modified by replacing CTP (cytidine-5’-triphosphate) with 5mCTP (Trilink biotechnologies, N-1014) in an overnight incubation at 37 °C. DNA was degraded with 2 U of RQ1 DNase (Promega, M6101) for 30 minutes at 37 °C. RNA was purified using a RNeasy MinElute Cleanup Kit (Qiagen, 74204) following the manufacturer’s protocol, eluting with 50 μL RNase-free H2O. For different mRNA modifications, chemically modified nucleotides were completely substituted for their unmodified counterparts while synthesizing the mRNA.
RNA capping and tailing
25 μg RNA was resuspended in 15 μL RNase-free H2O and denatured at 65 °C for 5 minutes, and immediately placed on ice. RNA was capped using the Vaccinia Capping System (New England BioLabs, M2080S) in 50 μL reaction per the manufacturer’s protocol and incubated at 37 °C for 30 minutes. Poly(A) tails were added using E. coli Poly(A) Polymerase (New England BioLabs, M0276S) by adding 10 μL 10X PAP Reaction Buffer, 10 μL 10 mM ATP, 5 μL (25 U) E. Coli PAP, and 25 μL RNase-free H2O and incubated at 37 °C for 30 minutes. Reactions were stopped with the addition of 100 μL of RNA binding buffer (Zymo Research, R1013-2-25). mRNA was purified using a Zymo RNA Clean & Concentrator Kit (Zymo Research, R1018) per the manufacturer’s protocol. Quality control testing of mRNA was conducted using a Bioanalyzer (Agilent 2100 Bioanalyzer; Agilent Technologies)
RNA extraction and cDNA synthesis
30 mg of disrupted frozen tissue was resuspended in TRIzol™ Reagent (Thermo Fisher Scientific, 15596026); total RNA was extracted per the manufacturer’s protocol. 2 μg of extracted RNA was treated with 1U RQ1 DNase, 1X RQ1 DNase buffer, and 20 U RNase inhibitor (New England Biolabs, M0314S) for 30 minutes at 37 °C to remove any remaining DNA in solution. The reaction was terminated by adding 1 μL Stop solution and incubated for 10 minutes at 65 °C. 1 μL Oligo dT (Integrated DNA Technologies, 51-01-15-05) was added to each reaction and denatured for 5 minutes at 70 °C, and then immediately placed on ice. Reverse transcription of the DNase-treated RNA was carried out in a 20 μL reaction using 1 μL of GoScript Reverse Transcriptase (Promega, A5003) containing a final concentration of 1X GoScript Reaction Buffer, 2.5 mM MgCl2, 0.5 mM dNTPs using the following cycling conditions: 25 °C for 5 minutes, 42 °C for 1 hour, and 70 °C for 15 minutes.
Barcoded mRNA (b-mRNA) library preparation
Library templates were prepared via two stages of PCR. In the first stage, adapters were added to the cDNA using the following primers:
Luc_Seq_US1:
5’- AGACGTGTGCTCTTCCGATCTGGACGAGGTGCCTAAAGGAC
NeoR_Seq_US2:
5’- ACACGACGCTCTTCCGATCTGCCCAGTCATAGCCGAATAG
PCR was carried out in 1X Phusion HF buffer containing a final concentration of 0.5 μM Luc_Seq_US1, 0.5 μM NeoR_Seq_US2, 200 μM dNTPs, and 0.4 U Phusion High-Fidelity DNA Polymerase. Templates were denatured at 98 °C for 30 seconds followed by 16 cycles of: 98 °C for 10 seconds, 65 °C for 30 seconds, 72 °C for 2 minutes followed by a final 10-minute extension at 72°C with an expected product size of 218bp. Templates were purified using 1.8 volumes of Mag-Bind TotalPure NGS beads (Omega Biotek, M1378–00), followed by two 80% ethanol washes and elution in 20 μL TE.
In the second stage, Illumina primers were added to the cDNA using the following primers from a previous study62:
Forward (Index-Base):
5’-AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCT
Reverse (Universal):
5’- TGACTGGAGTTCAGACGTGTGCTCTTCCGATCT
Miseq primers (Table S1):
5’- CAAGCAGAAGACGGCATACGAGAT[index]GTGACTGGAGTTCAGACGTGTGCTCT TCCGATCT
cDNA was denatured at 98 °C for 30 seconds followed by 16 cycles of 98 °C for 10 seconds, 65 °C for 30 seconds, 72 °C for 2 minutes followed by a final 10-minute extension at 72 °C with an expected size of 301bp. PCR products were purified using a 1.8X volume ratio of Mag-Bind TotalPure NGS beads (Omega Biotek, M1378–00), followed by two 80% ethanol washes and eluted in 20 μL TE. The purified products were kept frozen until deep sequencing.
b-DNA library preparation
b-DNA design parameters were adopted from a previous report62. b-DNA consisted of 61 nucleotide single-stranded DNA with three consecutive phosphorothioate bonds at each end. The barcode region was comprised of 10 nucleotides in the center of the oligonucleotide. An additional 10 random nucleotides were included at 3′ of the barcode region. The 5′ and 3′ ends of each b-DNA contained priming sites for Illumina adapters. A full list of b-DNA sequences can be found in Table S3. Desalted oligonucleotides were ordered from Integrated DNA Technologies. To extract DNA from a tissue sample, approximately 30 mg of disrupted frozen tissue was resuspended in lysis buffer66 that contained 100 mM Tris-HCl (Fisher Scientific, 50155887), 5 mM EDTA (Fisher Scientific, 50997738), 0.2% SDS (Fisher Scientific, 507513793), 200 mM NaCl (Fisher Scientific, S318100), and 0.2 mg/mL proteinase K (Thermo Fisher Scientific, PI17916). Extracted DNA was further purified by Zymo Oligo Clean and Concentrator columns (Zymo Research, D4060) according to the manufacturer’s instructions. b-DNA amplification was conducted by PCR using the following recipe: 5 μL 5× HF Phusion buffer, 0.5 μL 10 mM dNTPs, 0.25 μL Phusion High-Fidelity DNA Polymerase (Thermo Fisher Scientific, F530S), 1.18 μL extracted DNA, 0.5 μL 5 μM reverse (universal), 0.5 μL 5 μM Miseq primer (Table S1), 0.5 μL 0.5 μM forward (Index-base), 2 μL DMSO, and 15.25 μL H2O. PCR cycling conditions were 98 °C for 12 seconds, 67 °C for 22 seconds, and 72 °C for 28 seconds, for a total of 35 cycles. Primer sequences were shown below:
Forward (Index-Base):
5’- AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCT
Reverse (Universal):
5’- TGACTGGAGTTCAGACGTGTGCTCTTCCGATCT
Miseq primers (Table S1):
5’- CAAGCAGAAGACGGCATACGAGAT[index]GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT
PCR products were run by gel electrophoresis on 1.4% agarose (Universal Medical, IB70060) in Tris-acetate-EDTA buffer (Fisher Scientific, 24710030). Amplified b-DNA (144bp) was excised from the gel, pooled, and purified by Zymo Gel Extraction columns (Zymo Research, D4001) according to the manufacturer’s instructions. The purified products were kept frozen until deep sequencing was performed.
Lipid nanoparticle (LNP) formulation
LNPs were formulated by mixing an aqueous phase containing mRNA or DNA with an ethanol phase containing ionizable lipids and excipients using a microfluidic chip device54. Specifically, the ethanol phase contained a mixture of an ionizable lipid (C12–200, synthesized as previously described67), 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE, Avanti Polar Lipids, 850725P), cholesterol (Sigma-Aldrich, C8667), and 1,2-dimyristoylsn-glycero-3-phosphoethanolamine-N-[methoxy(polyethyleneglycol)-2000] (ammonium salt) (C14-PEG 2000, Avanti Polar Lipids, 880150P) at predetermined molar ratios shown in Table 1. High-performance liquid chromatography (HPLC) and mass spectrometry data for the ionizable lipid were shown in Fig. S4. The aqueous phase was prepared in 10 mM citrate, pH 3.0 buffer (Teknova, Q2445) with either in-house synthesized b-mRNA, Luciferase mRNA (Trilink Biotechnologies), or b-DNA (Integrated DNA Technologies). Syringe pumps were used to perfuse the ethanol and aqueous phases at a 3:1 ratio through the microfluidic device54. The resulting LNPs were dialyzed against PBS in a 20,000 MWCO cassette at room temperature for 2 hours and then extruded through a 0.22 μm sterile filter (Genesee Scientific, 25243).
LNP characterization
DNA or mRNA concentration in LNP formulations was determined using a NanoDrop Spectrophotometer (Thermo Fisher Scientific). To calculate mRNA encapsulation efficiency within LNPs, a modified Quant-iT RiboGreen RNA assay (Thermo Fisher Scientific, R11490) was used as previously described26. LNP hydrodynamic diameter and polydispersity (PDI) were measured using a Zetasizer Nano ZS machine (Malvern Instrument). For analysis of LNP structure using cryogenic-transmission electron microscopy (Cryo-TEM), LNP samples were prepared in a vitrification system (25°C, ~100% humidity). A 3 μL sample of LNP solution was dropped on a lacey copper grid coated with a continuous carbon film and blotted to remove excess sample without damaging the carbon layer. A grid was mounted on a Gatan 626 single tilt cryogenic-holder equipped in the TEM column. Images of LNP samples were recorded on an UltraScan 1000 CCD camera (Gatan).
In vitro mRNA delivery
bEnd.3 mouse cerebral cortex endothelial cells (ATCC) were maintained at 37°C and 5% CO2 in high glucose Dulbecco’s Modified Eagles Medium (Thermo Fisher) supplemented with 10% fetal bovine serum (by volume), 20 U/mL penicillin and 20 U/mL streptomycin. Cells were seeded in black 48-well plates at a density of 30,000 cells per well. After 24 hours, cells were treated with LNPs encapsulating different b-mRNA (modified with either pseudouridine (ψ) or 5-methylcytosine (m5C)) or commercially available Trilink-mRNA at different concentration (10 nM, 25 nM, or 50 nM). After 48 hours transfection68–70, cells were washed with PBS and incubated with D-luciferin (150μg/mL). Subsequently, luciferase activity was measured using an IVIS imaging system (PerkinElmer).
Animal experiments
All procedures were performed under an animal protocol approved by the University of Pennsylvania Institutional Animal Care and Use Committee (IACUC). To evaluate b-mRNA or b-DNA delivery, 8-week-old female C57BL/6 mice (Charles River Labs, 18–21 g) were injected intravenously via the tail vein with a pool of different barcoded LNPs at 0.25 μg b-mRNA or 1 μg b-DNA per formulation. To quantify mRNA delivery and luciferase in vivo, mice were injected intravenously via the tail vein with LNPs containing 5 μg of either mRNA coding for luciferase (Trilink Biotechnologies) or b-mRNA coding for luciferase. For all experiments, tissues were harvested 4 hours post-injection. To evaluate b-mRNA delivery or b-DNA delivery, tissues were snap-frozen in liquid nitrogen, disrupted into powder using a Geno/Grinder (SPEX Sample Prep), and stored in a −80 °C freezer. To evaluate luciferase expression, mice were administered via an intraperitoneal injection of 130 μL of D-luciferin (30 mg/mL in PBS) 15 minutes before they were sacrificed. Luminescence of harvested organs (liver, spleen, lymph node, lungs, heart, brain, pancreas and, kidneys) was analyzed using an IVIS imaging system (PerkinElmer) and quantified using Living Image Software (PerkinElmer). To assess human erythropoietin (EPO) expression in vivo, mice were administered intravenously with LNPs encapsulating 5 μg of EPO mRNA (Trilink Biotechnologies). 4 hours after the injection, whole blood was collected from the saphenous vein and centrifuged at 10,000 relative centrifugal force (RCF) for 10 minutes to separate out the red blood cells. The resulting serum supernatant was collected. Subsequently, serum EPO levels were measured using an enzyme-linked absorbent assay (ELISA, Biolegend, 442907) following the manufacturer’s protocol.
Deep sequencing and barcode delivery quantification
All deep-sequencing runs were performed using multiplexed runs on Illumina MiSeq (Illumina). PCR product pools were quantitated using the KAPA Library Quantification Kit for next generation sequencing. PCR product pools were loaded onto flow cells at 4 nM concentration. Python scripts were written to quantify barcodes from Illumina fastq files.
b-mRNA delivery or b-DNA delivery of a specific barcode to a certain tissue was calculated according to the following 3 steps: (i) dividing the number of sequencing reads of one barcode delivered by a single LNP formulation by the total amount of reads from all barcodes delivered by all LNPs in a specific tissue; (ii) dividing the number of sequencing reads of the same barcode (utilized in (i)) by the total amount of reads from all barcodes of all LNPs in the non-injected LNP pool. (iii) dividing the results from (i) by the results from (ii). By using this quantification method, the delivery of different LNP formulations within the same organ can be compared, but the delivery of the same LNP formulation across different organs cannot be compared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors acknowledge support from a Burroughs Wellcome Fund Career Award at the Scientific Interface (CASI), a US National Institutes of Health (NIH) Director’s New Innovator Award (DP2 TR002776), a grant from the American Cancer Society (129784-IRG-16-188-38-IRG), an Abramson Cancer Center (ACC)-School of Engineering and Applied Sciences (SEAS) Discovery Grant (P30 CA016520), and a 2018 AACR- Bayer Innovation and Discovery Grant, Grant Number 18-80-44-MITC (to M.J.M.). R.S.R. is supported by an NIH T32 multidisciplinary training grant (T32 HL007954). M.M.B. is supported by a Tau Beta Pi Graduate Research Fellowship.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Oberli MA; Reichmuth AM; Dorkin JR; Mitchell MJ; Fenton OS; Jaklenec A; Anderson DG; Langer R; Blankschtein D, Lipid nanoparticle assisted mRNA delivery for potent cancer immunotherapy. Nano letters 2016, 17 (3), 1326–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cheng Q; Wei T; Jia Y; Farbiak L; Zhou K; Zhang S; Wei Y; Zhu H; Siegwart DJ, Dendrimer - Based Lipid Nanoparticles Deliver Therapeutic FAH mRNA to Normalize Liver Function and Extend Survival in a Mouse Model of Hepatorenal Tyrosinemia Type I. Advanced Materials 2018, 1805308. [DOI] [PubMed] [Google Scholar]
- 3.Luo X; Li B; Zhang X; Zhao W; Bratasz A; Deng B; McComb D; Dong Y, Dual-functional lipid-like nanoparticles for delivery of mRNA and MRI contrast agents. Nanoscale 2017, 9 (4), 1575–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang R; Billingsley MM; Mitchell MJ, Biomaterials for vaccine-based cancer immunotherapy. Journal of Controlled Release 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Riley RS; June CH; Langer R; Mitchell MJ, Delivery technologies for cancer immunotherapy. Nature Reviews Drug Discovery 2019, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hajj KA; Whitehead KA, Tools for translation: non-viral materials for therapeutic mRNA delivery. Nature Reviews Materials 2017, 2 (10), 17056. [Google Scholar]
- 7.Mukalel AJ; Riley RS; Zhang R; Mitchell MJ, Nanoparticles for Nucleic Acid Delivery: Applications in Cancer Immunotherapy. Cancer letters 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pardi N; Hogan MJ; Porter FW; Weissman D, mRNA vaccines—a new era in vaccinology. Nature Reviews Drug Discovery 2018, 17 (4), 261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kowalski PS; Rudra A; Miao L; Anderson DG, Delivering the Messenger: Advances in Technologies for Therapeutic mRNA Delivery. Molecular Therapy 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bettinger T; Carlisle RC; Read ML; Ogris M; Seymour LW, Peptide-mediated RNA delivery: a novel approach for enhanced transfection of primary and post-mitotic cells. Nucleic acids research 2001, 29 (18), 3882–3891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sahin U; Karikó K; Türeci Ö, mRNA-based therapeutics—developing a new class of drugs. Nature reviews Drug discovery 2014, 13 (10), 759. [DOI] [PubMed] [Google Scholar]
- 12.Zhang R; Ulery BD, Synthetic vaccine characterization and design. Journal of Bionanoscience 2018, 12 (1), 1–11. [Google Scholar]
- 13.Islam MA; Reesor EK; Xu Y; Zope HR; Zetter BR; Shi J, Biomaterials for mRNA delivery. Biomaterials science 2015, 3 (12), 1519–1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weissman D; Karikó K, mRNA: fulfilling the promise of gene therapy. Molecular Therapy 2015, 23 (9), 1416–1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Weide B; Carralot J-P; Reese A; Scheel B; Eigentler TK; Hoerr I; Rammensee H-G; Garbe C; Pascolo S, Results of the first phase I/II clinical vaccination trial with direct injection of mRNA. Journal of immunotherapy 2008, 31 (2), 180–188. [DOI] [PubMed] [Google Scholar]
- 16.Weide B; Pascolo S; Scheel B; Derhovanessian E; Pflugfelder A; Eigentler TK; Pawelec G; Hoerr I; Rammensee H-G; Garbe C, Direct injection of protamine-protected mRNA: results of a phase 1/2 vaccination trial in metastatic melanoma patients. Journal of immunotherapy 2009, 32 (5), 498–507. [DOI] [PubMed] [Google Scholar]
- 17.Kranz LM; Diken M; Haas H; Kreiter S; Loquai C; Reuter KC; Meng M; Fritz D; Vascotto F; Hefesha H, Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature 2016, 534 (7607), 396. [DOI] [PubMed] [Google Scholar]
- 18.Alipour M; Hosseinkhani S; Sheikhnejad R; Cheraghi R, Nano-biomimetic carriers are implicated in mechanistic evaluation of intracellular gene delivery. Scientific reports 2017, 7, 41507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Alipour M; Majidi A; Molaabasi F; Sheikhnejad R; Hosseinkhani S, In vivo tumor gene delivery using novel peptideticles: pH - responsive and ligand targeted core–shell nanoassembly. International journal of cancer 2018, 143 (8), 2017–2028. [DOI] [PubMed] [Google Scholar]
- 20.Whitehead KA; Dorkin JR; Vegas AJ; Chang PH; Veiseh O; Matthews J; Fenton OS; Zhang Y; Olejnik KT; Yesilyurt V, Degradable lipid nanoparticles with predictable in vivo siRNA delivery activity. Nature communications 2014, 5, 4277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Smith CE; Zain R, Therapeutic oligonucleotides: state of the art. Annual review of pharmacology and toxicology 2019, 59, 605–630. [DOI] [PubMed] [Google Scholar]
- 22.Fenton OS; Kauffman KJ; Kaczmarek JC; McClellan RL; Jhunjhunwala S; Tibbitt MW; Zeng MD; Appel EA; Dorkin JR; Mir FF, Synthesis and biological evaluation of ionizable lipid materials for the in vivo delivery of messenger RNA to B lymphocytes. Advanced Materials 2017, 29 (33), 1606944. [DOI] [PubMed] [Google Scholar]
- 23.Fenton OS; Kauffman KJ; McClellan RL; Appel EA; Dorkin JR; Tibbitt MW; Heartlein MW; DeRosa F; Langer R; Anderson DG, Bioinspired alkenyl amino alcohol ionizable lipid materials for highly potent in vivo mRNA delivery. Advanced materials 2016, 28 (15), 2939–2943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hajj KA; Ball RL; Deluty SB; Singh SR; Strelkova D; Knapp CM; Whitehead KA, Branched - Tail Lipid Nanoparticles Potently Deliver mRNA In Vivo due to Enhanced Ionization at Endosomal pH. Small 2019, 1805097. [DOI] [PubMed] [Google Scholar]
- 25.Dahlman JE; Barnes C; Khan OF; Thiriot A; Jhunjunwala S; Shaw TE; Xing Y; Sager HB; Sahay G; Speciner L, In vivo endothelial siRNA delivery using polymeric nanoparticles with low molecular weight. Nature nanotechnology 2014, 9 (8), 648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dong Y; Love KT; Dorkin JR; Sirirungruang S; Zhang Y; Chen D; Bogorad RL; Yin H; Chen Y; Vegas AJ, Lipopeptide nanoparticles for potent and selective siRNA delivery in rodents and nonhuman primates. Proceedings of the National Academy of Sciences 2014, 111 (11), 3955–3960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Akinc A; Zumbuehl A; Goldberg M; Leshchiner ES; Busini V; Hossain N; Bacallado SA; Nguyen DN; Fuller J; Alvarez R, A combinatorial library of lipid-like materials for delivery of RNAi therapeutics. Nature biotechnology 2008, 26 (5), 561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cheng Q; Wei T; Jia Y; Farbiak L; Zhou K; Zhang S; Wei Y; Zhu H; Siegwart DJ, Dendrimer - Based Lipid Nanoparticles Deliver Therapeutic FAH mRNA to Normalize Liver Function and Extend Survival in a Mouse Model of Hepatorenal Tyrosinemia Type I. Advanced Materials 2018, 30 (52), 1805308. [DOI] [PubMed] [Google Scholar]
- 29.Siegwart DJ; Whitehead KA; Nuhn L; Sahay G; Cheng H; Jiang S; Ma M; Lytton-Jean A; Vegas A; Fenton P, Combinatorial synthesis of chemically diverse core-shell nanoparticles for intracellular delivery. Proceedings of the National Academy of Sciences 2011, 108 (32), 12996–13001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Granot Y; Peer D In Delivering the right message: Challenges and opportunities in lipid nanoparticles-mediated modified mRNA therapeutics—An innate immune system standpoint, Seminars in immunology, Elsevier: 2017; pp 68–77. [DOI] [PubMed] [Google Scholar]
- 31.Cheng X; Lee RJ, The role of helper lipids in lipid nanoparticles (LNPs) designed for oligonucleotide delivery. Advanced drug delivery reviews 2016, 99, 129–137. [DOI] [PubMed] [Google Scholar]
- 32.Mui BL; Tam YK; Jayaraman M; Ansell SM; Du X; Tam YYC; Lin PJ; Chen S; Narayanannair JK; Rajeev KG, Influence of polyethylene glycol lipid desorption rates on pharmacokinetics and pharmacodynamics of siRNA lipid nanoparticles. Molecular Therapy-Nucleic Acids 2013, 2, e139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wilhelm S; Tavares AJ; Dai Q; Ohta S; Audet J; Dvorak HF; Chan WC, Analysis of nanoparticle delivery to tumours. Nature reviews materials 2016, 1 (5), 16014. [Google Scholar]
- 34.Akinc A; Querbes W; De S; Qin J; Frank-Kamenetsky M; Jayaprakash KN; Jayaraman M; Rajeev KG; Cantley WL; Dorkin JR, Targeted delivery of RNAi therapeutics with endogenous and exogenous ligand-based mechanisms. Molecular Therapy 2010, 18 (7), 1357–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ball RL; Bajaj P; Whitehead KA, Oral delivery of siRNA lipid nanoparticles: fate in the GI tract. Scientific reports 2018, 8 (1), 2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yan Y; Liu L; Xiong H; Miller JB; Zhou K; Kos P; Huffman KE; Elkassih S; Norman JW; Carstens R, Functional polyesters enable selective siRNA delivery to lung cancer over matched normal cells. Proceedings of the National Academy of Sciences 2016, 113 (39), E5702–E5710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Paunovska K; Sago CD; Monaco CM; Hudson WH; Castro MG; Rudoltz TG; Kalathoor S; Vanover DA; Santangelo PJ; Ahmed R, A direct comparison of in vitro and in vivo nucleic acid delivery mediated by hundreds of nanoparticles reveals a weak correlation. Nano letters 2018, 18 (3), 2148–2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang Y-SS; Atukorale PU; Moynihan KD; Bekdemir A; Rakhra K; Tang L; Stellacci F; Irvine DJ, High-throughput quantitation of inorganic nanoparticle biodistribution at the single-cell level using mass cytometry. Nature communications 2017, 8, 14069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yaari Z; Da Silva D; Zinger A; Goldman E; Kajal A; Tshuva R; Barak E; Dahan N; Hershkovitz D; Goldfeder M, Theranostic barcoded nanoparticles for personalized cancer medicine. Nature communications 2016, 7, 13325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dahlman JE; Kauffman KJ; Xing Y; Shaw TE; Mir FF; Dlott CC; Langer R; Anderson DG; Wang ET, Barcoded nanoparticles for high throughput in vivo discovery of targeted therapeutics. Proceedings of the National Academy of Sciences 2017, 114 (8), 2060–2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mullard A, DNA tags help the hunt for drugs. Nature Publishing Group: 2016. [DOI] [PubMed] [Google Scholar]
- 42.Sago CD; Lokugamage MP; Paunovska K; Vanover DA; Monaco CM; Shah NN; Castro MG; Anderson SE; Rudoltz TG; Lando GN, High-throughput in vivo screen of functional mRNA delivery identifies nanoparticles for endothelial cell gene editing. Proceedings of the National Academy of Sciences 2018, 115 (42), E9944–E9952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Viger-Gravel J; Schantz A; Pinon AC; Rossini AJ; Schantz S; Emsley L, Structure of Lipid Nanoparticles Containing siRNA or mRNA by Dynamic Nuclear Polarization-Enhanced NMR Spectroscopy. The Journal of Physical Chemistry B 2018, 122 (7), 2073–2081. [DOI] [PubMed] [Google Scholar]
- 44.Halder K; Benzler M; Hartig JS, Reporter assays for studying quadruplex nucleic acids. Methods 2012, 57 (1), 115–121. [DOI] [PubMed] [Google Scholar]
- 45.McKinlay CJ; Benner NL; Haabeth OA; Waymouth RM; Wender PA, Enhanced mRNA delivery into lymphocytes enabled by lipid-varied libraries of charge-altering releasable transporters. Proceedings of the National Academy of Sciences 2018, 115 (26), E5859–E5866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li B; Dong Y, Preparation and optimization of lipid-like nanoparticles for mRNA delivery In RNA Nanostructures, Springer: 2017; pp 207–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li B; Luo X; Deng B; Giancola JB; McComb DW; Schmittgen TD; Dong Y, Effects of local structural transformation of lipid-like compounds on delivery of messenger RNA. Scientific reports 2016, 6, 22137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Miller JB; Zhang S; Kos P; Xiong H; Zhou K; Perelman SS; Zhu H; Siegwart DJ, Non - viral CRISPR/Cas gene editing in vitro and in vivo enabled by synthetic nanoparticle co - delivery of Cas9 mRNA and sgRNA. Angewandte Chemie International Edition 2017, 56 (4), 1059–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Karikó K; Muramatsu H; Welsh FA; Ludwig J; Kato H; Akira S; Weissman D, Incorporation of pseudouridine into mRNA yields superior nonimmunogenic vector with increased translational capacity and biological stability. Molecular therapy 2008, 16 (11), 1833–1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Karikó K; Buckstein M; Ni H; Weissman D, Suppression of RNA recognition by Toll-like receptors: the impact of nucleoside modification and the evolutionary origin of RNA. Immunity 2005, 23 (2), 165–175. [DOI] [PubMed] [Google Scholar]
- 51.Warren L; Manos PD; Ahfeldt T; Loh Y-H; Li H; Lau F; Ebina W; Mandal PK; Smith ZD; Meissner A, Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mRNA. Cell stem cell 2010, 7 (5), 618–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kormann MS; Hasenpusch G; Aneja MK; Nica G; Flemmer AW; Herber-Jonat S; Huppmann M; Mays LE; Illenyi M; Schams A, Expression of therapeutic proteins after delivery of chemically modified mRNA in mice. Nature biotechnology 2011, 29 (2), 154. [DOI] [PubMed] [Google Scholar]
- 53.Kauffman KJ; Dorkin JR; Yang JH; Heartlein MW; DeRosa F; Mir FF; Fenton OS; Anderson DG, Optimization of lipid nanoparticle formulations for mRNA delivery in vivo with fractional factorial and definitive screening designs. Nano letters 2015, 15 (11), 7300–7306. [DOI] [PubMed] [Google Scholar]
- 54.Chen D; Love KT; Chen Y; Eltoukhy AA; Kastrup C; Sahay G; Jeon A; Dong Y; Whitehead KA; Anderson DG, Rapid discovery of potent siRNA-containing lipid nanoparticles enabled by controlled microfluidic formulation. Journal of the American Chemical Society 2012, 134 (16), 6948–6951. [DOI] [PubMed] [Google Scholar]
- 55.Hajj KA; Ball RL; Deluty SB; Singh SR; Strelkova D; Knapp CM; Whitehead KA, Branched - Tail Lipid Nanoparticles Potently Deliver mRNA In Vivo due to Enhanced Ionization at Endosomal pH. Small 2019, 15 (6), 1805097. [DOI] [PubMed] [Google Scholar]
- 56.DeRosa F; Guild B; Karve S; Smith L; Love K; Dorkin J; Kauffman K; Zhang J; Yahalom B; Anderson D, Therapeutic efficacy in a hemophilia B model using a biosynthetic mRNA liver depot system. Gene therapy 2016, 23 (10), 699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Macdougall IC; Tucker B; Thompson J; Tomson CR; Baker LR; Raine AE, A randomized controlled study of iron supplementation in patients treated with erythropoietin. Kidney international 1996, 50 (5), 1694–1699. [DOI] [PubMed] [Google Scholar]
- 58.Coleman T; Brines M, Science review: recombinant human erythropoietin in critical illness: a role beyond anemia? Critical Care 2004, 8 (5), 337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ball RL; Hajj KA; Vizelman J; Bajaj P; Whitehead KA, Lipid nanoparticle formulations for enhanced co-delivery of siRNA and mRNA. Nano letters 2018. [DOI] [PubMed] [Google Scholar]
- 60.Kulkarni JA; Myhre JL; Chen S; Tam YYC; Danescu A; Richman JM; Cullis PR, Design of lipid nanoparticles for in vitro and in vivo delivery of plasmid DNA. Nanomedicine: Nanotechnology, Biology and Medicine 2017, 13 (4), 1377–1387. [DOI] [PubMed] [Google Scholar]
- 61.Kulkarni JA; Darjuan MM; Mercer JE; Chen S; van der Meel R; Thewalt JL; Tam YYC; Cullis PR, On the Formation and Morphology of Lipid Nanoparticles Containing Ionizable Cationic Lipids and siRNA. ACS nano 2018, 12 (5), 4787–4795. [DOI] [PubMed] [Google Scholar]
- 62.Dahlman JE; Kauffman KJ; Xing Y; Shaw TE; Mir FF; Dlott CC; Langer R; Anderson DG; Wang ET, Barcoded nanoparticles for high throughput in vivo discovery of targeted therapeutics. Proceedings of the National Academy of Sciences 2017, 201620874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kenworthy AK; Hristova K; Needham D; McIntosh TJ, Range and magnitude of the steric pressure between bilayers containing phospholipids with covalently attached poly (ethylene glycol). Biophysical journal 1995, 68 (5), 1921–1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sago CD; Lokugamage MP; Islam FZ; Krupczak BR; Sato M; Dahlman JE, Nanoparticles that deliver RNA to bone marrow identified by in vivo directed evolution. Journal of the American Chemical Society 2018, 140 (49), 17095–17105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Paunovska K; Da Silva Sanchez AJ; Sago CD; Gan Z; Lokugamage MP; Islam FZ; Kalathoor S; Krupczak BR; Dahlman JE, Nanoparticles Containing Oxidized Cholesterol Deliver mRNA to the Liver Microenvironment at Clinically Relevant Doses. Advanced Materials 2019, 1807748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Laird PW; Zijderveld A; Linders K; Rudnicki MA; Jaenisch R; Berns A, Simplified mammalian DNA isolation procedure. Nucleic acids research 1991, 19 (15), 4293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Love KT; Mahon KP; Levins CG; Whitehead KA; Querbes W; Dorkin JR; Qin J; Cantley W; Qin LL; Racie T, Lipid-like materials for low-dose, in vivo gene silencing. Proceedings of the National Academy of Sciences 2010, 107 (5), 1864–1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sayers E; Peel S; Schantz A; England R; Beano M; Bates S; Desai A; Puri S; Ashford M; Jones A, Endocytic Profiling of Cancer Cell Models Reveals Critical Factors Influencing Lipid Nanoparticle Mediated mRNA Delivery and Protein Expression. Molecular Therapy 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yasar H; Biehl A; De Rossi C; Koch M; Murgia X; Loretz B; Lehr C-M, Kinetics of mRNA delivery and protein translation in dendritic cells using lipid-coated PLGA nanoparticles. Journal of nanobiotechnology 2018, 16 (1), 72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Van Tendeloo VF; Ponsaerts P; Lardon F; Nijs G; Lenjou M; Van Broeckhoven C; Van Bockstaele DR; Berneman ZN, Highly efficient gene delivery by mRNA electroporation in human hematopoietic cells: superiority to lipofection and passive pulsing of mRNA and to electroporation of plasmid cDNA for tumor antigen loading of dendritic cells. Blood 2001, 98 (1), 49–56. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.